

Abstract
Background:
The line probe assay (LPA) is one of the most accurate diagnostic tools for detection of different Mycobacterium species. Several commercial kits based on the LPA for detection of Mycobacterium species are currently available. Because of their high cost, especially for underdeveloped and developing countries, and the discrepancy of non-tuberculous mycobacteria (NTM) prevalence across geographic regions, it would be reasonable to consider the development of an in-house LPA. The aim of this study was to develop an LPA to detect and differentiate mycobacterial species and to evaluate the usefulness of PCR-LPA for direct application on clinical samples.
Methods:
One pair of biotinylated primers and 15 designed DNA oligonucleotide probes were used based on multiple aligned internal transcribed spacer (ITS) sequences. Specific binding of the PCR-amplified products to the probes immobilized on nitrocellulose membrane strips was evaluated by the hybridization method. Experiments were performed three times on separate days to evaluate the assay’s repeatability. The PCR-LPA was evaluated directly on nine clinical samples and their cultivated isolates.
Results:
All 15 probes used in this study hybridized specifically to ITS sequences of the corresponding standard species. Results were reproducible for all the strains on different days. Mycobacterium species of the nine clinical specimens and their cultivated isolates were correctly identified by PCR-LPA and confirmed by sequencing.
Conclusion:
In this study, we describe a PCR-LPA that is readily applicable in the clinical laboratory. The assay is fast, cost-effective, highly specific, and requires no radioactive materials.
Key Words: Diagnosis, Line Probe Assay (LPA), Mycobacterium Infection, Tuberculosis
Introduction
Mycobacterium tuberculosis, the causative agent of ‘tuberculosis,’ is an ancient human pathogen that has re-emerged as a major health problem worldwide. According to World Health Organization (WHO) reports, about one-third of world's population has been infected with M. tuberculosis. In 2017, 1.3 million people died from tuberculosis (TB) and about 54 million TB-related deaths were recorded during 2000-2017 (1). This disease is a life-threatening infection and can easily be transmitted through respiratory droplets. Therefore, rapid and accurate diagnosis of TB is critical for appropriate therapy and to prevent its spread within the community.
In recent years, the prevalence of non-tuberculous mycobacteria (NTM) infections has increased significantly. Some NTM infections, especially M. chelonae, M. avium complex (MAC), and M. fortuitum, are rapidly increasing worldwide (2). Unlike M. tuberculosis, NTMs are often isolated from natural resources such as water and soil. Therefore, infections with these organisms are almost inevitable, especially in aged and immunodeficient populations (3). Although many NTMs cause the same pulmonary disease as TB, the drug type, dose, and treatment regimens used to treat them differ from those of TB (4). For example, the erm 38, erm 39, and erm 41 genes in M. smegmatis, M. fortuitum, and M. abscessus complex, respectively, induce resistance to macrolides and may lead to treatment failure (5-7) or presence of various secretion systems in different mycobacterium species affects the virulence, viability and pathogenicity (8).
Rapid identification and differentiation of Mycobacterium species are critical ensure the appropriate therapies. Traditional strategies based on culture methods and phenotypic characteristics such as colony morphology, growth rate, pigment production, and biochemical characteristics are time-consuming, difficult to interpret, and unreliable (9). Several molecular techniques including polymerase chain reaction (PCR), restriction fragment length polymorphism (RFLP) and real-time PCR have been developed as rapid tests for detection of Mycobacterium species; however, none of these techniques can simultaneously differentiate a large number of Mycobacterium species in the clinical laboratory. Therefore, a highly discriminatory method is needed for laboratory diagnoses and epidemiological studies. The line probe assay (LPA) is a diagnostic tool based on the detection of a specific RNA or DNA sequence by hybridizing the complementary strand of a nucleotide probe to a part of the target sequence (10). This method has considerably improved the accuracy and rapidity of Mycobacterium species. differentiation (11).
Several target genes or sequences are currently used to differentiate Mycobacterium species; these include 16S rRNA (12), 16S-23S rRNA or internal transcribed spacer (ITS) (13), 23S rRNA (14), recA (15), rpoB (15), dnaj (16), secA1 (17), dnaA (18), gyrB (19), sod (15), and hsp65 genes (20). Among these, the ITS has been found to have an outstanding potential for this purpose both because of its variations in size and sequence between species, and because it contains both variable and highly conserved regions. Previous studies have also indicated that the ITS has good reproducibility and high variation in mycobacteria (13, 21).
Several LPA-based commercial kits for detection of Mycobacterium species are available. Because of their high cost, especially for underdeveloped and developing countries, and the discrepancy of NTM prevalence across geographic regions, it would be reasonable to consider the development of an in-house LPA for rapid identification and differentiation of Mycobacterium species.
The main aim of the present study was to design an in-house and cost-effective LPA-based method targeting the ITS to identify prevalent NTM species. The method was additionally evaluated for direct application to DNA from clinical specimens and compared to that extracted from bacterial colonies from the same specimen.
