

Abstract
Human dihydrolipoamide dehydrogenase (hLADH, hE3) deficiency (OMIM# 246900) is an often prematurely lethal genetic disease usually caused by inactive or partially inactive hE3 variants. Here we report the crystal structure of wild-type hE3 at an unprecedented high resolution of 1.75 Å and the structures of six disease-causing hE3 variants at resolutions ranging from 1.44 to 2.34 Å. P453L proved to be the most deleterious substitution in structure as aberrations extensively compromised the active site. The most prevalent G194C-hE3 variant primarily exhibited structural alterations close to the substitution site, whereas the nearby cofactor-binding residues were left unperturbed. The G426E substitution mainly interfered with the local charge distribution introducing dynamics to the substitution site in the dimer interface; G194C and G426E both led to minor structural changes. The R460G, R447G and I445M substitutions all perturbed a solvent accessible channel, the so-called H+/H2O channel, leading to the active site. Molecular pathomechanisms of enhanced reactive oxygen species (ROS) generation and impaired binding to multienzyme complexes were also addressed according to the structural data for the relevant mutations. In summary, we present here for the first time a comprehensive study that links three-dimensional structures of disease-causing hE3 variants to residual hLADH activities, altered capacities for ROS generation, compromised affinities for multienzyme complexes and eventually clinical symptoms. Our results may serve as useful starting points for future therapeutic intervention approaches.
Introduction
Dihydrolipoamide dehydrogenase (LADH, E3, EC 1.8.1.4) is a homodimeric flavin-disulfide oxidoreductase. As the common third component of the mitochondrial alpha-keto-glutarate dehydrogenase (KGDHc), pyruvate dehydrogenase (PDHc) and branched chain alpha-keto acid dehydrogenase (BCKDHc) multienzyme complexes (1–4), E3 plays crucial roles in metabolism by catalyzing the oxidation of the dihydrolipoamide (DHLA) cofactor covalently linked to the E2 components of these complexes and generating NADH (Supplementary Material, Fig. S1). This oxidation is essential to re-establish initial conditions in the complexes after each turnover thereby enabling cycling, and its failure would halt the enzymatic processing ability after a single catalytic event. E3 is also part of the glycine cleavage system (5). In addition to its physiological importance, the human E3 (hE3) also possesses moonlighting functions, mainly as an enzyme not part of any multienzyme complex (6,7). The pathologically most important such activity of hE3 is likely to be the production of reactive oxygen species (ROS) (Supplementary Material, Fig. S1), predominantly as part of the hKGDHc (8–10). KGDHc proved to be a major contributor to mitochondrial oxidative stress when its ROS generation via E3 becomes significant under pathologically relevant conditions, such as low pH and elevated NADH/NAD+ ratio (9,11,12). As a selectively delivered exogenous ROS-generating drug candidate, the hE3 was very recently reported to induce cancer cell death (13).
The course of the catalytic action (14–16) and the structure [(17, 18) and references therein] of E3 have been extensively studied in different species. The monomeric hE3 is ~50 kDa in size and comprises four domains: a FAD-binding domain (residues 1–149; numbering follows the mature hE3 sequence), a NAD+/NADH-binding domain (residues 150–282), a central domain (residues 283–350) and an interface domain (residues 351–474) [Supplementary Material, Fig. S2 (18,19)]. The hE3 is an obligate homodimer as both active sites require residues from the other monomer. A key feature is the non-covalently bound FAD prosthetic group that divides the active site into two compartments by its isoalloxazine ring: the oxidized/reduced lipoamide (LA) binding site with a redox-active disulfide bond (Cys45-Cys50) and a catalytic base (His452′; ′ stands for the adjacent monomer) on the si face and the NAD+/NADH-binding site on the re face (14). The kinetics of the hE3 reaction suggest a ping-pong mechanism while, due to the active site configuration, the two half-reactions of the physiological reaction are spatially separated (14,18,20,21). Structures for NAD+- and NADH-complexed hE3 were already reported (19); however, the exact binding mode of dihydrolipoamide/lipoamide is still obscure (22–24). In addition to the two substrate-binding channels/pockets, there also exists a third important channel in the protein referred to as the H+/H2O channel. This channel is the direct continuation of the LA-binding channel and was proposed to play important roles in catalysis by providing an outlet for H2O molecules upon the entry of DHLA to its binding site (23) and/or for H+/H3O+ (25) that is being released upon reduction of NAD+.
Pathogenic mutations of the DLD gene (RefSeq NM_000108) result in hE3 variants affecting several central metabolic pathways simultaneously and leading to the often prematurely lethal human disease referred to as E3 deficiency (OMIM# 246900) (26–29); the most prevalent disease-causing substitution is G194C (c.685G>T, its highest carrier frequency is 1:94 in the Ashkenazi Jewish population) (26,30–34). Typical clinical phenotypes of E3 deficiency are lactate acidosis, hypoglycemia, hyperammonaemia, encephalopathy, neurological deficit, Leigh syndrome, liver dysfunction, hypertrophic cardiomyopathy and/or muscle weakness [(26–29) and references therein]. In the 14 disease-causing enzyme variants reported to date (Fig. 1), the catalytic activity is compromised to variable degrees. The respective substitutions/deletion may affect the active site [G101del (c.405_407delAGG), P453L (c.1463C>T)], the cofactor-binding regions [I12T (c.140T>C), K37E (c.214A>G), G194C (c.685G>T), I318T (c.1058T>C), M326V (c.1081A>G), I358T (c.1178T>C)] or the dimer interface region [E340K (c.1123G>A), G426E (c.1382G>A), D444V (c.1436A>T), I445M (c.1440A>G), R460G (c.1483A>G), R447G (c.1444A>G)] [(29,35) and references therein]. The severity of the clinical manifestations of E3 deficiency, however, generally do not correlate well with the loss in LADH function (26,31), hence other auxiliary biochemical mechanisms must also contribute to the pathogenesis. Enhanced ROS generation was proposed to be a contributing mechanism as selected disease-causing variants, namely the P453L-, G194C-, D444V- and E340K-hE3, displayed augmented ROS-generating capacities as compared to the wild-type enzyme, in vitro (12). Additionally, D444V-, E340K-, R447G- and R460G-hE3 were reported to cause oxidative damage to the LA cofactors of the PDHc and KGDHc in a yeast model; an identical phenomenon was also observed in D444V-homozygous human fibroblasts (36). Neither the loss in LADH activity nor the enhanced ROS-producing capacity shows direct correlation with the localization of the pathogenic substitution (12). Selected disease-causing substitutions led to losses in the affinity for the hPDHc (37,38), which account for the impaired overall PDHc activities measured in respective patient samples or after assembling the relevant enzyme complexes in vitro (37).
Figure 1.
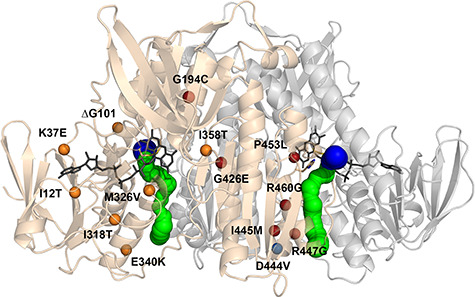
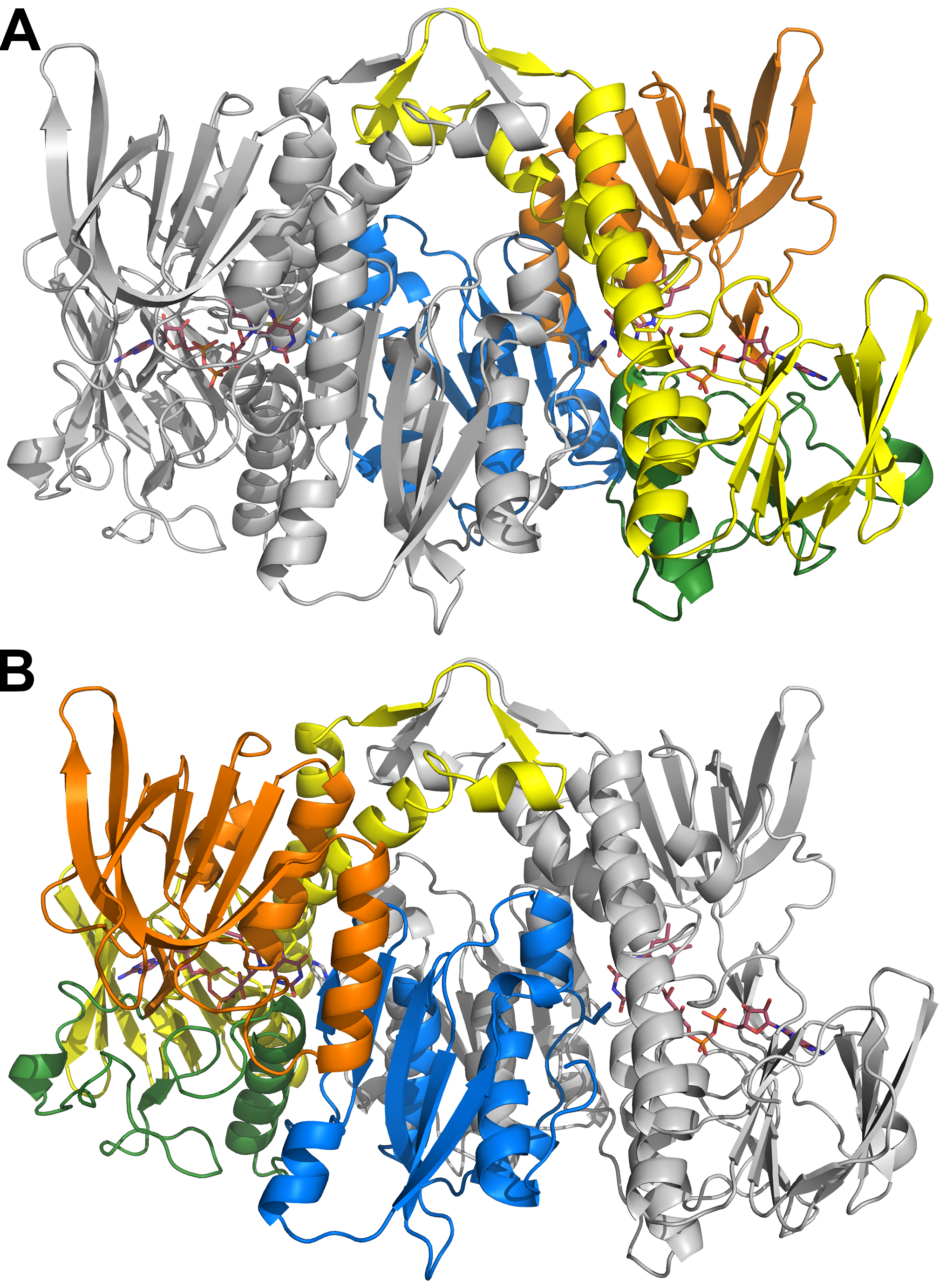
Structure of the hE3 obligate homodimer at 1.75 Å resolution (PDB ID: 6I4Q) with the 14 disease-causing substitution/deletion sites reported to date. The pathogenic substitution/deletion sites are represented in a single hE3 monomer. Spheres designate the Cα atoms of the respective residues and are color coded as follows: variants investigated in the present study—red; variant structure was reported earlier—blue; no variant structure is available—orange. Solvent accessible channels leading to the active sites were computed by Caver applying the dominant Glu332 and Arg460 conformations (see text). The LA-binding and H+/H2O channel segments are blue and green, respectively. Monomers A and B are related by a two-fold symmetry axis and colored beige and grey, respectively. The FAD prosthetic groups, the Cys45-Cys50 disulfide bonds and the His452′ residues are depicted as sticks.
The first crystal structure of a disease-causing variant of hE3 (D444V-hE3, PDB ID: 5J5Z) was reported recently from Semmelweis (18). It was concluded that the D444V substitution induced no significant rearrangements in the overall protein structure, instead it led to local structural alterations in the solvent accessible continuation of the LA-binding channel (the H+/H2O channel, see above). Most of the pathogenic dimer interface substitutions of hE3 (D444V, I445M, R447G, R460G) reside in the spatial vicinity of this H+/H2O channel, suggesting its possible involvement in each of the molecular pathomechanisms. D444V-hE3, as well as nine additional disease-causing variants were examined by H/D-exchange mass spectrometry (HDX-MS) against hE3 and conclusions were drawn regarding the respective structural alterations (relevant at the temperature of the measurements, 25°C) (39). Previously, molecular dynamics (MD) simulation was also applied for analyzing the above variants (40,41).
