

Abstract
AMP-activated protein kinase (AMPK) is essential for maintaining energy balance and has a crucial role in various inflammatory pathways. In this study, AMPK levels positively correlated with many inflammatory indexes in rheumatoid arthritis (RA) patients, especially in the affected synovium. In RA sera, a positive correlation between phosphorylated (p-)AMPK-α1 levels and DAS28 (disease activity score 28) activity (r = 0.270, p < 0.0001) was found. Similarly, a positive correlation was observed between AMPK-α1 and tumor necrosis factor α (TNF-α) levels (r = 0.460, p = 0.0002). Differentially expressed genes between osteoarthritis (OA) and RA synovium from NCBI GEO profiles and our RNA sequencing data suggested activation of metabolic pathways specific to RA-fibroblast-like synoviocytes (FLSs). AMPK-α1 was highly expressed in the synovium of RA but not OA patients. An AMPK activator, metformin, inhibited FLS proliferation at higher but not lower concentrations, whereas the inhibitor dorsomorphin promoted the proliferation of RA-FLSs. Interestingly, both metformin and dorsomorphin inhibited the migration of RA-FLSs. After metformin treatment, expression of interleukin 6 (IL-6), TNF-α, and IL-1β were significantly downregulated in RA-FLSs; however, increased expression of p-AMPK-α1, protein kinase A (PKA)-α1, and HAPLN1 (hyaluronan and proteoglycan link protein 1) was observed. Increased levels of HAPLN1 in RA-FLSs by an AMPK activator could potentially be beneficial in protecting the joints. Hence, our results demonstrate the potential of an AMPK activator as a therapeutic for RA.
Keywords: rheumatoid arthritis, disease activity, metabolism, AMP-activated protein kinase, fibroblast-like synoviocyte, metformin, hyaluronan and proteoglycan link protein 1, therapeutic target
Graphical Abstract
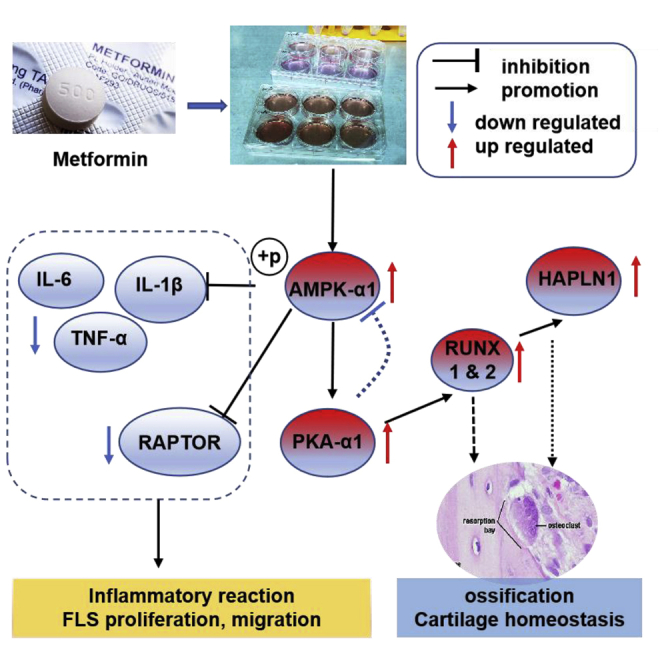
AMPK levels positively correlated with inflammatory indexes in RA. Differentially expressed genes in the synovium indicate activation of FLS-specific metabolic pathways. An AMPK activator, metformin, has concentration-dependent effects on FLS proliferation. Metformin treatment significantly downregulated pro-inflammatory cytokines in FLS but upregulated HAPLN1 expression, which could potentially protect the joints.
Introduction
Rheumatoid arthritis (RA) is a chronic, systemic autoimmune disease affecting joints, which leads to pain, swelling, and deformity, causing considerable morbidity and mortality worldwide.1,2 During RA development the synovium transforms from a quiescent state with a relatively acellular structure to a hyperplastic and invasive state infiltrated with many inflammatory cells. In this context, fibroblast-like synoviocytes (FLSs) are increasingly recognized as key effector cells.3 The activated FLSs are hyperproliferative and resistant to apoptosis with an increased capacity for migration, causing pannus formation and destruction of joint cartilage. Production of inflammatory factors such as tumor necrosis factor α (TNF-α) and interleukin 17 (IL-17), chemokines, and matrix-degrading molecules by FLSs contributes to their invasiveness.4 Additionally, RA-FLSs are considered to form an inflammatory memory response, accounting for disease flare-ups.5 Thus, FLSs could be a potential therapeutic target in RA.
Growing evidence shows the importance of metabolic variations in the pathogenesis of several autoimmune diseases. Inflammation-associated macrophages and T helper 17 (Th17) cells display a shift toward enhanced glucose uptake, glycolysis, and use of the pentose phosphate pathway. In contrast, anti-inflammatory cells such as M2 macrophages, regulatory T cells (Tregs), and quiescent memory T cells exhibit lower glycolytic rates but with higher levels of oxidative metabolism.6 RA patients were reported to have an increased risk for insulin resistance. The glucose and lipid metabolic disturbances are strongly associated with the degree of systemic inflammation.7 Under pro-inflammatory conditions, glycolysis is activated in RA-FLSs and blocking it might be beneficial to attenuate inflammation.8 Metformin, an AMP-activated protein kinase (AMPK) activator, inhibited experimental arthritis through reciprocal regulation of the Treg/Th17 cell balance.9,10 Similarly, metformin inhibited the growth of cancer cells in vitro;11 however, the action of metformin on RA-FLSs is not yet clear.
AMPK is a highly conserved metabolic fuel gauge present in eukaryotes, which senses changes in the intracellular AMP/ATP ratio in response to energy deprivation by regulating mitochondrial biogenesis.12 AMPK plays an important role in several inflammatory pathways. It controls transcriptional regulation of autophagy and lysosomal genes,13 promotes autophagy through phosphorylation of Unc-51 such as autophagy-activating serine/threonine protein kinase Ulk1,14 attenuates CD40-mediated inflammatory activities and Toll-like receptor (TLR)-induced inflammatory functions, and decreases the capacity of antigen-presenting cells.15 Activation of AMPK alters metabolism and pathogenic pathways, which may promote or inhibit inflammation.16
Herein, we report a positive correlation between AMPK levels and disease activity in RA patients. An AMPK activator, metformin, inhibited proliferation, migration, and activation of RA-FLSs. Metformin treatment inhibited the expression of pro-inflammatory cytokines IL-6, TNF-α, and IL-1β, while increasing the phosphorylated (p-)AMPK-α1, protein kinase A α1 (PKA-α1), and hyaluronan and proteoglycan link protein 1 (HAPLN1) expression. Such increased expression of HAPLN1 in RA-FLSs might potentially be beneficial in protecting joints.
Results
Serum p-AMPK-α1 Levels Are Positively Correlated with Disease Activity in RA
To investigate the difference in AMPK levels between RA and osteoarthritis (OA) patients and to detect the presence of correlation between AMPK levels and RA disease activity, we determined AMPK-α1 and p-AMPK-α1 levels using 20 OA and 61 RA patients having different disease activities. No significant differences exist in AMPK-α1 levels between OA and RA patients (Figure 1A). However, p-AMPK-α1 levels were higher in OA compared to RA patients, who had lower disease activity (p = 0.002) (Figure 1B). This significance level was increased when the values for p-AMPK-α1 levels were log transformed to get a normal distribution (p < 0.0001). Interestingly, after log transformation, RA patients having high disease activity had significantly higher levels of p-AMPK-α1 compared to patients having low disease activity (p = 0.007) (Figure 1C). In addition, p-AMPK-α1 levels were positively correlated with DAS28 (disease activity score 28) scores (r = 0.270, 95% confidence interval [CI]: 0.142–0.492, p < 0.0001) and C-reactive protein (CRP) levels (r = 0.259, 95% CI: 0.009–0.478, p = 0.042) (Figures 1D and 1E). However, such a correlation did not exist with erythrocyte sedimentation rate (ESR) levels (Figure 1F).
