

Abstract
In this study, we applied a series of genetic modifications to wild-type S. cerevisiae strain BY4741 to address the bottlenecks in the l-tyrosine pathway. A tyrosine ammonia-lyase (TAL) gene from Rhodobacter capsulatus, which can catalyze conversion of l-tyrosine into p-coumaric acid, was overexpressed to facilitate the analysis of l-tyrosine and test the strain’s capability to synthesize heterologous derivatives. First, we enhanced the supply of precursors by overexpressing transaldolase gene TAL1, enolase II gene ENO2, and pentafunctional enzyme gene ARO1 resulting in a 1.55-fold increase in p-coumaric acid production. Second, feedback inhibition of 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase and chorismate mutase was relieved by overexpressing the mutated feedback-resistant ARO4K229L and ARO7G141S, and a 3.61-fold improvement of p-coumaric acid production was obtained. Finally, formation of byproducts was decreased by deleting pyruvate decarboxylase gene PDC5 and phenylpyruvate decarboxylase gene ARO10, and p-coumaric acid production was increased 2.52-fold. The best producer—when TAL1, ENO2, ARO1, ARO4K229L, ARO7G141S, and TAL were overexpressed, and PDC5 and ARO10 were deleted—increased p-coumaric acid production by 14.08-fold (from 1.4 to 19.71 mg L−1). Our study provided a valuable insight into the optimization of l-tyrosine metabolic pathway.
Electronic supplementary material
The online version of this article (10.1007/s13205-020-02223-3) contains supplementary material, which is available to authorized users.
Keywords: l-Tyrosine, Metabolic engineering, p-Coumaric acid, Saccharomyces cerevisiae
Introduction
The plant secondary metabolites flavonoids, stilbenoids and alkaloids have attracted increasing attention due to their pharmaceutical and nutritional applications (Akinwumi et al. 2018; Chougule et al. 2011; Yao et al. 2004). They are mainly obtained by extraction from plants and the extraction is energy intensive, inefficient and not environmentally friendly (Donnez et al. 2009; Sato et al. 2007; Silva et al. 2017). Heterologous biosynthesis in engineered microbes may be a good choice to achieve low consumption of energy and high yield of these secondary metabolites (Xu et al. 2013). To maximize product yield, two general strategies are often used: optimization of heterologous pathways; and improvement of plant secondary metabolite precursors in host cells. Because l-tyrosine is a common precursor for many plant secondary metabolites, it is vital to optimize its metabolic pathway.
Saccharomyces cerevisiae is often chosen as the microbial host for the production of heterologous compounds, due to its safe use status in food industry and in pharmaceutical biotechnology, its high amenability to genetic manipulation, and its eukaryotic nature, which may be helpful for the functional expression of plant-derived enzymes, such as cytochrome P450 enzymes (Borodina and Nielsen 2014; Jiang and Morgan 2004; Krivoruchko and Nielsen 2015). CEN.PK and BY4741 are the two most common hosts for producing plant secondary metabolites, such as resveratrol and vanillin-β-glucoside (Li et al. 2015; Liu et al. 2017; Strucko et al. 2015). Strucko et al. compared CEN.PK and S288c (parental strain of BY4741) for the production of vanillin glucoside, and found that the production of vanillin-β-glucoside in S288c was tenfold higher than that in CEN.PK under the continuous cultivation condition. Thus, we chose BY4741 as the host for optimizing l-tyrosine metabolic pathway in this study.
In S. cerevisiae, l-tyrosine synthesis starts with the shikimate pathway, a common pathway for synthesis of all three aromatic amino acids, which includes seven enzymatic reactions to synthesize chorismate (Braus 1991). First, one of the two 3-deoxy-d-arabino-heptulosonate-7-phosphate (DAHP) synthase isozymes, Aro3p and Aro4p, which are feedback-inhibited by l-phenylalanine and l-tyrosine, respectively, catalyzes the condensation of phosphoenolpyruvate (PEP) and erythrose 4-phosphate (E4P) to form DAHP (Paravicini et al. 1989). Then, DAHP is converted into 5-enolpyruvyl-shikimate-3-phosphate by a pentafunctional enzyme Aro1p, which catalyzes five reactions (Duncan et al. 1988; Graham et al. 1993). Finally, chorismate synthase Aro2p catalyzes the formation of chorismic acid, the last precursor common to all three aromatic amino acids (Braus 1991). Synthesis of prephenate (PPA), the last common precursor of both l-tyrosine and l-phenylalanine, is catalyzed by chorismate mutase Aro7p, whose activity is stimulated by l-tryptophan and inhibited by l-tyrosine (Hartmann et al. 2003; Luttik et al. 2008). Then, PPA is converted into p-hydroxyphenylpyruvate (HPP) by prephenate dehydrogenase Tyr1p, and l-tyrosine is obtained by reversible transamination of aromatic aminotransferase I Aro8p and aromatic aminotransferase II Aro9p (Iraqui et al. 1998; Karsten et al. 2011; Ohashi et al. 2017; Romagnoli et al. 2015). Meanwhile, HPP can also be converted into byproduct p-hydroxy-acetaldehyde by pyruvate decarboxylase Pdc5p or/and phenylpyruvate decarboxylase Aro10p (Fig. 1) (Choo et al. 2018; Vuralhan et al. 2005).
Fig. 1.

Schematic illustration of the p-coumaric acid biosynthetic pathway in S. cerevisiae. G3P glyceraldehyde 3-phosphate, S7P sedoheptulose-7-phosphate, 2-PG 2-phosphoglycerate, F6P fructose 6-phosphate, E4P erythrose 4-phosphate, PEP phosphoenolpyruvate, DAHP 3-deoxy-d-arabinoheptulosonic acid-7-phosphate, DHQ 3-dehydroquinate, DHS 3-dehydroshikimate, SHIK shikimate, S3P shikimate 3-phosphate, EPSP 5-enolpyruvyl-shikimate 3-phosphate, CHA chorismic acid, PPA prephenate, HPPp-hydroxyphenylpyruvate, l-Trpl-tryptophan, l-Phel-phenylalanine, l-Tyrl-tyrosine, p-PACp-hydroxy-acetaldehyde, TAL tyrosine ammonia-lyase, p-CAp-coumaric acid. Genes in blue represent overexpression, while in red indicate deletion, the star represents allosteric regulation
In the last decade, there have been several efforts to optimize l-tyrosine biosynthesis in S. cerevisiae, and research has generally focused on both eliminating the feedback inhibition of key enzymes and preventing the formation of byproducts. Overexpression of DAHP synthase gene ARO4K229L and chorismate mutase gene ARO7G141S could produce a 200-fold increase in extracellular aromatic amino acids compared to the reference strain and an increment of 4.5-fold of the flux through the aromatic amino acid biosynthesis pathway (Luttik et al. 2008). A triple knockout of the phenylpyruvate decarboxylase genes ARO10, PDC5, and PDC6 could prevent the formation of byproduct phenylethanol, thus increasing the flux through the l-tyrosine biosynthesis pathway (Koopman et al. 2012). A p-coumaric acid overproducing platform strain was obtained by overexpressing feedback-resistant mutants of ARO4 and ARO7, E. coli shikimate kinase II gene (aro L) and tyrosine ammonia-lyase (TAL) gene from Flavobacterium johnsoniae in a Δpdc5Δaro10 background strain (Rodriguez et al. 2015). Gold et al. combined localized pathway engineering with global engineering of central metabolism to develop a strain that accumulated intracellular l-tyrosine up to 520 μmol g−1 dry cell weight or 192 mM in the cytosol (Gold et al. 2015).