We report here the high-resolution crystal structures of six disease-causing variants of hE3 including four dimer interface variants (G426E-, I445M-, R447G- and R460G-hE3), an active site variant (P453L-hE3) and a variant that affects the cofactor-binding region (G194C-hE3). Additionally, a wild-type hE3 structure with much improved resolution is also reported. Based on the detailed analysis of the structures, an individual molecular pathomechanism is proposed for each disease-causing variant investigated.
Results
In the present study, crystal structures of hE3 and six of its disease-causing variants were determined in the resolution range of 1.44–2.34 Å. Data and model statistics together with PDB accession codes are summarized in Table 1. The proteins were all crystallized under similar conditions resulting in nearly identical crystal packing and hence good comparability. Neither NAD+/NADH nor any lipoyl derivative was given as substrate in the course of purification or crystallization. All the protein variants were purified in the presence of excess FAD to form the holoenzymes, and the prosthetic group could indeed be observed in all the structures determined. Except for P453L-hE3, a single dimer formed the asymmetric unit (ASU).
Table 1.
Data collection, processing and refinement statistics*
| Structure | hE3 | G194C-hE3 | P453L-hE3 | R447G-hE3 | R460G-hE3 | I445M-hE3 | G426E-hE3 |
|---|---|---|---|---|---|---|---|
| PDB ID | 6I4Q | 6I4P | 6I4Z | 6I4S | 6I4R | 6I4T | 6I4U |
| Data collection and processing | |||||||
| Wavelength (Å) | 0.9184 | 0.9184 | 0.9184 | 0.9184 | 0.9184 | 0.9184 | 0.9184 |
| Resolution range (Å) | 46.00–1.75 (1.85–1.75) |
42.98–1.60 (1.69–1.60) |
45.82–2.34 (2.43–2.34) |
45.51–1.75 (1.81–1.75) |
46.11–1.44 (1.52–1.44) |
45.6–1.82 (1.93–1.82) |
42.56–1.84 (1.95–1.84) |
| Space group | P 21 21 2 | P 21 21 2 | P 1 | P 21 21 2 | P 21 21 2 | P 21 21 2 | P 21 21 2 |
| Unit cell parameters a, b, c (Å) |
119.41, 169.94, 61.56 | 119.49, 169.42, 61.87 | 55.56, 123.39, 158.41 | 117.27, 169.04, 60.85 | 118.38, 169.64, 62.01 | 118.92, 169.00, 60.83 | 118.74, 168.66, 61.04 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 106.0, 91.3, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Total reflections | 827 399 (127246) |
1 051 475 (141600) | 817 849 (38849) |
800 678 (130782) |
1 487 502 (226718) |
716 118 (110546) |
711 417 (114374) |
| Unique reflections | 125 138 (19837) | 162 966 (23841) | 164 477 (10787) | 121 942 (19452) | 225 933 (34983) | 108 394 (16652) | 106 853 (16793) |
| Completeness (%) | 98.1 (97.4) | 97.4 (89.1) | 96.6 (85.6) | 99.1 (98.9) | 99.0 (95.5) | 98.5 (94.5) | 99.2 (97.5) |
| I/σI | 10.72 (0.55) | 11.26 (0.57) | 7.45 (1.22) | 14.49 (1.49) | 12.12 (0.62) | 11.05 (1.39) | 13.27 (1.24) |
| Wilson B-factor (Å2) | 40.24 | 33.73 | 40.21 | 29.41 | 28.65 | 36.4 | 35.85 |
| CC1/2 (%) | 99.9 (63.2) | 99.9 (66.9) | 99.3 (60.2) | 99.9 (69.0) | 99.9 (59.7) | 99.9 (84.0) | 99.9 (80.7) |
| Refinement | |||||||
| Reflections used in refinement | 124 949 | 162 789 | 164 476 | 121 929 | 225 005 | 108 233 | 106 812 |
| Reflections used for R-free | 2071 | 2085 | 2465 | 2099 | 2348 | 2093 | 2094 |
| R-work | 0.2052 | 0.1873 | 0.2272 | 0.1784 | 0.1714 | 0.1909 | 0.1838 |
| R-free | 0.2238 | 0.2115 | 0.2490 | 0.1944 | 0.1888 | 0.2103 | 0.2006 |
| No. of non-H atoms | 7904 | 7990 | 29 093 | 8056 | 8292 | 7732 | 7719 |
| Protein | 7183 | 7224 | 28 082 | 7164 | 7200 | 7159 | 7205 |
| Ligands | 184 | 179 | 559 | 156 | 179 | 162 | 167 |
| Water | 537 | 587 | 452 | 736 | 913 | 411 | 347 |
| RMS (bonds) (Å) | 0.012 | 0.012 | 0.012 | 0.009 | 0.006 | 0.006 | 0.006 |
| RMS (angles) (°) | 1.19 | 1.28 | 1.26 | 1.35 | 1.23 | 1.16 | 1.15 |
| Ramachandran favored (%) | 98.21 | 98.63 | 97.93 | 98.42 | 98.31 | 98.10 | 98.42 |
| Ramachandran allowed (%) | 1.79 | 1.37 | 2.07 | 1.58 | 1.69 | 1.90 | 1.58 |
| Ramachandran outliers (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Rotamer outliers (%) | 1.43 | 2.18 | 3.76 | 1.30 | 1.42 | 2.48 | 2.07 |
| Average B-factor | 49.15 | 42.17 | 58.35 | 28.28 | 30.43 | 43.31 | 41.50 |
| Macromolecules | 49.01 | 41.96 | 58.64 | 27.67 | 29.49 | 43.47 | 41.66 |
| Ligands | 61.36 | 47.45 | 54.40 | 34.68 | 32.45 | 42.84 | 41.59 |
| Solvent | 46.84 | 43.15 | 45.04 | 32.83 | 37.50 | 40.73 | 38.11 |
| Number of TLS groups | 15 | 12 | 53 | 13 | 12 | 16 | 9 |
| Molprobity results | |||||||
| Clashscore | 0.88 (99th percentile) |
2.62 (99th percentile) |
8.13 (97th percentile) |
2.66 (99th percentile) |
2.22 (99th percentile) |
2.65 (99th percentile) |
3.18 (98th percentile) |
| Molprobity score | 0.89 (100th percentile) |
1.31 (96th percentile) |
1.90 (94th percentile) |
1.14 (99th percentile) |
1.11 (98th percentile) |
1.35 (97th percentile) |
1.35 (98th percentile) |
*Statistics for the highest-resolution shell are shown in parentheses.
The 1.75 Å resolution structure of hE3
The hE3 structure of the yet highest (1.75 Å) resolution (Fig. 2) was analyzed for novel structural features and in particular areas of specific interest relevant for the pathogenic mutations studied here. The two monomers in the ASU are related by non-crystallographic two-fold axis symmetry and form a 3894 Å2 interface area (interactions of the homodimer interface are listed in Supplementary Material, Table S1). The overall organization of the active site (shown in Fig. 3A), involving the Cys45-Cys50 pair, the FAD isoalloxazine ring, His452′ and its H-bonding partner Glu457′, was unaltered as compared with the previously published hE3 structures (18,19,37,42,43). The Cys45-Cys50 disulfide bond was modeled in a partially reduced form (Fig. 3A).
Figure 2.
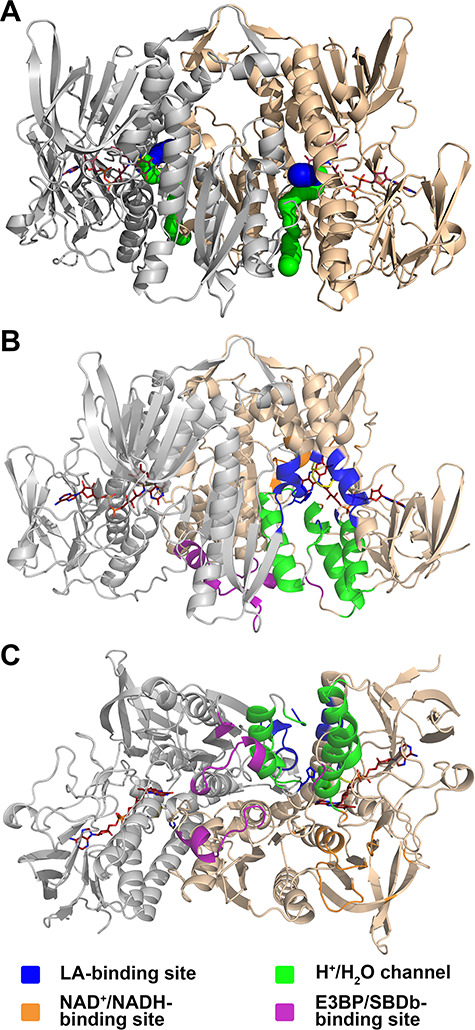
Functionally important regions in hE3. (A) The hE3 structure (PDB ID: 6I4Q) is represented in a similar way, but in a different orientation, to Fig. 1 for reference to B and C (colors and all representations are as for Fig. 1). (B and C) Functionally important regions in hE3 under investigation in this study are represented in two orientations in monomer A. Selected residues from the adjacent monomer also contribute to the formation of the active site, the LA-binding site and the H+/H2O channel. These residues are not distinguished by coloring in this figure (see also Supplementary Material, Fig. S6).
Figure 3.
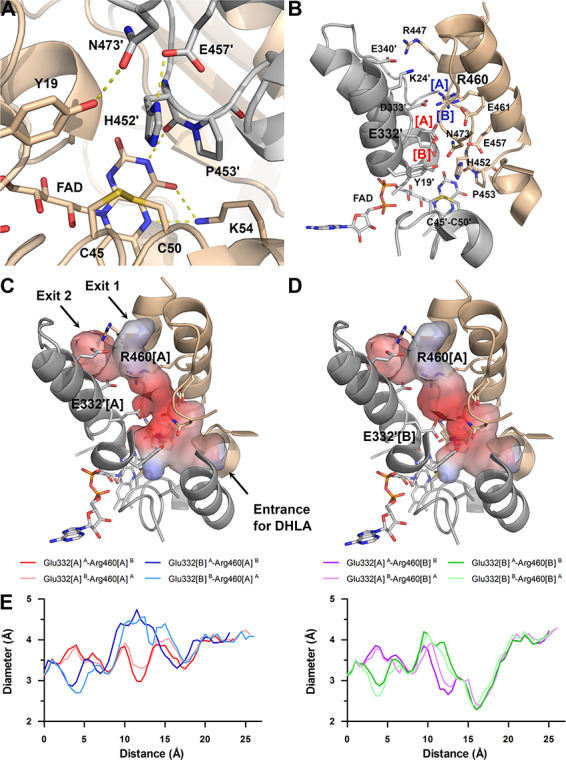
Active site and its accessibility in the hE3 structure (PDB ID: 6I4Q). (A) Close-up view on the active site: Cys45-Cys50, FAD and His452′ (all depicted as sticks). Chains A and B are colored beige and grey, respectively. H-bonds crucial for catalytic activity are shown as yellow dashed lines. (B) Secondary structural elements that encompass the LA-binding and H+/H2O channels as well as polar/protonatable side chains along the H+/H2O channel are displayed together with the active site in chain B. Alternative conformers are designated with square brackets. Glu332[A] was dominant in both chains, whereas Arg460[A] and [B] were dominant in chains A and B, respectively. (C) Channel surface potentials for Arg460[A]-Glu332′[A]. (D) Channel surface potentials for Arg460[A]-Glu332′[B]. For (C) and (D), surface potentials are displayed on an arbitrary scale (red: negative, blue: positive). The Pro16-Gly29 helix is partially omitted for clarity and thus Lys24 (contributing to surface polarity) is also omitted. Orientations in panels C and D are slightly different relative to panel B. (E) H+/H2O channel diameter along the channel axis as a function of distance from the active site is plotted with respect to the Glu332 and Arg460 alternative conformers. All combinations with Arg460[A] and [B] are in the left and right plots, respectively; A and B in superscript designate the chains.
The active sites are located on the si faces of the FAD isoalloxazine rings and each of them is accessible through an LA-binding substrate channel, as well as a more hydrophilic H+/H2O channel from a nearly perpendicular direction (Figs. 2A and 3B–D). Both types of channels are located in the dimer interface region while several inter-subunit interactions involve residues of the channel-forming secondary structural elements. The LA-binding channel is framed by Pro16, Tyr19, Leu46, Ile51, Ala98-Ile103, Leu106, Ala386′, Ala389′-Ser392′, Lys395′ and Asn473′-Phe474′, whose side chains secure a highly hydrophobic environment for LA (Figs. 2B–C and 3C–D). Leu46 adopted an intermediate state between two conformations (not shown), which were reported earlier (18,19,37,42,43); the high B-factor also suggested a flexible side chain that likely only becomes fixed upon binding LA.