Figure 1.

Expression of Serum AMPK-α1 and p-AMPK-α1 Levels in RA and OA Patients
(A) AMPK-α1 levels between RA (n = 61) and OA (n = 20) patients having moderate to high disease activity were shown, but no statistical significance between groups was observed. (B) Serum p-AMPK-α1 level was higher in OA compared to RA patients having lower disease activity. Log-transformed p-AMPK-α1 values were significantly higher in OA than RA patients having lower disease activity. (C) In RA, patients with higher disease activity had significantly higher levels of log-transformed p-AMPK-α1 levels compared to patients with lower disease activity. (D and E) Log p-AMPK-α1 levels significantly correlated with DAS28 (D) and CRP (E) levels. (F) Correlation with ESR levels was not statistically significant. (G and H) Increased expression of IL-17 (G) and TNF-α (H) was observed in RA than OA patients. (I) Levels of AMPK-α1 moderately correlated with TNF-α levels. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
IL-17 and TNF-α levels were reported to have a correlation with RA activity and thus were currently selected as therapy targets.17,18 We analyzed the presence of possible correlation between AMPK levels with IL-17 and TNF-α in the serum samples. The results demonstrated an increased expression of both of the inflammatory cytokines in RA patients compared with OA patients, and AMPK-α1 levels were moderately correlated with TNF-α levels (r = 0.46, 95% CI: 0.241–0.640, p = 0.0002) (Figures 1G–1I). However, no correlation exists between log-transformed p-AMPK-α1 and IL-17 or TNF-α levels (Figure S1). Although AMPK is a key regulator of glucose metabolism,19 and RA and other autoimmune diseases are associated with an increased risk of diabetes mellitus,20 no differences in glucose levels between groups were observed in our study (Figure S1).
AMPK Levels Were More Significantly Present in RA Synovial Samples
Since AMPK has an anti-inflammatory effect in many inflammation-related diseases,21,22 and our data showed a mild positive correlation with disease activity, we next evaluated the data available in NCBI GEO profiles (GEO: GSE12021, GSE55235,23 and GSE5545724) for AMPK levels from RA and OA patients’ synovial tissue samples. In total, 119 differentially expressed genes (DEGs) intersected in all three datasets (Data S1), which demonstrated the high consensus existing between results from different experiments. Although AMPK was not directly detected in the analysis, one of the most obviously changed pathways was the metabolic pathway consisting of nine genes (Figure 2A). Since AMPK has a controlling function in metabolism,13,25 we focused on its expression at both protein and mRNA levels in the synovium samples from OA and RA patients. The immunohistochemical (IHC) staining revealed a higher level of p-AMPK-α1 expression in RA than in OA synovium (Figure 2B). Similarly, relative expression levels of AMPK-α1, AMPK-α2, and AMPK-γ3 genes were higher in synovium of RA than OA patients (Figure 2C).
Figure 2.

Changes in the Metabolic Pathway in RA Compared to OA Synovium
(A) In total, 119 DEGs between RA and OA patients’ synovium intersected in all the three datasets (NCBI GEO profiles; GEO: GSE12021, GSE55235, and GSE55457). Among them, changes in the metabolic pathway-related nine genes were more obvious. (B) IHC staining for p-AMPK-α1 was more intense in RA (n = 20) than in OA (n = 17) synovium. (C) Relative expression levels of AMPK-α1, AMPK-α2, and AMPK-γ3 genes were higher in RA (n = 10) than in OA (n = 9) synovium. ∗p < 0.05.
Metabolism Variations in FLSs
Glucose and phospholipid metabolism as well as bioactive lipids are involved in the activation of FLSs. These metabolic changes contribute to FLS activation and invasive potential, leading to joint destruction.26 p-AMPK-α1 expressed in the proliferating FLSs was identified using IHC staining and confirmed by immunofluorescence studies (Figure S2). Since FLS activation is crucial in RA pathogenesis,27 we analyzed the differences in the expression of genes between RA and OA FLSs using a high-throughput RNA sequencing method. We identified 111 DEGs, and the expression levels of 95 genes were upregulated and 16 genes were downregulated in RA compared with OA FLSs (Figure 3A; for all DEGs present in the individual samples, see Data S2). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis suggested involvement of metabolic and glycerolipid metabolism pathways with nine genes closely related to AMPK regulation (Figures 3A and 3B). Among them, diacylglycerol kinase gamma (DGKG) and prostaglandin D2 synthase (PTGDS) expressions were downregulated, while the expression levels of the other seven genes, that is, lipase G (LIPG), heparanase (HPSE), glycerol-3-phosphate acyltransferase 2 (GPAT2), phospholipase A2 group VII (PLA2G7), choline dehydrogenase (CHDH), ST6 β-galactoside α-2,6-sialyltransferase 2 (ST6GAL2), and ST8 α-N-acetyl-neuraminide α-2,8-sialyltransferase 5 (ST8SIA5), were upregulated in RA compared to OA FLSs. Interestingly, HAPLN1 expression was highly upregulated in RA-FLSs (Figure 3A).
Figure 3.

DEGs in FLSs Identified by mRNA High-Throughput Sequencing Method
(A) In total, 111 DEGs were identified in RA-FLSs compared to OA samples (n = 3/group). Expression of 95 genes was upregulated compared to the downregulation in the expression of 16 genes. Of all of the nine DEGs related to metabolism, expression levels of DGKG and PTGDS genes were downregulated, while the expression of LIPG, HPSE, GPAT2, PLA2G7, CHDH, ST6GAL2, and ST8SIA5 genes were upregulated in RA-FLSs compared to OA samples. HAPLN1 expression was most obviously upregulated in RA-FLSs. (B) KEGG pathway enrichment analysis demonstrated that the metabolic pathway is one of the most obviously changed pathways.
Metformin Affects FLS Proliferation
Based on our above results and prior knowledge on anti-inflammatory functions of AMPK,14,15 we deduced that upregulation of AMPK-α1 expression in RA synovium might be due to the ongoing inflammation stress in the joints. So, we used metformin28 and dorsomorphin29 as an AMPK activator and inhibitor, respectively, to evaluate their effects on FLS proliferation.
FLSs were cultured with different concentrations of metformin in six-well plates and observed between 0 and 72 h under an inverted microscope for their viability. A 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide (MTT) assay was performed to confirm the cellular viability of FLSs after metformin treatment. The results demonstrated inhibition of FLS proliferation by metformin at 5 and 10 mM concentrations. However, at a lower concentration, metformin promoted FLS proliferation (Figure 4A), which was further confirmed by reducing the concentration (Figure 4B), demonstrating a dose-dependent effect of metformin on FLS proliferation. In contrast, dorsomorphin promoted FLS proliferation significantly at a 5–10 μM concentration (Figure 4C).
Figure 4.