Materials and methods
Strains and clinical specimens
In this study, 15 Mycobacterium and 10 non-mycobacterial species were examined (Table 1). Moreover, the clinical specimens and their isolates from culture medium were collected from Pasteur Institute of Iran during six months to evaluate the results of our developed LPA in a pilot study. The clinical samples were positive for acid-fast bacilli and the cultivated isolates were negative for niacin (i.e., the patients were suspected of NTM infection). The samples and isolates were evaluated in parallel by PCR-LPA and sequencing.
Table 1.
Bacteria and reference strains used in this study
| Bacterial species | Reference strains* |
|---|---|
| Mycobacterial species | |
| M. abscessus | DSMZ 44196T |
| M. avium | ATCC 25291 |
| M. avium subsp. silvaticum | DSMZ 44175 |
| M. bovis | Clinical isolate |
| M. chelonae | Environmental isolate |
| M. fortuitum | TMC 1530 |
| M. fortuitum subsp. acetamidolyticum | ATCC 35391 |
| M. gordonae | Clinical isolate |
| M. intracellulare | ATCC 13950 |
| M. kansasii | TMC 1204 |
| M. marinum | Environmental isolate |
| M. scrofulaceum | ATCC 35785 |
| M. simiae | Clinical isolate |
| M. tuberculosis | H37Rv |
| M. ulcerans | ATCC 35840 |
| Non-mycobacterial species | |
| Acinetobacter baumannii | NCTC 13304 |
| Corynebacterium diphtheriae | Clinical isolate |
| Escherichia coli | ATCC25922 |
| Enterococcus faecalis | ATCC29212 |
| Klebsiella pneumonia subsp. pneumonia | ATCC700603 |
| Listeria monocytogenes | ATCC7644 |
| Pseudomonas aeruginosa | ATCC27853 |
| Salmonella enteritidis | RITCC1624 |
| Salmonella enteric serovar Typhimurium | ATCC14028 |
| Staphylococcus aureus | ATCC25923 |
ATCC, American Type Culture Collection; DSMZ, Deutsche Sammlung von MikroorganismenundZellkulturen GmbH; TMC, Trudeau Mycobacterial Culture; NCTC, The National Collection of Type Cultures; RITCC, Razi Institute Type Culture Collection.
Probe and primer design
The 55 mycobacteria ITS sequences were obtained from the NCBI GenBank database (Table 2). Multiple alignment analyses of the sequences were performed using BioEdit software (Ibis Biosciences, Carlsbad, CA, USA) and species-specific probes were selected from mycobacterial polymorphic and conserved regions to ensure probe specificity. Gene Runner software (Hastings Software, Colorado, USA) was used to compute the probe parameters. Then, selected oligonucleotide probes were analyzed with Basic Local Alignment Search Tool (BLAST) searches at the nucleotide collection database to determine the specificity of the selected regions. Primers Sp1 (5’-ACC TCC TTT CTA AGG AGCACC-3’) and Sp2 (5’-GAT GCT CGC AAC CAC TAT YCA-3’), targeting the Mycobacterium ITS region were used from the previous study (13). Primer Sp2 was labeled with biotin at the 5’ end and a poly-(T) tail was added at the 3’ end.
Table 2.
GenBank sequence identification numbers (accession number) for Mycobacterium species
| Gene | Mycobacterial Species | Accession Number |
|---|---|---|
| ITS (internal transcribed spacer) | M. tuberculosis complex | L15623, DQ133991, AJ315568, L26330, AJ315570, AJ315571, L26328, AJ315569, L26329 |
| M. chelonea | AJ291583, AJ291584, AJ314873, AJ314874, AF144327 | |
| M. abscessus | AJ314869, AJ291580, AJ314871, AJ314870 | |
| M. gordonae | AJ315574, AJ315575 | |
| M. ulcerans | X99217 | |
| M. marinum | Y14185, AJ315572, AJ315573, GU827996 | |
| M. simiae | Y14187, Y14186, Y14188, Z46426 | |
| M. intracellularae | X74057, Z46425, Z46423, AJ306711, AJ314865, AJ314864 | |
| M. avium | AJ314863, L15620, AJ314862, Z46422, Z46421, X74054 | |
| M. scrofluaceum | L15622, AJ314884 | |
| M. kansasii | L42262, L42263, L42264, X97632 | |
| M. fortuitum | AJ291593, AJ291592, AJ291587, AJ291588, AJ291589, AJ291590, AJ291591, AF144326 |
DNA extraction and PCR amplification
DNA was extracted from samples with the boiling method (22). Briefly, samples were washed two times with TE buffer (10 mM Tris-HCl pH 7.6, 1 mM EDTA), resuspended in 50 µl of TE buffer, and then boiled for 30 min in a water bath. The samples were then centrifuged at 10,000×g for 10 min. The supernatant was used as the template for the PCR. PCRs were performed in 25 µl volumes containing 1 µl each of forward and reverse primers (10 pmol), 12.5 µl PCR Master Mix (Parstous biotechnology, Iran), and 1 or 10 µl of DNA samples (1 µl for the cultivated samples and 10 µl for the clinical samples) in a Mastercycler Gradient (ASTEC, Japan). The reaction scheme was as follows: an initial denaturation step at 95 °C for 5 min, 40 cycles with the following three steps: denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s, and extension at 72 °C for 30 s, and a final extension step at 72 °C for 10 min. The PCR products were analyzed by electrophoresis on 2% agarose gels stained with Green Viewer™. The 200-300 bp amplicons were visualized on a UV-transilluminator. For each PCR run, sterile water and 1 ng of M. tuberculosis H37Rv genomic DNA were used instead of template as negative and positive controls, respectively.
