

Abstract
Virtual memory T (TVM) cells are antigen-naïve CD8+ T cells that exist in a semi-differentiated state and exhibit marked proliferative dysfunction in advanced age. High spare respiratory capacity (SRC) has been proposed as a defining metabolic characteristic of antigen-experienced memory T (TMEM) cells, facilitating rapid functionality and survival. Given the semi-differentiated state of TVM cells and their altered functionality with age, here we investigate TVM cell metabolism and its association with longevity and functionality. Elevated SRC is a feature of TVM, but not TMEM, cells and it increases with age in both subsets. The elevated SRC observed in aged mouse TVM cells and human CD8+ T cells from older individuals is associated with a heightened sensitivity to IL-15. We conclude that elevated SRC is a feature of TVM, but not TMEM, cells, is driven by physiological levels of IL-15, and is not indicative of enhanced functionality in CD8+ T cells.
Subject terms: Lymphocyte activation, Cytotoxic T cells
Fatty acid oxidation (FAO) is thought to contribute to high spare respiratory capacity (SRC), which in turn affects CD8+ T cell function. Here, the authors show that ex vivo virtual memory T cells (and not antigen experienced memory T cells) have high SRC, a metabolic state that it is affected by ageing and IL-15 signalling and not directly by FAO.
Introduction
Previous studies have highlighted that the metabolic phenotype of CD8+ T cells dramatically impacts their functional and survival capacities (reviewed in refs. 1,2). True naive (TN) cells are quiescent and predominantly utilise oxidative phosphorylation (OXPHOS) to meet their low energy demands. In contrast, effector (TEFF) cells undergo transcriptional reprogramming to upregulate aerobic glycolysis after TCR stimulation. Conventional memory (TMEM) cells revert to the predominant utilisation of OXPHOS but are characterised by a higher mitochondrial energy reserve, known as spare respiratory capacity (SRC)3–5. SRC is the difference between basal and maximal oxygen consumption rates (OCR)4,5 and it reflects the mitochondrial capacity that a cell holds in reserve, which may mitigate stress from sudden increases in energy demand. Increased SRC has been proposed to mediate both enhanced T cell functionality, in the form of metabolic memory that confers immediate responsiveness after secondary antigen exposure5, and the increased longevity of TMEM cells5,6.
The greater SRC observed in TMEM cells was in turn associated with increased mitochondrial load and, in particular, a distinct fused mitochondrial morphology when compared to TN and TEFF cells5,6. Mitochondrial fusion was causally linked with TMEM formation and function, since deletion of the inner mitochondrial membrane fusion protein, Opa1, abrogated the development of TMEM cells after infection, while promoting mitochondrial fusion in TEFF cells conferred a memory phenotype6. More recently, high SRC was shown to partition preferentially with a subset of long-lived TMEM cells known as central memory (TCM) cells, rather than short-lived effector memory (TEM) cells. Enforcing glycolysis, rather than OXPHOS, in CD8+ TMEM cells limited their ability to survive and establish the long-lived TCM population7. Importantly, many studies on metabolic characteristics of TMEM cells have utilised memory phenotype cells generated in vitro in the context of high levels of IL-15 (refs. 3–6,8), but the specific impact of IL-15 on these metabolic characteristics has not been well defined.
TVM cells are a subset of antigen-naive, semi-differentiated CD8+ T cells. They are generated in neonatal mice9,10 independently of antigen exposure, as evidenced by their presence in germ-free mice, antigen-free mice and CD8+ T cell populations specific for viral antigens in naive mice11–14. Common γ (γc) chain cytokine signalling is thought to drive the semi-differentiated phenotype of TVM cells, likely via homoeostatic proliferation, with IL-15 transpresentation by CD8α+ dendritic cells (DCs) required for their generation11,15, and they appear to develop from T cells with modestly self-reactive TCRs13,14,16–18. Although antigenically naive, TVM cells are functionally distinct from true naive T (TN) cells, as TVM engage proliferation and cytokine production more rapidly upon TCR stimulation and can also respond to cytokine stimulation17,19. TVM cells are also phenotypically distinct from both TN and TMEM cells, with high levels of CD44, a classical marker of activation, but low levels of CD49d, which is only upregulated upon strong cognate antigen encounter11,20. Of note, the high level expression of CD44 and CD62L on TVM cells has resulted in their frequent misclassification as central memory (TCM) cells, if CD49d expression is not assessed20.
While TVM cells exhibit augmented function in the young, ageing dramatically undermines the functionality of both TVM and TMEM subsets in vitro and in vivo17,21,22. Moreover, while TN cells decline substantially in number and proportion, both TVM and TMEM cells accumulate with advanced age17,20,23,24, with the accumulation of TVM cells partially dependent on type I IFN-related signalling pathways25. Overall, it is unclear whether the metabolic profile of CD8+ T cell subsets changes with age and whether metabolic changes reflect their capacity for function and survival over the lifespan.
Given that TVM cells span the phenotypic and functional divide between TN and TMEM cells, we aim to dissect the metabolic characteristics and associated mitochondrial features of this unique T cell subset to determine whether these features are indicative of their function and survival capacity during ageing. We define TMEM cells as only those that have encountered antigen, which necessitates a reanalysis of the metabolic, survival, and functional characteristics of bona fide TMEM cells. Collectively, this study refines our understanding of how the metabolic state impacts on CD8+ T cell function and longevity, which is ultimately key to augmenting or suppressing T cell function using clinical interventions.
Results
TVM and cells from aged mice have increased spare respiratory capacity
We sought to understand whether the basal metabolic phenotype of TVM cells is more closely aligned with the TN population or shares characteristics with conventional TMEM cells, such as increased mitochondrial load and SRC, and to understand how these profiles change with age. We, therefore, undertook a comprehensive mitochondrial and metabolic analysis for each of these subsets isolated from the spleens of naive young and aged specific-pathogen-free (SPF) mice.
The basal mitochondrial metabolic profile of young and aged mouse CD8+ T cells subsets was determined by performing a Mito Stress test, using a Seahorse XFe96 Bioanalyser, on sorted TN (CD44lo), TVM (CD44hiCD49dlo) and TMEM (CD44hiCD49dhi) CD8+ cells directly ex vivo. In this assay, oxygen consumption rate (OCR) is tracked in the basal state (OCRBas) and then during treatment with various mitochondrial inhibitors to enable measurement of maximal OCR (OCRMax), with the difference between the OCRBas and OCRMax representing SRC. TN cells from young and aged mice had comparable OCR profiles, resulting in comparable SRC for these subsets (Fig. 1a, b). TVM cells from young mice had significantly higher OCR than TN cells, at both OCRBas and, more noticeably, at OCRMax, resulting in significantly increased SRC (Fig. 1a, b), consistent with a memory-like phenotype. The TVM cells from aged mice also had consistently higher OCR than those from young mice leading to substantially higher SRC (Fig. 1a, b). Strikingly, TMEM cells from young mice did not exhibit higher SRC than TN cells, but rather had the lowest SRC of all subsets (Fig. 1a, b), in contrast to previous reports4. While a significant increase in SRC was also observed in TMEM cells with age (Fig. 1a, b), it remained significantly lower than the SRC observed for TVM cells from aged mice. These data suggest that high SRC is not a hallmark of TMEM cells but instead defines TVM cells, and that ageing drives an increase in SRC in all memory phenotype CD8+ T cells.
Fig. 1. TVM cells have high SRC and CIV, which increases with age.