Effect of AMPK Modulators on RA-FLS Proliferation
(A) An MTT assay was used to determine the effects of metformin on RA-FLS viability. Metformin inhibited RA-FLS proliferation at 5 and 10 mM, whereas at 1 mM it has an opposite effect compared to the control group (n = 5). (B) This result was confirmed by using even lower concentration of metformin (n = 5). (C) Dorsomorphin promoted FLS proliferation significantly at 5 and 10 μM compared to the control group after culturing for 36 h (n = 5). A representative example from three independent experiments is shown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Metformin Inhibited FLS Migration
Based on the concentration gradient of metformin for its inhibitory effects on FLS proliferation, we selected metformin and dorsomorphin at a 5 μM concentration to study their effect on FLS migration by testing wound repair rate in a scratch test and transwell assay. Interestingly, both metformin and dorsomorphin inhibited FLS migration significantly (Figures 5A and 5B). Earlier, dorsomorphin was reported to inhibit the migration of certain cancer cells, and this phenomenon was explained by AMPK-independent mechanisms.29
Figure 5.

AMPK Modulators Inhibited Migration of RA-FLSs
(A) A scratch test was performed to evaluate the effects of metformin and dorsomorphin on FLS migration, represented as scratch repair rate (%). Both the AMPK activator (metformin) and inhibitor (dorsomorphin) inhibited FLS migration significantly (n = 3). (B) A transwell assay confirmed the effects of metformin and dorsomorphin on FLS migration after treatment for 24 h (n = 3). A representative example from three independent experiments is shown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Metformin Inhibited Inflammatory Cytokines by Promoting AMPK Levels
Results from semi-quantification of mRNA levels in FLSs after metformin treatment by qRT-PCR showed a significant increase in AMPK-α1 levels, which was confirmed by western blot (Figures 6A and 6D). Similarly, the mRNA levels of PKA-α1 were also found to be increased after metformin treatment. PKA-α is a regulatory subunit of the cyclic AMP (cAMP)-dependent protein kinases involved in cAMP-mediated signaling events in the cells, and a mutual promotion effect between AMPK and PKA-α was reported earlier.29 AMPK also phosphorylates the mammalian target of rapamycin complex 1 (mTORC1) subunit, regulatory associated protein of mTOR (RAPTOR), which is essential for AMPK function as a metabolic checkpoint.30 The AMPK-mTOR (RAPTOR) pathway was earlier reported to regulate cell growth in response to nutrient and insulin levels.31 Although RAPTOR and mTOR did not have any significant changes after metformin treatment at mRNA levels, a negative correlation between RAPTOR and AMPK-α1 expression was detected (r = −0.470, 95% CI: −0.682 to −0.185, p = 0.002, n = 40) (Figure 6B). Western blot results confirmed the downregulated RAPTOR levels in FLSs after metformin treatment.
Figure 6.

Metformin Inhibited Inflammatory Cytokines by Promoting AMPK
Expression levels of inflammatory cytokines were measured after culturing RA-FLSs with normal saline or metformin for 36 h (n = 3). (A) Compared to controls, expression levels of AMPK-α1 and PKA-α1 genes were significantly increased while the expression of IL-6 was significantly decreased in metformin-treated RA-FLSs. (B and C) Negative correlation of AMPK-α1 with RAPTOR (B) and IL-6 (C) gene expression. (D) Western blot analysis of IL-1β and TNF-α levels. (E) Negative correlation between p-AMPK-α1 and TNF-α levels. A representative example from three independent experiments is shown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
The role of IL-6 in the pathogenesis of joint and systemic inflammation in RA has been clearly demonstrated,32 and IL-6 inhibitor has been used for the treatment of RA with favorable outcomes.33 Our results confirmed AMPK-dependent effects of metformin on IL-6 gene expression,34,35 and a significant negative correlation between AMPK-α1 and IL-6 gene expressions was observed (r = −0.422, 95% CI: −0.672 to −0.0865, p = 0.016, n = 32) (Figures 6A and 6C). Similarly, upon metformin treatment in RA-FLSs, IL-1β was found to be downregulated, and the TNF-α level had a significant negative correlation (r = −0.825, 95% CI: −0.967 to −0.288, p = 0.012, n = 8) with p-AMPK-α1 level (Figures 6D and 6E).
Metformin Promoted HAPLN1 Expression in FLSs
Interestingly, after treating FLSs with metformin, an upregulation of mRNA and protein levels of HAPLN1 expression was observed (Figures 7A and 7B). HAPLN1 mRNA was significantly positively correlated with AMPK-α1 mRNA (r = 0.560, 95% CI: 0.308–0.738, p < 0.0001, n = 32, Figure 7C) as well as at protein levels (r = 0.565, 95% CI: 0.139–0.789, p = 0.007, n = 19, Figure 7D). HAPLN1 was reported as one of the distinctive genes expressed in RA-FLSs correlating with the disease activity.36 However, the effects of metformin on AMPK-α1 expression and subsequent modulation of HAPLN1 have not been reported earlier.
Figure 7.

Metformin Promoted HAPLN1 in the PKA-α1/RUNX1/RUNX2 Pathway
Relative HAPLN1 expression levels were measured after culturing RA-FLSs with normal saline or metformin for 36 h (n = 3). (A and B) HAPLN1 was increased significantly at both mRNA (A) and protein (B) levels, whereas RUNX1 and RUNX2 were not regulated upon AMPK activation. (C and D) Positive correlation between HAPLN1 and AMPK-α1 at both mRNA (C) and protein (D) levels. (E and F) Positive correlation of HAPLN1 with PKA-α1 (E) and RUNX1 (F) genes. A representative example from three independent experiments is shown. ∗p < 0.05.
In granulosa cells, HAPLN1 was proposed to be promoted through the PKA-RUNX1/RUNX2 pathway.37 In our study, PKA-α1, RUNX1, and RUNX2 expressions were significantly upregulated in metformin treated FLSs (Figure 7A). HAPLN1 showed a positive correlation with PKA-α1 (r = 0.616, 95% CI: 1.528–3.638, p < 0.006, n = 40, Figure 7E) and RUNX2 (r = 0.547, 95% CI: 0.291–0.729, p < 0.0002, n = 40, Figure 7F). Note that the RUNX family is essential for the maturation of osteoblasts and it has an important role in the intra-membranous and endochondral ossification processes.38,39
Discussion
AMPK is one of the most important energy sensors identified so far that can regulate the cellular metabolism. Similarly, AMPK inhibits a broad range of inflammatory reactions through multiple biological pathways by regulating immune cell metabolism and functions.40,41 In our current study, no obvious differences in the serum levels of AMPK-α1 and p-AMPK-α1 between OA and RA patients were observed. However, AMPK-α1 and log(p-AMPK-α1) levels were moderately positively correlated with disease activity in RA patients. Moreover, p-AMPK-α1 expression was higher in RA than in OA synovium samples. However, no big differences in AMPK or p-AMPK levels between OA and RA patients’ sera were observed, except in OA and low-activity RA patients. We suppose that insufficient inflammation in the joints has failed to cause big changes in the sera, which also led to no obvious differences in serum glucose levels between the groups. At the same time, this might also be due to the number of samples used in this study. Alternatively, in the synovium of joints where all the inflammatory reactions occur,42 an upregulation of AMPK is more clearly distinct, although a cause-and-effect relationship between AMPK and RA is not yet clear. Possibly, inflammation-induced stress in the synovium could lead to an increase in AMPK secretion level.
Although metformin as an AMPK activator had inhibitory potential at higher concentrations, it promoted FLS proliferation at lower concentrations in our study, which is in disagreement with the study of Chen et al.,43 who had reported inhibitory capacity of metformin even at very low concentrations (5–60 μM). Since the dose differences used in both of the studies were very high, a difference in metformin preparations obtained from different manufacturers may not be the major cause. It is plausible that differences in the source of FLSs, patient characteristics, genetic variations, and treatment conditions could have contributed to these contradictory observations. Most publications reported the inhibitory effect of metformin when mainly using cancer cells.44,45 An analogy to our observation is the use of methotrexate or radiation, which can be used to treat cancer while being carcinogenic. In fact, pharmacological concentrations of metformin at low or high doses affect cells by different mechanisms.46 At a higher concentration (5 mM), it was proposed to inhibit the respiratory chain complex 1 in intact hepatocytes, causing an increase in the AMP/ATP ratio.47 However, more studies are needed to clarify this issue.