Previous approaches to engineering l-tyrosine overproduction in S. cerevisiae were designed for only a few candidates of the l-tyrosine pathway at a time. Therefore, one pathway bottleneck might be eliminated using these approaches; while, another bottleneck might be introduced somewhere else within this pathway. In this study, we systematically analyzed nine genes necessary for production of l-tyrosine: seven for the l-tyrosine biosynthetic pathway and two for overproducing E4P and PEP. To facilitate the analysis of l-tyrosine and evaluate the strains as a platform for the synthesis of plant secondary metabolites derived from l-tyrosine, the TAL from Rhodobacter capsulatus was employed to catalyze the conversion of l-tyrosine into p-coumaric acid (Kyndt et al. 2002). We modified not only the shikimate pathway but also the carbon flux to PEP and E4P. Our study provided a new strategy for the optimization of l-tyrosine metabolic pathway, obtained a l-tyrosine high-producing strain with inheritable stability and biosecurity, and developed a platform for the study of plant secondary metabolites deriving from l-tyrosine.
Materials and methods
Strains, plasmids, media, and growth conditions
All strains and plasmids used in this study are listed in Table 1.
Table 1.
Strains and plasmids used
| Strains and plasmids | Relevant characteristics | Source |
|---|---|---|
| E. coli strain | ||
| E. coli DH5α | F-ϕ80dlacZΔM15 endAI recAI hsd RI7(rK−mK+) supE44 thi-1 relAI Δ(lacZYA-argF)U169 gyrA96 deoR. Host strain to amplify plasmid DNA | Stratagene |
| S. cerevisiae strains | ||
| BY4741 | MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0 | EUROSCARF, Frankfurt, Germany |
| NK-L71 | BY4741; PGPD1-ENO2; PGPD1-TAL1 | Mao et al. (2017) |
| NK-L107 | NK-L71; PGPD1-ARO1 | Mao et al. (2017) |
| NKA2 | NK-L107; PTEF1-ARO4fbr-TADH1 | This study |
| NKA3 | NK-L107; PPGK1-ARO7fbr-TCYC1 | This study |
| NKA4 | NK-L107; PTEF1-ARO4fbr-TADH1; PPGK1-ARO7fbr-TCYC1 | This study |
| NKA5 | NKA4; pdc5Δ::loxP | This study |
| NKA6 | NKA4; aro10Δ::loxP | This study |
| NKA7 | NKA4; pdc5Δ::loxP; aro10Δ::loxP | This study |
| NKA8 | NKA7; PTEF1-TYR1-TADH1 | This study |
| NKA9 | NKA7; PTEF1-ARO2-TADH1 | This study |
| BY4741T | BY4741; pLC-m7 | This study |
| NK-L71T | NK-L71; pLC-m7 | This study |
| NKA1T | NK-L107; pLC-m7 | This study |
| NKA2T | NKA2; pLC-m7 | This study |
| NKA3T | NKA3; pLC-m7 | This study |
| NKA4T | NKA4; pLC-m7 | This study |
| NKA5T | NKA5; pLC-m7 | This study |
| NKA6T | NKA6; pLC-m7 | This study |
| NKA7T | NKA7; pLC-m7 | This study |
| NKA8T | NKA8; pLC-m7 | This study |
| NKA9T | NKA9; pLC-m7 | This study |
| Plasmids | ||
| pUG72 | Template for the loxP-URA3-loxP cassette, Ampr | Hegemann and Heick (2011) |
| pSH62 | HIS3, Cre expression vector | Hegemann and Heick (2011) |
| pLC41 | 2 μm ori, HIS3, PTEF1-TADH1, PPGK1-TCYC1, Ampr | This lab |
| pLC-m1 | pSP-G1::PTEF1-ARO4fbr-TADH1, PPGK1-ARO7fbr-TCYC1 | Mao et al. (2017) |
| pLC-m2 | pLC42::PTEF1-ARO2-TADH1, PPGK1-TCYC1 | Mao et al. (2017) |
| pLC-m3 | pLC42::PTEF1-TYR1-TADH1, PPGK1-TCYC1 | Mao et al. (2017) |
| pLC-m7 | pLC41::PTEF1-TADH1, PPGK1-TAL-TCYC1 | Mao et al. (2017) |
Escherichia coli DH5α competent cells were purchased from ComWin Biotech Company (Beijing, China) and used for bacterial transformation and propagation. They were grown at 37 °C in Luria–Bertani broth (1% NaCl, 1% tryptone and 0.5% yeast extract) supplemented with ampicillin (100 mg L−1) to select positive E. coli transformants (Liu et al. 2014).
Wild-type S. cerevisiae strain BY4741 (MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0) was obtained from Euroscarf (Frankfurt, Germany) and grown at 30 °C in synthetic complete (SC) medium (2% glucose, 0.67% yeast nitrogen base without amino acids and 0.13% amino acid mixture). Drop-out media (SC-Ura and SC-His), prepared using single drop-out mixture of amino acids (SD/ − uracil or SD/ − histidine), were used in the transformation and complementation experiments. The SG medium (2% galactose, 0.67% yeast nitrogen base without amino acids and 0.13% amino acid mixture) was used to induce expression of Cre recombinase in the yeast transformants. All solid media used in this study contained 2% agar.
For each engineered strain, three parallel correct transformants were picked and inoculated in 5 mL of SC-His liquid medium at 30 °C with 200-rpm agitation until cell density was saturated, and then cells were subcultured into 20 mL of SC-His medium in three 100-mL shaking flasks with a starting OD600 of 0.1. The cells in shaking flasks were cultivated at 30 °C and 200-rpm agitation for 120 h. Experimental samples were withdrawn every 12 h for OD600 measurements and product quantification.
Overexpression of ARO4fbr, ARO7fbr, ARO2 and TYR1
The ARO4fbr overexpression strain was constructed by replacing ARO4 with the URA3 and PTEF1-ARO4fbr-TADH1 cassettes in strain NK-L107. A detailed overexpression route is illustrated in Fig. S1. The complete overexpression cassette was derived from four individual parts as follows. Part 1 consisted of a 679-bp upstream homologous sequence of ARO4 and a 40–60-bp sequence homologous to the 5′-terminus of part 2. In part 2, a 40–60-bp sequence homologous to the 3′-terminus of part 1, the URA3 cassette and a 40–60-bp sequence homologous to the 5′-terminus of part 3 were included. Part 3 comprised a 40–60-bp sequence homologous to the 3′-terminus of part 2, the PTEF1-ARO4fbr-TADH1 cassette and a 40–60-bp sequence homologous to the 5′-terminus of part 4. In part 4, a 40–60-bp sequence homologous to the 3′-terminus of part 3 and a 660-bp downstream homologous sequence of ARO4 were included.
The four parts were generated by PCR amplification using primers and templates listed in Table 2: S. cerevisiae genomic DNA was the PCR template of parts 1 and 4; plasmid pUG72 was used for amplifying part 2 (Hegemann and Heick 2011); and part 3 was amplified from plasmid pLC-m1 (Fig. S1) (Mao et al. 2017). After purification, the four parts were co-transformed into yeast strain utilizing a lithium acetate procedure described previously (Gietz and Woods 2002), and then the intact overexpression cassette was generated through recombination between the two 40 and 60-bp overlapping regions by means of the homologous recombination machinery of S. cerevisiae. More precisely, the upstream and downstream homologous sequences of parts 1 and 2 were used for targeted homologous recombination. Transformants were selected on SC-Ura yeast synthetic drop-out media and confirmed by PCR. The correct transformants were transformed with plasmid pSH62, and URA3 selection marker was looped out by Cre recombinase expression induced by galactose (Hegemann and Heick 2011; Sauer 1987). The same method was used for overexpression of ARO7fbr, ARO2 and TYR1. The original genes ARO7, ARO2 and TYR1 were replaced by PPGK1-ARO7fbr-TCYC1, PTEF1-ARO2-TADH1 and PTEF1-TYR1-TADH1 cassettes, amplified from plasmids pLC-m1, pLC-m2 and pLC-m3, respectively (Fig. S2) (Mao et al. 2017).