Line probe assay
The LPA was modified from a previous publication (23). Briefly, 10 µl of 20 pmol/µl species-specific probes in TE buffer were immobilized onto a nitrocellulose membrane (pore size 0.45 μm; Amersham Pharmacia Biotech) as 16 parallel lines using a Bio-Dot SF microfiltration apparatus (Bio-Rad, USA), with the first line as a positive control containing a biotinylated PCR product. The strips were incubated for 1 hour at 80 °C to fix the probes on the strips. Equal volumes (20 µl) of biotinylated PCR product (50 ng/µl) and denaturation solution (400 mM NaOH + 10 mM EDTA) were mixed in a microtube and incubated at 37 °C for 15 min. At the same time, 2 ml of hybridization buffer containing 1.8 ml of 2X SSC, 0.1% SDS, and 200 µl of 10% bovine serum albumin (BSA) were added to each strip at room temperature. Then, 2 ml of 60 °C hybridization buffer containing the denatured PCR product was added to the strip and incubated at 59 °C for 30 min with shaking in a ProBlot 12 hybridization oven (Labnet International, Woodbridge, NJ). The strips were washed once in 2 ml of 2X hybridization solution for 1 min at room temperature and once in 0.1X SSC for 5 min at 62 °C. The strips were then incubated with 2 ml of a 1:1000-diluted alkaline phosphatase-labeled streptavidin (Sigma) in Tris-buffered saline at pH 8 on a rotating platform at room temperature for 30 min. The strips were then washed in 1X SSC for 10 min at room temperature. Finally, 2 ml of 5-bromo-4-chloro-3-indolylphosphate and nitroblue-tetrazolium (BCIP/NBT, Sigma) were added to the strips and incubated with shaking at room temperature for 10 min in the dark. The color reactions occurred and the strip results were visually interpreted using a probe alignment guide, which shows the position of the probes in each line. To evaluate the repeatability or intra-assay precision, PCR-LPA was performed three times on separate days.
Nucleotide sequence analysis
To confirm the LPA results, PCR products of cultivated samples were sequenced (Macrogen, Seoul, South Korea) in one direction using SP1as the primer. The obtained ITS nucleotide sequences were then analyzed by BLAST searches of the NCBI GenBank database.
Results
Designed specific probes: The 55 mycobacteria ITS sequences were evaluated to select the best conserved region from each species. It was essential to design more than one probe for some of the species, such as M. fortuitum and M. kansasii. Some species, including M. ulceranse and M. marinum were not differentiable by ITS target; therefore, one genus-specific and fifteen species-specific probes for the 12 Mycobacterium species were designed after consideration of length and melting temperature. These probes were 19-23 bp, which was suitable for the hybridization procedure (Table 3). The optimal melting temperatures (Tm) were 60.6-62.5 °C (Tm difference ≤ 1.9 °C) and the optimal free energy values (ΔG) for hairpin and self-dimer were less than -2.2 and 5.3 kcal/mol, respectively (Table 3). The selected probes were checked with the nucleotide collection database and it was confirmed that the designed probes and non-related bacterial sequences were not homologous.
Table 3.