Our results are in accordance with metformin effects on the activation of AMPK and in attenuating inflammation.48 We have observed downregulation of IL-6 gene expression, as well as TNF-α and IL-1β protein levels, after metformin treatment. In collagen antibody-induced arthritis (CAIA), an animal model to study the effector phase of RA,49 metformin treatment significantly improved arthritis score, with reduced bone destruction, inflammatory cytokines production, and retinoic acid-related orphan receptor γt (RORγt)-expressing T cells associated with the AMPK/mTOR-mediated inhibition of signal transducer and activator of transcription 3 (STAT3) signaling.10 This favorable AMPK/mTOR/STAT3-dependent effect of metformin on Th17-mediated inflammation was confirmed in collagen-induced arthritis (CIA), the classical model of RA.9 IL-6 is important for both joint destruction and systemic disease manifestations.50 Several studies demonstrated that AMPK activation suppresses nuclear factor κB (NF-κB) signaling through its downstream mediators SIRT1, forkhead box O (FoxO) family, and peroxisome proliferator-activated receptor γ co-activator 1α,51 which in turn could suppress the expression of NF-κB-dependent inflammatory factors such as IL-6, IL-1β and TNF-α. Earlier it was shown that in macrophage cell lines, constitutively expressed AMPK reduced LPS-stimulated IκBα (inhibitor of NF-κB) degradation and p65 binding to the IL-6 promoter.52 Hence, metformin effects on the expression of inflammatory cytokines might be the inhibitory molecular mechanism of metformin on the activation, proliferation, and migration of RA-FLSs.
Our RNA sequencing results demonstrated an upregulation of HAPLN1 expression in RA-FLSs, which mimics the condition of cancer cells in proliferation and migration, leading to cartilage destruction.27 HAPLN1 is considered as a oncogene, and it is significantly elevated at both the RNA and protein levels in mesenchymoma tissues.53 HAPLN1 reappears in aggressive hepatocytes expressing cytoplasmic β-catenin and stem cell markers, and it is associated with poor disease outcomes.54 HAPLN1 is also a susceptibility gene for lung cancer.55 In RA, Urano et al.56 studied the correlation between HAPLN1 gene polymorphism and spinal degenerative changes in 622 post-menopausal Japanese women and showed that mutations in the specific HAPLN1 loci are related to spinal degeneration. Furthermore, genome-wide association analysis reported HAPLN1 as one of the important susceptibility genes for ankylosing spondylitis in the Han population.57 In RA-FLSs, expression of the HAPLN1 gene was positively correlating with CRP that reflects disease activity.36 This evidence led us to postulate HAPLN1 as a pathogenic factor in RA-FLSs. In addition, PKA-α, which exerts a negative effect on AMPK activation,58 was found to be upregulated by metformin treatment. Interestingly, we have identified upregulation of HAPLN1 expression on AMPK activation, which showed a positive correlation with AMPK-α1 and PKA-α1. HAPLN1 interacts with the globular domains of hyaluronic acid and proteoglycans to form stable ternary complexes in various extracellular matrices. Its main biological function is to maintain the stable aggregation of hyaluronic acid and proteoglycan monomers in the extracellular cartilage matrix,53 leading to stabilization of the binding interactions between hyaluronic acid and chondroitin sulfate, which contribute to the compression resistance in the joints.56 Perinatal mice containing targeted mutations in the HAPLN1 gene developed lethal cartilage dysplasia, suggesting the essential role of HAPLN1 as a regulator of cartilage homeostasis and formation.59 Based on the anti-inflammatory effects of AMPK, and its positive correlation with HAPLN1, together with the critical role of HAPLN1 reported in cartilage homeostasis,37 we propose the AMPK-HAPLN1 axis as a possible pathway for metformin functions on FLSs, which could benefit RA patients.
In summary, metformin as the AMPK activator inhibited proliferation and migration of RA-FLSs. Metformin also affected the expression of many inflammatory and metabolic molecules. Surprisingly, at lower concentrations, metformin increased the proliferation of RA-FLSs. Considering the dosage used in the in vitro studies, it is most likely that metformin may not be used as an effective drug for the treatment of RA patients. However, it is clear that the AMPK pathway is a valuable target for treatment for RA, and developing new drugs targeting AMPK activation might be more beneficial for protecting the joints in RA patients. In addition, increased HAPLN1 levels after AMPK activation observed in our study might open up a new research direction to find drugs to treat RA, which needs to be addressed with more functional experiments with HAPLN1.
Materials and Methods
Ethics Approval
This study was approved by the Ethics Committee of Integrated Traditional Chinese and Western Medicine Hospital, Southern Medical University, China (approval no. NFZXYEC-2017-002). Informed consent was obtained from all participants in accordance with the Declaration of Helsinki.
Patients’ Samples
We enrolled 61 RA patients with comprehensive medical records from the Rheumatology Department, Integrated Traditional Chinese and Western Medicine Hospital, Southern Medical University, China, between October 2017 to December 2018, including 17 males and 44 females. The disease activity was defined by the DAS28-ESR scores: DAS28-ESR > 5.1, high activity; 3.2 < DAS28 ≤ 5.1, moderate activity; and DAS28 ≤ 3.2, low activity. Twenty OA patients were enrolled as the control group. Patients with other autoimmune diseases, acute inflammation, fever, thyroid disease, diabetes, and liver and kidney diseases were excluded from the study. Patients’ sera were collected for ELISA assays. Patients’ general information and laboratory tests, including DAS28, ESR, CRP, and blood glucose, were collected from the hospital information system (HIS). General information on patients recruited is given in Table 1.
Table 1.
General Information of Patients Recruited or This Study
| Patients’ Details, Who Donated Sera | ||||
|---|---|---|---|---|
| General Information | RA (n = 61) |
OA (n = 20) | ||
| High Activity (n = 22) | Moderate Activity (n = 20) | Low Activity (n = 19) | ||
| Male/female | 7:14 | 5:15 | 5:14 | 8:12 |
| DAS28-ESR | 5.58 ± 0.50∗∗∗∗ | 4.24 ± 0.55∗∗∗∗ | 2.44 ± 0.43∗∗∗∗ | NA |
| Age (years) | 52.3 ± 12.5 | 48.5 ± 14.4 | 52.2 ± 12.2 | 64.2 ± 13.7 |
| Patients’ Details, Who Donated Synovium for qPCR Studies | ||||
| General Information | RA (n = 10) | OA (n = 9) | ||
| Male/female | 2:8 | 5:4 | ||
| Age (years) | 54.2 ± 8.65 | 66.6 ± 8.61∗∗ | ||
| ESR (mm/h) | 86.5 ± 50.5 | 37.8 ± 29.3∗∗ | ||
| CRP (mg/L) | 19.1 ± 13.63 | 5.13 ± 7.19∗ | ||
| Patients’ Details, Who Donated Synovium for IHC Staining Studies | ||||
| General Information | RA (n = 20) | OA (n = 17) | ||
| Male/female | 8:12 | 5:12 | ||
| Age (years) | 50.5 ± 13.5 | 60.6 ± 8.9∗∗∗ | ||
| ESR (mm/h) | 79.4 ± 46.9 | 39.4 ± 26.1∗∗∗ | ||
| CRP (mg/L) | 27.3 ± 4.70 | 8.70 ± 4.30∗ | ||
NA, not applicable. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Enzyme-Linked Immunosorbent Assay
All blood samples were centrifuged after standing at room temperature for 2 h at 1,500 rpm for 10 min. The serum was collected with 1.5-mL Eppendorf (EP) tubes. AMPK-α1, p-AMPK-α1, TNF-α, and IL-17 levels were analyzed using enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s protocols (R&D Systems, USA). The ELx808 absorbance microplate reader was used to read the optical density (OD) value at 450 nm. The samples’ concentrations were calculated based on the standard curve.