a Oxygen consumption rate (OCR) across time for sorted TN, TVM and TMEM cells from the spleens of naive young and aged SPF mice. Arrows indicate the addition of mitochondrial inhibitors (oligomycin; FCCP; antimycin A/rotenone) or timepoints for assessment of SRC (OCRBas, OCRMax) (n = 2–4, 5 experimental replicates). b Change in OCR from OCRBas to OCRMax (SRC) for each sorted subset (n = 2–4, 5 experimental replicates). c Electron microscope images of sorted cells directly ex vivo, scale bar indicates 0.2 µm (1 experimental replicate). d Confocal microscopy of sorted cells directly ex vivo, green fluorescence is Cytochrome C staining, scale bar indicates 2 µm, which was used to define (e), predominant mitochondrial morphology (fused, intermediate or fragmented) for 140 cells per subset and f average mitochondrial footprint per cell as calculated from confocal images (3 experimental replicates). g BN-PAGE and Blot for Cox5a from ETC CIV, with bands for CIV, CII, CIII2, CV and the CI/CIII2/CIV supercomplex indicated, alongside the Coomassie stained blot (3 experimental replicates). h Oxygen consumption rate (OCR) across time for sorted TVM cells from young mice, with high (200 µM) or low (5 µM) dose Etomoxir, (n = 3, 3 experimental replicates). Shown is mean ± standard error of the mean (SEM). NS indicates not significant, * indicates p ≤ 0.05, ** indicates p ≤ 0.01, unpaired t test.
Correlation of SRC mitochondrial characteristics
Increased SRC is often thought to reflect quantitative and/or qualitative changes in the mitochondria themselves, including (i) denser mitochondrial cristae, promoting processivity and efficiency of oxygen consumption by the electron transport chain (ETC)3, (ii) a fused mitochondrial morphology6 and (iii) increased mitochondrial load or volume5. To assess these mitochondrial characteristics, TN, TVM and TMEM cells from young and aged mice were sorted and mitochondria were imaged directly ex vivo. There were no obvious differences in the size, density or morphology of mitochondrial cristae across cell types or ages, by electron microscopy (Fig. 1c). When mitochondrial fusion was scored by confocal microscopy (Fig. 1d), an intermediate or fused morphology was observed only in a minority of cells, even for TMEM cells (Fig. 1e), suggesting that mitochondrial fusion is not required for TMEM cell maintenance. In addition, no TVM cells from young mice were observed with a fused morphology (Fig. 1e), despite their high SRC. Strikingly, there was a substantial increase in mitochondrial fusion with age across all cell types, with this effect being most apparent in TVM cells (Fig. 1e). Finally, mitochondrial footprint per cell as a measure of mitochondrial load was highest in TVM and TMEM cells and it increased significantly with age in TN cells and TVM cells (Fig. 1f).
Generally, mitochondrial cristae morphology, fusion or volume correlated poorly with the high SRC observed selectively in the TVM cell subset. To obtain a more direct measure of Electron Transport Chain (ETC) capacity, the levels of mitochondrial ETC Complex IV (CIV; Cox5a) from a defined number of each cell subset was quantitated directly via blue-native PAGE and immunoblotting. Although not an absolute correlation, the amount of CIV appeared to correlate better with SRC than mitochondrial load or morphology; namely CIV was increased in TVM compared to TN cells from young mice, and age-related increases in CIV were most marked in the TVM population (Fig. 1g). Collectively, our analyses of mitochondrial load and morphology suggested that they were broadly predictive of age-related increases in SRC. Expression levels of ETC CIV appeared to most accurately predict cellular SRC across age and subsets.
To provide a mechanistic basis for increased mitochondrial load/activity in TVM cells from aged mice, RNA-Seq data previously generated from TN, TVM, and TMEM subsets from young and aged mice17 was interrogated for transcripts involved in mitochondrial biogenesis, mitophagy, and mitochondrial fusion or fission. Across all T cell subsets there was an age-dependent decrease in Atg101 and Ulk1 transcripts, which are critical for mitophagy (Supplementary Fig. 1a). There was a corresponding increase in PGC-1α transcripts (Ppargcla) with age in both memory phenotype populations, which was particularly striking in the TVM subset (Supplementary Fig. 1a). Analysis of transcript levels associated with mitochondrial fusion (Mfn1, Mfn2, Opa1) or fission (Dnm1l) revealed minimal to no change across T cell subsets or with aging (Supplementary Fig. 1c). Collectively, whilst true delineation of the impact of mitochondrial dynamics on mitochondrial load requires more detailed biochemical analyses, these transcriptional data highlight that there may be a decrease in mitochondrial degradation and an increase in biogenesis with age, particularly in the TVM subset, which may drive the observed increase in mitochondrial load and SRC.
No evidence of FAO fuelling high SRC in TVM cells
High SRC in TMEM cells was previously thought to be fuelled by fatty acid oxidation (FAO), a mechanism largely defined using etomoxir to inhibit carnitine palmitoyltransferase I (Cpt1), which is a rate-limiting enzyme for FAO3. However, it was recently demonstrated that the high concentration of etomoxir used in these studies also inhibited other components of OXPHOS to reduce SRC26–28. To examine the impact of etomoxir on high SRC in TVM cells, the drug was incorporated into the Mito Stress assay with young mouse TVM cells at either a high concentration (200 µM) or a low concentration (5 µM), the latter of which is predicted to maintain specificity for Cpt1 (ref.27). A substantial decrease in OCRMax was observed with the high concentration of etomoxir, but there was only a very modest decrease in OCRMax with the low concentration (Fig. 1h). This suggests that FAO, via Cpt1, does not facilitate the high SRC observed in TVM cells.
We next investigated the possibility that glycolysis was required to fuel the high SRC observed in TVM cells, most likely via the production of pyruvate29. The Mito Stress assay was performed on TVM cells from naive mice with the addition of 2-Deoxy-D-glucose (2-DG), a glucose analogue that inhibits glycolysis. The addition of 2-DG had a minimal effect on OCR (Supplementary Fig. 2a), similar to the addition of low dose etomoxir (Fig. 1h). By contrast, 2-DG addition dramatically reduced ECAR, confirming its effective inhibition of glycolysis (Supplementary Fig. 2b). These data suggest that the high basal SRC observed in TVM cells is not exclusively dependent on either FAO or glycolysis, but may be fuelled by a substrate generated independently of both pathways.
Virus infection drives increased SRC in TVM cells
High SRC was not observed in TMEM cells in this study (Fig. 1a, b), but these TMEM cells were isolated out of naive SPF mice and were likely generated in response to commensal or low-pathogenicity organisms and in conditions of low inflammation. To assess SRC in infection-generated TMEM cells, mice were infected with influenza A virus (IAV) and TMEM cells specific for tetrameric H-2Db loaded with NP366, PA224 and PB1-F262 epitopes (TMEM (IAV) cells) were isolated 20 days later. Both TMEM and TMEM (IAV) cells exhibited a similarly low SRC (Fig. 2a, b). Interestingly, TMEM (IAV) cells consistently exhibited substantially higher basal and maximal extracellular acidification rates (ECARBas and ECARMax), compared to TN and TMEM cells (Fig. 2c). These data illustrate that TMEM and TMEM (IAV) cells are metabolically distinct with regard to glycolytic, but not OXPHOS, capacity.
Fig. 2. TVM cells increase SRC with recent infection.

a OCR for sorted TN and TMEM cells from the spleens of uninfected young SPF mice or Tetramer+ TMEM (IAV) cells from IAV-infected mice (20 days post infection) and b change in OCR for each sorted subset (n = 4–2, 3 experimental replicates). c ECAR for sorted TN and TMEM cells from young uninfected mice or Tetramer+ TMEM (IAV) cells from IAV-infected mice (20 days post infection), with ECARBas and ECARMax indicated (n = 2–4, 3 experimental replicates). d OCR for sorted TN and TVM cells from young uninfected mice or TVM (IAV) cells from IAV-infected mice (20 days post infection) and e change in OCR for each sorted subset (n = 4–5, 3 experimental replicates). f ECAR for sorted TN and TVM cells from young uninfected mice or IAV-infected mice (20 days post infection) (n = 5, 3 experimental replicates). Shown is mean ± SEM. NS indicates not significant, * indicates p ≤ 0.05, ** indicates p ≤ 0.01, unpaired t test.
Strikingly, recent IAV infection caused a substantial elevation in the SRC of TVM cells (Fig. 2d, e), without any shift in glycolytic capacity (Fig. 2f). These data demonstrate that infection, like ageing, leads to an environment that augments SRC selectively in TVM cells (and thus in an antigen-independent manner), and reinforce that high SRC is not a canonical feature of TMEM cells, even those induced by infection.