The H+/H2O channel is ~26 Å long and surrounded by four helices (Pro16-Gly29, Leu327-Gly344, Glu443′-Val448′ and His452′-Phe468′) and part of the C-terminal loop (Ser471′-Asn473′) (Figs. 2B–C and 3B–D). This channel possesses an exit to the protein surface divided by the Glu340-Arg447′ salt bridge. Hereinafter, the shorter and straighter path leading to the protein surface (the path with Exit 1 in Fig. 3C) will be considered as the H+/H2O channel, if not stated otherwise. The H+/H2O channel surface is more hydrophilic than the one in the LA-binding channel due to the charged side chains of Lys24, Glu332, Asp333, Glu340, Glu443′, Arg447′, Glu457′, Arg460′ and Glu461′ (Fig. 3B). Alternative conformations for Glu332 and Arg460 were evident only in the current hE3 structure, owing to the higher resolution. Glu332, which together with Glu457′ and Glu461′, create a highly negative environment in the entry of the channel, either extended toward Arg460′ or aligned with the catalytic His452′ (Fig. 3B–D). Dynamics of Glu332 is also justified by the flexibility of the sole interacting partner Arg460′ (see Supplementary Material, Table S1). Arg460′ greatly contributes to the integrity of the channel by forming a salt bridge with Asp333. The differing dominant conformations of Arg460 in the two chains also reflect the considerable dynamics of this residue. The mobile Glu332 and Arg460′ side chains jointly regulate the inner diameter of the channel (Fig. 3E), while Glu332 also significantly affects the local charge distribution (Fig. 3C and D). The channel in its entirety is widest with Glu332[A]-Arg460′[A]; Glu332[A] is forming the 3–3.2 Å wide bottlenecks of the channels together with either Glu457′ and Asn473′ or Glu461′ and Arg460′[A] in the hE3 dimer. Transition of Glu332[A] to Glu332[B] generates a new bottleneck formed by Tyr19, Glu332[B] and His452′ with a 2.7–2.9 Å diameter. Transition from Arg460′[A] to Arg460′[B] significantly decreases the inner diameter in the vicinity to 2.3–2.4 Å, while the new bottleneck is formed by Arg460′[B], Leu464′ and Ile472′, regardless of the Glu332 conformation.
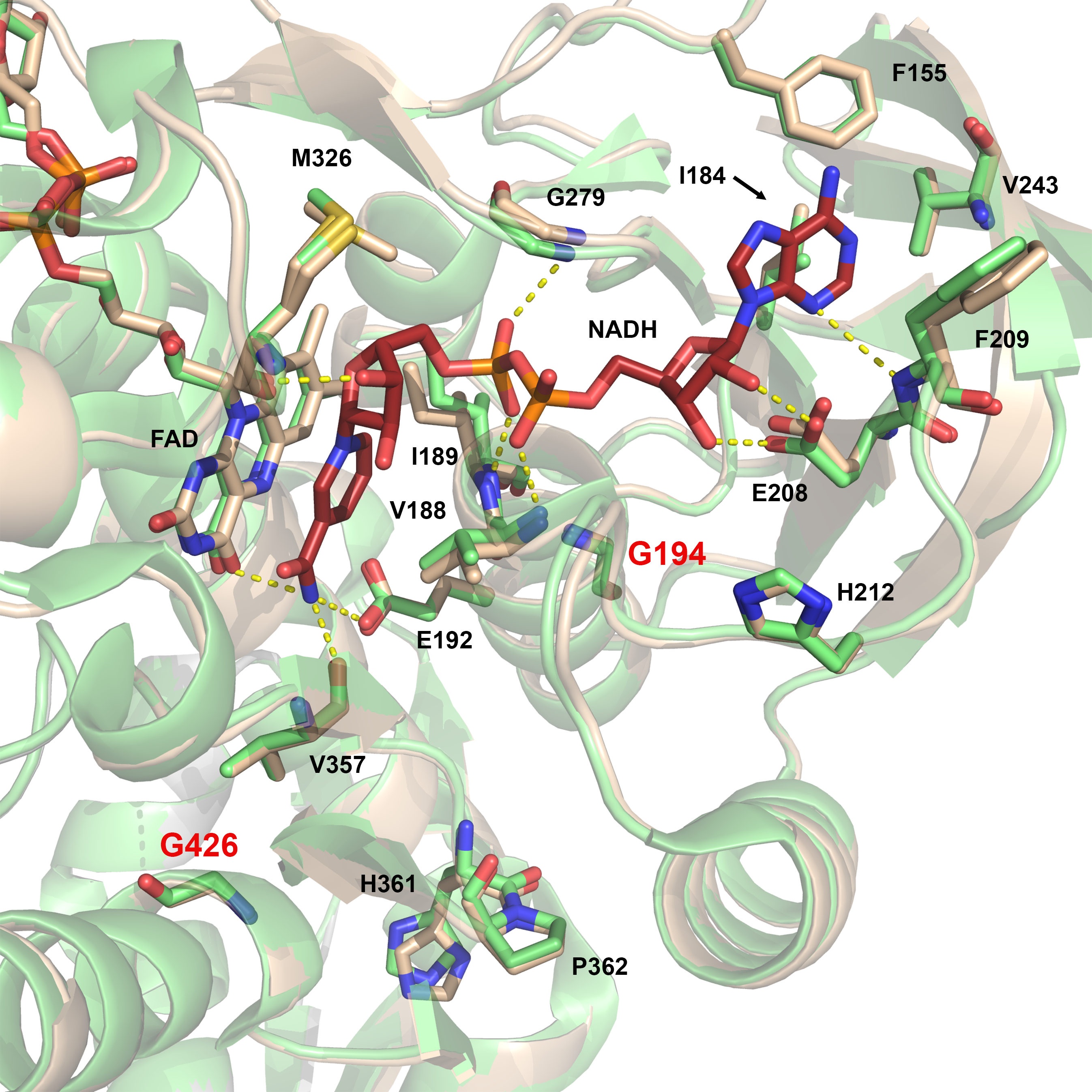
The NAD+/NADH-binding region is located on the re face of the FAD isoalloxazine ring and involves the N-terminus of the Val188-Gly201 helix and the preceding loop on the Ile184-Glu192 segment, as well as other loops (Glu208-Gly215, Cys277-Arg280) and four rather uninvolved amino acids (Phe155, Val243, Met326, Val357), according to hE3 structures determined in the presence of NAD+ and NADH by others before (19) (Supplementary Material, Fig. S3). The neutral, hydrophobic binding site in oxidized hE3 promotes NADH binding, as pointed out by Brautigam et al. (19).
The hE3 binds to complex-specific hE2 subunits in the hKGDHc and hBCKDHc, and to a non-catalytic, E2-homologous E3-binding protein (E3BP, also known as Protein X) in the hPDHc. The overlapping interfaces to E3BP (37,43) and hBCKDHc-E2 (42) are dynamic in nature (18) and comprise Glu437, Tyr438, Glu443, Asp444 (and Glu439 for E3BP) as interacting residues. This region is heavily involved in crystal contacts in the present hE3 structure (Supplementary Material, Fig. S4). Relevant to the pathogenic D444V-hE3 variant (18) and E2/E3BP–hE3 subcomplex formation (37,42,43) (a ‘subcomplex’ being a complex of two or more enzyme components in an incomplete multienzyme complex), a three-party stacking interaction was formed by Tyr438, Tyr438′ and a symmetry-related Arg307 in the present hE3 structure (Supplementary Material, Fig. S4B). The same type of interdimer interaction (crystallization artifact) was observed earlier in hE3 (19). Due to this stacking interaction, the rather open conformation of the tyrosines seen in two of the four dimers in our previous hE3 structure (18) (PDB ID: 5NHG) was not observed in the present hE3 structure, consequently H-bonding between Tyr438 and Asp444′ could only be formed one way (see Supplementary Material, Table S1). Importantly, the Tyr438-Tyr438′ stacking is stabilized by E3BP and SBDb in a similar fashion (37,42,43). Two surface loops (Gly344-Val354, Lys410-Arg414) also contribute to the interaction with E3BP, via Val347, His348, Thr412 and Asp413 (37,43), which were again affected by crystal contacts mostly in one of the two chains (Supplementary Material, Fig. S4B and C). The only E3BP-binding residue not affected by such interactions was Arg447. For SBDb-binding, only Asp413 and Arg414 are recruited from the above two surface loops (42) and they were also involved in crystal contacts. Since most of these crystal contacts were also formed in the variant structures, the respective structural alterations often could not be addressed.
Disease-causing hE3 variants
Active site variant: P453L-hE3
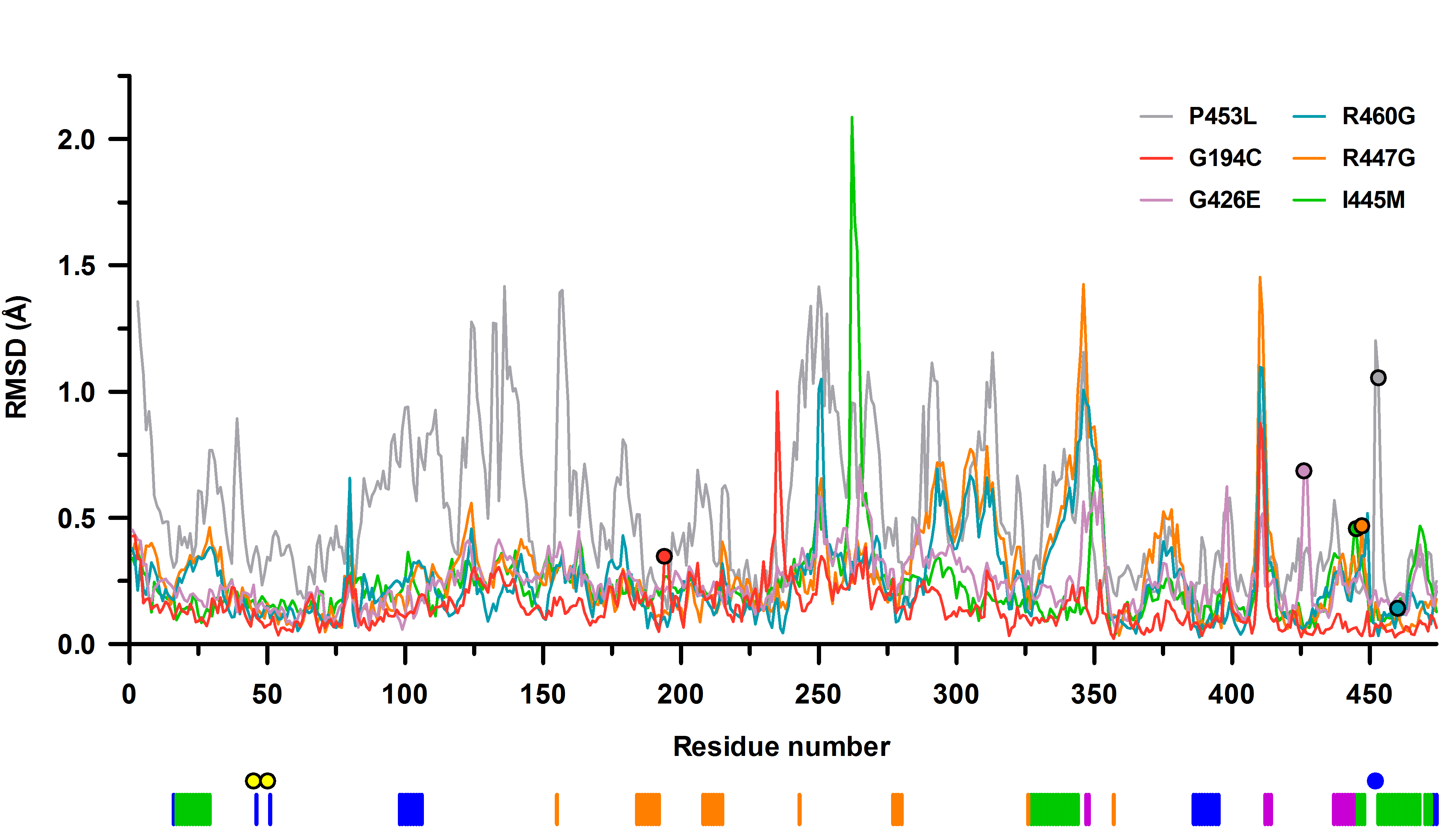
The P453L-hE3 structure was refined to 2.34 Å resolution with eight monomers in the ASU (chains A–H). Superimposition of all four P453L-hE3 dimers to hE3 yielded mean root-mean-square deviation (RMSD) values of 0.52 (Cα) and 1.19 Å (side chain, non-H atoms).