Analysis of Differentially Expressed Genes between RA and OA Synovium
We analyzed the data of synovial tissues from RA and OA patients obtained from NCBI GEO profiles (GEO: GSE12021,23 GSE55235, and GSE5545724). DEGs were selected based on the results having multiples of difference (|log2fold change [FC]| > 1) and significance level (adjusted p value, false discovery rate [FDR] < 0.05). DEGs between RA and OA patients intersecting in all three datasets were selected for enrichment analysis by submitting to the KOBAS 3.0 web tool (http://kobas.cbi.pku.edu.cn/) for KEGG pathway enrichment analysis.
Isolation and Culture of FLSs
FLSs were derived from synovial tissue specimens harvested from three patients by needle arthroscopy. FLSs were isolated by enzyme digestion and subsequently cultured in Dulbecco’s modified essential medium (DMEM) containing 10% fetal bovine serum (FBS, Invitrogen) and antibiotics (penicillin and streptomycin) at 37°C with 5% CO2. Cells cultured between passages 4 and 9 were used for this study. Cells were frozen with cell freezing medium and stored in a −80°C freezer until used.
High-Throughput mRNA Sequencing
High-throughput RNA sequencing was performed with FLSs from RA and OA patients (n = 3/group) (for patients’ information, see Table 2). All the patients were free from other systemic and immune-related diseases. DEGs were selected based on the results having multiples of difference (|log2FC| > 1) and significance level (adjusted p value, FDR < 0.05). Gene expression differences between RA and OA FLSs were investigated using KEGG pathway enrichment analysis.
Table 2.
Patients’ General Information
| Case No. | Sex | Age (Years) | Disease Course (Years) | ESR (mm/h; Normative Value, 0–20) | CRP (mg/L; Normal Value, 0–6) | Albumin/Globulin (Normal Value, 1.2–2.5) |
|---|---|---|---|---|---|---|
| OA1 | female | 48 | 0.9 | 45 | 2.27 | 1.5 |
| OA2 | male | 54 | 2 | 5 | 1.05 | 1.6 |
| OA3 | female | 55 | 10 | 80 | 5.7 | 1.6 |
| RA1 | female | 51 | 5 | 150 | 29.84 | 0.5 |
| RA2 | female | 49 | 6 | 50 | 35.42 | 1.2 |
| RA3 | female | 41 | 2 | 100 | 13.18 | 1.1 |
Immunohistochemical Studies
Synovium samples were collected from arthroscopic surgery done with 20 RA and 17 OA patients for therapeutic purposes (general information is given in Table 1). Streptavidin-biotin-based immunoperoxidase staining for p-AMPK-α1 was performed using IHC as follows: formalin-fixed, paraffin-embedded synovium specimens were cut into 4-μm-thick sections, deparaffinized, and treated with 0.3% hydrogen peroxide to block endogenous peroxidase activity and 5% non-fat dry milk protein as the blocking agent. Antigen retrieval was done by immersing the sections in boiling 0.01 M sodium citrate buffer (pH 6.0) for 10 min. Rabbit anti-human p-AMPK-α1 antibodies (Proteintech, Chicago, IL, USA) were added as primary antibodies at the dilution of 1:400 for 2 h and incubated for 37°C. Biotin-conjugated goat anti-rabbit antibodies (ZSGB-Bio, Beijing, P.R. China), streptavidin-peroxidase conjugate, and diaminobenzidine were used as the detecting system.
qRT-PCR
Synovium specimens from 10 RA and 9 OA patients obtained by knee arthroscopy (for patients’ general information, see Table 1). Specimens were immersed in TRIzol reagent (Thermo Scientific, USA) after removing the adipose tissue under aseptic conditions and stored at −20°C until used. The mRNA expression levels of AMPK-α1, AMPK-α2, AMPK-γ1, and AMPK-γ3 genes were evaluated using qRT-PCR. Briefly, total RNA was extracted from individual synovium samples using RNAzol BD (Molecular Research Center, Cincinnati, OH, USA) following the manufacturer’s protocol. The extracted total RNA was reverse transcribed into cDNA using PrimeScript RT master mix (Takara Bio, Shiga, Japan). qRT-PCR analysis was conducted using the Applied Biosystems ViiA 7 real-time PCR system (Wcgene Biotechnology, Shanghai, China). The reaction mixture contained 5 μL of 2× TB Green Premix Ex Taq II (Takara Bio, Shiga, Japan), 3 μL of nuclease-free water, 1 μL of cDNA, 0.4 μL of each gene-specific primer, and 0.2 μL of ROX reference dye. The target mRNA was amplified separately in duplicates. The expression levels of the target mRNAs relative to GAPDH expression were calculated with the ΔCT method. The primers for amplifying the target mRNAs are listed in Table 3.
Table 3.
Sequences of qRT-PCR Primers
| Primer Names | Sense Primers (5′→3′) | Antisense Primers (5′→3′) |
|---|---|---|
| AMPK-α1 | TTGAAACCTGAAAATGTCCTGCT | GGTGAGCCACAACTTGTTCTT |
| AMPK-α2 | ATGCCAAGATAGCCGATTTCG | TGCTGCATAATTTGGAGATCCG |
| AMPK-γ1 | CTGCACCGCTACTATAAATCAGC | AGTCCTGGAGATACACCTCTCT |
| AMPK-γ3 | CCTGGGAGTGTGAGCTAGAAG | GTTTCCGCAGTTCGTCATCC |
| IL-6 | CCTGAACCTTCCAAAGATGGC | TTCACCAGGCAAGTCTCCTCA |
| PKA-α | AGCCCACTTGGATCAGTTTGA | GTTCCCGGTCTCCTTGTGT |
| RAPTOR | GCAGATTTGCCAACTATCTTCGG | CAGCGGTAAAAGTGTCCCCTG |
| HAPLN1 | TCTGGTGCTGATTTCAATCTGC | TGCTTGGATGTGAATAGCTCTG |
| RUNX1 | TCTTCACAAACCCACCGCAA | CTGCCGATGTCTTCGAGGTTC |
| RUNX2 | TCAACGATCTGAGATTTGTGGG | GGGGAGGATTTGTGAAGACGG |
| GAPDH | ACAACTTTGGTATCGTGGAAGG | GCCATCACGCCACAGTTTC |
To evaluate the effects of metformin on FLSs, relative mRNA expression levels of IL-6, AMPK-α1, PKA-α, RAPTOR, mTOR, HAPLN1, RUNX1, and RUNX2 genes were measured by qPCR. Each experiment was done in triplicate. FLS samples digested using 0.25% pancreatin were transferred to two 96-well plates (105 cells/well). After 18 h of culture, the FLSs were treated either with metformin (5 mM) or saline. The target mRNA was amplified in duplicates, and its expression level relative to GAPDH was calculated using the ΔΔCT method. Primers are listed in Table 3.