Conventionally defined TCM cells are predominantly TVM Cells
Recently, high SRC was shown to partition preferentially with the long-lived central memory (TCM; CD44hiCD62Lhi) subset of TMEM, rather than short-lived effector memory (TEM; CD44hiCD62Llo) cells7. In that study, TCM cells appear to have been defined as CD44hiCD62Lhi CD8+ T cells obtained from mice after acute lymphocytic choriomeningitis virus (LCMV) infection, which would include TVM cells20. To determine the extent to which metabolic characteristics of TCM cells have been conflated with those of TVM cells, we assessed the proportion of classically defined TCM cells (CD44hiCD62Lhi) that were actually TVM cells (CD44hiCD62LhiCD49dlo) in naive young, naive aged or LCMV-infected mice. In young and aged naive mice, the vast majority (≥85%) of TCM cells were found to be CD49dlo and therefore TVM cells (Fig. 3a). Even 40 days after acute LCMV infection, which induces a substantial CD8+ T cell response and establishes robust antigen-specific memory populations30, over 60% of TCM cells were found to be TVM cells (Fig. 3a). This highlights the possibility that CD8+ T cell populations previously defined as TCM cells, from young, aged or infected mice, may have been predominantly comprised of TVM cells.
Fig. 3. TVM cells comprise the majority of the CD44hiCD62Lhi TCM cell population.

a Histograms for CD49d expression on CD44hiCD62Lhi CD8+ T cells (TCM cells) from naive young mice, naive aged mice and young mice after infection with LCMV (40 days post infection), with bar graphs showing the proportion of TCM cells that are TVM cells (n = 4–5). b Overlays of CD44hiCD49dlo TVM cells on total CD8+ T cells with TEM/TCM cell gating (CD44hiCD62Llo and CD44hiCD62Lhi, respectively). c Overlays of CD44hiCD49dhi TMEM cells on total CD8+ T cells. d Representative dot plots identifying IAV-specific tetramer+ CD8+ T cells that are CD62Lhi (TCM cells) or CD62Llo (TEM cells) (60 days post infection), with bar graphs of the average frequency of tetramer+ CD8+ T cells that are in each subset (n = 5). e OCR for sorted TN and TVM cells from young uninfected mice and sorted TCM and TEM cells from LCMV-infected mice (60 days post infection) and f change in OCR for each sorted subset (n = 5–6, 3 experimental replicates). Data from a–d are representative of at least 2 individual experiments. Shown is mean ± SEM. * indicates p ≤ 0.05, ** indicates p ≤ 0.01, unpaired t test.
The reciprocal analysis was also performed to determine the distribution of sorted TVM (CD44hiCD49dlo) and TMEM (CD44hiCD49dhi) cells from naive young, naive aged or LCMV-infected mice across classical TCM (CD44hiCD62Lhi) and effector memory (TEM; CD44hiCD62Llo) gates. When TVM cells were overlaid onto CD44/CD62L plots, they distributed predominantly to the TCM gate (~80%; Fig. 3b). TMEM cells were predominantly distributed to the TEM gate (49-95%; Fig. 3c), although a substantial proportion was found in the TCM gate in naive young mice. We also found that TMEM (IAV) cells were predominantly TEM cells (88.93 ± 6.67%) with some TCM cells (11.07 ± 6.67%) (Fig. 3d).
To definitively determine the relative SRC of TCM and TEM cells alongside TVM cells, we performed the Seahorse Mito Stress assay on TN and TVM cells isolated from naive SPF mice and TEM and TCM cells isolated from LCMV-infected mice at 60 days post infection. As previously observed, TVM cells had a significantly higher SRC than all other subsets (Fig. 3e, f). In addition, we found that TEM cells had a significantly lower SRC than all other subsets, and TCM cells had a modestly higher SRC than TN cells (Fig. 3e, f). This comprehensively demonstrates that high SRC is not a defining feature of conventional TMEM cells, in particular the TEM cell subset, but is instead characteristic of TVM cells.
SRC correlates with markers of survival, not functionality
High SRC had been proposed as a TMEM cell characteristic facilitating both their enhanced functionality and long-term survival4,5. Accordingly, we aimed to determine if high SRC in TVM cells and the SRC increase with age correlated with enhanced CD8+ T cell functionality or survival.
Three key functions of CD8+ T cells during an immune response are proliferation, cytokine production (particularly IFN-γ) and cytotoxicity. We recently found that TVM cells from aged individuals, with the highest SRC, have very low TCR-driven proliferative capacity, although the few that can respond still produce IFN-γ17. To further evaluate the functionality of CD8+ T cell subsets, their cytotoxic capacity immediately ex vivo was assessed. Sorted TN, TVM and TMEM cells from young and aged OT-I mice were used in an in vitro cytotoxicity assay with ovalbumin-loaded splenocytes as targets. In young mice, TVM cells were substantially more cytotoxic than TN cells and equivalent to TMEM cells (Fig. 4a). With increasing age, the cytotoxicity of TN cells remained low while the cytotoxicity of TVM and TMEM cells declined substantially (Fig. 4a). These data, along with our previous work17, demonstrate that TVM and TMEM cells decline in cytotoxic and proliferative capacity with age, despite their increasing SRC.
Fig. 4. SRC correlates with Bcl-2 expression but not with CD8+ T cell functionality.

a Percent lysis of OVA-loaded targets by sorted TN, TVM and TMEM cells from young or aged OT-I mice (n = 3–8). * Indicates p ≤ 0.05, ** indicates p ≤ 0.01, unpaired t test, data are representative of at least 3 individual experiments. Simple linear regression analyses of mean SRC from Fig. 1b against b the mean number of divisions of sorted CD8+ T cells following 60 h TCR stimulation from ref. 17, c the average percent lysis from (a), d the average proportion of sorted CD8+ T cells making IFN-γ at 36 h after TCR stimulation from ref. 17, and e the median fluorescence intensity (MFI) of Bcl-2 expression from ref. 17.
To formally test whether the capacity for any CD8+ T cell function (proliferation, cytokine production or cytotoxicity) correlated with SRC, linear regression analyses were performed across the cell types and ages. There was no significant correlation observed for any of the functions (Fig. 4b–d), comprehensively demonstrating that high SRC does not necessarily indicate enhanced CD8+ T cell function, particularly in CD8+ T cells from aged individuals.
Our previous work indicated that TVM cells from aged individuals, which exhibit the highest SRC, had a survival advantage following adoptive transfer17, which appeared to parallel expression of the anti-apoptotic protein, Bcl-2. Linear regression analysis of Bcl-2 expression vs SRC across the cell types and ages revealed a significant positive correlation (Fig. 4e), highlighting that SRC is associated with Bcl-2 expression as a surrogate for survival capacity in CD8+ T cells.
Increased TVM cell IL-15 signalling drives increased SRC
Given the strong correlation of Bcl-2 with SRC, mediators of Bcl-2 expression were assessed to define the drivers of high SRC in TVM cells with age. The cytokine, IL-15 (reviewed in ref. 31), was a strong candidate as it has been shown to promote survival of memory phenotype CD8+ T cells through induction of STAT5 phosphorylation (pSTAT5) and expression of Bcl-2 (ref. 32). To understand how IL-15 signalling might change with age in the different subsets, we assessed the expression of cytokine receptor subunits and downstream signalling in TN, TVM and TMEM cells from young and aged mice.
Expression of IL-15Rβ was low on TN cells, modestly higher on TMEM cells, and markedly and significantly higher on TVM cells from young mice (5-fold, p < 0.0001; Fig. 5a). In addition, while neither TN nor TMEM cells exhibited substantial age-related changes in expression, TVM cells exhibited a marked increase in IL-15Rβ expression with age (Fig. 5a). When each subset was sorted and stimulated with soluble IL-15, the intensity of pSTAT5 tracked with receptor expression levels, with TVM cells from young mice exhibiting high pSTAT5 that increased further with age (Fig. 5b). Together, these data indicate that, of all CD8+ T cell subsets, TVM cells exhibit the greatest sensitivity to IL-15 signalling and this sensitivity increases with age.
Fig. 5. High IL-15 sensitivity in TVM cells increases with age and mediates increased SRC.