Sequences and the parameters of oligonucleotide probes targeting the Mycobacterium ITS. *
| Species | Sequence | Length (mer)/Position | Tm(°C) | Self-dimer ΔG (kcal/mol) | Hairpin ΔG (kcal/mol) |
|---|---|---|---|---|---|
| M. tuberculosis complex | ACTTGTTCCAGGTGTTGTCCCAC | 23/134-157 | 61.5 | 1.8 | 1.1 |
| M. chelonea | CAGCCGAATGAGCTTGGGAA | 20/16-36 | 62.0 | -1.3 | -0.6 |
| M. abscessus | TCCCAGTCGAATGAACTAGGGAA | 23/13-36 | 61.8 | -2.8 | -2.2 |
| M. gordonae | CGTGAGGGGTCATCGTCTGTAG | 22/42-64 | 60.6 | 1.5 | 2.4 |
| M. ulcerans/marinum | ACATCTCTGTTGGTTTCGGGATG | 23/124-147 | 61.8 | 0.0 | 0.6 |
| M. simiae | CCGACTTCGGTTGAAGTGGTGT | 22/134-156 | 62.5 | -3.3 | -0.8 |
| M. intracellularae | GTCGATCCGTGTGGAGTCCCT | 21/133-154 | 61.9 | -1.8 | 1.2 |
| M. intracellularae/avium | CCGTGTGGAGTCCCTCCATCT | 21/139-160 | 61.9 | -3.2 | -2.2 |
| M. scrofluaceum | CAACACTCGGCTCGTTCTGAGT | 22/124-146 | 60.6 | 0.5 | -0.2 |
| M. kansasii (KAN1) | GGACTTGTCTGGGTCGTTGTCC | 22/133-155 | 61.7 | -1.0 | -0.4 |
| M. kansasii (KAN2) | TCGGGCTCTGTTCGAGAGTTGT | 22/129-151 | 62.5 | -1.8 | -1.0 |
| M. fortuitum (F1) | CCACGAAAAGGGTTGAGACACTG | 23/8-31 | 62.5 | NA | NA |
| M. fortuitum (F2) | ACGTTGAGCCGCAAGGGAT | 19/92-111 | 61.2 | -1.2 | 0.6 |
| M. fortuitum (F3) | CGTAAATCGCGGGATCAGCT | 20/29-49 | 61.5 | -5.3 | 2.8 |
The parameters were determined at 330 mM salt concentration. NA, not applicable; ΔG, Gibbs free energy.
PCR-LPA
All Mycobacterium species ITS fragments were PCR-amplified with the SP1 and SP2-biotinylated primers. The PCRs were performed at an annealing temperature of 59 °C and produced single bands ranging from 220 to 300 bp for the different Mycobacterium species (Fig. 1). In this study, the faster-growing Mycobacterium species, such as M. fortuitum, produced larger amplicons (300 bp) than the slower-growing species. The Sp1 and Sp2 genus-specific primers were shown to be specific for mycobacteria as they failed to amplify any DNA fragments from bacteria other than Mycobacterium. All the immobilized oligonucleotide probes on the membranes hybridized with their relevant biotinylated Mycobacterium PCR products. The specificity of the LPA was 100% using DNA extracted from mycobacterial and non-mycobacterial reference strains (Table 1 and Fig. 2). LPA results were reproducible for all the strains on different days. However, there are several considerations about our PCR-LPA: 1) to detect different M. kansasii and M. fortuitum types, two and three different probes, respectively, were required, 2) M. marinum and M. ulceranse were not differentiable due to 100% sequence homology in their ITS regions, 3) the M. tuberculosis complex members cannot be differentiated from each other, and 4) both M. avium and M. intracellulare were detected by one INT/AVprobe and were differentiated from each other using an AV probe that detects only M. avium (Fig. 2).
Fig. 1.
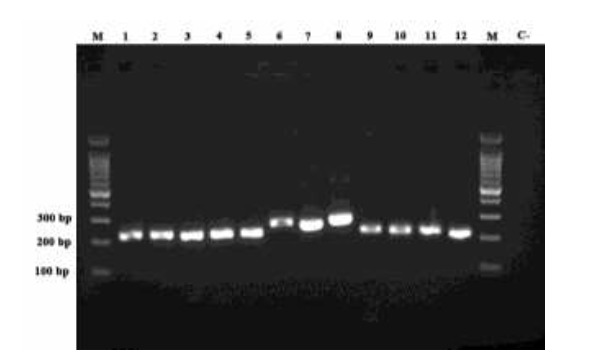
Agarose gel electrophoresis of ITS-PCR products for standard Mycobacterium species. Lane 1: M. tuberculosis H37Rv, Lane 2: M. simiae (clinical isolate), Lane 3: M. intracellulare ATCC 13950, Lane 4: M. avium ATCC 25291, Lane 5: M. kansasii TMC 1204, Lane 6: M. chelonae (environmental isolate), Lane 7: M. abscessus DSMZ 44196T, Lane 8: M. fortuitum TMC 1530, Lane 9: M. scrofulaceum ATCC 35785, Lane10: M. ulcerans ATCC 35840, Lane 11: M. marinum (environmental isolate), Lane 12: M. gordonae (clinical isolate). Lane M: 100 bp DNA marker; Lane C-: negative control.
Fig. 2.