Table 2.
Primers used
| Primers | Sequences (5′-3′) | Applications |
|---|---|---|
| The overexpression of mutated DAHP synthase ARO4 and chorismate mutase ARO7 to avoid feedback inhibition | ||
| 1 | CTGCACAACCCACATAATTC | Amplify ARO4 upstream homologous region for ARO4fbr overexpression from S. cerevisiae genome, forward primer |
| 2 | CGACCTGCAGCGTACGAAGCTTCAGCTGGAGAGACTCTTCTAGTTATTCTTAATTAC | Amplify ARO4 upstream homologous region for ARO4fbr overexpression from S. cerevisiae genome, reverse primer |
| 3 | GTAATTAAGAATAACTAGAAGAGTCTCTCCAGCTGAAGCTTCGTACGCTGCAGGTCG | Amplify URA3 selection marker for ARO4fbr overexpression from pUG72, forward primer |
| 4 | CATCTTTGTAAAACTTCGATACCACACACGCGGCCGCATAGGCCACTAGTGGATCTG | Amplify URA3 selection marker for ARO4fbr overexpression from pUG72, reverse primer, reverse primer |
| 5 | CAGATCCACTAGTGGCCTATGCGGCCGCGTGTGTGGTATCGAAGTTTTACAAAGATG | Amplify PTEF1-ARO4fbr-TADH1 DNA fragment for ARO4fbr overexpression from pLC-m1, forward primer |
| 6 | CATCCACGAAATCAGTGAAAGGTCAGCGCACACACCATAGCTTCAAAATGTTTCTAC | Amplify PTEF1-ARO4fbr-TADH1 DNA fragment for ARO4fbr overexpression from pLC-m1, reverse primer |
| 7 | GTAGAAACATTTTGAAGCTATGGTGTGTGCGCTGACCTTTCACTGATTTCGTGGATG | Amplify ARO4 downstream homologous region for ARO4fbr overexpression from S. cerevisiae genome, forward primer |
| 8 | CCTATTTCTGGTACTGGCTTTTC | Amplify ARO4 downstream homologous region for ARO4fbr overexpression from S. cerevisiae genome, reverse primer |
| 9 | GATCGTATATGACACAACGAGG | Amplify ARO7 upstream homologous region for ARO7fbr overexpression from S. cerevisiae genome, forward primer |
| 10 | CGACCTGCAGCGTACGAAGCTTCAGCTGATCTTATACCAATTTTATGCAGGATGCTG | Amplify ARO7 upstream homologous region for ARO7fbr overexpression from S. cerevisiae genome, reverse primer |
| 11 | CAGCATCCTGCATAAAATTGGTATAAGATCAGCTGAAGCTTCGTACGCTGCAGGTCG | Amplify URA3 selection marker for ARO7fbr overexpression from pUG72, forward primer |
| 12 | GATAAGACCCCATTCTTTGAAGGTACTTCCCGGCCGCATAGGCCACTAGTGGATCTG | Amplify URA3 selection marker for ARO7fbr overexpression from pUG72, reverse primer, reverse primer |
| 13 | CAGATCCACTAGTGGCCTATGCGGCCGGGAAGTACCTTCAAAGAATGGGGTCTTATC | Amplify PPGK1-ARO7fbr-TCYC1 DNA fragment for ARO7fbr overexpression from pLC-m1, forward primer |
| 14 | GTGGAATTGTTACCGTGATAGCCTTCATGCCTTCGAGCGTCCCAAAACCTTCTCAAGC | Amplify PPGK1-ARO7fbr-TCYC1 DNA fragment for ARO7fbr overexpression from pLC-m1, reverse primer |
| 15 | GCTTGAGAAGGTTTTGGGACGCTCGAAGGCATGAAGGCTATCACGGTAACAATTCCAC | Amplify ARO7 downstream homologous region for ARO7fbr overexpression from S. cerevisiae genome, forward primer |
| 16 | GTCATCGACTTTGTCATTGCC | Amplify ARO7 downstream homologous region for ARO7fbr overexpression from S. cerevisiae genome, reverse primer |
| The knockout of ARO10 (phenylpyruvate decarboxylase) and PDC5 (pyruvate decarboxylase) to avoid production of aromatic alcohols and direct the pathway flux to aromatic amino acids | ||
| 17 | CCTCTTCTTCTTGTGTGTTTAAGC | Amplify ARO10 upstream homologous region for ARO10 knockout from S. cerevisiae genome, forward primer |
| 18 | CGACCTGCAGCGTACGAAGCTTCAGCTGGCTTAAGGGAGTTTCTTTGTTATCTTG | Amplify ARO10 upstream homologous region for ARO10 knockout from S. cerevisiae genome, reverse primer |
| 19 | CAAGATAACAAAGAAACTCCCTTAAGCCAGCTGAAGCTTCGTACGCTGCAGGTCG | Amplify URA3 selection marker for ARO10 knockout from pUG72, forward primer |
| 20 | CACACGATAGGAATGACAGAAAAAAGCGGCCGCATAGGCCACTAGTGGATCTG | Amplify URA3 selection marker for ARO10 knockout from pUG72, reverse primer |
| 21 | CAGATCCACTAGTGGCCTATGCGGCCGCTTTTTTCTGTCATTCCTATCGTGTG | Amplify ARO10 downstream homologous region for ARO10 knockout from S. cerevisiae genome, forward primer |
| 22 | CGAAGTATTTCGGACTCTTTCTTC | Amplify ARO10 downstream homologous region for ARO10 knockout from S. cerevisiae genome, reverse primer |
| 23 | GTTGAAAATGACGACGAGCCTG | Amplify PDC5 upstream homologous region for PDC5 knockout from S. cerevisiae genome, forward primer |
| 24 | CGACCTGCAGCGTACGAAGCTTCAGCTGGTGTAATAAGAAAGAGAGGAAAGGAC | Amplify PDC5 upstream homologous region for PDC5 knockout from S. cerevisiae genome, reverse primer |
| 25 | GTCCTTTCCTCTCTTTCTTATTACACCAGCTGAAGCTTCGTACGCTGCAGGTCG | Amplify URA3 selection marker for PDC5 knockout from pUG72, forward primer |
| 26 | CCTAAACATCTATAACCTTCAAAAGCGGCCGCATAGGCCACTAGTGGATCTG | Amplify URA3 selection marker for PDC5 knockout from pUG72, reverse primer |
| 27 | CAGATCCACTAGTGGCCTATGCGGCCGCTTTTGAAGGTTATAGATGTTTAGG | Amplify PDC5 downstream homologous region for PDC5 knockout from S. cerevisiae genome, forward primer |
| 28 | CTCTTGCTTGTTCGGAGTTC | Amplify PDC5 downstream homologous region for PDC5 knockout from S. cerevisiae genome, reverse primer |
| Overexpression of l-tyrosine pathway genes ARO2 and TYR1 | ||
| 29 | CGTGAACACGTACACTATACG | Amplify ARO2 upstream homologous region for ARO2 overexpression from S. cerevisiae genome, forward primer |
| 30 | CGACCTGCAGCGTACGAAGCTTCAGCTGCGATGAGAATAACGCTTAGATGATGCCG | Amplify ARO2 upstream homologous region for ARO2 overexpression from S. cerevisiae genome, reverse primer |
| 31 | CGGCATCATCTAAGCGTTATTCTCATCGCAGCTGAAGCTTCGTACGCTGCAGGTCG | Amplify URA3 selection marker for ARO2 overexpression from pUG72, forward primer |
| 32 | CATCTTTGTAAAACTTCGATACCACACACGCGGCCGCATAGGCCACTAGTGGATCTG | Amplify URA3 selection marker for ARO2 overexpression from pUG72, reverse primer, reverse primer |
| 33 | CAGATCCACTAGTGGCCTATGCGGCCGCGTGTGTGGTATCGAAGTTTTACAAAGATG | Amplify PTEF1-ARO2-TADH1 DNA fragment for ARO2 overexpression from pLC-m2, forward primer |
| 34 | CATAACTCTTGAGGGGTTTTGTTTCTATCGCACACACCATAGCTTCAAAATGTTTCTAC | Amplify PTEF1-ARO2-TADH1 DNA fragment for ARO2 overexpression from pLC-m2, reverse primer |