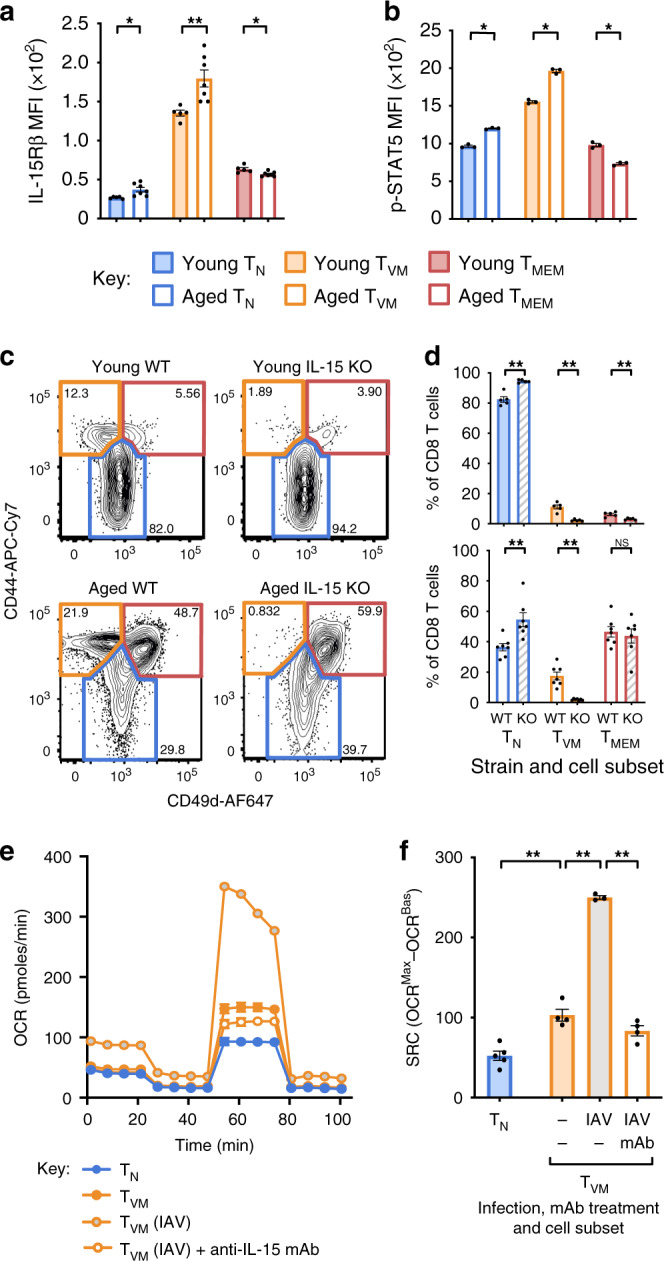
a IL-15Rβ MFI directly ex vivo from individual mice (n = 5–7) or b pSTAT5 MFI after 15 min of stimulation with IL-15 in vitro on sorted TN, TVM and TMEM cells from young or aged mice (n = 3). c Representative dot plots for CD8+ T cells gated on TN (CD44lo), TVM (CD44hiCD49dlo) and TMEM (CD44hiCD49dhi) cells (frequency of CD8+ T cells indicated) and d frequency of CD8+ T cells in each subset in WT or IL-15 KO mice (n = 5 for WT and 7 for KO). e OCR for sorted TN, TVM and TMEM cells from young uninfected, IAV-infected and IAV-infected/IL-15 neutralising mAb treated mice and f change in OCR for each sorted subset. Shown is mean ± SEM. * Indicates p ≤ 0.05, ** indicates p ≤ 0.01, unpaired t test, data for a, b, e, f are representative of at least 3 individual experiments.
IL-15 is regarded as a critical cytokine for both TVM cells and TMEM cells as evidenced by the fact that IL-15 knockout (KO) mice lose approximately half of their memory phenotype (CD44+) cells33 and the TVM cell population fails to develop in young mice in the absence of IL-15 or IL-15Rα14,15. To ascertain whether the IL-15Rβ expression and IL-15 responsiveness of CD8+ T cell subsets reflects their relative dependence on IL-15, we assessed the development and persistence of each subset in young and aged IL-15 KO mice. TVM cells were absent in young IL-15 KO mice, consistent with previous studies, and TVM cells were also absent in aged mice (Fig. 5c, d). Of note, the TMEM cell population appears to be relatively intact in aged mice (Fig. 5c, d). This highlights that IL-15 is absolutely essential for the development of TVM cells, but is dispensable for the generation and maintenance of many TMEM cells, in aged mice.
IL-15 is produced at low levels in steady state but can be dramatically upregulated in DCs during infection in response to type I IFN signalling34. To test whether production of IL-15 during infection was leading to the observed increase in SRC in TVM cells (Fig. 2d, e), we administered an IL-15 neutralising monoclonal antibody (mAb) to young mice infected with IAV. The mAb was administered at days 0 and 3 after infection and SRC was assessed at day 14. Abrogating IL-15 signalling during infection was sufficient to abrogate the infection-driven increase in SRC in TVM cells (Fig. 5e, f). Collectively, these data highlight that the metabolic profile, and in particular elevated SRC, that was previously associated with TMEM cell function and longevity, appears to be a direct function of IL-15 exposure and independent of TMEM phenotype. In addition, the sensitivity of TVM cells to IL-15 appears to account for the high SRC observed in these cells directly ex vivo from young mice and the increase in SRC in TVM cells with age.
Elevated IL-15Rβ and SRC in older human CD8+ T cells
To determine whether our findings in mice were relevant to humans, we first analysed expression of IL-15Rβ on young adult (20–30 yo) and older (60–80 yo) human CD8+ T cells. We found significantly higher expression of IL-15Rβ on TVM cells compared to TN cells in young adults, and this expression increased significantly with age across all subsets (Fig. 6a, b), similar to our observations in mice. Moreover, we observed a trend toward increased SRC in total CD8+ T cells with advanced age (p = 0.1) (Fig. 6c, d). Given our previous description of diminished proliferative capacity in CD8+ T cells from older humans35, these data suggest a similar lack of correlation between SRC and functionality in human CD8+ T cells. Moreover, these data indicate a correlation between elevated SRC and an age-related increase in IL-15 sensitivity of human CD8+ T cells.
Fig. 6. Increased IL-15Rβ expression and SRC in older human CD8+ T cells.

a IL-15Rβ MFI directly ex vivo on CD8 T cell subsets from individual young (20–30 yo) or older (60–80 yo) adult human donors (n = 6–7) and b Representative histograms of IL-15Rβ expression, with expression on CD4+ T cells used as a control. c OCR for enriched CD8+ T cells from young or older human donors and d change in OCR for young or older human donors. This experiment was performed once. Bars or datapoints represent mean ± SEM. * Indicates p ≤ 0.05, ** indicates p ≤ 0.01, unpaired t test.
Discussion
By detailed dissection of the metabolic phenotype of TVM cells compared with TN and TMEM cells, we have demonstrated that high SRC correlates best with IL-15 sensitivity and Bcl-2 expression, and it is therefore selectively associated with the superior survival capacity of TVM cells. In contrast, neither elevated SRC nor substantial IL-15 dependence were characteristics of conventional antigen-driven memory CD8+ T cells, and SRC did not associate with multiple measures of T cell functionality across various cell types and ages. Our data, therefore, illustrate that previous work defining high SRC as a characteristic of TMEM cells, and TCM cells in particular, is likely due to the conflation of these populations with TVM cells, as well as the use of high levels of IL-15 to generate the memory-phenotype CD8+ T cells studied. As previously mentioned, TVM cells cannot be distinguished from TCM cells without inclusion of the marker for CD49d20. The original study proposing SRC as a characteristic of TMEM cells either isolated CD8+CD44hiCD62Lhi cells ex vivo from Listeria monocytogenes infected mice or generated cells in vitro using high levels of exogenous IL-15 (ref. 4). Subsequent work proposed that high SRC partitioned preferentially with the TCM cell population but TCM cells in this study also appeared to be identified as CD8+CD44hiCD62Lhi cells from LCMV-infected mice7. Based on our findings, we contend that these in vivo strategies would have included a substantial population (60–80% of sorted cells) of TVM cells (Fig. 3a), while the in vitro strategy, which has been widely used to define phenotypic, functional and metabolic characteristics of memory cells3–6,8,36,37, relies on robust IL-15 signalling to drive a memory-like phenotype. Recent work has suggested that IL-15 signalling has direct effects on metabolic profiles, with vaccine-induced effector CD8+ T cells shown to depend predominantly on IL-15 for increased SRC38, and another study showing increased expression of Bcl-2, increased IL-15Rβ and metabolic adaptations in memory CD8+ T cells (CD44hi) from aged mice, again incorporating TVM cells23. IL-15 signalling is also known to promote OXPHOS in other cell types, with IL-15 overexpression driving oxidative metabolism in both adipose tissue and skeletal muscle39,40. Thus, we contend that the selective sensitivity of TVM cells to IL-15 is the basis for the uniquely elevated SRC in those cells. Our data suggest that physiological IL-15 signalling can drive high SRC in TVM cells, and that this can be further increased with recent infection and increasing age. Accordingly, we propose that high SRC is an indicator of recent IL-15 signalling in T cells, which itself appears to be a defining characteristic of naturally generated TVM cells, rather than TMEM or TCM cells.