MTT Assay
An MTT assay was used to ascertain the effects of metformin and dorsomorphin on FLS viability at different concentrations. FLS samples digested using 0.25% pancreatin were transferred to two 96-well plates with 3–5 × 103 cells/well. After 24 h of culture, the FLSs were either treated with AMPK agonist metformin (0, 1, 5, and 10 mM) or the antagonist dorsomorphin (0, 2.5, 5, and 10 μM). Six replicates were used at each time point. At different time intervals (0, 12, 24, 48, and 72 h), MTT solution (5 mg/mL, Sigma-Aldrich, St. Louis, MO, USA) was added, followed by a 4-h incubation period. Then, the culture medium was aspirated and 150 μL of dimethyl sulfoxide (DMSO) was added to each well to dissolve the formazan crystals. OD was measured at 490 nm using a Universal Microplate Reader (BioTek Instruments, Winooski, VT, USA). Data curves were plotted with OD values on the y axis and time intervals on the x axis. Each experiment was done in triplicate.
Scratch Test
A scratch test was performed to evaluate the effects of metformin and dorsomorphin on FLS migration viability. Perpendicular and horizontal coordinates were outlined with intervals of 0.5–1.0 cm on the back of six-well plates with the adjusted density of 105 cells/well. After incubation for 18 h, all cells were adhered to the plate, and then a vertical scratch was made on the cell layer. FLSs in logarithmic growth phase were treated using RPMI 1640 medium containing metformin (5 mM), dorsomorphin (5 μM), or normal saline. The cells were further incubated in DMEM medium with 5% CO2 at 37°C. The plates were imaged at 0, 6, 12, 24, and 36 h using an inverted microscope. Scratch width was measured at each time interval using the ruler present in the Olympus CellSens standard microscope (Olympus, Tokyo, Japan). Each experiment was done in triplicate. Scratch repair rate was calculated using the following formula: wound repair rate = [(initial scratch width − final scratch width)/initial scratch width] × 100%.
Transwell Assay
A transwell assay was performed to evaluate the effects of metformin and dorsomorphin on FLS migration capacity. After incubation with metformin (5 mM), dorsomorphin (5 μM), or normal saline for 24 h, FLSs were re-suspended and diluted to a concentration of 5 × 104/mL in serum-free medium. The cell suspension (200 μL) was added to the upper chamber coated with Matrigel (BD Bioscience) diluted in DMEM (1:3), while 500 μL of DMEM containing 10% FBS was added in the lower chamber. Of note, the transwell insert consists of an 8.0-μm-thick polyester membrane. After incubation for 12 h at 37°C, the cells in the upper chamber were discarded and, from the lower chamber, were fixed using 4% paraformaldehyde for 25 min and stained with 0.1% crystal violet (Beyotime, USA) for 15 min. Next, mean numbers of migrated FLSs in five random fields under a microscope were counted. Each experiment was done in triplicate.
Automated Electrophoresis Western Blot Analysis
FLSs treated with metformin (5 mM) or saline for 36 h were examined for relative changes in p-AMPK-α1, IL-1β, TNF-α, HAPLN1, and TUBULIN-β3 levels by automated electrophoresis followed by western blot analysis. FLSs were seeded at 1–2 × 105 cells per well in six-well plates. Medium was replaced in the wells with fresh medium containing metformin (5 mM) or saline for 36 h. After aspiration of the medium, cell monolayers were rinsed with 1 mL of ice-cold PBS and lysed in 80 μL of lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% [v/v] Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate) supplemented with fresh 1 mM Na3VO4 and 1 mM dithiothreitol containing 1× protease inhibitor cocktail (Roche Applied Science, Basel, Switzerland). Lysates were pre-cleared by centrifugation at 18,000 × g for 15 min at 4°C. Supernatants were collected and the protein concentrations were measured using a Bradford assay (Thermo Fisher Scientific, MA, USA). Lysates were adjusted to 5 mg/mL protein concentration. Capillary electrophoresis-western blot analysis was carried out using reagents provided in the kit and following instructions in the user manual (Wes, ProteinSimple, San Jose, CA, USA). Briefly, 5.6 μL of the cell lysate was mixed with 1.4 μL of fluorescent master mix and heated at 95°C for 5 min. The samples, blocking reagent, wash buffer, antibodies binding to tublin, p-AMPK-α1, IL-1β, TNF-α, and hapln1 (1:100, R&D Systems), secondary antibodies, and chemiluminescent substrate were dispensed into the microplates. The electrophoretic separation and immunodetection were performed automatically using default settings. Data were analyzed using in-built Compass software for SW 4.0. The truncated and full length of target protein intensities (area under the curve) were normalized to those of the tubulin peak (control). In most of the figures, electropherograms are represented as pseudo-blots, generated using Compass software.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 7.0 software. All data are given as mean ± SD. Differences between two groups were evaluated for statistical significance using a Student’s t test. One-way ANOVA with a Tukey’s multiple comparison test was used to evaluate the differences among three or more groups. Correlations were evaluated using a linear regression and correlation test. p <0.05 was considered as statistically significant.
Author Contributions
Yong C. and C.X. designed the study. Yong C., F.Q., and B.Y. performed most of the experiments and collected the data. Yong C. and Yanjuan C. analyzed the data and wrote the manuscript. F.F.Z. provided all of the clinical samples and collected data from RA patients. X.Z. did the bioinformatics analysis. K.S.N. and C.X. participated in the discussion and reviewed the original data and manuscript. All authors have read and approved the manuscript for publication.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank staff in the Rheumatology Department, including Dr. Yuanyi, Dr. Ensheng Chen, Dr. Yanan Bi, and Dr. Yanyancao, for their help in providing necessary clinical samples. The research secretary, Dr. Xiaofeng Zhao, helped in securing ethics approvals. This work was funded by National Natural Science of China (NSFC), (project no. 81673723), and by the Guangdong Medical Research Fund Project, China (project no. A2019053).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.05.008.
Contributor Information
Kutty Selva Nandakumar, Email: nandakumar@smu.edu.cn.
Changhong Xiao, Email: chnghngxiao@aliyun.com.