| 35 | GTAGAAACATTTTGAAGCTATGGTGTGTGCGATAGAAACAAAACCCCTCAAGAGTTATG | Amplify ARO2 downstream homologous region for ARO2 overexpression from S. cerevisiae genome, forward primer |
| 36 | CATTGATGTTACCTTCACCAAGG | Amplify ARO2 downstream homologous region for ARO2 overexpression from S. cerevisiae genome, reverse primer |
| 37 | CCCATCTTTGAAAACTCTAACG | Amplify TYR1 upstream homologous region for TYR1 overexpression from S. cerevisiae genome, forward primer |
| 38 | CGACCTGCAGCGTACGAAGCTTCAGCTGGTTTATCAAGTGGATATGCTGTCCTCTTTC | Amplify TYR1 upstream homologous region for TYR1 overexpression from S. cerevisiae genome, reverse primer |
| 39 | GAAAGAGGACAGCATATCCACTTGATAAACCAGCTGAAGCTTCGTACGCTGCAGGTCG | Amplify URA3 selection marker for TYR1 overexpression from pUG72, forward primer |
| 40 | CATCTTTGTAAAACTTCGATACCACACACGCGGCCGCATAGGCCACTAGTGGATCTG | Amplify URA3 selection marker for TYR1 overexpression from pUG72, reverse primer, reverse primer |
| 41 | CAGATCCACTAGTGGCCTATGCGGCCGCGTGTGTGGTATCGAAGTTTTACAAAGATG | Amplify PTEF1-TYR1-TADH1 DNA fragment for TYR1 overexpression from pLC-m3, forward primer |
| 42 | CGATAATCGCGGTAGTGAGCATTACACGCACACACCATAGCTTCAAAATGTTTCTAC | Amplify PTEF1-TYR1-TADH1 DNA fragment for TYR1 overexpression from pLC-m3, reverse primer |
| 43 | GTAGAAACATTTTGAAGCTATGGTGTGTGCGTGTAATGCTCACTACCGCGATTATCG | Amplify TYR1 downstream homologous region for TYR1 overexpression from S. cerevisiae genome, forward primer |
| 44 | CGACCAGGATCCATATATC | Amplify TYR1 downstream homologous region for TYR1 overexpression from S. cerevisiae genome, reverse primer |
| Quantitative real-time PCR | ||
| 45 | GTGAACAATTACACAGCTCCTATAG | Amplify partial cDNA of S. cerevisiae ALG9 gene for real-time PCR, forward primer |
| 46 | CCTATGATTATCTGGCAGCAGGAAAG | Amplify partial cDNA of S. cerevisiae ALG9 gene for real-time PCR, reverse primer |
| 47 | CTGGTGATTTCGGCTCTATTGCCAAG | Amplify partial cDNA of S. cerevisiae TAL1 gene for real-time PCR, forward primer |
| 48 | GGTGGTCTTACCATGCTTCTTACCG | Amplify partial cDNA of S. cerevisiae TAL1 gene for real-time PCR, reverse primer |
| 49 | GACTTGTCTAAGTCCAAGACCTCTCC | Amplify partial cDNA of S. cerevisiae ENO2 gene for real-time PCR, forward primer |
| 50 | CATGGCTTCAGCGAAGGTCTTAGC | Amplify partial cDNA of S. cerevisiae ENO2 gene for real-time PCR, reverse primer |
| 51 | CCTGGAATTCAAGGCTTCTTTGCCAG | Amplify partial cDNA of S. cerevisiae ARO1 gene for real-time PCR, forward primer |
| 52 | CGCTACCATAACCGTATCACGAGTAC | Amplify partial cDNA of S. cerevisiae ARO1 gene for real-time PCR, reverse primer |
| 53 | CGCTAACGGTGAAAACGCCATTACC | Amplify partial cDNA of S. cerevisiae ARO4 gene for real-time PCR, forward primer |
| 54 | CGTCTTCAGTAGTTTCCCAACCTATAC | Amplify partial cDNA of S. cerevisiae ARO4 gene for real-time PCR, reverse primer |
| 55 | GAGAGGTCGCATTTCGCCACATG | Amplify partial cDNA of S. cerevisiae ARO7 gene for real-time PCR, forward primer |
| 56 | CAGGTGATTCGAATCTTCTGATGCG | Amplify partial cDNA of S. cerevisiae ARO7 gene for real-time PCR, reverse primer |
| 57 | GGTCATTTGGTATCCAGGCAAAGCG | Amplify partial cDNA of S. cerevisiae TYR1 gene for real-time PCR, forward primer |
| 58 | CTCGATTTCCGGCAGCTTACAACTC | Amplify partial cDNA of S. cerevisiae TYR1 gene for real-time PCR, reverse primer |
| 59 | GCTTCAGGTGCCATTGCTGAGAAG | Amplify partial cDNA of S. cerevisiae ARO2 gene for real-time PCR, forward primer |
| 60 | CCTGGTGATGGTGTTCAACAGATGC | Amplify partial cDNA of S. cerevisiae ARO2 gene for real-time PCR, reverse primer |
Deletion of ARO10 and PDC5
The method of single gene deletion was similar to that of gene overexpression. The gene knockout cassette had three parts (Fig. S3). In part 1, a 600–1000-bp upstream homologous sequence of the target gene and a 40–60-bp sequence homologous to the 5′-terminus of part 2 were included. Part 2 consisted of a 40–60-bp sequence homologous to the 3′-terminus of part 1, the URA3 cassette and a 40–60-bp sequence homologous to the 5′-terminus of part 3. Part 3 comprised a 40–60-bp sequence homologous to the 3′-terminus of part 2 and a 600–1000-bp downstream homologous sequence of the target gene. Parts 1 and 3 were amplified from S. cerevisiae genomic DNA, and plasmid pUG72 used as the PCR template for part 2 (Hegemann and Heick 2011). The three knockout fragments were transformed into S. cerevisiae and transformants were selected on SC-Ura yeast synthetic drop-out media. The target gene was replaced by a URA3 cassette. Then, the Cre/lox P system was again used to remove the URA3 cassette (Sauer 1987). All primers used are listed in Table 2. The double-deletion strain was obtained by sequential deletion of PDC5 and ARO10.
Codon optimization, synthesis, and overexpression of TAL
The TAL from R. capsulatus was codon optimized and synthesized for S. cerevisiae by Genewiz (Su Zhou, China), assembled under PGK1 promoter into pLC41 vector (Mao et al. 2017). The resulting plasmid named pLC-m7 was transformed into strain BY4741 and its derivative strains to generate a series of strains producing p-coumaric acid. Descriptions of strains and plasmids used in this study are summarized in Table 1.
Real time quantitative PCR (RT-PCR)
Strains were cultured at 30 °C in SC-His medium to exponential phase. The mRNAs were extracted using a RNApure yeast kit (CWBIO, Beijing, China) and reverse transcribed into cDNAs using a superRT cDNA kit (CWBIO). The gene relative expression levels were quantified by RT-qPCR using Hieff™ qPCR SYBR green master mix (Yeasen, Shanghai, China). The gene ALG9, which had a relatively stable expression level, was used as the reference gene. All of the primers used are listed in Table 2 and the data were analyzed using threshold cycle (2−∆∆CT) method. Each experiment was performed in triplicate.