In hE3, the conserved Pro453 in the cis configuration is essential for proper orientation of the adjacent catalytic His452 (there is an H-bond between His452 and FAD′, see Supplementary Material, Table S2 and Fig. 3A). In the absence of the cis-proline, the active site is significantly distorted in P453L-hE3 (Fig. 4B and C); however, residue level RMSDs indicate that the structural changes are not limited to the active site (Fig. 4E). In P453L-hE3, the catalytic His452 is in a trans peptide bond with Leu453 that indeed led to the displacement of His452, hence the crucial interaction(s) with FAD′ (and Glu457) could not develop (see Fig. 4B and C and Supplementary Material, Table S2). Glu457 was also misplaced due to the lack of stabilization by His452. A slight displacement of Tyr19′ and the altered conformation of Glu461 also yielded a lost H-bond between Tyr19′ and Asn473 (Fig. 4B), which might also adversely affect the active site (see Discussion).
Figure 4.
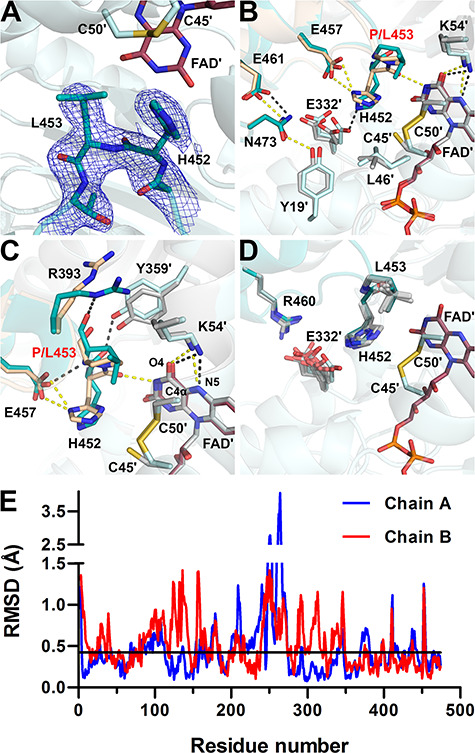
Active site structure in P453L-hE3 (PDB ID: 6I4Z). (A) (2mFo-DFc) composite omit map contoured at 1.5σ is shown as blue mesh for the mutated residue and adjacent amino acids for the Ala451-Thr454 segment in chain A. Electron density clearly supports the presence of the P453L substitution and trans peptide bond between His452 and Leu453 in P453L-hE3. Residues in chains A and B are dark and light teal, respectively, the stick representation of FAD is colored raspberry. (B and C) Active site area in P453L-hE3 (A-B dimer, dark/light teal; FAD′: raspberry; H-bonds: grey) and hE3 (beige/grey; FAD′: grey; H-bonds: yellow) in chain B. Average distances for Lys54(NZ)-FAD(O4) in hE3: 2.66 Å, P453L-hE3: 2.91 Å, while for Lys54(NZ)-FAD(N5) in hE3: 3.18 Å, P453L-hE3: 3.26 Å. (D) Conformational dynamics of His452, Leu453, Arg460, and Glu332′ in the eight P453L-hE3 monomers. (E) Average RMSD values for main chain atoms are plotted as a function of residue number in chains A and B upon A-B dimer superimposition of P453L-hE3 and hE3. The black horizontal line indicates average RMSD for main chain atoms (0.42 Å) after pairwise superimpositions of the four P453L-hE3 dimers.
The P453L substitution also triggered changes near the FAD isoalloxazine ring. The Tyr359′ side chain moved away from the isoalloxazine ring, likely due to the significant displacement of Arg393 (see Fig. 4C). The Arg393 is no longer involved in inter-subunit interactions with Glu363′ and Glu427′ (not shown), but extends toward Lys54′ that is essential in stabilizing FAD′ via H-bonding. The Lys54′ side chain was significantly altered in only one monomer; however, it displayed the greatest RMSD and electron density ambiguity among the variants. The Lys37 and Asn38 side chains moved away from the FAD adenosine ribose (Supplementary Material, Table S2) further weakening the FAD–protein interaction. Among residues implicated in NAD+/NADH-binding, only the Glu208-Gly215 peptide and Val243 were misplaced (Fig. 4E and Supplementary Material, Fig. S6).
Selected residues framing the LA-binding site and the H+/H2O channel were also affected: the Ala98-Leu106 helix at the entrance of the LA-binding site and the C-terminal ends of the Val20-Gly29 and Leu327-Gly344 helices at the exit of the H+/H2O channel were displaced to varying degrees (Fig. 4E and Supplementary Material, Fig. S6), likely modulating the accessibility of the active site. Glu332′, near the active site, exhibits a rather smeared dynamics (Fig. 4D), perhaps only because of the lower resolution. The displaced and mostly flipped His452 imidazole ring is at times in H-bonding distance to Glu332′[A]. The interaction was lost between Glu332′ and Arg460 along the H+/H2O channel as Arg460 could exclusively be modeled in a single conformation (Arg460[A] in hE3). In general, the H+/H2O channel became narrower around the active site, whose vicinity always comprises the bottleneck, as a result of the significantly displaced His452 and Leu46′. The bottleneck is often composed of Tyr19′ and Glu332′ and/or His452. The above effects and the often significantly displaced H+/H2O channel axis suggest that the channel was adversely affected.
Surface residues involved in E2/E3BP-hE3 subcomplex formation were modified to a rather small degree (primarily Val347, His348, Glu437 and Tyr438 were affected, see Fig. 4E and Supplementary Material, Fig. S6), which likely led to the decrease in affinity for E3BP (38).
NAD+/NADH-binding domain variant: G194C-hE3
The structure of this variant, refined to 1.69 Å resolution, is remarkably similar to hE3 (RMSD for Cα: 0.17 Å, for side chain non-H atoms: 0.66 Å; dimer fitting). The substitution perturbs a highly conserved helix (Val188-Gly201) whose N-terminus provides residues to the NAD+/NADH-binding site (Fig. 5 and Supplementary Material, Fig. S3). Although, conformational changes occurred in the neighboring β-sheets (especially for Val204, Val213, Phe235; see Fig. 5C), the NAD+/NADH-binding residues suffered no significant structural changes directly. Cys194 and Ser195 were both modeled with two alternative conformations in chain A suggesting a certain degree of mobility gained in the region, an effect that might propagate to the NAD+/NADH-binding site via the helix dipole moments.
Figure 5.
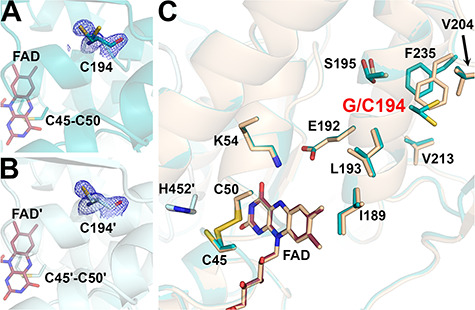
Structure of the G194C-hE3 variant (PDB ID: 6I4P). (A and B) Cys194 is shown in (2mFo-DFc) composite omit map (2.0σ) in chains A and B, respectively. Based on indubitable difference densities, Cys194 could only be modeled with two alternative conformers in chain A (A). Active site disulfide and FAD (raspberry) are represented with sticks. (C) Active and substitution sites are shown in the superimposed structures of G194C-hE3 (dark teal; FAD: raspberry) and hE3 (beige; FAD: beige) in chain A. Cartoons are semitransparent and peptide segment 160–180 was removed for clarity.
The G194C substitution induced no direct alterations to the active site, the LA-binding site or the FAD-binding residues. The dimer interface suffered no significant change in area (3822 Å2 versus 3894 Å2 in hE3) losing only a single interaction (Asp333-Arg460′[A], due to the absence of Arg460′[A] from hE3, not shown). This latter effect is the sole alteration in the H+/H2O channel (the Glu332 side chain remained mobile, the geometry and composition of the bottleneck was unchanged). As for the E3BP/SBDb-binding residues, only Thr412 and its surrounding peptide segment (Lys410-Thr412) were displaced, primarily in chain B (Supplementary Material, Fig. S6).
Dimer interface variants
R460G-hE3
This structure was refined to 1.44 Å resolution; RMSD for Cα: 0.26 Å, for side chain non-H atoms: 0.67 Å, relative to the hE3 dimer. Neither the inter-subunit nor the FAD-binding interactions (Supplementary Material, Table S1 and S2, respectively) were significantly affected by the substitution with the exception of those that directly involve Arg460 (especially the salt bridge with Asp333′). Despite affecting the interface region, this substitution caused no dissociation to monomers; the buried area of the interface is 3735 Å2 (versus 3894 Å2 in hE3). In hE3, the Arg460-Asp333′ salt bridge links two helices that surround the H+/H2O channel while Arg460 also forms interaction with Glu332′ (see above). Upon the substitution, the Asp333′ side chain conformation became destabilized and could only be modeled with two conformers (different dominant conformers in the two monomers) (Fig. 6A and B). Furthermore, the main chains of an entire H+/H2O channel forming helix (Leu327-Gly344) and the subsequent loop (Gly345-Cys353) were also slightly displaced (Fig. 6B and Supplementary Material, Fig. S6) affecting not only the integrity of the H+/H2O channel, but also two residues (Val347, His348) participating in E3BP binding (this effect is mostly hidden in chain A where the surface residues between Ala343 and His348 are involved in crystal contacts). Additionally, in the absence of Arg460, the negative charge of Asp333′ is not neutralized by any electrostatic interaction, which accounts for the increased negative surface potential of the H+/H2O channel locally (Fig. 6D). Glu332′ adopts only a single conformation in R460G-hE3 that leads to decreased dynamics and reduced negative polarity in the initial segment of the H+/H2O channel. Glu332′ contributes to the formation of the 3.0–3.2 Å bottleneck together with Glu457 and Asn473. Disappearance of the Arg460 side chain significantly widens the H+/H2O channel locally, in the void volume H2O molecules could be modeled (Fig. 6A and C). Solvent accessibility of the peptide segment connecting Arg447 on the surface with His452 in the active site increased (Fig. 6A and C). The His450 imidazole ring became tilted likely due to the displacement of the Cys449-His450 peptide bond. The Glu443-His450 segment was slightly shifted (Fig. 6C and Supplementary Material, Fig. S6) affecting Glu443, Asp444 and Arg447, all being E3BP/SBDb-binding residues. A more distant surface loop (Lys410-Arg414) that comprises three additional E3BP/SBDb-binding residues (Thr412, Asp413, Arg414) was also displaced affecting primarily Thr412 in chain B (see Supplementary Material, Fig. S6). Near the active site, modulating the diameters of both the LA-binding and H+/H2O channels increased flexibility (alternative conformers) of the Leu46 side chain was observed (not shown). Catalytic residues (His452, Cys45, Cys50, Glu457) were not affected directly by the substitution.
Figure 6.
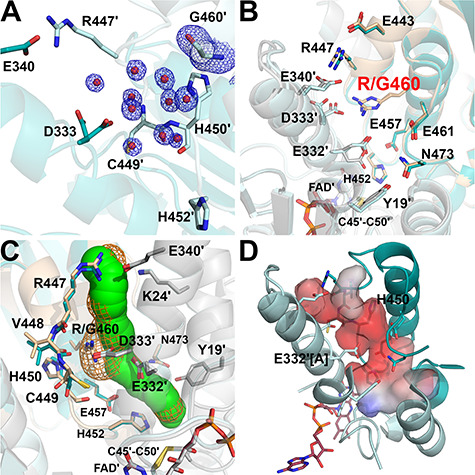
Structure of the R460G-hE3 variant (PDB ID: 6I4R). (A) (2mFo-DFc) composite omit map (1.5σ) confirms the R460G substitution [Arg side chain was replaced with H2O (red spheres)] in R460G-hE3. The two chains are colored with two shades of teal. (B) H+/H2O channel and relevant polar side chains are shown (with the Pro16-Gly29 helix partially omitted, see Fig. 3C and D) in the superimposed R460G-hE3 (dark/light teal; FAD′: raspberry) and hE3 (beige/grey; FAD′: beige) structures. Glu332 was modeled with a single conformer in R460G-hE3. (C) H+/H2O channels computed by Caver are shown in the superimposed hE3 (green) and R460G-hE3 (orange mesh) structures; only dominant conformers were considered for calculation. (D) Surface polarity analysis of the LA-binding and H+/H2O channels in R460G-hE3 is presented using an arbitrary scale (red: negative, blue: positive); orientation and secondary structural elements are as for hE3 in Fig. 3C and D.