This observation also raises the question of whether other well-accepted characteristics of TMEM populations are primarily canonical TVM cell characteristics20, particularly with regard to IL-15. The dependence on IL-15 for survival is a well-accepted characteristic of antigen-experienced TMEM cells, alongside increased levels of IL15Rβ, and sensitivity to IL-15 stimulation33,41. Here, we demonstrate that TVM cells express higher levels of receptor, are more sensitive to IL-15 signalling, and are entirely dependent on IL-15 for their generation and maintenance, while TMEM cells are surprisingly resistant to a lack of IL-15. Furthermore, IL-15 has been traditionally regarded as a key mediator of CD4 help for antigen-specific CD8+ T cell responses, with CD4 T cells providing help by engaging through CD40L/CD40 interactions with DCs to amplify their IFN-induced production of IL-15 (ref. 42). The provision of CD4 help, including type I IFN and IL-15, during an antigen-specific CD8+ T cell response limits CD8+ T cell contraction, resulting in a memory population responsive to secondary antigenic stimulation41. However, these cytokines also directly expand memory-phenotype populations in an antigen non-specific manner34,43 and are crucial for generation of TVM cell populations14,15. To better delineate the qualitative and quantitative impact of IL-15 on antigen-experienced TMEM vs antigen-naive TVM cells, it is imperative to perform more nuanced analyses using bona fide antigen-experienced TMEM cell populations. This distinction is more than semantic; it is essential to accurately define the fundamental effects of IL-15 on T cell populations and to determine whether treatments using IL-15 or downstream targets will primarily act upon antigen-induced memory T cell populations or for the improved recruitment of antigenically naive CD8+ T cells following primary antigen encounter.
Of note, this data highlights that high SRC is not necessarily linked to superior CD8+ T cell function. Previously, high SRC was proposed to facilitate accelerated proliferation and increased cytokine production with in vitro generated TMEM cells5. We and others have previously demonstrated that proliferative capacity is highest in TVM cells, followed by TN and TMEM cells directly ex vivo in young individuals9,17,19,22, which tracks with their SRC. However, any correlation breaks down with ageing, as proliferative capacity is highest in TN cells and relatively poor in TVM and TMEM cells17,22 and the latter two populations exhibit elevated SRC. We propose that this lack of correlation between SRC and function in aging is compounded by an age-related defect in TCR signalling that uncouples metabolic potential in resting cells from metabolic engagement and functionality after activation. Dysfunctional TCR-driven proliferation in TVM cells from aged individuals and dysregulation of signalling cascades downstream of TCR engagement are evident with age17,22,44,45. This dysregulation may prevent TVM cells from efficiently engaging any increased mitochondrial reserve upon TCR signalling. To test this paradigm further, it would be informative to assess SRC in other T cell subsets, such as stem cell memory T cells, which are highly proliferative, and resident memory T cells, which retain a level of proliferative capacity in situ and are highly IL-15 dependent. Unfortunately, the low frequency of these cells precludes their analysis using the current Seahorse technology.
Interestingly, our mouse data appears to partly contradict earlier work suggesting that human naive CD8+ T cells exhibit an age-related reduction in SRC, although an age-related increased mitochondrial load was observed46. This study used older individuals up to the age of 85 yo, which may be older than the equivalent mouse age of 18–20 mo characterised here. Thus, it is possible that the SRC is reduced in TN cells at extreme ages. Irrespective, our results in mice highlight that a dramatic metabolic shift occurs within the CD8+ T cell population from aged mice, coincident with our analyses of dysfunction and leading to a substantial increase in SRC in memory-phenotype cells. This also appears to be supported by our observation of elevated SRC in older human CD8+ T cells.
Our data, and others, suggests that high SRC might be a more relevant indicator of T cell longevity, rather than functionality. The elevated SRC observed in TVM cells is paralleled by our previous data suggesting these cells have a survival advantage both in vivo and in vitro13,17. Moreover, IL-15 is necessary for both elevated SRC and essential for TVM cell survival15. Finally, SRC has been found to be directly responsible for survival of myocytes under conditions of hypoxia or nutrient deprivation47. We propose that the differential sensitivity of distinct CD8+ T cell subsets to IL-15 exacerbates its effects in states of increased IL-15 production, such as infection or ageing. IL-15 is produced at low levels in the steady state but it can be dramatically upregulated in DCs during infection, in response to type I IFN signalling34. Type I IFN and IL-15 signalling may also increase with age, as a result of heightened inflammation, or inflammageing48. Increased IL-15 transcription has been observed in bone marrow49 and lymph node stromal cells with age50. Bcl-2 was similarly seen to be increased in CD8+ T cells that survived after transfer into a lymphopenic environment, which also correlated with metabolic shifts51. While SRC and IL-15 sensitivity are clearly linked, the mechanism by which IL-15 signalling drives increased SRC remains unclear. It may upregulate Bcl-2 - Bcl-2 family members have been shown to increase mitochondrial capacity by increasing CIV expression52 and inhibiting mitophagy53. It remains unclear, however, whether there is a causal link between SRC and survival or whether these are independently mediated by IL-15. Additionally, while our results show that IL-15 is necessary for high SRC in TVM cells, it may not be entirely sufficient, and additional signalling pathways may be required to co-ordinate with IL-15 to drive increased SRC.
In the T cell field, mitochondrial fusion has been largely associated with advantageous outcomes; namely elevated SRC and memory T cell formation3,6, although this association was not observed in this study, while a more fragmented (or fissed) mitochondria is associated with effector T cells6. However, while fission of mitochondria is essential for cytokinesis and for isolating dysfunctional mitochondria to allow autophagic degradation54, fusion of mitochondria is generally considered to reflect an age- or senescence-related stress response to promote survival of the cell by enabling functional complementation, whereby damaged or dysfunctional mitochondria fuse with intact mitochondria, to maintain ATP production and functionality55,56. We speculate that mitochondrial fusion in this biological scenario reflects an age-related functional complementation.
Collectively, our study underlines how CD8+ T cell subset function and survival is controlled by molecular and metabolic pathways, knowledge of which is essential for design of interventions that restore functionality or modify survival in T cells from aged individuals.
Methods
Mice
Female C57BL/6 mice and male CD45.1+ OT-I mice on a C57BL/6 background were bred and housed in specific-pathogen-free (SPF) conditions at the Monash Animal Research Platform (MARP) and Animal Research Facility (ARL) at Monash University. IL-15 knockout (KO) mice were housed in SPF conditions at the Biomedical Research Facility in the Department of Microbiology and Immunology (DMI) at the University of Melbourne. Young mice were defined as 2–3 months old (mo), and aged mice were ex-breeding stock that were 18–20 mo, unless otherwise noted in the figure legends. During tissue harvests, mice were examined for gross abnormalities, tumours, and enlarged lymph nodes and spleens, and excluded from analyses if these were evident. All animal experimentation was conducted following the Australian National Health and Medical Research Council Code of Practice for the Care and Use of Animals for Scientific Purposes guidelines for housing and care of laboratory animals and performed in accordance with Institutional regulations after pertinent review and approval by the University of Melbourne and Monash University Animal Ethics Committees.