High-performance liquid chromatograph (HPLC) analysis of products
Samples were centrifuged at 12,000g for 5 min and the supernatant was extracted and filtered through 0.22-μm pore-size polyethersulfone membrane syringe filters for HPLC analysis.
For quantification of p-coumaric acid, a HPLC coupled with an ultraviolet detector and an inertsil ODS-3/C18 column (250 mm × 4.6 mm, 5 μm) was used. Mobile phases A and B were composed of water (5% acetonitrile and 0.1% trifluoroacetic acid) and acetonitrile (0.1% trifluoroacetic acid), respectively. A gradient method was used with a flow rate of 1 mL min−1: 6–50% phase B for 15 min, 50–98% phase B for 15 min, 98% phase B for 3 min, 98–6% for 2 min and 6% phase B for an additional 5 min. The injection volume was 10 μL. Quantification was based on the peak areas of absorbance at 310 nm and retention time was 16.9 min.
Glucose was analyzed by a Waters Alliance 2695 HPLC (Waters, Milford, MA, USA) equipped with an isocratic pump, a refractive index detector and a Hitachi auto sampler. A Bio-Rad Aminex HPX-87H column (300 × 7.8 mm) was utilized at 65 °C with 5 mM H2SO4 as the mobile phase at a flow rate of 0.6 m L min−1.
Statistical analysis
Data were expressed as means ± standard errors. Student’s t test was used to compare the difference between engineered and parental strains. Statistical significance was p < 0.05.
Results
Effects of overexpression of enolase II, transaldolase, and pentafunctional enzyme Aro1p on p-coumaric acid production
Both E4P and PEP are the intermediate metabolites of the pentose phosphate and glycolytic pathways, respectively (Fig. 1). To enhance the supply of E4P and PEP, the transaldolase, which catalyzes conversion of sedoheptulose-7-phosphate and glyceraldehyde 3-phosphate into E4P and fructose 6-phosphate, and enolase II, which catalyzes the conversion of 2-phosphoglycerate into PEP, were simultaneously overexpressed through promoter replacement in situ in wild-type S. cerevisiae BY4741, generating the engineered strain NK-L71. Their original promoter was replaced by a strong GPD1 promoter, which was previously described by Mao et al. (2017). The promoter of Aro1p was also replaced by GPD1 promoter in the strain NK-L71 to yield an engineered strain NK-L107 (Fig. 2). Furthermore, p-coumaric acid production strains NK-L71T and NKA1T were constructed by introducing pLC-m7 into strains NK-L71 and NK-L107, respectively.
Fig. 2.

Schematic representation of the engineered strains NK-L71, NK-L107, NKA2, NKA3, NKA4, NKA5, NKA6, NKA7, NKA8 and NKA9. Promoters of TAL1, ENO2, and ARO1 were replaced with a strong constitutive promoter PGPD1; ARO4K229L was overexpressed under the strong constitutive promoter PTEF1 at the ARO4 locus; ARO7G141S was overexpressed under the strong constitutive promoter PPGK1 at the ARO7 locus; TYR1 and ARO2 were overexpressed under the strong constitutive promoter PTEF1 in situ, respectively
The RT-qPCR analysis indicated that replacement of the promoter could significantly up-regulate the gene expression levels (Fig. 3). Fermentations were performed and p-coumaric acid production was determined daily—peaking at 48 h and then decreasing gradually (Fig. S4). The reference strain BY4741T produced 1.40 mg L−1 of p-coumaric acid; the engineered strain NK-L71T overexpressing ENO2 and TAL1 produced 1.81 mg L−1; and the engineered strain NKA1T overexpressing ENO2, TAL1 and ARO1 increased p-coumaric acid production to 2.17 mg L−1, which was 1.55-fold higher than that of the reference strain (Fig. 4). Thus, enhancing supply of E4P and PEP, and overexpression of Aro1p, had a positive effect on production of p-coumaric acid. There were no significant differences in growth rates and glucose consumption rates between the reference strain and engineered strains (Figs. S5 and S6).
Fig. 3.

The RT-qPCR analysis of gene expression levels of the engineered strains compared with BY4741T strain. Average ± standard deviations were calculated from three biological replicates. mRNAs were extracted using a RNApure yeast kit (CWBIO, Beijing, China) and reverse transcribed into cDNAs using a superRT cDNA kit (CWBIO). The gene relative expression levels were quantified by RT-qPCR using Hieff™ qPCR SYBR green master mix (Yeasen, Shanghai, China). The gene ALG9 was used as the reference gene. Data were analyzed using threshold cycle (2−△△CT) method
Fig. 4.
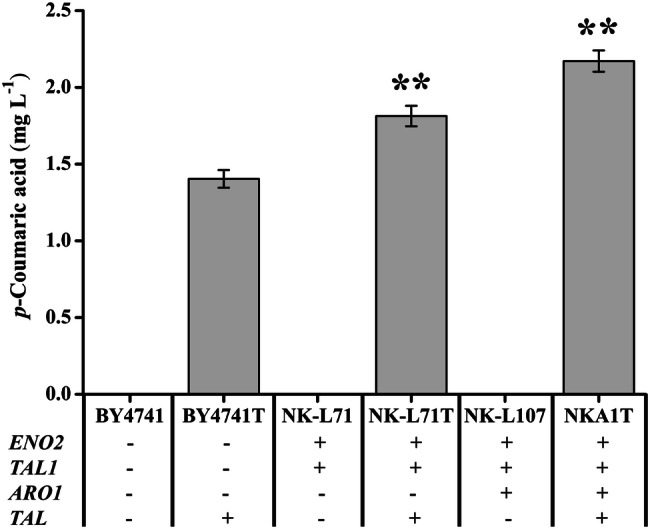
Effect of the overexpression of ENO2, TAL1, and ARO1 on p-coumaric acid production in strains overexpressing tyrosine ammonia-lyase RcTAL from Rhodobacter capsulatus. Strains were cultured in 20 mL of SC-His medium at 30 °C and 200 rpm, and p-coumaric acid production peaked at 48 h. Average ± standard deviations were calculated from three biological replicates. *p < 0.05, **p < 0.01 represent statistical significance compared with the BY4741T strain
p-Coumaric acid production after overexpression of feedback-resistant mutants of DAHP synthase and chorismate mutase
DAHP synthase, encoded by ARO4, and chorismate mutase, encoded by ARO7, are both feedback inhibited by l-tyrosine. Substitution of the lysine residue 229 by a leucine relieved the inhibiting effect of l-tyrosine, leading to a feedback-resistant DAHP synthase. A single serine-to-glycine substitution in Aro7p at position 114 resulted in a feedback-resistant chorismate mutase (Luttik et al. 2008). To increase the activity of Aro4p and Aro7p, we performed single overexpression of mutated feedback-resistant ARO4K229L, ARO7G141S, and simultaneous overexpression of ARO4K229L and ARO7G141S in background strain NK-L107, resulting in strains NKA2, NKA3, and NKA4, respectively (Fig. 2). By introducing plasmid pLC-m7 into strains NKA2, NKA3, and NKA4, respectively, we obtained three p-coumaric acid production strains NKA2T, NKA3T, and NKA4T.
After confirmation of gene expressions by RT-qPCR (Fig. 3), fermentations were performed. Unlike the reference strain NKA1T, the p-coumaric acid production of all three engineered strains peaked at 120 h (Fig. S4). The strain NKA2T with single overexpression of ARO4K229L produced 6.93 mg L−1 of p-coumaric acid, but the strain NKA3T with overexpression of ARO7G141S produced 2.82 mg L−1. The highest production (7.83 mg L−1) was achieved in the strain NKA4T with the combined overexpression of ARO4K229L and ARO7G141S (Fig. 5). These results showed that elimination of feedback inhibition had a positive effect on p-coumaric acid production, and co-expression of ARO4K229L and ARO7G141S enhanced the yield further. The engineered strains showed a slower growth rate and lower glucose consumption compared with the control strain (Figs. S5 and S6).