R447G-hE3
The R447G-hE3 structure was refined to 1.75 Å resolution; RMSD for Cα: 0.32 Å, for side chain non-H atoms: 0.71 Å, relative to the hE3 dimer. Inter-subunit and FAD-binding interactions (Supplementary Material, Table S1 and S2, respectively) were not affected significantly, except for the Arg447-Glu340′ salt bridge. Dissociation of the homodimer was not evident; the interface area is 3735 Å2 (versus 3894 Å2 in hE3). The Arg447-Glu340′ salt bridge, unlike the Arg460-Asp333′ one, resides in hE3 at the exit of the H+/H2O channel and splits the exit into two narrower outlets (18) (see Fig. 3C). Although Glu340′ is thus stabilized by no electrostatic interaction in this variant, its energetically favorable conformation was retained (Fig. 7B). The Ile337′-Asn352′ segment, which includes Glu340′, was however partially displaced (Fig. 7B and Supplementary Material, Fig. S6) underlying the importance of the Arg447-Glu340′ salt bridge in stabilizing one of the main H+/H2O channel-forming helices (Leu327′-Gly344′) and a random coil on the protein surface (Gly345′-Val354′) that binds to E3BP. The substitution widened the H+/H2O channel exit to be a funnel-like structure, while the channel length became 21 Å (versus ~26 Å in hE3). The negatively charged surface near the channel exit became even more negative by the dissociated Glu340′ (Fig. 7C and D). The geometry of the H+/H2O channel is primarily regulated by Glu332′, as no significant mobility was observed for Arg460. Similarly to hE3, Glu332′ was modeled with two alternative conformers (Fig. 7B). With Glu332′[A], a 3 Å bottleneck was formed by Glu332′[A], Glu457 and Asn473 (Fig. 7B and C). Upon Glu332′[B], the bottleneck (2.6 Å) is formed by Glu332′[B], Tyr19′ and His452 (Fig. 7B and C). To a lesser degree, a very narrow bottleneck (2.2 Å) was also formed by Arg460[B], Leu464 and Ile336′, similarly to hE3.
Figure 7.
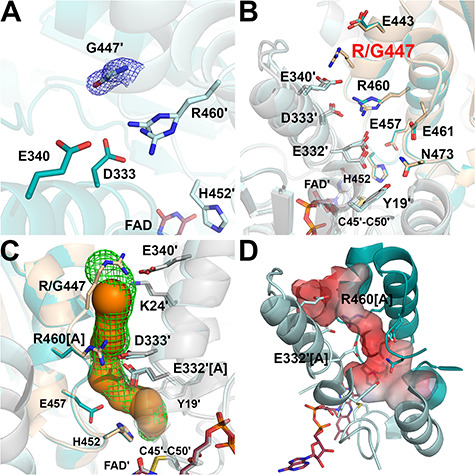
Structure of the R447G-hE3 variant (PDB ID: 6I4S). (A) (2mFo-DFc) composite omit map (1.5σ) confirms the substitution in R447G-hE3. The two chains are colored with two shades of teal. Arg460 was modeled with two alternative conformations in chain B. (B) H+/H2O channel and relevant polar side chains are shown (as for Fig. 6B) in R447G-hE3 (dark/light teal; FAD′: raspberry) and hE3 (beige/grey; FAD′: beige). (C) H+/H2O channels computed by Caver are shown in hE3 (green mesh) and R447G-hE3 (orange); only dominant conformers were considered for calculation. (D) Surface polarity analysis of the LA-binding and H+/H2O channels in R447G-hE3 is presented using an arbitrary scale (red: negative, blue: positive).
As for the E3BP/SBDb-binding residues: Arg447 was missing, Glu443, Val347, His348, Thr412 and Asp413 were partially displaced (see above and Supplementary Material, Fig. S6), while the other residues were stabilized by inter-subunit interactions and/or crystal contacts.
I445M-hE3
This structure was refined to 1.82 Å resolution; RMSD for Cα: 0.26 Å, for side chain non-H atoms: 0.74 Å, relative to the hE3 dimer. FAD-binding residues were not affected, whereas a few interactions of the dimer interface were perturbed (see below); the interface area is 3730 Å2 (versus 3894 Å2 in hE3).
In hE3, the residue Ile445 resides in a short helical segment (Glu443-Val448) at the dimer interface near the H+/H2O channel exit; it is of potential significance that Ile445′ is in close spatial proximity (Fig. 8A). Substitution with Met interfered little with the Glu443-Val448 helix, unlike with the adjacent His452-Phe468 helix; e.g. the side chains of Phe459 and Glu461 were rather significantly displaced (Fig. 8B). Arg460 and Glu332′ virtually lost their mobility and stabilize one another via H-bonding. The displacements of Glu461 (see above) and Ile472 (at the C-terminus, not shown) perturbed the H+/H2O channel axis, while the bottleneck was formed by Arg460, Ile472 and Glu461/Leu464 with a diameter of 2.1–2.5 Å. Leu46′ was modeled with two alternative conformers (not shown); therefore, it is capable of dynamically modulating the diameters of both the LA-binding and H+/H2O channels.
Figure 8.
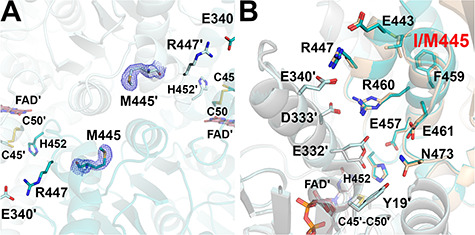
Structure of the I445M-hE3 variant (PDB ID: 6I4T). (A) (2mFo-DFc) composite omit map (2.0σ) confirms the substitution in I445M-hE3. (B) H+/H2O channel and relevant polar side chains are shown (as for Fig. 6B) in I445M-hE3 (dark/light teal; FAD′: raspberry) and hE3 (beige/grey; FAD′: beige). In I445M-hE3, both Glu332 and Arg460 were modeled with single conformations.
From among the E3BP/SBDb-binding residues, only Tyr438, Asp444, Glu437, Gly439, Glu443, Arg447 and Thr412 were somewhat displaced (Glu443 and Arg447 are shown in Fig. 8B; see also Supplementary Material, Fig. S6).
G426E-hE3
This structure was refined to 1.84 Å resolution; RMSD for Cα: 0.27 Å, for non-H side chain atoms: 0.80 Å, relative to the hE3 dimer. No inter-subunit or FAD-binding interactions were affected directly; the dimer interface is 3794 Å2 (versus 3894 Å2 in hE3).
In hE3, Gly426 resides near the active site (Supplementary Material, Fig. S3), adjacent to the N-terminus of the Ala425-Tyr438 helix, in the interface domain. In G426E-hE3, the active site and the LA-binding channel were unaffected. Glu substitution was placed into a region heavily involved in inter-subunit interactions and it obstructed the formation of the Glu427-Arg393′ and Glu363-Arg393′ salt bridges (see Supplementary Material, Table S1 and Fig. 9). The significantly displaced Arg393′ interacts with Glu426 in G426E-hE3; however, this newly formed interaction cannot completely stabilize Glu426, which exhibits two alternative conformations. The lack of interaction with Arg393′ and the dynamics of Glu426 led to a flexible Glu363. The alternative conformers of Glu426 and Glu363 appear to be coupled for steric reasons (Fig. 9B). Glu427 was not displaced. Glu363 dynamics perturbed His361, which is in a cis-peptide bond with Pro362 (not shown); integrity of this latter bonding appears to be essential for the formation of a tight β-turn close to the NAD+/NADH-binding site.
Figure 9.
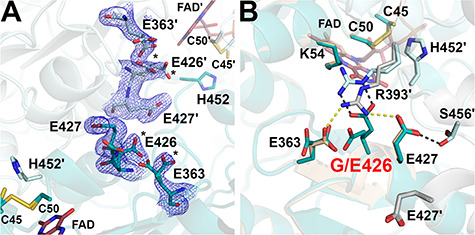
Structure of the G426E-hE3 variant (PDB ID: 6I4U). (A) (2mFo-DFc) composite omit map (1.5σ) confirms the substitution in G426E-hE3. Dominant conformers of Glu426 and Asp363 are labeled with asterisks (*) in both chains. Cys45-Cys50, His452 and FAD are also shown to demonstrate their proximity to the substitution site. (B) Close-up view on the substitution site in G426E-hE3 (dark/light teal; FAD: raspberry) and hE3 (beige/grey; FAD: beige). Disrupted (Glu363-Arg393′, Glu427-Arg393′) and newly-formed (Glu426-Arg393′, Glu427-Ser456′) intermonomeric interactions upon the substitution are represented as yellow and grey dashed lines, respectively.
The G426E substitution does not appear to be that directly associated with the H+/H2O channel. Similarly to hE3, the Glu332 and Arg460′ side chains exhibit two alternative conformations each, which appear to be coupled (not shown). Channel geometry is dynamically modulated by alternating between the Glu332[A]-Arg460′[B] and Glu332[B]-Arg460′[A] configurations leading to a bottleneck by either His452′, E457′ and N473′ (2.8 Å) or Tyr19, Glu332 and His452′ (3.1 Å) (not shown). Leu46 was modeled in a single conformation (not shown), which was also sampled in hE3 and most variants investigated here, and whose interpretation is hence inconclusive. Glu461 was partially displaced (not shown), which led to a narrower channel locally, similarly to I445M-hE3.
The 10/6 E3BP/SBDb-binding residues were least perturbed by this dimer interface substitution: only Thr412, Val347 and His348 were displaced to a rather small extent (Supplementary Material, Fig. S6).
Discussion
Similarly to D444V-hE3 (12,18), there were no significant overall structural changes detected in the disease-causing hE3 variants investigated here, relative to hE3. The greatest structural effects/alterations occurred in P453L-hE3 (see Supplementary Material, Fig. S6), which was shown earlier to be the most deleterious pathogenic variant (12,44). It appears that the evolutionarily well-conserved (14,17) and hence delicately orchestrated intrinsic, multivariant (29) mechanisms of the dihydrolipoamide dehydrogenase enzyme can easily be perturbed at times even by the slightest deformations in local structures. However, the substantial residual enzymatic activities remaining in some of the pathogenic hE3 variants here investigated (Table 2) necessitate a mechanistic competence at a significant level, thus structurally destructive changes perhaps should not even be expected in these variants. This may simply be because given the importance of hE3 in numerous, key metabolic pathways, patients with more dramatic, destructive mutations likely would not survive long enough for such mutations to be detected or recognized. Perhaps the flexibility and ability to overcome the changes imposed by the substitutions is necessitated in view of the variety of E2 components with which the E3 must partner. In other words, a substitution may be deleterious in one complex, but not in the others.
Table 2.
Structural and functional alterations in disease-causing hE3 variants studied
| Substitution | Affected region | Former functional studies | Present crystallograpic study | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Residual enzymatic activity (% of control) | Source | Genotype in patient | Reference | Major structural alterations | |||||
| E3 | PDHc | KGDHc | BCKDHc | ||||||
| P453L | Active site | 6 | – | – | – | F | P453L/K37E | (44) | Extensively perturbed active site including catalytic residues. |
| 4–9 | – | – | – | RP | – | (12) | |||
| ND | – | – | – | RP in yeast cell | – | (72) | |||
| G194C | NAD+/NADH-binding domain | 7–21M, 8–34F |
11–12M, 20F |
12–19M | – | M, F | G194C/G194C | (30–32,34) | Altered charge equilibrium and induced dynamics near the nicotinamide-binding site. |
| 8–20 | – | – | – | M | Y35X*/G194C | (30) | |||
| 10–30 | 69 | 44 | 58 | F | G194C/I12T | (26) | |||
| 47–100† | 29 | 61 | – | RP | – | (10,12,36,50) | |||
| G426E | Interface domain | – | 44 | 22 | 43 | F | I40fs*/G426E | (51) | |
| R460G | 1.5L, 14F | 26L, 11F | 20F | – | L, F | Y35X*/R460G | (52) | Perturbed H+/H2O channel forming residues (to various degrees) | |
| 10–91† | – | – | – | RP | – | (12,36–38,73) | |||
| R447G | 20 | 63 | 0 | 56 | F | R447G/R447G | (54) | ||
| 54–92† | – | – | – | RP | – | (36,37) | |||
| I445M | ND | 97‡ | – | – | F | I445M/I445M | (56) | ||
*Numbering reflects the immature protein sequence containing the 35 amino acid mitochondrial leader sequence.
†Enzymatic activities were measured under different assay conditions.
‡% of lower limit of control range.
Abbreviations:
ND = not detectable
F = fibroblasts
RP = recombinant protein
M = muscle homogenate
L = lymphocytes
fs = frame shift
hE3 structure at 1.75 Å resolution reveals dynamics in the H+/H2O-channel
Owing to the highest resolution for hE3 here achieved, new features of the hE3 structure were revealed. Although, this structure has not changed our previous understanding of the overall mechanism of action, the newly revealed features shed light on a few rather delicate details in the LADH reaction mechanism. Several alternative conformations and their occupancies could be determined in functionally relevant regions, which provided a better description especially of the H+/H2O channel dynamics. The H+/H2O channel is a solvent accessible channel that leads to the active site and through which H2O [17] and/or H+/H3O+ [19] can potentially leave the active site upon the entry and oxidation of the DHLA substrate. Most importantly, Glu332′ and Arg460 were found to dynamically control the geometry and polarity of the channel by adopting two alternative conformations (see Fig. 3B–E). Given the both physically and chemically dynamic characteristics of H+-translocation in a channel, likely requiring a series of acceptor/donor interactions, the newly revealed features of the protonatable and protonated side chains of Glu332′ and Arg460, respectively, are likely of significance. The H+/H2O channel is of special interest since several disease-causing dimer interface substitutions of hE3 are associated with this channel (see below) (18).