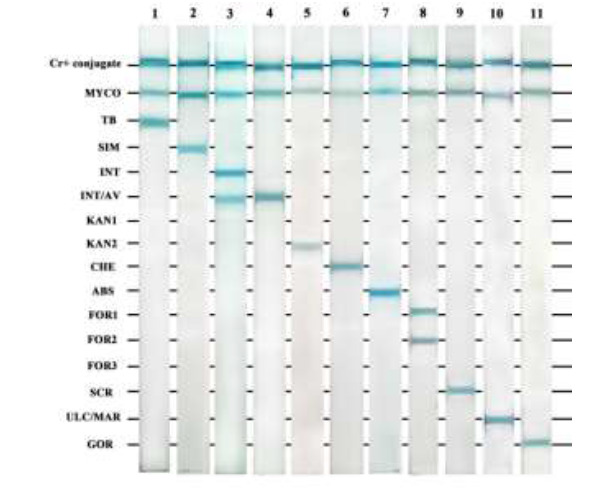
LPAs for standard Mycobacterium species. Strip 1: M. tuberculosis H37Rv, strip 2: M. simiae (clinical isolate), strip 3: M. intracellulare ATCC 13950, strip 4: M. avium ATCC 25291, strip 5: M. kansasii TMC 1204, strip 6: M. chelonae (environmental isolate), strip 7: M. abscessus DSMZ 44196T, strip 8: M. fortuitum TMC 1530, strip 9: M. scrofulaceum ATCC 35785, strip 10: M. ulcerans ATCC 35840 and M. marinum (environmental isolate), strip 11: M. gordonae (clinical isolate). Briefly, species-specific probes in TE buffer were immobilized onto a nitrocellulose membrane (pore size 0.45 μm; Amersham Pharmacia Biotech) as 16 parallel lines. The membrane was cut into strips and biotinylated PCR products were denatured, added to the membranes, and incubated at 59 ºC for 30 min. The strips were then incubated with a 1:1000-diluted alkaline phosphatase-labeled streptavidin in TBS at pH 8 on a rotating platform at room temperature for 30 min. The strips were washed and BCIP/NBT was added and incubated with shaking at room temperature for 10 min in the dark. The color reactions occurred and the results were visually interpreted with a probe alignment guide, which shows the position of the probes in each line.
PCR-LPA on clinical specimens and mycobacterial isolates
Considering the time constraints in this study and the lower prevalence of NTMs than tuberculosis in this region, nine clinical specimens and their relevant isolates from culture media were defined as NTMs initially, and examined by PCR-LPA in the pilot study. Our findings were then compared with the sequencing results. Clinical specimens 7 and 9 were both shown to contain two different NTM species by gel electrophoresis (Fig. 3). Two different types of M. fortuitum were confirmed by PCR-LPA in specimen 7; while specimen 9 was found by PCR-LPA to be co-infected with M. simiae and M. fortuitum. NTM species in all the other clinical specimens were identified as M. fortuitum. The PCR-LPA results of the clinical specimens and their relevant clinical isolates were identical. The PCR-LPA and sequence results were also identical except for the two mixed-infection specimens in which the sequencing results were not interpretable. For this reason, these two specimens were re-amplified, the PCR products purified using a gel extraction kit (GeNet Bio, Korea), and the purified PCR products sequenced. The results showed that the two species extracted from sample 7 belong to different types of M. fortuitum, and sample 9 contained both M. fortuitum and M. simiae, which confirmed the PCR-LPA result.
Fig. 3.
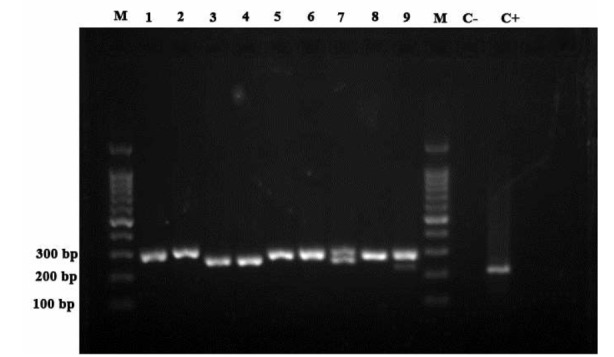
Agarose gel electrophoresis of ITS-PCR products for cultivated samples. Lanes 1-8: M. fortuitum, Lane 9: M. fortuitum+ M. simiae, Lane M, 100 bp DNA marker; Lane C-: negative control; Lane C+: positive control.
Discussion
Due to the increase in Mycobacterium infections, rapid diagnosis and differentiation of Mycobacterium species is crucial for successful treatment. Traditional methods usually take at least four weeks to give results, while molecular methods can be an appropriate alternative for rapid diagnosis and differentiation of Mycobacterium species. Although the Mycobacterium genome contains more than 4000 bp, a few genes can be candidate targets for molecular diagnosis of these bacteria. Some of the most prevalent NTM species lack unique sequences within the candidate genome region to be differentiated from each other, due to the high genomic similarity among the Mycobacterium species (24).
Most of the PCR assays used to detect mycobacteria identify a single or limited number of Mycobacterium species (25); therefore, these methods seem to be inefficient for identifying Mycobacteria at the species level. Restriction fragment length polymorphism is a reliable method for differentiation of mycobacteria (26), but interpretation of RFLP results is actually quite difficult because the band sizes can be different between some species and emerge the new patterns that have not yet been reported (13). Furthermore, RFLP is unreliable in detection of mixed cultures and is not applicable to clinical samples. Real-time PCR is also costly and requires considerable expertise. Although sequencing is the gold standard for mycobacteria species detection, it cannot differentiate mixed culture samples or co-infection with more than one NTM species, as it requires a high-quality, pure, PCR product. The advantages and disadvantages of various molecular techniques are summarized in Table 4.