Fig. 5.
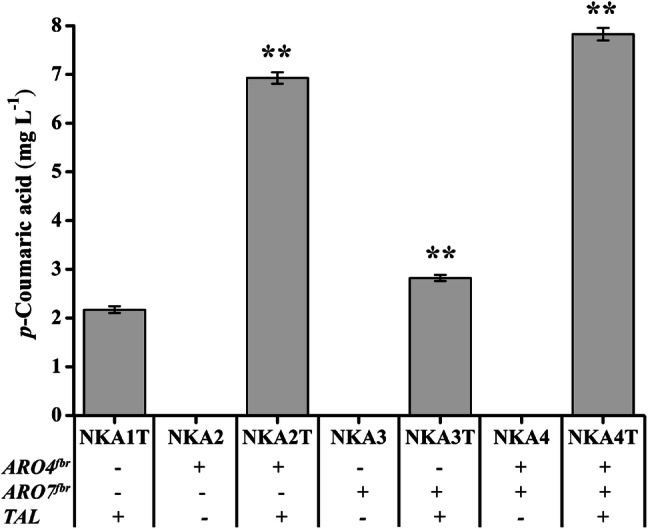
Production of p-coumaric acid upon overexpression of ARO4fbr and ARO7fbr in strains with overexpression of ENO2, TAL1, ARO1 and RcTAL. Strains were cultured in 20 mL of SC-His medium at 30 °C and 200 rpm. p-Coumaric acid of NKA1T strain peaked at 48 h and of NKA2T, NKA3T, and NKA4 peaked at 120 h. Average ± standard deviations were calculated from three biological replicates. *p < 0.05, **p < 0.01 represent statistical significance compared with the NKA1T strain
Effect of elimination of competing phenylpyruvate decarboxylase activity on p-coumaric acid production
To reduce the diversion of carbon flux into the Ehrlich pathway, and further improve p-coumaric acid production, the PDC5 and ARO10 were deleted in background strain NKA4, generating the PDC5 knockout strain NKA5, ARO10 knockout strain NKA6, and double-knockout strain NKA7 (Δpdc5 and Δaro10) (Fig. 2). The three engineered strains were transformed with plasmid pLC-m7, resulting in three p-coumaric acid production strains NKA5T, NKA6T, and NKA7T, respectively.
All three p-coumaric acid production strains had the highest production at 120 h (Fig. S4). The p-coumaric acid production of single-deletion strains NKA5T and NKA6T increased to 11.75 and 13.10 mg L−1, respectively, and were correspondingly 1.50- and 1.67-fold higher than that of the control strain NKA4T. The double deletion of PDC5 and ARO10 resulted in the highest production (19.71 mg L−1), indicating that deletion of the two phenylpyruvate decarboxylase genes PDC5 and ARO10 had a synergetic relationship in improving p-coumaric acid production (Fig. 6). A slight reduction of growth rate and glucose consumption occurred in the engineered strains (Figs. S5 and S6).
Fig. 6.
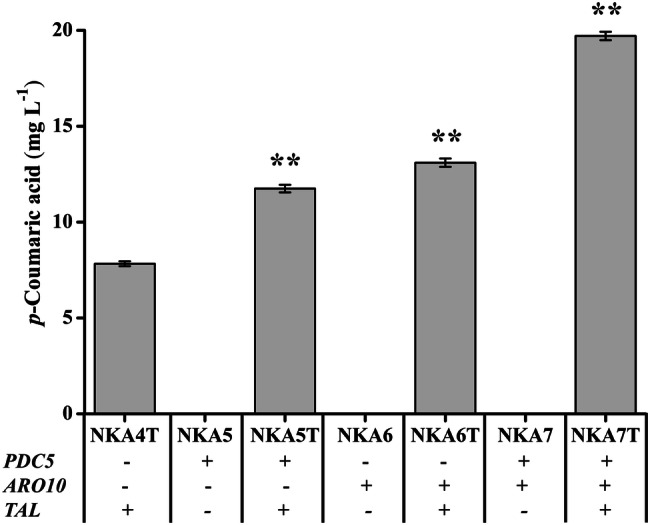
Production of p-coumaric acid upon deletion of PDC5 and ARO10 in strains with overexpression of ENO2, TAL1, ARO1, ARO4fbr, ARO7fbr and RcTAL. Strains were cultured in 20 mL of SC-His medium at 30 °C and 200 rpm, and p-coumaric acid production peaked at 120 h. Average ± standard deviations were calculated from three biological replicates. *p < 0.05, **p < 0.01 represent statistical significance compared with the NKA4T strain
Overexpression of chorismate synthase and prephenate dehydrogenase
To find flux-controlling steps and further improve p-coumaric acid production, TYR1 (prephenate dehydrogenase) and ARO2 (chorismate synthase) were separately overexpressed in background strain NKA7 to yield strains NKA8 and NKA9, accordingly (Fig. 2). Then, plasmid pLC-m7 was transformed into these two strains, generating p-coumaric acid production strains NKA8T and NKA9T. The RT-qPCR assay revealed that the expression levels of ARO2 and TYR1 were significantly improved through genetic modification. However, overexpression of these two enzymes resulted in lower p-coumaric acid production than in the control strain (Fig. 7).
Fig. 7.
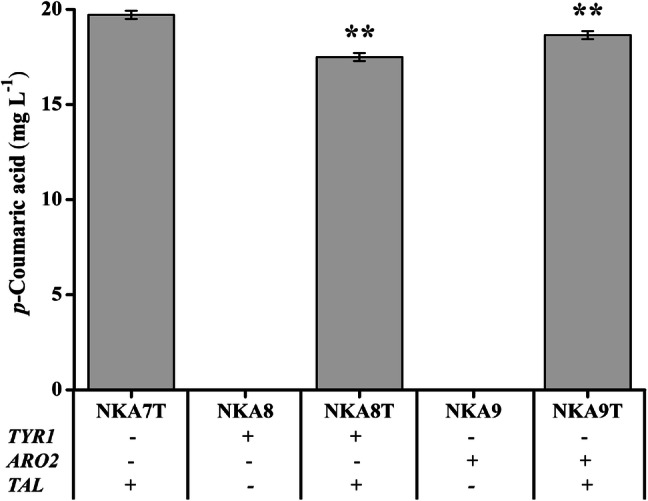
Effect of overexpression of TYR1 or ARO2 on p-coumaric acid production in strains overexpressing ENO2, TAL1, ARO1, ARO4fbr, ARO7fbr, and RcTAL and deletion of PDC5 and ARO10. Strains were cultured in 20 mL of SC-His medium at 30 °C and 200 rpm, and p-coumaric acid production peaked at 120 h. Average ± standard deviations were calculated from three biological replicates. *p < 0.05, **p < 0.01 represent statistical significance compared with the NKA7T strain
Comparison of p-coumaric acid highest-producing strain and wild-type strain
Through optimization of the l-tyrosine pathway above, we obtained strain NKA7T, which was the highest producer of p-coumaric acid. Strain NKA7T had a slightly reduced growth rate in 48 h, but the final OD value of NKA7T was a little higher than that of BY4741T (Fig. 8a). The glucose consumption of NKA7T was slightly lower than that of BY4741T in 12 h and glucose was entirely consumed after 24 h in the fermentation process of NKA7T and BY4741T (Fig. 8b). Production of p-coumaric acid was determined daily, and peaked at 48 h in BY4741T; however, p-coumaric acid concentration kept increasing until day 120 h for NKA7T (Fig. 8c). The p-coumaric acid production of NKA7T reached 19.71 mg L−1, which was 14.08-fold higher than that of the wild-type BY4741T.