The higher resolution hE3 structure provided no additional details of potential mechanistic significance regarding the active site, the FAD prosthetic group and the LA- and NAD+/NADH-binding sites.
The hE3 residues that are involved in tethering to hPDHc and hBCKDHc (37,38,42,43) were found to be in either close spatial proximity to or interacting with symmetry-related residues. This resulted in limited opportunities for evaluating local dynamics and hence significant novel conclusions could not be drawn regarding this region.
The P453L substitution in the active site led to significant local and relayed structural alterations
The cis peptide bond of His452-Pro453 is crucial for the proper configuration of the active site in hE3 enabling His452(O) to form an H-bond with FAD′(N3) (18,19). Indispensability of this cis configuration was previously confirmed by substituting Pro453 with Val, an amino acid with a similar size, but propensity for trans peptide bonding, which resulted in a near complete loss in enzyme activity (45). According to the P453L-hE3 crystal structure, this pathogenic substitution indeed resulted in trans peptide bonding for His452-Leu453, as was predicted before (19), also triggering a significant remodeling of the active site to which the bigger size of the Leu side chain also contributed. The conformation and local environment of the catalytic His452 was significantly altered according to the crystal structure. Loss of the interaction with Glu457 likely perturbed the pKa of the His452 imidazole ring (22), thus His452 might have become less capable of properly promoting H+-shuffling in the catalytic mechanism (14,22). In the active site, the Tyr19′-Asn473 H-bond also became significantly weaker, or at times even entirely lost, which could contribute to the enhanced C-terminus flexibility observed before by HDX-MS at 25°C (39). The C-terminus (Gly469-Phe474) creates a physical barrier between the LA-binding and H+/H2O channels, hence its enhanced dynamics simultaneously perturbs both channels. A few E3BP-binding residues were also slightly displaced, which likely account for the 4-fold increase in Kd of the P453L-hE3–E3BP subcomplex (38); however, the primary reason for the severely reduced overall PDHc and KGDHc activities most likely is the significantly compromised LADH activity (29).
The substitution was previously found to perturb FAD-binding resulting in 34% FAD loss (12), which was supported by the loss of a few FAD-stabilizing interactions according to the P453L-hE3 structure. The loss of the His452(O)-FAD′(N3) H-bond likely also affects the FAD redox potential. Lys54′ forms H-bonds with the isoalloxazine O4 and N5 atoms, modulating the FAD′ redox potential and stabilizes the thiolate-FAD intermediate in the catalytic mechanism in hE3 (46). In the variant, the bulkier side chain of Arg393 moving closer to Leu453 and Lys54′, and the altered Tyr359′ side chain conformation likely all contributed to the slight displacement of Lys54′. This displacement, however, interfered little with the above-mentioned H-bonds with FAD′ (Fig. 4C). The flexibility/exposure of the Ile35-Asp70 segment was also altered according to HDX-MS (39). The above structural alterations, which all took place near the FAD N5-C4α locus (Fig. 3C), where superoxide formation likely occurs (47), could explain the dramatically increased rate of superoxide/H2O2 generation detected for P453L-hE3 (12). Since properly positioned and positively charged side chains around the FAD N5-C4α locus were reported to be important for oxygen reactivity (48,49), it can be hypothesized that the structural alterations regarding Lys54 and its local environment might also contribute to the significantly elevated ROS-generating capacity of P453L-hE3.
Substitutions in the spatial vicinity of the nicotinamide-binding site led to minor structural changes
Despite affecting two different domains in hE3, both the G194C and G426E substitutions take place in the direct proximity of the nicotinamide-binding site (see Supplementary Material, Fig. S3). Since these two replacements induced strikingly similar structural changes, we discuss them together below.
The cofactor- (NAD+/NADH/FAD) and LA-binding residues as well as the active site exhibited no significant structural alterations relative to hE3 in either G194C-hE3 or G426E-hE3. Therefore, the respective residues could not be directly associated with the FAD loss (12), increased KM for NAD+/NADH (26,50), loss in enzyme activity (36,50) [although in our hands there was essentially none (12), see Table 2], and enhanced ROS generation capacity (12), all measured for G194C-hE3, or with the moderately compromised overall multienzyme complex activities detected for G426E-hE3 (51) (neither LADH activity nor ROS production capacity has been reported in the literature for the isolated G426E-hE3). In a previous study, NADH binding displaced (primarily) the N-terminus of the Val188-Gly201 helix (NAD+ binding was not favored by the oxidized enzyme) (19); however, those displaced residues (Val188, Ile189) were not affected either in the crystal structures [or even in the H/D-exchange experiments for G194C-hE3 (39)]. The G194C and G426E substitutions introduced relatively large, polar side chains, where had been none, which, however, greatly interfered with the local charge distribution and even led to increased dynamics near the nicotinamide-binding pocket, according to the crystal structures.
Despite the small conformational changes of a few H+/H2O channel-forming residues, the H+/H2O channel appeared to be mostly unaltered in both G194C-hE3 and G426E-hE3. The E3BP/SBDb-binding residues were also the least perturbed by these substitutions.
In the H/D-exchange analysis of G194C-hE3 at 25°C, flexibility/solvent exposure alterations were observed in those regions where perturbation could indeed account for the biochemical data (39). Therefore, a plausible interpretation of both HDX-MS and crystallographic results is that minor perturbations of the local structures and dynamics that manifest themselves only at near physiological temperatures occur upon the G194C substitution. This rather modest structural deviation is also represented in the generally mild clinical phenotypes associated with this variant (29). HDX-MS analysis has not been reported in the literature for the isolated G426E-hE3. G426E-hE3 is another pathogenic hE3 variant that presents with relatively mild clinical pictures (51), hence the minor structural deviations displayed by the G426E-hE3 crystal structure relative to hE3 are also well justified.
Interface variants R460G-hE3, R447G-hE3 and I445M-hE3 primarily perturb the H+/H2O channel
Arg447, Arg460, Asp444 and Glu340 are highly conserved residues in the dimer interface and play key roles in the stabilization of the hE3 homodimer. Pathogenic substitutions of these residues (R447G, R460G, D444V and E340K) led to very severe, but remarkably different clinical manifestations and often to premature death (31,52–54). The pathomechanism of action for all the above variants was proposed to be the dissociation of the functional homodimer (19,31,51); however, dissociation to monomers as a possible pathomechanism for E3-deficiency was later ruled out experimentally (11,12,37,39). Crystal structures of R460G-hE3, R447G-hE3 and I445M-hE3 support these latter results. In these variants, LADH activity was compromised to various degrees (Table 2), but diaphorase activity, whose mechanism is analogous to the one of superoxide generation by hE3 (29,55), was enhanced (I445M-hE3 was not measured) (36); ROS-generating capacity measured directly for R460G-hE3 showed a decrease relative to hE3 (12). R460G-hE3 and R447G-hE3 caused oxidative damage to the LA cofactors of KGDHc and PDHc in a yeast model (36). Since R460G-hE3, R447G-hE3 and I445M-hE3 displayed no enhanced ROS generation in a direct assay, no structural conclusions were sought in this regard.
In D444V-hE3, the altered catalytic and ROS-generating activities were attributed to perturbation of the geometry and polarity of the H+/H2O channel (18). Glu340, Arg447 and Arg460 are even more closely associated with the structural integrity of the H+/H2O channel than Asp444; therefore, it was hypothesized that the pathogenic substitutions of all of these residues ought to cause structural alterations to the channel. The R460G and R447G substitutions indeed perturbed the geometry and polarity of the H+/H2O channel according to the crystal structures (no crystal structure is available yet for E340K-hE3). Local changes induced by the elimination of the Arg460 side chain, a highly conserved side chain in its position (52), are in accord with HDX-MS results, where perturbation was detected for the Ile445-Ala458 segment (39). However, the crystal structure uncovered that neither the catalytic His452 nor any of the active site residues including Glu457 were affected by the R460G substitution. Substitution of Arg447 with Gly primarily affected the H+/H2O channel exit, leading to a funnel-like cavity and the overall negative surface potential, which increased locally. The R460G and R447G substitution sites are distant from the active site and led to no displacements of catalytic or FAD/NAD+/NADH-binding residues.
Therefore, similarly to D444V-hE3 (18), the compromised activities due to the R460G and R447G substitutions are primarily proposed to be the results of H+/H2O channel perturbations, namely loss of a charged residue within the channel and at the channel exit, respectively. This is reflected in a decreased dynamics of some charged amino acid side chains (Glu332 in R460G-hE3 and Arg460 in R447G-hE3), as well as altered charge distributions along the H+/H2O channel. All of these changes presumably lead to certain degrees to impairment in H+/H2O translocation from the active site toward the protein surface, which could account for the decreased enzymatic activities of R460G-hE3 and R447G-hE3.
The I445M substitution also took place in a H+/H2O channel-forming helix near the protein surface but without leading to the loss of a charged residue in contrast to R460G-hE3 and R447G-hE3. Despite its location, this substitution induced no significant structural rearrangements in the H+/H2O channel according to the I445M-hE3 crystal structure. The most prominent alterations are the apparently complete losses in mobility for Glu332′ and Arg460. Since no significant alterations could be observed in the active site and for the cofactor-binding residues, and because HDX-MS demonstrated an increased flexibility/exposure of the C-terminus (39) that simultaneously affects the LA-binding and H+/H2O channels, the above mentioned H+/H2O channel perturbations are proposed to have key roles in the molecular pathogenesis. The rather minor structural deviations from hE3 observed correlate well with the relatively mild clinical manifestations, which present despite the virtually undetectable LADH activity and the significantly reduced overall hPDHc activity in clinical samples (56) (Table 2).
The R447G and R460G substitutions both lowered affinity for E3BP by orders of magnitude (37,38). This effect also contributed to the severely compromised overall hPDHc activities measured for the hPDHc-R447G-hE3 and hPDHc-R460G-hE3 variants (37,52,54). Relevant biochemical data have not yet been reported in the literature for I445M-hE3. The R447G substitution disrupts a direct interaction to E3BP (37,43). Unfortunately, the other E3BP/SBDb-binding residues were often involved in crystal contacts in both the R460G-hE3 and R447G-hE3 structures, therefore only limited conclusions could be drawn regarding these residues based solely on the crystallographic data. HDX-MS suggested before that E3BP/SBDb-binding residues Glu437-Gly439 and Arg447 are perturbed in R460G-hE3, whereas Glu437-Gly439 and Val347-His348 (and Arg447) are perturbed in R447G-hE3, relative to hE3. These data are in accord with residue Arg447 in R460G-hE3 and residues Val347-His348 in R447G-hE3 being displaced, relative to hE3, in the relevant crystal structures. The above findings likely account for the significantly decreased affinities of R460G-hE3 and R447G-hE3 measured before for E3BP.
Conclusions
Detailed structural analysis of hE3 and six of its disease-causing variants (primarily the dimer interface variants) shed light on the key role of H+/H2O channel dynamics in the catalytic activity of hE3. The flexible Glu332 and Arg460′ side chains, along with other polar/protonatable side chains in the channel likely contribute to H+/H2O elimination in the course of the catalytic cycle. The generalized pathomechanism proposed earlier for most disease-causing dimer interface variants (18) was confirmed by the (D444V-hE3) R460G-hE3, R447G-hE3 and I445M-hE3 crystal structures. The molecular pathomechanisms of these variants appear to be based primarily on H+/H2O channel perturbations, but in distinct ways. The P453L-hE3 variant exhibited the most deleterious structural aberrations, as was expected from the severe clinical outcomes. G426E-hE3 and G194C-hE3 displayed the least deviations in structure, which is in accord with the respective (mild) clinical manifestations. These long-awaited, high-resolution crystal structures of disease-causing hE3 variants will serve as platforms for further structural investigations of these variants and perhaps structure-based drug design.
Materials and Methods
Chemicals
All chemicals were purchased in molecular biology grade from Sigma-Aldrich (St. Louis, MO, USA), unless stated otherwise.