Table 4.
Characteristics of various molecular methods for detection and differentiation of Mycobacterium species.
| Method | Time | Cost | Clinical or Cultivated samples | Difficulty | Detection of multiple-species | Results |
|---|---|---|---|---|---|---|
| PCR | 3 h | Low | Both | Requires specific target for each species | + (Limited) | Electrophoresis |
| RFLP | up to 2 days | Low | Cultivated | Difficulty in interpretation | - | Electrophoresis |
| Real time PCR | 3 h | High | Both | Requires considerable expertise | + | Computer analysis |
| LPA (In this study) | 7 h | 6-7 € | Both | Requires considerable expertise | + (12 species) | visually |
| Commercial LPA | 7 h | High | Both | Requires expertise and extra instrument | + (the number of species) | visually |
| sequencing analysis | Up to 1 weeks | High | Both | Requires high quality and pure product | - | BLAST search |
PCR: Polymerase chain reaction, RFLP: Restriction fragment length polymorphism, LPA: Line probe assay
Due to disadvantages of these diagnostic techniques, use of a molecular method based on reverse hybridization can be useful in detecting and differentiating Mycobacterium species. PCR-LPA is a reverse hybridization technique that enables differentiation of Mycobacterium species. Line probe assays are faster than traditional methods, more accurate than chromatographic tests, and less expensive than sequencing for diagnosis of mycobacteria (13). However, LPA is a PCR-based method; low DNA and high inhibitor levels in clinical specimens can lead to false-negative results, especially in smear-negative specimens (27, 28).
Therefore, cultivation of bacteria from clinical specimens and testing on colonies is the most appropriate NTM identification method. However, a major drawback with mycobacterial culture is the overgrowth of one or more NTM species, which may mask the growth of undergrown species in the cases of co-infections.
In our study, ITS showed a strong potential for discriminating closely-related Mycobacterium species. Mycobacterial ITS sequence analyses in different Mycobacterium species revealed that the 16S-23S spacer lengths of the faster-growing Mycobacteria are longer than those of the slower-growing species (Fig. 1). Moreover, the differences in the various Mycobacterium species ITS lengths have made it easy to detect mixed cultures or co-infections by electrophoresis of PCR products. We found more than one species in two samples and one specie in each of the other seven samples (Fig. 3). Therefore, the ITS target can be useful for diagnostic purposes.
To date, several lung infections have been reported with more than one species of NTM or more than one genotype of a specific species; however, information on such infections is limited. The highest incidence of simultaneous infection with two NTM species is related to M. avium complex and M. abscessus co-infection (29), but in this study, we found a co-infection with M. simiae and M. fortuitum, and a mixed infection with different M. fortuitum genotypes PCR-LPA analysis indicated that discrimination between M. marinum and M. ulcerans, which have high sequence homology, is not possible. Interestingly, PCR-LPA was able to discriminate between M. chelonae and M. abscessus, with two nucleotide differences in their ITS sequences, and between M. avium and M. intracellulare, which cannot be distinguished by chromatographic methods (13).
In our pilot study of nine clinical samples and their cultured isolates, M. fortuitum was the most common mycobacterial species in the sample collection region of Tehran, Iran. Consistent with our results, Irandoost and colleagues reported the same species as the most prevalent in this region (30). Of the 6800 patients with suspected tuberculosis, 64 were positive for NTM, and 40 (62.5%) of these cases were identified as M. fortuitum by PCR-RFLP (30). The low incidence of NTMs in this region and the few positive samples is one of the limitations of our study.
Our finding showed that this method can detect Mycobacterium species in clinical samples as accurately as culture or sequencing methods (Fig. 4). Although the LPA results from clinical samples showed weaker band intensity than those from cultured isolates, we had no problems with the result interpretations.
Fig. 4.
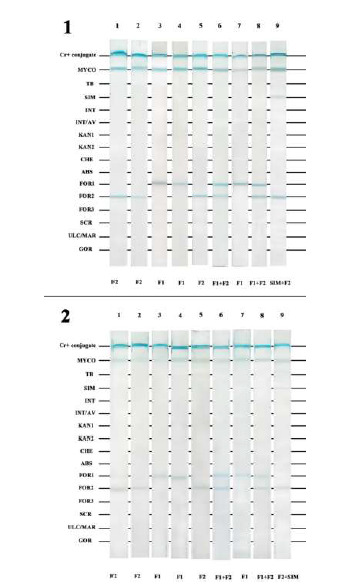
1) LPA results for the nine cultivated samples. 2) LPA results for the nine clinical samples. Lanes 1-8: various M. fortuitum types; Lane 9: M. simiae+M. fortuitum. Briefly, species-specific probes in TE buffer were immobilized onto a nitrocellulose membrane as 16 parallel lines. The membrane was cut into strips and biotinylated PCR products were denatured, added to the membranes, and incubated at 59 °C for 30 min. The strips were then incubated with a 1:1000-diluted alkaline phosphatase-labeled streptavidin in TBS at pH 8 on a rotating platform at room temperature for 30 min. The strips were washed and BCIP/NBT was added and incubated with shaking at room temperature for 10 min in the dark. The color reactions occurred and the results were visually interpreted with a probe alignment guide, which shows the position of the probes in each line.