Supplemental Information
DEGs between RA and OA Synovium Based on Intersection of NCBI GEO Profiles
DEGs between RA and OA FLSs by mRNA Sequencing
References
- 1.Calabresi E., Petrelli F., Bonifacio A.F., Puxeddu I., Alunno A. One year in review 2018: pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2018;36:175–184. [PubMed] [Google Scholar]
- 2.Fazal S.A., Khan M., Nishi S.E., Alam F., Ashraf G.M. A clinical update and global economic burden of rheumatoid arthritis. Endocr. Metab. Immune Disord. Drug Targets. 2018;18:98–109. doi: 10.2174/1871530317666171114122417. [DOI] [PubMed] [Google Scholar]
- 3.Bergström B., Lundqvist C., Vasileiadis G.K., Carlsten H., Ekwall O., Ekwall A.H. The rheumatoid arthritis risk gene AIRE is induced by cytokines in fibroblast-like synoviocytes and augments the pro-inflammatory response. Front. Immunol. 2019;10:1384. doi: 10.3389/fimmu.2019.01384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzalez-Rey E., Gonzalez M.A., Varela N., O’Valle F., Hernandez-Cortes P., Rico L., Büscher D., Delgado M. Human adipose-derived mesenchymal stem cells reduce inflammatory and T cell responses and induce regulatory T cells in vitro in rheumatoid arthritis. Ann. Rheum. Dis. 2010;69:241–248. doi: 10.1136/ard.2008.101881. [DOI] [PubMed] [Google Scholar]
- 5.Ospelt C., Reedquist K.A., Gay S., Tak P.P. Inflammatory memories: is epigenetics the missing link to persistent stromal cell activation in rheumatoid arthritis? Autoimmun. Rev. 2011;10:519–524. doi: 10.1016/j.autrev.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Ghesquière B., Wong B.W., Kuchnio A., Carmeliet P. Metabolism of stromal and immune cells in health and disease. Nature. 2014;511:167–176. doi: 10.1038/nature13312. [DOI] [PubMed] [Google Scholar]
- 7.Arias de la Rosa I., Escudero-Contreras A., Rodríguez-Cuenca S., Ruiz-Ponce M., Jiménez-Gómez Y., Ruiz-Limón P., Pérez-Sánchez C., Ábalos-Aguilera M.C., Cecchi I., Ortega R. Defective glucose and lipid metabolism in rheumatoid arthritis is determined by chronic inflammation in metabolic tissues. J. Intern. Med. 2018;284:61–77. doi: 10.1111/joim.12743. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Carbonell R., Divakaruni A.S., Lodi A., Vicente-Suarez I., Saha A., Cheroutre H., Boss G.R., Tiziani S., Murphy A.N., Guma M. Critical role of glucose metabolism in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol. 2016;68:1614–1626. doi: 10.1002/art.39608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Son H.J., Lee J., Lee S.Y., Kim E.K., Park M.J., Kim K.W., Park S.H., Cho M.L. Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediators Inflamm. 2014;2014:973986. doi: 10.1155/2014/973986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang K.Y., Kim Y.K., Yi H., Kim J., Jung H.R., Kim I.J., Cho J.H., Park S.H., Kim H.Y., Ju J.H. Metformin downregulates Th17 cells differentiation and attenuates murine autoimmune arthritis. Int. Immunopharmacol. 2013;16:85–92. doi: 10.1016/j.intimp.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 11.Mallik R., Chowdhury T.A. Metformin in cancer. Diabetes Res. Clin. Pract. 2018;143:409–419. doi: 10.1016/j.diabres.2018.05.023. [DOI] [PubMed] [Google Scholar]
- 12.Cantó C., Gerhart-Hines Z., Feige J.N., Lagouge M., Noriega L., Milne J.C., Elliott P.J., Puigserver P., Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herzig S., Shaw R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018;19:121–135. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J., Kundu M., Viollet B., Guan K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carroll K.C., Viollet B., Suttles J. AMPKα1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J. Leukoc. Biol. 2013;94:1113–1121. doi: 10.1189/jlb.0313157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guma M., Sanchez-Lopez E., Lodi A., Garcia-Carbonell R., Tiziani S., Karin M., Lacal J.C., Firestein G.S. Choline kinase inhibition in rheumatoid arthritis. Ann. Rheum. Dis. 2015;74:1399–1407. doi: 10.1136/annrheumdis-2014-205696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang X., Yuan Y., Pan Z., Ma Y., Wu M., Yang J., Han R., Chen M., Hu X., Liu R. Elevated circulating IL-17 level is associated with inflammatory arthritis and disease activity: a meta-analysis. Clin. Chim. Acta. 2019;496:76–83. doi: 10.1016/j.cca.2019.06.026. [DOI] [PubMed] [Google Scholar]
- 18.van Mulligen E., de Jong P.H.P., Kuijper T.M., van der Ven M., Appels C., Bijkerk C., Harbers J.B., de Man Y., Molenaar T.H.E., Tchetverikov I. Gradual tapering TNF inhibitors versus conventional synthetic DMARDs after achieving controlled disease in patients with rheumatoid arthritis: first-year results of the randomised controlled TARA study. Ann. Rheum. Dis. 2019;78:746–753. doi: 10.1136/annrheumdis-2018-214970. [DOI] [PubMed] [Google Scholar]
- 19.Yamauchi T., Kamon J., Minokoshi Y., Ito Y., Waki H., Uchida S., Yamashita S., Noda M., Kita S., Ueki K. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 20.Solomon D.H., Love T.J., Canning C., Schneeweiss S. Risk of diabetes among patients with rheumatoid arthritis, psoriatic arthritis and psoriasis. Ann. Rheum. Dis. 2010;69:2114–2117. doi: 10.1136/ard.2009.125476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samimi Z., Kardideh B., Zafari P., Bahrehmand F., Roghani S.A., Taghadosi M. The impaired gene expression of adenosine monophosphate-activated kinase (AMPK), a key metabolic enzyme in leukocytes of newly diagnosed rheumatoid arthritis patients. Mol. Biol. Rep. 2019;46:6353–6360. doi: 10.1007/s11033-019-05078-x. [DOI] [PubMed] [Google Scholar]
- 22.Thornton C.C., Al-Rashed F., Calay D., Birdsey G.M., Bauer A., Mylroie H., Morley B.J., Randi A.M., Haskard D.O., Boyle J.J., Mason J.C. Methotrexate-mediated activation of an AMPK-CREB-dependent pathway: a novel mechanism for vascular protection in chronic systemic inflammation. Ann. Rheum. Dis. 2016;75:439–448. doi: 10.1136/annrheumdis-2014-206305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huber R., Hummert C., Gausmann U., Pohlers D., Koczan D., Guthke R., Kinne R.W. Identification of intra-group, inter-individual, and gene-specific variances in mRNA expression profiles in the rheumatoid arthritis synovial membrane. Arthritis Res. Ther. 2008;10:R98. doi: 10.1186/ar2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woetzel D., Huber R., Kupfer P., Pohlers D., Pfaff M., Driesch D., Häupl T., Koczan D., Stiehl P., Guthke R., Kinne R.W. Identification of rheumatoid arthritis and osteoarthritis patients by transcriptome-based rule set generation. Arthritis Res. Ther. 2014;16:R84. doi: 10.1186/ar4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marcinko K., Steinberg G.R. The role of AMPK in controlling metabolism and mitochondrial biogenesis during exercise. Exp. Physiol. 2014;99:1581–1585. doi: 10.1113/expphysiol.2014.082255. [DOI] [PubMed] [Google Scholar]
- 26.Bustamante M.F., Garcia-Carbonell R., Whisenant K.D., Guma M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res. Ther. 2017;19:110. doi: 10.1186/s13075-017-1303-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bartok B., Firestein G.S. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol. Rev. 2010;233:233–255. doi: 10.1111/j.0105-2896.2009.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duca F.A., Côté C.D., Rasmussen B.A., Zadeh-Tahmasebi M., Rutter G.A., Filippi B.M., Lam T.K.T. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med. 2015;21:506–511. doi: 10.1038/nm.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dasgupta B., Seibel W. Compound C/dorsomorphin: its use and misuse as an AMPK inhibitor. Methods Mol. Biol. 2018;1732:195–202. doi: 10.1007/978-1-4939-7598-3_12. [DOI] [PubMed] [Google Scholar]