Mutagenesis and protein isolation
Site-directed mutagenesis on the hE3-coding pET-52b(+)-based vector plasmid (DNA 2.0, Menlo Park, CA, USA) was carried out using the QuikChange II mutagenesis kit (Stratagene, Cedar Creek, TX, USA). hE3 and its disease-causing variants were isolated using the BL21(DE3)/pET-52b(+) expression system, a single Strep-tag fused to the N-termini of the proteins, and affinity chromatography (57,58), as published before (39). Protein sequences were all verified by MS analysis (data not shown).
Protein crystallization
Protein samples after purification were extensively buffer exchanged into 100 mm Tris–HCl, 150 mm NaCl, 1 mm EDTA, pH 8.0 prepared in ultrapure water and filtered at 0.22 μm. Crystallization trials were carried out applying the sitting drop vapor diffusion method and using preformulated screening solutions (Hampton Research, Aliso Viejo, CA, USA). The 1 μl protein samples were mixed with equal amounts of the reservoir solutions in 24-well plates (Hampton Research) and equilibrated against the reservoir solutions (500 μl) at 20°C. Crystal Screen #39 [Hampton Research, 2 M (NH4)2SO4, 2 v/v% PEG 400, 0.1 M Hepes, pH 7.5] provided three-dimensional, well-diffracting crystals for R447G-, I445M- and G426E-hE3. Further optimization of the above condition was required for hE3 and G194C-, R460G- and P453L-hE3. The compositions of the crystallization solutions that resulted in the best diffracting crystals for these variants were: 2 M (NH4)2SO4, 2 v/v% PEG 400, 0.1 M Bis-Tris (pH 6.9) for G194C-hE3; 2 M (NH4)2SO4, 2 v/v% PEG 400, 0.1 M Bis-Tris (pH 6.5) for R460G-hE3; 2 M (NH4)2SO4, 1.5 v/v% PEG 400, 0.1 M Hepes (pH 7.3) for P453L-hE3; 2 M (NH4)2SO4, 1.5 v/v% PEG 400, 0.1 M Bis-Tris (pH 6.9) for hE3; stock solutions for the optimizations [1 M Hepes (pH 7.5), 1 M Bis-Tris (pH 6.5), 3.5 M (NH4)2SO4, 100 v/v% PEG 400] were purchased from Hampton Research, while pH was adjusted using 5 M NaOH or HCl.
Crystals were flash frozen and stored in liquid nitrogen until data collection; no cryoprotection was used.
Structure determination and analysis
Diffraction data were collected on the BL14.1 beamline at the BESSY II electron storage ring operated by the Helmholtz-Zentrum Berlin (HZB, Berlin-Adlershof, Germany) (59). Diffraction data were processed on site using XDSAPP 2.0 (60–62). The variants, except for P453L-hE3, and hE3 all crystallized in the P21212 space group; P453L-hE3 crystallized in the P1 space group. All the structures were determined by molecular replacement using Molrep (63) from the CCP4 program suite (64); as a search model, chain A of a previously determined hE3 structure (PDB ID: 1ZMD) was used. Molrep identified two monomers in the ASU for hE3 and R447G-, I445M-, G426E-, G194C-, R460G-hE3, while eight monomers were found in the ASU of the P453L-hE3 structure. For all the structures, rigid body refinement was followed by repeated cycles of restrained refinement in Refmac5 (65) and model building in Coot (66). Final refinements were always carried out using phenix.refine in Phenix (67) applying automatically assigned TLS groups (for translation, libration and screw-rotation of the groups of atoms to approximate anisotropy), occupancy refinement of side chain atoms in alternative conformations, noncrystallographic symmetry (NCS) torsion-angle restraints and real-space refinement; hydrogen atoms in riding positions proved to be beneficial to the refinements and were not deleted from the final models. Final model validation was carried out using the Molprobity server (68). All the structures have been deposited to the Protein Data Bank (PDB; for PDB IDs, see Table 1).
Least squares fitting of the variant structures onto the wild-type structure and calculations of the overall and residue-level RMSD values were performed using ProFit (69). Dimer interfaces and protein–FAD interactions were characterized using the online PISA server (70) and the CONTACT program of CCP4. The LA-binding and H+/H2O channels were examined by the Caver Analyst 2.0 program (71); the starting point for tunnel calculation was always the center of gravity between the Cys45(SG; in disulfide bond) and His452′(NE2) atoms in the active site, except occasionally for P453L-hE3, where the two atoms were Cys45(SG) and His452′(CE1) in case the His452′ imidazole ring was flipped. Surface polarity analysis was performed in PyMol. All the structural comparisons were performed relative to the present hE3 structure, if not stated otherwise.
Accession numbers
The coordinates and corresponding structure factor amplitudes of the structures of the wild-type hE3, as well as the P453L-, G194C-, R460G-, R447G-, I445M- and G426E-hE3 variants have been deposited in the RCSB Protein Data Bank, with accession numbers 6I4Q, 6I4Z, 6I4P, 6I4R, 6I4S, 6I4T and 6I4U, respectively.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank HZB and Advanced Photon Source (APS)/Argonne National Laboratory (Lemont, IL, USA) for allocation of synchrotron radiation beamtime.
Conflict of interest statement. None declared.
Funding
National Institutes of Health [GM050380 and R15-GM116077 to F.J., GM117357 to W.M. and R01GM121469 to W.F.]; Department of Veterans Affairs [Merit Review Awards] to W.F.; Hungarian Academy of Sciences (HASc) [02001]; Hungarian Scientific Research Fund [OTKA 112230]; Hungarian Brain Research Program [KTIA_13_NAP-A-III/6. and 2017-1.2.1-NKP-2017-00002]; Hungarian Higher Education Institution Excellence Program [FIKP 61822 64860 EATV], all to V.A.-V.; EMBO [short-term fellowship]; Fulbright Commission [research fellowship]; HASc [two Bolyai fellowships]; Semmelweis University [young investigator grant]; Chemical Works of Gedeon Richter Plc. [young investigator grant]; Helmholtz Zentrum Berlin (HZB) [181-06410-ST]; Hungarian Higher Education Institution Excellence Program [FIKP 61826 690289 EATV], all to A.A.; HZB [162-04087-ST] and the European Union [EFOP-3.6.3-VEKOP-16-2017-00009], both to S.E.
References
- 1. Pettit F.H. and Reed L.J. (1967) Alpha-keto acid dehydrogenase complexes. 8. Comparison of dihydrolipoyl dehydrogenases from pyruvate and alpha-ketoglutarate dehydrogenase complexes of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A., 58, 1126–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reed L.J. and Oliver R.M. (1968) The multienzyme alpha-keto acid dehydrogenase complexes. Brookhaven Symp. Biol., 21, 397–412. [PubMed] [Google Scholar]
- 3. Reed L.J. (1974) Multienzyme complexes. Acc. Chem. Res., 7, 40–46. [Google Scholar]
- 4. Zhou J., Yang L., Ozohanics O., Zhang X., Wang J., Ambrus A., Arjunan P., Brukh R., Nemeria N.S. and Furey W. (2018) A multipronged approach unravels unprecedented protein–protein interactions in the human 2-oxoglutarate dehydrogenase multienzyme complex. J. Biol. Chem., 293, 19213–19227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kikuchi G. and Hiraga K. (1982) The mitochondrial glycine cleavage system—unique features of the glycine decarboxylation. Mol. Cell. Biochem., 45, 137–149. [DOI] [PubMed] [Google Scholar]
- 6. Klyachko N.L., Shchedrina V.A., Efimov A.V., Kazakov S.V., Gazaryan I.G., Kristal B.S. and Brown A.M. (2005) pH-dependent substrate preference of pig heart lipoamide dehydrogenase varies with oligomeric state—response to mitochondrial matrix acidification. J. Biol. Chem., 280, 16106–16114. [DOI] [PubMed] [Google Scholar]
- 7. Babady N.E., Pang Y.P., Elpeleg O. and Isaya G. (2007) Cryptic proteolytic activity of dihydrolipoamide dehydrogenase. Proc. Natl. Acad. Sci. U. S. A., 104, 6158–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Starkov A.A., Fiskum G., Chinopoulos C., Lorenzo B.J., Browne S.E., Patel M.S. and Beal M.F. (2004) Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci., 24, 7779–7788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tretter L. and Adam-Vizi V. (2004) Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J. Neurosci., 24, 7771–7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ambrus A., Nemeria N.S., Torocsik B., Tretter L., Nilsson M., Jordan F. and Adam-Vizi V. (2015) Formation of reactive oxygen species by human and bacterial pyruvate and 2-oxoglutarate dehydrogenase multienzyme complexes reconstituted from recombinant components. Free Radic. Biol. Med., 89, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ambrus A., Tretter L. and Adam-Vizi V. (2009) Inhibition of the alpha-ketoglutarate dehydrogenase-mediated reactive oxygen species generation by lipoic acid. J. Neurochem., 109, 222–229. [DOI] [PubMed] [Google Scholar]
- 12. Ambrus A., Torocsik B., Tretter L., Ozohanics O. and Adam-Vizi V. (2011) Stimulation of reactive oxygen species generation by disease-causing mutations of lipoamide dehydrogenase. Hum. Mol. Genet., 20, 2984–2995. [DOI] [PubMed] [Google Scholar]
- 13. Dayan A., Fleminger G. and Ashur-Fabian O. (2019) Targeting the Achilles’ heel of cancer cells via integrin-mediated delivery of ROS-generating dihydrolipoamide dehydrogenase. Oncogene, 38, 5050–5061. [DOI] [PubMed] [Google Scholar]
- 14. Williams C.H., Jr. (1992) Lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase—a family of flavoenzyme transhydrogenases In Müller F. (ed), Chemistry and Biochemistry of Flavoenzymes. CRC Press, Boca Raton, FL, Vol. 3, pp. 121–211. [Google Scholar]
- 15. Gazaryan I.G., Krasnikov B.F., Ashby G.A., Thorneley R.N.F., Kristal B.S. and Brown A.M. (2002) Zinc is a potent inhibitor of thiol oxidoreductase activity and stimulates reactive oxygen species production by lipoamide dehydrogenase. J. Biol. Chem., 277, 10064–10072. [DOI] [PubMed] [Google Scholar]
- 16. Moxley M.A., Beard D.A. and Bazil J.N. (2014) A pH-dependent kinetic model of dihydrolipoamide dehydrogenase from multiple organisms. Biophys. J., 107, 2984–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chandrasekhar K., Wang J.J., Arjunan P., Sax M., Park Y.H., Nemeria N.S., Kumaran S., Song J.Y., Jordan F. and Furey W. (2013) Insight to the interaction of the dihydrolipoamide acetyltransferase (E2) core with the peripheral components in the Escherichia coli pyruvate dehydrogenase complex via multifaceted structural approaches. J. Biol. Chem., 288, 15402–15417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Szabo E., Mizsei R., Wilk P., Zambo Z., Torocsik B., Weiss M.S., Adam-Vizi V. and Ambrus A. (2018) Crystal structures of the disease-causing D444V mutant and the relevant wild type human dihydrolipoamide dehydrogenase. Free Radic. Biol. Med., 124, 214–220. [DOI] [PubMed] [Google Scholar]
- 19. Brautigam C.A., Chuang J.L., Tomchick D.R., Machius M. and Chuang D.T. (2005) Crystal structure of human dihydrolipoamide dehydrogenase: NAD(+)/NADH binding and the structural basis of disease-causing mutations. J. Mol. Biol., 350, 543–552. [DOI] [PubMed] [Google Scholar]
- 20. Massey V., Gibson Q.H. and Veeger C. (1960) Intermediates in the catalytic action of lipoyl dehydrogenase (diaphorase). Biochem. J., 77, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reed J.K. (1973) Studies on the kinetic mechanism of lipoamide dehydrogenase from rat liver mitochondria. J. Biol. Chem., 248, 4834–4839. [PubMed] [Google Scholar]
- 22. Kim H. and Patel M.S. (1992) Characterization of 2 site specifically mutated human dihydrolipoamide dehydrogenases (His-452-Gln and Glu-457-Gln). J. Biol. Chem., 267, 5128–5132. [PubMed] [Google Scholar]
- 23. de Kok A., Berg A., van Berkel W., Fabisz-Kijowska A., Westphal A., van den Akker F., Mattevi A. and Hol W.G.J. (1994) The pyruvate dehydrogenase complex from Azotobacter vinelandii In K. Yagi (ed.):Flavins and Flavoproteins 1993. Walter de Gruyter, Berlin, Germany, pp. 535–544. [Google Scholar]
- 24. Raddatz G. and Bisswanger H. (1997) Receptor site and stereospecifity of dihydrolipoamide dehydrogenase for R- and S-lipoamide: a molecular modeling study. J. Biotechnol., 58, 89–100. [DOI] [PubMed] [Google Scholar]