Fig. 8.

The comparison of wild-type strain and p-coumaric acid highest-producing strain. Strains were cultured in 20 mL of SC-His medium at 30 °C and 200 rpm for 5 days: a cell growth, b glucose consumption, and c formation of p-coumaric acid. Average ± standard deviations were calculated from three biological replicates. *p < 0.05, **p < 0.01 represent statistical significance
Discussion
We performed a series of genetic modifications to wild-type S. cerevisiae strain BY4741 to direct the carbon flux to l-tyrosine, and then introduced the heterologous TAL as a test of the metabolic engineering targets for improving carbon flux through the l-tyrosine pathway and the strain’s ability to synthesize heterologous metabolites derived from l-tyrosine. Meanwhile, the effect of optimizing l-tyrosine metabolic pathway on strain growth and glucose consumption was analyzed.
The carbon flux to E4P and PEP was optimized by overexpressing TAL1 and ENO2, respectively, and a 1.29-fold improvement of p-coumaric acid was obtained. Overexpression of ARO1 was effective in increasing p-coumaric acid production. This is consistent with the results of Rodriguez et al., in which p-coumaric acid production increased significantly with overexpression of ARO1 (Rodriguez et al. 2015). The overexpression of feedback-resistant ARO4K229L and ARO7G141S had a positive effect on p-coumaric acid production. The overexpression of ARO4K229L led to a 3.19-fold improvement compared to the control strain, but only 1.30-fold production was achieved by overexpressing ARO7G141S. This was expected, because overexpression of feedback-resistant mutant of DAHP synthase was previously reported to have a greater effect on l-tyrosine yield than overexpression of feedback-resistant mutant of chorismate mutase (Luttik et al. 2008). Koopman et al. reported that eliminating feedback inhibition not only increased the production of naringenin, a derivative of l-tyrosine, but also led to increased phenylethanol production, the major byproduct of l-tyrosine biosynthesis (Koopman et al. 2012). Therefore, we made the double deletion of PDC5 and ARO10, and this resulted in strongly increased p-coumaric acid production. This is also supported by research of Koopman et al., who significantly improved production of naringenin by a triple deletion of phenylpyruvate decarboxylases (Koopman et al. 2012).
The purpose of our study was the optimization of the l-tyrosine metabolic pathway in S. cerevisiae. We optimized the shikimate pathway and the carbon flux to E4P and PEP. The simultaneous overexpression of ARO4K229L and ARO7G141S led to a 3.61-fold improvement in p-coumaric acid production, but the same genetic modification by Rodriguez et al. only led to a 1.93-fold improvement (Rodriguez et al. 2015). Production of p-coumaric acid with double deletion of PDC5 and ARO10 showed a 2.52-fold increase in our study, which was higher than the 2.29-fold improvement reported by Rodriguez et al. when using the same genetic modification. Moreover, optimization of the l-tyrosine metabolic pathway in our research finally led to a 14.08-fold increase in p-coumaric acid, which was also higher than 7.90-fold reported by Rodriguez et al. These results might be due to the overexpression of TAL1 and ENO2 in our research, which increased the carbon flux to E4P and PEP, respectively.
The overexpression of TYR1 had a negative effect on p-coumaric acid production, which coincided with the result of Rodriguez et al. (Rodriguez et al. 2015). They also reported that overexpression of ARO1 could significantly improve p-coumaric acid production, but the p-coumaric acid titer of a strain simultaneously overexpressing ARO1 and ARO2 was very similar to that of the strain overexpressing ARO1 alone (Rodriguez et al. 2015). In our research, the combined overexpression of ARO1 and ARO2 produced less p-coumaric acid compared with the strain overexpressing ARO1 alone, which was not consistent with the result of Rodriguez et al. This result may be due to the different background strains we used compared with Rodriguez et al., and, in addition, the two strains were also modified differently. Our background strain was S. cerevisiae BY4741 and the modified strain before ARO2 overexpression was NKA7. However, the background strain used by Rodriguez et al. was S. cerevisiae CEN.PK102-5B and the modified strain was ST3213 (aro10Δ pdc5Δ ARO4K229L ARO7G141S). The reduction of p-coumaric acid production indicated that these two enzymes may not be bottlenecks for the l-tyrosine biosynthesis pathway in the engineered strain NKA7T; consequently, there may be other bottlenecks in the l-tyrosine pathway.
In conclusion, by optimizing supply of precursors, eliminating feedback inhibition and decreasing production of l-tyrosine byproducts, we obtained a series of strains producing p-coumaric acid. Among these strains, NKA7T was the highest producing, and the titer was 14.08-fold higher than that of the BY4741T (Fig. S7). The growth rate and glucose uptake rate of NKA7T showed only slight decreases compared with the wild-type strain. All of the genetic modifications of the l-tyrosine pathway were carried on chromosomes to ensure genetic stability, and selection marker gene was removed using the Cre/lox P system to ensure biosecurity, making this a good platform strain to produce l-tyrosine and research secondary metabolites derived from l-tyrosine.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by the Tianjin Key Research Program of Application Foundation and Advanced Technology [17JCZDJC32200]; the National Natural Science Foundation of China [31770102]; the Sino-Swiss Scientific and Technological Cooperation Project supported by the Ministry of Science and Technology of China [2015DFG32140].
Author contributions
All authors contributed to the conception and design of the study, performing experimental assessments, drafting the articles, and revising it. All authors have contributed, seen and approved the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Contributor Information
Haijin Xu, Email: xuhaijin@aliyun.com.
Mingqiang Qiao, Email: qiaomq@nankai.edu.cn.