Here we described a PCR-LPA method that is readily applicable for respiratory specimens in clinical laboratories. We compared the PCR-LPA results from clinical and cultivated samples from the same patients. Our PCR-LPA, based on the ITS target, differentiated 12 mycobacterial species and genotype using 15 probes. The speed of the assay is similar to commercial kits with high specificity. Moreover, the cost of this test is far less than that of the commercial kits; each test cost 6-7€ (Table 4). We routinely performed the PCR amplification in less than 3 h, followed by the line probe assay in 4 h.
PCR-LPA identified mixed cultures or co-infections in clinical samples that cannot be differentiated by PCR or nucleotide sequence analysis. Therefore, we recommend integrating PCR-LPA into the routine microbiological laboratory practice for diagnosis of mycobacterial infections, especially by its direct application on clinical samples.
Acknowledgements
This is part of Reza Kamali Kakhki’s Ph.D thesis (Medical Bacteriology). We thank Dr. Abolfazl Fateh from Department of Mycobacteriology and Pulmonary Research, Pasteur Institute of Iran, Tehran, for his generous cooperation in clinical sample collection. This project was supported by a grant from Mashhad University of Medical Sciences, Mashhad, Iran (Grant No. 941795). The authors declare that they have no competing interests.
References
- 1.World Health Organization. Global tuberculosis report 2018. Geneva:World Health Organization. 2018 Licence: CC BY-NCSA 3.0 IGO.2018.
- 2.Marras TK, Chedore P, Ying AM, Jamieson F. Isolation prevalence of pulmonary non-tuberculous mycobacteria in Ontario 1997-2003. Thorax. 2007 doi: 10.1136/thx.2006.070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khoor A, Leslie KO, Tazelaar HD, Helmers RA, Colby TV. Diffuse pulmonary disease caused by nontuberculous mycobacteria in immunocompetent people (hot tub lung). Am J Clin Pathol. 2001;115(5):755–62. doi: 10.1309/JRDC-0MJV-ACA3-2U9L. [DOI] [PubMed] [Google Scholar]
- 4.Winthrop KL, McNelley E, Kendall B, Marshall-Olson A, Morris C, Cassidy M, et al. Pulmonary nontuberculous mycobacterial disease prevalence and clinical features: an emerging public health disease. Am J Respir Crit Care Med. 2010;182(7):977–82. doi: 10.1164/rccm.201003-0503OC. [DOI] [PubMed] [Google Scholar]
- 5.Nash KA. Intrinsic macrolide resistance in Mycobacterium smegmatis is conferred by a novel erm gene, erm (38). . Antimicrob Agents Chemother. 2003;47(10):3053–60. doi: 10.1128/AAC.47.10.3053-3060.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nash KA, Brown-Elliott BA, Wallace RJ. A novel gene, erm (41), confers inducible macrolide resistance to clinical isolates of Mycobacterium abscessus but is absent from Mycobacterium chelonae. Antimicrob Agents Chemother. 2009;53(4):1367–76. doi: 10.1128/AAC.01275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nash KA, Zhang Y, Brown-Elliott BA, Wallace Jr RJ. Molecular basis of intrinsic macrolide resistance in clinical isolates of Mycobacterium fortuitum. J Antimicrob Chemother. 2005;55(2):170–7. doi: 10.1093/jac/dkh523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamali-Kakhki R, Aryan E. Mycobacterial Type VII Secretion System: A Possible Novel Target for Anti-Tuberculosis Drugs. . Pharmacologyonline. 2016;30:1–10. [Google Scholar]
- 9.Palomino J. Nonconventional and new methods in the diagnosis of tuberculosis: feasibility and applicability in the field. . Eur Respir J. 2005;26(2):339–50. doi: 10.1183/09031936.05.00050305. [DOI] [PubMed] [Google Scholar]
- 10.Neonakis IK, Gitti Z, Krambovitis E, Spandidos DA. Molecular diagnostic tools in mycobacteriology. J Microbiol Methods. 2008;75(1):1–11. doi: 10.1016/j.mimet.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 11.Nathavitharana RR, Cudahy PG, Schumacher SG, Steingart KR, Pai M, Denkinger CM. Accuracy of line probe assays for the diagnosis of pulmonary and multidrug-resistant tuberculosis: a systematic review and meta-analysis. Eur Respir J. 2017;49(1):1601075. doi: 10.1183/13993003.01075-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han XY, Pham AS, Tarrand JJ, Sood PK, Luthra R. Rapid and accurate identification of mycobacteria by sequencing hypervariable regions of the 16S ribosomal RNA gene. Am J Clin Pathol. 2002;118(5):796–801. doi: 10.1309/HN44-XQYM-JMAQ-2EDL. [DOI] [PubMed] [Google Scholar]