- 30.Gwinn D.M., Shackelford D.B., Egan D.F., Mihaylova M.M., Mery A., Vasquez D.S., Turk B.E., Shaw R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim Y.J., Lee J., Song M.K., Han T., Ryu J.C. Valproic acid inhibits cell size and cell proliferation by AMPK-mediated mTOR signaling pathway in JEG-3 cells. Biochip J. 2013;7:267–277. [Google Scholar]
- 32.Caiello I., Minnone G., Holzinger D., Vogl T., Prencipe G., Manzo A., De Benedetti F., Strippoli R. IL-6 amplifies TLR mediated cytokine and chemokine production: implications for the pathogenesis of rheumatic inflammatory diseases. PLoS ONE. 2014;9:e107886. doi: 10.1371/journal.pone.0107886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ogata A., Kato Y., Higa S., Yoshizaki K. IL-6 inhibitor for the treatment of rheumatoid arthritis: a comprehensive review. Mod. Rheumatol. 2019;29:258–267. doi: 10.1080/14397595.2018.1546357. [DOI] [PubMed] [Google Scholar]
- 34.Kim Y.D., Kim Y.H., Cho Y.M., Kim D.K., Ahn S.W., Lee J.M., Chanda D., Shong M., Lee C.H., Choi H.S. Metformin ameliorates IL-6-induced hepatic insulin resistance via induction of orphan nuclear receptor small heterodimer partner (SHP) in mouse models. Diabetologia. 2012;55:1482–1494. doi: 10.1007/s00125-012-2494-4. [DOI] [PubMed] [Google Scholar]
- 35.Liu G., Wu K., Zhang L., Dai J., Huang W., Lin L., Ge P., Luo F., Lei H. Metformin attenuated endotoxin-induced acute myocarditis via activating AMPK. Int. Immunopharmacol. 2017;47:166–172. doi: 10.1016/j.intimp.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Galligan C.L., Baig E., Bykerk V., Keystone E.C., Fish E.N. Distinctive gene expression signatures in rheumatoid arthritis synovial tissue fibroblast cells: correlates with disease activity. Genes Immun. 2007;8:480–491. doi: 10.1038/sj.gene.6364400. [DOI] [PubMed] [Google Scholar]
- 37.Liu J., Park E.S., Curry T.E., Jr., Jo M. Periovulatory expression of hyaluronan and proteoglycan link protein 1 (Hapln1) in the rat ovary: hormonal regulation and potential function. Mol. Endocrinol. 2010;24:1203–1217. doi: 10.1210/me.2009-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jung Y.J., Bae H.S., Ryoo H.M., Baek S.H. A novel RUNX2 mutation in exon 8, G462X, in a patient with cleidocranial dysplasia. J. Cell. Biochem. 2018;119:1152–1162. doi: 10.1002/jcb.26283. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X., Liu Y., Wang X., Sun X., Zhang C., Zheng S. Analysis of novel RUNX2 mutations in Chinese patients with cleidocranial dysplasia. PLoS ONE. 2017;12:e0181653. doi: 10.1371/journal.pone.0181653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Astakhova A., Chistyakov D., Thomas D., Geisslinger G., Brüne B., Sergeeva M., Namgaladze D. Inhibitors of oxidative phosphorylation modulate astrocyte inflammatory responses through AMPK-dependent Ptgs2 mRNA stabilization. Cells. 2019;8:1185. doi: 10.3390/cells8101185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lyons C.L., Roche H.M. Nutritional modulation of AMPK-impact upon metabolic-inflammation. Int. J. Mol. Sci. 2018;19:3092. doi: 10.3390/ijms19103092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hammer H.B., Michelsen B., Sexton J., Haugen I.K., Provan S.A., Haavardsholm E.A., Uhlig T., Kvien T.K. Swollen, but not tender joints, are independently associated with ultrasound synovitis: results from a longitudinal observational study of patients with established rheumatoid arthritis. Ann. Rheum. Dis. 2019;78:1179–1185. doi: 10.1136/annrheumdis-2019-215321. [DOI] [PubMed] [Google Scholar]
- 43.Chen K., Lin Z.W., He S.M., Wang C.Q., Yang J.C., Lu Y., Xie X.B., Li Q. Metformin inhibits the proliferation of rheumatoid arthritis fibroblast-like synoviocytes through IGF-IR/PI3K/AKT/m-TOR pathway. Biomed. Pharmacother. 2019;115:108875. doi: 10.1016/j.biopha.2019.108875. [DOI] [PubMed] [Google Scholar]
- 44.Yamashita T., Kato K., Fujihara S., Iwama H., Morishita A., Yamana H., Kobayashi K., Kamada H., Chiyo T., Kobara H. Anti-diabetic drug metformin inhibits cell proliferation and tumor growth in gallbladder cancer via G0/G1 cell cycle arrest. Anticancer Drugs. 2020;31:231–240. doi: 10.1097/CAD.0000000000000870. [DOI] [PubMed] [Google Scholar]
- 45.Aljofan M., Riethmacher D. Anticancer activity of metformin: a systematic review of the literature. Future Sci. OA. 2019;5:FSO410. doi: 10.2144/fsoa-2019-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He L., Wondisford F.E. Metformin action: concentrations matter. Cell Metab. 2015;21:159–162. doi: 10.1016/j.cmet.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 47.El-Mir M.Y., Nogueira V., Fontaine E., Avéret N., Rigoulet M., Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 48.Sciannimanico S., Grimaldi F., Vescini F., De Pergola G., Iacoviello M., Licchelli B., Guastamacchia E., Giagulli V.A., Triggiani V. Metformin: up to date. Endocr. Metab. Immune Disord. Drug Targets. 2020;20:172–181. doi: 10.2174/1871530319666190507125847. [DOI] [PubMed] [Google Scholar]
- 49.Nandakumar K.S., Holmdahl R. Antibody-induced arthritis: disease mechanisms and genes involved at the effector phase of arthritis. Arthritis Res. Ther. 2006;8:223. doi: 10.1186/ar2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Md Yusof M.Y., Emery P. Targeting interleukin-6 in rheumatoid arthritis. Drugs. 2013;73:341–356. doi: 10.1007/s40265-013-0018-2. [DOI] [PubMed] [Google Scholar]
- 51.Salminen A., Hyttinen J.M., Kaarniranta K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on healthspan and lifespan. J. Mol. Med. (Berl.) 2011;89:667–676. doi: 10.1007/s00109-011-0748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang Z., Kahn B.B., Shi H., Xue B.Z. Macrophage α1 AMP-activated protein kinase (α1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J. Biol. Chem. 2010;285:19051–19059. doi: 10.1074/jbc.M110.123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dudhia J. Aggrecan, aging and assembly in articular cartilage. Cell. Mol. Life Sci. 2005;62:2241–2256. doi: 10.1007/s00018-005-5217-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mebarki S., Désert R., Sulpice L., Sicard M., Desille M., Canal F., Dubois-Pot Schneider H., Bergeat D., Turlin B., Bellaud P. De novo HAPLN1 expression hallmarks Wnt-induced stem cell and fibrogenic networks leading to aggressive human hepatocellular carcinomas. Oncotarget. 2016;7:39026–39043. doi: 10.18632/oncotarget.9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones C.C., Bradford Y., Amos C.I., Blot W.J., Chanock S.J., Harris C.C., Schwartz A.G., Spitz M.R., Wiencke J.K., Wrensch M.R. Cross-cancer pleiotropic associations with lung cancer risk in African Americans. Cancer Epidemiol. Biomarkers Prev. 2019;28:715–723. doi: 10.1158/1055-9965.EPI-18-0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Urano T., Narusawa K., Shiraki M., Sasaki N., Hosoi T., Ouchi Y., Nakamura T., Inoue S. Single-nucleotide polymorphism in the hyaluronan and proteoglycan link protein 1 (HAPLN1) gene is associated with spinal osteophyte formation and disc degeneration in Japanese women. Eur. Spine J. 2011;20:572–577. doi: 10.1007/s00586-010-1598-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin Z., Bei J.X., Shen M., Li Q., Liao Z., Zhang Y., Lv Q., Wei Q., Low H.Q., Guo Y.M. A genome-wide association study in Han Chinese identifies new susceptibility loci for ankylosing spondylitis. Nat. Genet. 2011;44:73–77. doi: 10.1038/ng.1005. [DOI] [PubMed] [Google Scholar]
- 58.Cao J., Meng S., Chang E., Beckwith-Fickas K., Xiong L., Cole R.N., Radovick S., Wondisford F.E., He L. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK) J. Biol. Chem. 2014;289:20435–20446. doi: 10.1074/jbc.M114.567271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Watanabe H., Yamada Y. Mice lacking link protein develop dwarfism and craniofacial abnormalities. Nat. Genet. 1999;21:225–229. doi: 10.1038/6016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DEGs between RA and OA Synovium Based on Intersection of NCBI GEO Profiles
DEGs between RA and OA FLSs by mRNA Sequencing