- 25. de Kok A. and van Berkel W.J.H. (1996) Lipoamide dehydrogenase. In Patel M.S., Roche T.E. and Harris R.A. (eds.), Alpha-Keto Acid Dehydrogenase Complexes. Birkhäuser, Basel, Switzerland, pp. 53–70. [Google Scholar]
- 26. Cameron J.M., Levandovskiy V., MacKay N., Raiman J., Renaud D.L., Clarke J.T.R., Feigenbaum A., Elpeleg O. and Robinson B.H. (2006) Novel mutations in dihydrolipoamide dehydrogenase deficiency in two cousins with borderline-normal PDH complex activity. Am. J. Med. Genet., 140A, 1542–1552. [DOI] [PubMed] [Google Scholar]
- 27. Quinonez S.C., Leber S.M., Martin D.M., Thoene J.G. and Bedoyan J.K. (2013) Leigh syndrome in a girl with a novel DLD mutation causing E3 deficiency. Pediatr. Neurol., 48, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Quinonez S.C. and Thoene J.G. (2014) In Pagon R.A., Adam M.P., Ardinger H.H. et al. (eds), GeneReviews® [Internet] 1993–2016. University of Washington, Washington, Seattle, pp. 1–37. [Google Scholar]
- 29. Ambrus A. and Adam-Vizi V. (2018) Human dihydrolipoamide dehydrogenase (E3) deficiency: novel insights into the structural basis and molecular pathomechanism. Neurochem. Int., 117, 5–14. [DOI] [PubMed] [Google Scholar]
- 30. Shaag A., Saada A., Berger I., Mandel H., Joseph A., Feigenbaum A. and Elpeleg O.N. (1999) Molecular basis of lipoamide dehydrogenase deficiency in Ashkenazi Jews. Am. J. Med. Genet., 82, 177–182. [PubMed] [Google Scholar]
- 31. Shany E., Saada A., Landau D., Shaag A., Hershkovitz E. and Elpeleg O.N. (1999) Lipoamide dehydrogenase deficiency due to a novel mutation in the interface domain. Biochem. Biophys. Res. Commun., 262, 163–166. [DOI] [PubMed] [Google Scholar]
- 32. Hong Y.S., Korman S.H., Lee J., Ghoshal P., Qu Q., Barash V., Kang S., Oh S., Kwon M., Gutman A. et al. (2003) Identification of a common mutation (Gly194Cys) in both Arab Moslem and Ashkenazi Jewish patients with dihydrolipoamide dehydrogenase (E3) deficiency: possible beneficial effect of vitamin therapy. J. Inherit. Metab. Dis., 26, 816–818. [DOI] [PubMed] [Google Scholar]
- 33. Sansaricq C., Pardo S., Balwani M., Grace M. and Raymond K. (2006) Biochemical and molecular diagnosis of lipoamide dehydrogenase deficiency in a North American Ashkenazi Jewish family. J. Inherit. Metab. Dis., 29, 203–204. [DOI] [PubMed] [Google Scholar]
- 34. Brassier A., Ottolenghi C., Boutron A., Bertrand A.M., Valmary-Degano S., Cervoni J.P., Chretien D., Arnoux J.B., Hubert L., Rabier D. et al. (2013) Dihydrolipoamide dehydrogenase deficiency: a still overlooked cause of recurrent acute liver failure and Reye-like syndrome. Mol. Genet. Metab., 109, 28–32. [DOI] [PubMed] [Google Scholar]
- 35. Ambrus A. (2019) An updated view on the molecular pathomechanisms of human dihydrolipoamide dehydrogenase deficiency in light of novel crystallographic evidence. Neurochem. Res., in press, DOI: 10.1007/s11064-11019-02766-11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vaubel R.A., Rustin P. and Isaya G. (2011) Mutations in the dimer interface of dihydrolipoamide dehydrogenase promote site-specific oxidative damages in yeast and human cells. J. Biol. Chem., 286, 40232–40245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brautigam C.A., Wynn R.M., Chuang J.L., Machius M., Tomchick D.R. and Chuang D.T. (2006) Structural insight into interactions between dihydrolipoamide dehydrogenase (E3) and E3 binding protein of human pyruvate dehydrogenase complex. Structure, 14, 611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Patel M.S., Korotchkina L.G. and Sidhu S. (2009) Interaction of E1 and E3 components with the core proteins of the human pyruvate dehydrogenase complex. J. Mol. Catal. B: Enzym., 61, 2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ambrus A., Wang J.J., Mizsei R., Zambo Z., Torocsik B., Jordan F. and Adam-Vizi V. (2016) Structural alterations induced by ten disease-causing mutations of human dihydrolipoamide dehydrogenase analyzed by hydrogen/deuterium-exchange mass spectrometry: implications for the structural basis of E3 deficiency. Biochim. Biophys. Acta, 1862, 2098–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ambrus A. and Adam-Vizi V. (2013) Molecular dynamics study of the structural basis of dysfunction and the modulation of reactive oxygen species generation by pathogenic mutants of human dihydrolipoamide dehydrogenase. Arch. Biochem. Biophys., 538, 145–155. [DOI] [PubMed] [Google Scholar]
- 41. Ambrus A., Mizsei R. and Adam-Vizi V. (2015) Structural alterations by five disease-causing mutations in the low-pH conformation of human dihydrolipoamide dehydrogenase (hLADH) analyzed by molecular dynamics—implications in functional loss and modulation of reactive oxygen species generation by pathogenic hLADH forms. Biochem. Biophys. Rep., 2, 50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brautigam C.A., Wynn R.M., Chuang J.L., Naik M.T., Young B.B., Huang T.H. and Chuang D.T. (2011) Structural and thermodynamic basis for weak interactions between dihydrolipoamide dehydrogenase and subunit-binding domain of the branched-chain alpha-ketoacid dehydrogenase complex. J. Biol. Chem., 286, 23476–23488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ciszak E.M., Makal A., Hong Y.S., Vettaikkorumakankauv A.K., Korotchkina L.G. and Patel M.S. (2006) How dihydrolipoamide dehydrogenase-binding protein binds dihydrolipoamide dehydrogenase in the human pyruvate dehydrogenase complex. J. Biol. Chem., 281, 648–655. [DOI] [PubMed] [Google Scholar]
- 44. Liu T.C., Kim H., Arizmendi C., Kitano A. and Patel M.S. (1993) Identification of two missense mutations in a dihydrolipoamide dehydrogenase-deficient patient. Proc. Natl. Acad. Sci. U. S. A., 90, 5186–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim H. (2006) Examination of the importance of Pro-453 in human dihydrolipoamide dehydrogenase predicted from the three-dimensional structure. Bull. Korean Chem. Soc., 27, 819–820. [Google Scholar]
- 46. Liu T.C., Hong Y.S., Korotchkina L.G., Vettakkorumakankav N.N. and Patel M.S. (1999) Site-directed mutagenesis of human dihydrolipoamide dehydrogenase: role of lysine-54 and glutamate-192 in stabilizing the thiolate-FAD intermediate. Protein Expr. Purif., 16, 27–39. [DOI] [PubMed] [Google Scholar]
- 47. Bando Y. and Aki K. (1991) Mechanisms of generation of oxygen radicals and reductive mobilization of ferritin iron by lipoamide dehydrogenase. J. Biochem., 109,450–454. [DOI] [PubMed] [Google Scholar]
- 48. Mattevi A. (2006) To be or not to be an oxidase: challenging the oxygen reactivity of flavoenzymes. Trends Biochem. Sci., 31, 276–283. [DOI] [PubMed] [Google Scholar]
- 49. Gadda G. (2012) Oxygen activation in flavoprotein oxidases: the importance of being positive. Biochemistry, 51, 2662–2669. [DOI] [PubMed] [Google Scholar]
- 50. Yuan L., Cho Y.-J. and Kim H.-J. (2008) Characterization of two naturally occurring mutations close to cofactors in human dihydrolipoamide dehydrogenase. Bull. Korean Chem. Soc., 29, 2327–2328. [Google Scholar]
- 51. Carrozzo R., Torraco A., Fiermonte G., Martinelli D., Di Nottia M., Rizza T., Vozza A., Verrigni D., Diodato D., Parisi G. et al. (2014) Riboflavin responsive mitochondrial myopathy is a new phenotype of dihydrolipoamide dehydrogenase deficiency. The chaperon-like effect of vitamin B2. Mitochondrion, 18, 49–57. [DOI] [PubMed] [Google Scholar]
- 52. Hong Y.S., Kerr D.S., Craigen W.J., Tan J., Pan Y.Z., Lusk M. and Patel M.S. (1996) Identification of two mutations in a compound heterozygous child with dihydrolipoamide dehydrogenase deficiency. Hum. Mol. Genet., 5, 1925–1930. [DOI] [PubMed] [Google Scholar]
- 53. Hong Y.S., Kerr D.S., Liu T.C., Lusk M., Powell B.R. and Patel M.S. (1997) Deficiency of dihydrolipoamide dehydrogenase due to two mutant alleles (E340K and G101del)—analysis of a family and prenatal testing. Biochim. Biophys. Acta, 1362, 160–168. [DOI] [PubMed] [Google Scholar]
- 54. Odievre M.H., Chretien D., Munnich A., Robinson B.H., Dumoulin R., Masmoudi S., Kadhom N., Rötig A., Rustin P. and Bonnefont J.P. (2005) A novel mutation in the dihydrolipoamide dehydrogenase E3 subunit gene (DLD) resulting in an atypical form of alpha-ketoglutarate dehydrogenase deficiency. Hum. Mutat., 25, 323–324. [DOI] [PubMed] [Google Scholar]
- 55. Massey V. (1960) The identity of diaphorase and lipoyl dehydrogenase. Biochim. Biophys. Acta, 37, 314–322. [DOI] [PubMed] [Google Scholar]
- 56. Quintana E., Pineda M., Font A., Vilaseca M.A., Tort F., Ribes A. and Briones P. (2010) Dihydrolipoamide dehydrogenase (DLD) deficiency in a Spanish patient with myopathic presentation due to a new mutation in the interface domain. J. Inherit. Metab. Dis., 33, S315–S319. [DOI] [PubMed] [Google Scholar]
- 57. Ambrus A., Torocsik B. and Adam-Vizi V. (2009) Periplasmic cold expression and one-step purification of human dihydrolipoamide dehydrogenase. Protein Expr. Purif., 63, 50–57. [DOI] [PubMed] [Google Scholar]
- 58. Ambrus A., Torocsik B. and Adam-Vizi V. (2009) Refolding of the human dihydrolipoamide dehydrogenase. Biochem. Eng. J., 45, 120–125. [Google Scholar]
- 59. Mueller U., Förster R., Hellmig M., Huschmann F.U., Kastner A., Malecki P., Pühringer S., Röwer M., Sparta K., Steffien M. et al. (2015) The macromolecular crystallography beamlines at BESSY II of the Helmholtz-Zentrum Berlin: current status and perspectives. Eur. Phys. J. Plus, 130, 141–151. [Google Scholar]
- 60. Kabsch W. (2010) XDS. Acta Crystallogr., Sect D, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Krug M., Weiss M.S., Heinemann U. and Mueller U. (2012) XDSAPP: a graphical user interface for the convenient processing of diffraction data using XDS. J. Appl. Cryst., 45, 568–572. [Google Scholar]
- 62. Sparta K.M., Krug M., Heinemann U., Mueller U. and Weiss M.S. (2016) XDSAPP2.0. J. Appl. Cryst., 49, 1085–1092. [Google Scholar]
- 63. Vagin A. and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr., Sect D, 66, 22–25. [DOI] [PubMed] [Google Scholar]
- 64. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A. et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect D, 67,235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Murshudov G.N., Vagin A.A. and Dodson E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect D, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- 66. Emsley P., Lohkamp B., Scott W.G. and Cowtan K. (2010) Features and development of coot. Acta Crystallogr., Sect D, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. (2010) PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr., Sect D, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S. and Richardson D.C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect D, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Martin A.C.R., in press Available from: http://www.bioinf.org.uk/sotware/profit/.
- 70. Krissinel E. and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol., 372, 774–797. [DOI] [PubMed] [Google Scholar]
- 71. Jurcik A., Bednar D., Byska J., Marques S.M., Furmanova K., Daniel L., Kokkonen P., Brezovsky J., Strnad O., Stourac J. et al. (2018) CAVER analyst 2.0: analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories. Bioinformatics, 34,3586–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lanterman M.M., Dickinson J.R. and Danner D.J. (1996) Functional analysis in Saccharomyces cerevisiae of naturally occurring amino acid substitutions in human dihydrolipoamide dehydrogenase. Hum. Mol. Genet., 5, 1643–1648. [DOI] [PubMed] [Google Scholar]
- 73. Yuan L. and Kim H. (2010) Characterization of a naturally occurring mutation (Arg-460 to Gly) close to FAD in human dihydrolipoamide dehydrogenase. Bull. Korean Chem. Soc., 31, 3511–3512. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.