Seahorse Mito Stress assay
A Seahorse XFe96 Bioanalyser (Agilent) was used to determine OCR and ECAR for sorted CD8+ T cell subsets. Sorted cells were washed in assay media (XF Base media (Agilent) with glucose (10 mM), sodium pyruvate (1 mM) and L-glutamine (2 mM) (Gibco), pH 7.4 at 37 °C) before being plated onto Seahorse cell culture plates coated with Cell-Tak (Corning) at 2×105 cells per well. After adherence and equilibration, cell OCR and ECAR was measured during a Seahorse Mito Stress assay (Agilent), with addition of oligomycin (1 µM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP; 1.2 µM) and antimycin A and rotenone (0.5 μM each)). To assess pathways contributing to SRC, we added etomoxir at either 200 or 5 μM (Fig. 1h) or 500 mM of 2-deoxy-D-glucose (Supplementary Fig. 2), after FCCP and prior to antimycin A/rotenone. Assay parameters were as follows: 3 min mix, no wait, 3 min measurement, repeated 3–4 times at basal and after each addition. SRC was calculated as oxygen consumption rate (OCR) at maximum rate (OCRMax) − OCR in basal state (OCRBas).
Electron microscopy
To prepare for transmission electron microscopy, at least 1 × 106 sorted CD8+ TN, TVM, and TMEM cells from young and aged mice were fixed in 2.5% glutaraldehyde in 100 mM sodium cacodylate buffer for 2 h, washed in cacodylate buffer, and post-fixated (1% osmium tetroxide, 1.5% potassium ferricyanide, 65 mM sodium cacodylate buffer) for 1 h before storage in cacodylate buffer. Samples were serially dehydrated with increasing concentrations of ethanol, then propylene oxide before being embedded in serial ratios (3:1, 2:1, 1:1, 1:2 and 1:3) of propylene oxide:epon araldite resin, with samples in 100% epon araldite resin polymerised at 60˚C for 48 h. Embedded blocks were then microtomed into 60 nm sections that were stained with 2.5% uranyl acetate for 15 min then Reynold’s lead citrate for 3 min. Sections were imaged on a JEOL JEM-1400 electron microscope operated at 80 kV using a sCMOS Matataki Flash camera.
Confocal microscopy
To prepare for confocal microscopy, at least 1 × 106 sorted TN, TVM, and TMEM cells from young and aged mice were seeded on 0.1% gelatin-coated Fluorodishes (World Precision Instruments), allowed to settle, adhered for 30 min and fixed with 4% (w/v) paraformaldehyde in PBS (pH 7.4) for 10 min. After permeabilisation with 0.5% (w/v) Triton X-100 in PBS, cells were incubated with primary antibody against Cytochrome C (CytC, mouse monoclonal, BD Biosciences, 556432; 1:500 in 3% BSA-1xPBS) for 60 min at room temperature. The primary antibody was labelled with Alexa-Fluor-488 conjugated anti-mouse-IgG (Molecular Probes, 1:500 in 3% BSA-1xPBS). Hoechst 33258 (1 μg/ml) was used to stain nuclei.
Confocal microscopy was performed on a Leica TCS SP8 confocal microscope (405 nm, 488 nm, 552 nm, 647 nm; Leica Microsystems) equipped with HyD detectors using a 63×/1.40 NA oil immersion objective (HC PLAPO, CS2, Leica Microsystems). Microscopy data was recorded using the Leica LAS X Life software. Images in all experimental groups were obtained using the same settings. Z-sectioning was performed using 150-nm slices. Leica.lif files were converted to multi-colour.tiff composite stacks using custom-written Fiji/ImageJ macros (Version 1.52n).
Images were analysed using Fiji and custom-written macros. Mitochondrial network area (footprint) was evaluated using the MINA plugin for Fiji based on maximum intensity projections of 3D-image stacks. Mitochondrial organelle morphology quantitation into three subcategories (fragmented, intermediate, fused) was evaluated manually based on confocal z-stacks from three independent experiments totalling to 140 cells. All graphical representations and statistical analysis were carried out on Prism (v7.0a, GraphPad) using two-way ANOVA or Student’s t-tests. For representational figures, images were median filtered (1px) using ImageJ and Fiji.
Blue-native PAGE
To assess expression levels of ETC components, 5 × 105 sorted TN, TVM and TMEM cells from young and aged mice were solubilized in 1% digitonin solubilization buffer, subjected to BN-PAGE and transferred to PVDF membrane57 before immunoblotting for COX5A. Complex IV was detected using antibodies to COX5A (Santa Cruz sc-376907) and horseradish peroxidase coupled secondary antibodies and ECL chemiluminescent substrate (BioRad) were used for detection on a BioRad ChemiDoc XRS+ imaging system. The PVDF membrane was also stained with Coomassie Blue (50% methanol, 7% Acetic acid, 0.05% Coomassie Blue R) to assess relative protein loading.
Identification, sorting and phenotyping of T cell subsets
For identification and isolation of mouse TN, TVM, and TMEM cells, samples were processed and subsets were identified using the staining panel and gating strategy described previously17. For sorting, TN cells were defined as CD44lo (the bottom 30% of CD44 expression based on gating in a young, untreated control mouse); CD44int cells were not included in sorted populations.
For characterisation of surface expression of CD69, CD5, CD127 and CD122 on mouse subsets, the following panel was used: LIVE/DEAD Fixable AquaBlue Viability Dye (Life Technologies), anti-Dump (B220, CD4, CD11c, CD11b, F4/80, NK1.1)-FITC (BD Pharmingen; all 1:400), anti-CD8-PacBlue (53-6.7; BD Pharmingen; 1:200), anti-CD49d-AF647 (R1-2; Biolegend; 1:400), anti-CD44-APC-Cy7 (IM7; Biolegend; 1:400) and either anti-CD69:PE (H1:2F3; Biolegend; 1:400), anti-CD5:PE (53-7.3; BD Pharmingen; 1:200), anti-CD127:PE (A7R34; eBioscience; 1:400), or anti-CD122:PE (TM-β1; BD Pharmingen; 1:400).
For overlays of TCM/TEM gating with TN, TVM and TMEM subsets, the following panel was used: LIVE/DEAD Fixable Near IR Viability Dye (Life Technologies), anti-Dump (B220, CD4, CD11c, CD11b, F4/80, NK1.1; all 1:400)-FITC (BD Pharmingen), anti-CD8-BUV395 (53-6.7; BD Pharmingen; 1:400), anti-CD49d-AF647 (R1-2; Biolegend; 1:400), anti-CD44-PE-Cy7 (IM7; Biolegend; 1:1000) and anti-CD62L:BV605 (MEL-14; BD Pharmingen; 1:400). PE-labelled IAV-specific tetramers were included in Fig. 2 and Fig. 3d and were a pool of H2-Db-based tetramers loaded with NP366, PA224 and PB1-F262 epitopes.
IAV infection, LCMV infection and IL-15 neutralisation
For influenza A virus (IAV) infection, mice were anesthetized by isoflurane inhalation and infected intranasally with 1 × 104 plaque-forming units of the HKx31 (H3N2) IAV strain in 30 μL of PBS and spleens were harvested at indicated timepoints.
For lymphocytic choriomeningitis virus (LCMV) infection, mice were administered 3000 plaque-forming units (PFU) of LCMV (strain WE) intravenously and spleens were harvested at indicated timepoints.
For IL-15 neutralisation, 25 μg of an IL-15/Ra neutralising monoclonal antibody (mAb) (GRW15PLZ; eBioscience) was administered intraperitoneally on day 0 immediately prior to infection and at day 3 after IAV infection.