References
- Akinwumi BC, Bordun KAM, Anderson HD. Biological activities of stilbenoids. Int J Mol Sci. 2018;19(3):792. doi: 10.3390/Ijms19030792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodina I, Nielsen J. Advances in metabolic engineering of yeast Saccharomyces cerevisiae for production of chemicals. Biotechnol J. 2014;9(5):609–620. doi: 10.1002/biot.201300445. [DOI] [PubMed] [Google Scholar]
- Braus GH. Aromatic amino acid biosynthesis in the yeast Saccharomyces cerevisiae: a model system for the regulation of a eukaryotic biosynthetic pathway. Microbiol Rev. 1991;55(3):349–370. doi: 10.1128/MMBR.55.3.349-370.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo JH, Han C, Lee DW, Sim GH, Moon HY, Kim JY, Song JY, Kang HA. Molecular and functional characterization of two pyruvate decarboxylase genes, PDC1 and PDC5, in the thermotolerant yeast Kluyveromyces marxianus. Appl Microbiol Biotechnol. 2018;102(8):3723–3737. doi: 10.1007/s00253-018-8862-3. [DOI] [PubMed] [Google Scholar]
- Chougule M, Patel AR, Sachdeva P, Jackson T, Singh M. Anticancer activity of Noscapine, an opioid alkaloid in combination with Cisplatin in human non-small cell lung cancer. Lung Cancer. 2011;71(3):271–282. doi: 10.1016/j.lungcan.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnez D, Jeandet P, Clement C, Courot E. Bioproduction of resveratrol and stilbene derivatives by plant cells and microorganisms. Trends Biotechnol. 2009;27(12):706–713. doi: 10.1016/j.tibtech.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Duncan K, Edwards RM, Coggins JR. The Saccharomyces cerevisiae ARO1 gene. An example of the co-ordinate regulation of five enzymes on a single biosynthetic pathway. FEBS Lett. 1988;241(1–2):83–88. doi: 10.1016/0014-5793(88)81036-6. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Method Enzymol. 2002;350:87–96. doi: 10.1016/S0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- Gold ND, Gowen CM, Lussier FX, Cautha SC, Mahadevan R, Martin VJJ. Metabolic engineering of a tyrosine-overproducing yeast platform using targeted metabolomics. Microb Cell Fact. 2015;14:73. doi: 10.1186/s12934-015-0252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham LD, Gillies FM, Coggins JR. Over-expression of the yeast multifunctional arom protein. Biochim Biophys Acta. 1993;1216(3):417–424. doi: 10.1016/0167-4781(93)90009-3. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Schneider TR, Pfeil A, Heinrich G, Lipscomb WN, Braus GH. Evolution of feedback-inhibited beta/alpha barrel isoenzymes by gene duplication and a single mutation. Proc Natl Acad Sci USA. 2003;100(3):862–867. doi: 10.1073/pnas.0337566100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegemann JH, Heick SB. Delete and repeat: a comprehensive toolkit for sequential gene knockout in the budding yeast Saccharomyces cerevisiae. Methods Mol Biol. 2011;765:189–206. doi: 10.1007/978-1-61779-197-0_12. [DOI] [PubMed] [Google Scholar]
- Iraqui I, Vissers S, Cartiaux M, Urrestarazu A. Characterisation of Saccharomyces cerevisiae ARO8 and ARO9 genes encoding aromatic aminotransferases I and II reveals a new aminotransferase subfamily. Mol Gen Genet. 1998;257(2):238–248. doi: 10.1007/s004380050644. [DOI] [PubMed] [Google Scholar]
- Jiang H, Morgan JA. Optimization of an in vivo plant P450 monooxygenase system in Saccharomyces cerevisiae. Biotechnol Bioeng. 2004;85(2):130–137. doi: 10.1002/bit.10867. [DOI] [PubMed] [Google Scholar]
- Karsten WE, Reyes ZL, Bobyk KD, Cook PF, Chooback L. Mechanism of the aromatic aminotransferase encoded by the Aro8 gene from Saccharomyces cerevisiae. Arch Biochem Biophys. 2011;516(1):67–74. doi: 10.1016/j.abb.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Koopman F, Beekwilder J, Crimi B, van Houwelingen A, Hall RD, Bosch D, van Maris AJA, Pronk JT, Daran JM. De novo production of the flavonoid naringenin in engineered Saccharomyces cerevisiae. Microb Cell Fact. 2012;11:155. doi: 10.1186/1475-2859-11-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivoruchko A, Nielsen J. Production of natural products through metabolic engineering of Saccharomyces cerevisiae. Curr Opin Biotechnol. 2015;35:7–15. doi: 10.1016/j.copbio.2014.12.004. [DOI] [PubMed] [Google Scholar]
- Kyndt JA, Meyer TE, Cusanovich MA, Van Beeumen JJ. Characterization of a bacterial tyrosine ammonia lyase, a biosynthetic enzyme for the photoactive yellow protein. FEBS Lett. 2002;512(1–3):240–244. doi: 10.1016/S0014-5793(02)02272-X. [DOI] [PubMed] [Google Scholar]
- Li MJ, Kildegaard KR, Chen Y, Rodriguez A, Borodina I, Nielsen J. De novo production of resveratrol from glucose or ethanol by engineered Saccharomyces cerevisiae. Metab Eng. 2015;32:1–11. doi: 10.1016/j.ymben.2015.08.007. [DOI] [PubMed] [Google Scholar]
- Liu D, Li BZ, Liu H, Guo XJ, Yuan YJ. Profiling influences of gene overexpression on heterologous resveratrol production in Saccharomyces cerevisiae. Front Chem Sci Eng. 2017;11(1):117–125. doi: 10.1016/j.meteno.2015.09.001. [DOI] [Google Scholar]
- Liu QL, Liu HJ, Yang YY, Zhang XM, Bai YL, Qiao MQ, Xu HJ. Scarless gene deletion using mazF as a new counter-selection marker and an improved deletion cassette assembly method in Saccharomyces cerevisiae. J Gen Appl Microbiol. 2014;60(2):89–93. doi: 10.2323/jgam.60.89. [DOI] [PubMed] [Google Scholar]
- Luttik MA, Vuralhan Z, Suir E, Braus GH, Pronk JT, Daran JM. Alleviation of feedback inhibition in Saccharomyces cerevisiae aromatic amino acid biosynthesis: quantification of metabolic impact. Metab Eng. 2008;10(3–4):141–153. doi: 10.1016/j.ymben.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Mao JW, Liu QL, Song XF, Wang HSY, Feng H, Xu HJ, Qiao MQ. Combinatorial analysis of enzymatic bottlenecks of l-tyrosine pathway by p-coumaric acid production in Saccharomyces cerevisiae. Biotechnol Lett. 2017;39(7):977–982. doi: 10.1007/s10529-017-2322-5. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Chaleckis R, Takaine M, Wheelock CE, Yoshida S. Kynurenine aminotransferase activity of Aro8/Aro9 engage tryptophan degradation by producing kynurenic acid in Saccharomyces cerevisiae. Sci Rep. 2017;7(1):12180. doi: 10.1038/s41598-017-12392-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravicini G, Mosch HU, Schmidheini T, Braus G. The general control activator protein GCN4 is essential for a basal level of ARO3 gene expression in Saccharomyces cerevisiae. Mol Cell Biol. 1989;9(1):144–151. doi: 10.1128/MCB.9.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Kildegaard KR, Li MJ, Borodina I, Nielsen J. Establishment of a yeast platform strain for production of p-coumaric acid through metabolic engineering of aromatic amino acid biosynthesis. Metab Eng. 2015;31:181–188. doi: 10.1016/j.ymben.2015.08.003. [DOI] [PubMed] [Google Scholar]
- Romagnoli G, Knijnenburg TA, Liti G, Louis EJ, Pronk JT, Daran JM. Deletion of the Saccharomyces cerevisiae ARO8 gene, encoding an aromatic amino acid transaminase, enhances phenylethanol production from glucose. Yeast. 2015;32(1):29–45. doi: 10.1002/yea.3015. [DOI] [PubMed] [Google Scholar]
- Sato F, Inui T, Takemura T. Metabolic engineering in isoquinoline alkaloid biosynthesis. Curr Pharm Biotechnol. 2007;8(4):211–218. doi: 10.2174/138920107781387438. [DOI] [PubMed] [Google Scholar]
- Sauer B. Functional expression of the cre-lox site-specific recombination system in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1987;7(6):2087–2096. doi: 10.1128/MCB.7.6.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva S, Costa EM, Calhau C, Morais RM, Pintado ME. Anthocyanin extraction from plant tissues: a review. Crit Rev Food Sci Nutr. 2017;57(14):3072–3083. doi: 10.1080/10408398.2015.1087963. [DOI] [PubMed] [Google Scholar]
- Strucko T, Magdenoska O, Mortensen UH. Benchmarking two commonly used Saccharomyces cerevisiae strains for heterologous vanillin-β-glucoside production. Metab Eng Commun. 2015;2:99–108. doi: 10.1016/j.meteno.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuralhan Z, Luttik MA, Tai SL, Boer VM, Morais MA, Schipper D, Almering MJ, Kotter P, Dickinson JR, Daran JM, Pronk JT. Physiological characterization of the ARO10-dependent, broad-substrate-specificity 2-oxo acid decarboxylase activity of Saccharomyces cerevisiae. Appl Environ Microbiol. 2005;71(6):3276–3284. doi: 10.1128/AEM.71.6.3276-3284.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Bhan N, Koffas MAG. Engineering plant metabolism into microbes: from systems biology to synthetic biology. Curr Opin Biotech. 2013;24(2):291–299. doi: 10.1016/j.copbio.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Yao LH, Jiang YM, Shi J, Tomas-Barberan FA, Datta N, Singanusong R, Chen SS. Flavonoids in food and their health benefits. Plant Food Hum Nutr. 2004;59(3):113–122. doi: 10.1007/s11130-004-0049-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.