Cytotoxicity assays
Effector TN, TVM and TMEM cells were sorted from young and aged male CD45.1+ OT-I mice. Target splenocytes from male C57BL/6 mice were stained with intermediate or high concentrations of Cell Trace Violet (CTV, Molecular Probes) and left unloaded (CTV low) or loaded with Ovalbumin-derived peptide (SIINFEKL) at 0.1 µM (CTV high). Unloaded and Ova-loaded targets were mixed in equal proportions with beads (1:1:1) and then effector cells were added at ratio of 5:1 with Ova-loaded target cells. Cultures were incubated overnight at 37 °C in 5% CO2. To identify live cells and differentiate effector cells from target cells, the sample was stained with Propidium Iodide (Molecular Probes), anti-CD45.1:APC-Cy7 (A20; Biolegend; 1:400) and anti-CD45.2:PE (104; Biolegend; 1:400). The ratio of live loaded target cells to beads after incubation with each effector cell type was normalised back to the ratio of live loaded target cells to beads in samples that were not incubated with effectors, to calculate the % lysis for each effector cell type.
Phosphorylation assays
TN, TVM and TMEM cells were sorted and left unstimulated, or stimulated with IL-7 (10 ng/mL) or IL-15 (100 ng/mL) in cRPMI at 37˚C in 5% CO2 for 15 min. Cells were processed with Lyse/Fix solution and Perm Buffer II (BD Biosciences) before staining with anti-phospho-STAT5 (CST; 1:400) followed by phycoerythrin (PE)-conjugated anti-rabbit secondary mAb (CST; 1:500).
Human analyses
Human experimental work was conducted according to the Declaration of Helsinki Principles and to the Australian National Health and Medical Research Council (NHMRC) Code of Practice. Signed informed consent was obtained from all blood donors before the study. The study was approved by the University of Melbourne Human Ethics Committee (HREC 1443389). Donor details are shown in Table 1.
Table 1.
Human PBMC donor details.
| Young Adults (20–30 years old (yo)) | Older Adults (60–80 yo) | ||
|---|---|---|---|
| Sex | Age | Sex | Age |
| Female | 23 | Male | 69 |
| Female | 22 | Female | 74 |
| Female | 20 | Female | 70 |
| Male | 27 | Female | 74 |
| Male | 22 | NR | 79 |
| Female | 24 | NR | 79 |
| NR | 62 | ||
NR not recorded.
Briefly, PBMCs were defrosted and rested in complete RPMI overnight. One sample was taken and stained35 to identify CD8+ T cell subsets (TN, TVM, TCM, TEM and TEMRA) based on CD45RA, CD27, Pan-KIR and NKG2A expression, along with anti-human IL-15Rβ-BV421 (TU27; Biolegend; 1:200) and Propidium Iodide was substituted as the viability dye. The remaining sample was negatively enriched for CD8+ T cells using the human CD8+ T Cell Isolation Kit (Miltenyi Biotec) as per manufacturer’s instructions, plated at 2 × 105 cells per well and run in a standard Seahorse Mito Stress assay, as detailed above.
Statistical analyses
Data were analysed in Graphpad Prism (version 7.0a) using the unpaired, two-tailed t-test without correction for multiple comparisons, as indicated in figure legends.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We thank Adam Costin and Joan Clark for technical assistance in electron microscopy, and staff at Monash University Ramaciotti Centre for Cryo-Electron Microscopy, Monash University FlowCore, Monash Micro Imaging, and Monash University Animal Facilities. This work was supported by a Sylvia and Charles Viertel Senior Medical Research Fellowship, an Australian Research Council (ARC) Future Fellowship FT170100174, a National Health and Medical Research Council (NHMRC) Program grant APP1071916 (to N.L.L.), a Rebecca L. Cooper Foundation Medical Research Grant (to K.M.Q), a Viertel-Belberry Senior Medical Research Fellowship (to K.L.G.-J.), Monash Graduate Scholarship and Monash International Postgraduate Research Scholarship (to T.H.) a Monash University Biomedicine Discovery Scholarship (to L.C.) and funding from the Bonn and Melbourne Research and Graduate School (GRK2168). C.E.S. has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 792532 and University of Melbourne McKenzie Fellowship laboratory support. KK is supported by a NHMRC Senior Research Fellowship Level B (GNT#1102792).
Source data
Author contributions
K.M.Q. and N.L.L. designed the study, K.M.Q. performed the majority of experiments and analysed results, T.H. performed assays of T cell function, F.K. performed confocal microscopy, L.M.A. performed cytotoxicity assays, L.E.F. performed western blots, W.K.L. and G.R. performed electron microscopy, L.C., E.W-J., L.L., C.E.S, and K.K. contributed to the procurement and processing of samples, E.C.S. and M.J.D. optimised Seahorse XFe96 experiments, N.L.L., K.M.Q., M.T.R., K.G-J, L.M., and M.J.M. supervised the research, K.M.Q. and N.L.L. wrote the initial draft of the paper and all authors participated in writing the final paper.
Data availability
RNASeq data accessed during this study are available at GEO under the accession code GSE112304. All other data that support the findings of this study are available from the corresponding authors upon request. Raw data for all figures are provided in the Source data file. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer review reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Kylie M. Quinn, Email: Kylie.quinn@rmit.edu.au
Nicole L. La Gruta, Email: Nicole.la.gruta@monash.edu
Supplementary information
Supplementary information is available for this paper at 10.1038/s41467-020-16633-7.
References
- 1.Almeida L, Lochner M, Berod L, Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin. Immunol. 2016;28:514–524. doi: 10.1016/j.smim.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 2.van der Windt GJ, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012;249:27–42. doi: 10.1111/j.1600-065X.2012.01150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Sullivan D, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. 2014;41:75–88. doi: 10.1016/j.immuni.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Windt GJW, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Windt GJW, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc. Natl Acad. Sci. USA. 2013;110:14336–14341. doi: 10.1073/pnas.1221740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buck MD, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. 2016;166:63–76. doi: 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phan AT, et al. Constitutive glycolytic metabolism supports CD8(+) T cell effector memory differentiation during viral infection. Immunity. 2016;45:1024–1037. doi: 10.1016/j.immuni.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geltink, R. I. K. et al. Mitochondrial priming by CD28. Cell171, 385–397.e11(2017). [DOI] [PMC free article] [PubMed]
- 9.Akue AD, Lee J-Y, Jameson SC. Derivation and maintenance of virtual memory CD8 T cells. J. Immunol. 2012;188:2516–2523. doi: 10.4049/jimmunol.1102213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith NL, et al. Developmental origin governs CD8+ T cell fate decisions during infection. Cell. 2018;174:117–130.e114. doi: 10.1016/j.cell.2018.05.029. [DOI] [PubMed] [Google Scholar]
- 11.Haluszczak C, et al. The antigen-specific CD8+ T cell repertoire in unimmunized mice includes memory phenotype cells bearing markers of homeostatic expansion. J. Exp. Med. 2009;206:435–448. doi: 10.1084/jem.20081829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.La Gruta NL, et al. Primary CTL response magnitude in mice is determined by the extent of naive T cell recruitment and subsequent clonal expansion. J. Clin. Invest. 2010;120:1885–1894. doi: 10.1172/JCI41538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quinn KM, et al. Heightened self-reactivity associated with selective survival, but not expansion, of naïve virus-specific CD8+ T cells in aged mice. Proc. Natl Acad. Sci. USA. 2016;113:1333–1338. doi: 10.1073/pnas.1525167113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White JT, et al. Virtual memory T cells develop and mediate bystander protective immunity in an IL-15-dependent manner. Nat. Commun. 2016;7:11291. doi: 10.1038/ncomms11291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sosinowski T, et al. CD8alpha+ dendritic cell trans presentation of IL-15 to naive CD8+ T cells produces antigen-inexperienced T cells in the periphery with memory phenotype and function. J. Immunol. 2013;190:1936–1947. doi: 10.4049/jimmunol.1203149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drobek, A. et al. Strong homeostatic TCR signals induce formation of self-tolerant virtual memory CD8 T cells. EMBO J. 37, e98518(2018). [DOI] [PMC free article] [PubMed]
- 17.Quinn KM, et al. Age-related decline in primary CD8+ T cell responses is associated with the development of senescence in virtual memory CD8+ T cells. Cell Rep. 2018;23:3512–3524. doi: 10.1016/j.celrep.2018.05.057. [DOI] [PubMed] [Google Scholar]
- 18.Rudd BD, et al. Nonrandom attrition of the naive CD8+ T-cell pool with aging governed by T-cell receptor:pMHC interactions. Proc. Natl Acad. Sci. USA. 2011;108:13694–13699. doi: 10.1073/pnas.1107594108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J-Y, Hamilton SE, Akue AD, Hogquist KA, Jameson SC. Virtual memory CD8 T cells display unique functional properties. Proc. Natl Acad. Sci. USA. 2013;110:13498–13503. doi: 10.1073/pnas.1307572110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiu B-C, Martin BE, Stolberg VR, Chensue SW. Cutting edge: central memory CD8 T cells in aged mice are virtual memory cells. J. Immunol. 2013;191:5793–5796. doi: 10.4049/jimmunol.1302509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lanzer KG, Cookenham T, Reiley WW, Blackman MA. Virtual memory cells make a major contribution to the response of aged influenza-naïve mice to influenza virus infection. Immun. Ageing. 2018;15:17. doi: 10.1186/s12979-018-0122-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renkema KR, Li G, Wu A, Smithey MJ, Nikolich-Zugich J. Two separate defects affecting true naive or virtual memory T cell precursors combine to reduce naive T cell responses with aging. J. Immunol. 2013;192:151–159. doi: 10.4049/jimmunol.1301453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davenport B, et al. Aging of antiviral CD8+ memory T cells fosters increased survival, metabolic adaptations, and lymphoid tissue homing. J. Immunol. 2019;202:460–475. doi: 10.4049/jimmunol.1801277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goronzy JJ, Fang F, Cavanagh MM, Qi Q, Weyand CM. Naive T cell maintenance and function in human aging. J. Immunol. 2015;194:4073–4080. doi: 10.4049/jimmunol.1500046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinet V, et al. Type I interferons regulate eomesodermin expression and the development of unconventional memory CD8(+) T cells. Nat. Commun. 2015;6:7089. doi: 10.1038/ncomms8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Divakaruni AS, et al. Etomoxir inhibits macrophage polarization by disrupting CoA homeostasis. Cell Metab. 2018;28:490–503.e7. doi: 10.1016/j.cmet.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Connor RS, et al. The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Sci. Rep. 2018;8:6289. doi: 10.1038/s41598-018-24676-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raud B, et al. Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. 2018;28:504–515.e7. doi: 10.1016/j.cmet.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quinn KM, Palchaudhuri R, Palmer CS, La Gruta NL. The clock is ticking: the impact of ageing on T cell metabolism. Clin. Transl. Immunol. 2019;8:e01091. doi: 10.1002/cti2.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butz EA, Bevan MJ. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity. 1998;8:167–175. doi: 10.1016/s1074-7613(00)80469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raeber ME, Zurbuchen Y, Impellizzieri D, Boyman O. The role of cytokines in T-cell memory in health and disease. Immunol. Rev. 2018;283:176–193. doi: 10.1111/imr.12644. [DOI] [PubMed] [Google Scholar]
- 32.Lai YG, et al. IL-15 modulates the balance between Bcl-2 and Bim via a Jak3/1-PI3K-Akt-ERK pathway to promote CD8alphaalpha+ intestinal intraepithelial lymphocyte survival. Eur. J. Immunol. 2013;43:2305–2316. doi: 10.1002/eji.201243026. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy MK, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J. Exp. Med. 2000;191:771–780. doi: 10.1084/jem.191.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X, Sun S, Hwang I, Tough DF, Sprent J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. 1998;8:591–599. doi: 10.1016/s1074-7613(00)80564-6. [DOI] [PubMed] [Google Scholar]
- 35.Quinn KM, et al. Age-related decline in primary CD8(+) T cell responses is associated with the development of senescence in virtual memory CD8(+) T cells. Cell Rep. 2018;23:3512–3524. doi: 10.1016/j.celrep.2018.05.057. [DOI] [PubMed] [Google Scholar]
- 36.Manjunath N, et al. Effector differentiation is not prerequisite for generation of memory cytotoxic T lymphocytes. J. Clin. Invest. 2001;108:871–878. doi: 10.1172/JCI13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weninger W, Crowley MA, Manjunath N, von Andrian UH. Migratory properties of naive, effector, and memory CD8(+) T cells. J. Exp. Med. 2001;194:953–966. doi: 10.1084/jem.194.7.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klarquist, J. et al. Clonal expansion of vaccine-elicited T cells is independent of aerobic glycolysis. Science Immunol.3, eaas9822 (2018). [DOI] [PMC free article] [PubMed]
- 39.Barra NG, et al. Interleukin-15 Modulates Adipose Tissue by Altering Mitochondrial Mass and Activity. PLoS ONE. 2014;9:e114799–114722. doi: 10.1371/journal.pone.0114799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quinn LS, Anderson BG, Conner JD, Wolden-Hanson T. IL-15 overexpression promotes endurance, oxidative energy metabolism, and muscle PPARδ, SIRT1, PGC-1α, and PGC-1β expression in male mice. Endocrinology. 2013;154:232–245. doi: 10.1210/en.2012-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schluns KS, Williams K, Ma A, Zheng XX, Lefrançois L. Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J. Immunol. 2002;168:4827–4831. doi: 10.4049/jimmunol.168.10.4827. [DOI] [PubMed] [Google Scholar]
- 42.Greyer M, et al. T cell help amplifies innate signals in CD8(+) DCs for optimal CD8(+) T cell priming. Cell Rep. 2016;14:586–597. doi: 10.1016/j.celrep.2015.12.058. [DOI] [PubMed] [Google Scholar]
- 43.Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. 1996;272:1947–1950. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]
- 44.Lanna A, et al. A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat. Immunol. 2017;18:354–363. doi: 10.1038/ni.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lanna A, Henson SM, Escors D, Akbar AN. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014;15:965–972. doi: 10.1038/ni.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moskowitz DM, et al. Epigenomics of human CD8 T cell differentiation and aging. Sci. Immunol. 2017;2:eaag0192. doi: 10.1126/sciimmunol.aag0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfleger J, He M, Abdellatif M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015;6:e1835. doi: 10.1038/cddis.2015.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014;69:S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 49.Pangrazzi L, et al. Increased IL-15 production and accumulation of highly differentiated CD8+ effector/memory T cells in the bone marrow of persons with cytomegalovirus. Front. Immunol. 2017;8:715. doi: 10.3389/fimmu.2017.00715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cui G, et al. Characterization of the IL-15 niche in primary and secondary lymphoid organs in vivo. Proc. Natl Acad. Sci. USA. 2014;111:1915–1920. doi: 10.1073/pnas.1318281111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu A, et al. IL-15 signaling promotes adoptive effector T-cell survival and memory formation in irradiation-induced lymphopenia. Cell Biosci. 2016;6:30. doi: 10.1186/s13578-016-0098-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wali JA, et al. Loss of BIM increases mitochondrial oxygen consumption and lipid oxidation, reduces adiposity and improves insulin sensitivity in mice. Cell Death Differ. 2018;25:217–225. doi: 10.1038/cdd.2017.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hollville E, Carroll RG, Cullen SP, Martin SJ. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol. Cell. 2014;55:451–466. doi: 10.1016/j.molcel.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 54.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 55.Nakada K, et al. Inter-mitochondrial complementation: mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat. Med. 2001;7:934–940. doi: 10.1038/90976. [DOI] [PubMed] [Google Scholar]
- 56.Schon EA, Gilkerson RW. Functional complementation of mitochondrial DNAs: mobilizing mitochondrial genetics against dysfunction. Biochim. Biophys. Acta. 2010;1800:245–249. doi: 10.1016/j.bbagen.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 57.Formosa LE, et al. Characterization of mitochondrial FOXRED1 in the assembly of respiratory chain complex I. Hum. Mol. Genet. 2015;24:2952–2965. doi: 10.1093/hmg/ddv058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNASeq data accessed during this study are available at GEO under the accession code GSE112304. All other data that support the findings of this study are available from the corresponding authors upon request. Raw data for all figures are provided in the Source data file. Source data are provided with this paper.