- 13.Xiong L, Kong F, Yang Y, Cheng J, Gilbert GL. Use of PCR and reverse line blot hybridization macroarray based on 16S-23S rRNA gene internal transcribed spacer sequences for rapid identification of 34 mycobacterium species. . J Clin Microbiol. 2006;44(10):3544–50. doi: 10.1128/JCM.00633-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mäkinen J, Marjamäki M, Marttila H, Soini H. Evaluation of a novel strip test, GenoType Mycobacterium CM/AS, for species identification of mycobacterial cultures. Clin Microbiol Infect. 2006;12(5):481–3. doi: 10.1111/j.1469-0691.2006.01380.x. [DOI] [PubMed] [Google Scholar]
- 15.Adékambi T, Drancourt M. Dissection of phylogenetic relationships among 19 rapidly growing Mycobacterium species by 16S rRNA, hsp65, sodA, recA and rpoB gene sequencing. . Int J Syst Evol Microbiol. . 2004;54(6):2095–105. doi: 10.1099/ijs.0.63094-0. [DOI] [PubMed] [Google Scholar]
- 16.Takewaki S-I, Okuzumi K, Manabe I, Tanimura M, Miyamura K, Nakahara K-I, et al. Nucleotide sequence comparison of the mycobacterial dnaJ gene and PCR-restriction fragment length polymorphism analysis for identification of mycobacterial species. Int J Syst Bacteriol. 1994;44(1):159–66. doi: 10.1099/00207713-44-1-159. [DOI] [PubMed] [Google Scholar]
- 17.Zelazny AM, Calhoun LB, Li L, Shea YR, Fischer SH. Identification of Mycobacterium species by secA1 sequences. J Clin Microbiol. 2005;43(3):1051–8. doi: 10.1128/JCM.43.3.1051-1058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukai T, Miyamoto Y, Yamazaki T, Makino M. Identification of Mycobacterium species by comparative analysis of the dnaA gene. FEMS Microbiol Lett. . 2006;254(2):232–9. doi: 10.1111/j.1574-6968.2005.00031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dauendorffer J-N, Guillemin I, Aubry A, Truffot-Pernot C, Sougakoff W, Jarlier V. Identification of mycobacterial species by PCR sequencing of quinolone resistance-determining regions of DNA gyrase genes. J Clin Microbiol. 2003;41(3):1311–5. doi: 10.1128/JCM.41.3.1311-1315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim H, Kim S-H, Shim T-S, Kim M-n, Bai G-H, Park Y-G, et al. Differentiation of Mycobacterium species by analysis of the heat-shock protein 65 gene (hsp65). Int J Syst Evol Microbiol. . 2005;55(4):1649–56. doi: 10.1099/ijs.0.63553-0. [DOI] [PubMed] [Google Scholar]
- 21.Gray TJ, Kong F, Jelfs P, Sintchenko V, Chen SC. Improved Identification of Rapidly Growing Mycobacteria by a 16S–23S Internal Transcribed Spacer Region PCR and Capillary Gel Electrophoresis. PLoS One. 2014;9(7):e102290. doi: 10.1371/journal.pone.0102290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Afghani B, Stutman HR. Polymerase Chain Reaction for Diagnosis ofM. tuberculosis: Comparison of Simple Boiling and a Conventional Method for DNA Extraction. Biochem Mol Med. 1996;57(1):14–8. doi: 10.1006/bmme.1996.0003. [DOI] [PubMed] [Google Scholar]
- 23.Martin C, Roberts D, van der Weide M, Rossau R, Jannes G, Smith T, et al. Development of a PCR-based line probe assay for identification of fungal pathogens. . J Clin Microbiol. 2000;38(10):3735–42. doi: 10.1128/jcm.38.10.3735-3742.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Behr MA. Comparative genomics of mycobacteria: some answers, yet more new questions. Cold Spring Harb Perspect Med. 2015;5(2):a021204. doi: 10.1101/cshperspect.a021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chae H, Han SJ, Kim S-Y, Ki C-S, Huh HJ, Yong D, et al. Development of a one-step multiplex PCR assay for differential detection of major Mycobacterium species. J Clin Microbiol. 2017;55(9):2736–51. doi: 10.1128/JCM.00549-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roth A, Fischer M, Hamid ME, Michalke S, Ludwig W, Mauch H. Differentiation of phylogenetically related slowly growing mycobacteria based on 16S-23S rRNA gene internal transcribed spacer sequences. J Clin Microbiol. 1998;36(1):139–47. doi: 10.1128/jcm.36.1.139-147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kakhki RK, Neshani A, Sankian M, Ghazvini K, Hooshyar A, Sayadi M. The short-chain dehydrogenases/reductases (SDR) gene: A new specific target for rapid detection of Mycobacterium tuberculosis complex by modified comparative genomic analysis. . Infect Genet Evol. 2019 doi: 10.1016/j.meegid.2019.01.012. [DOI] [PubMed] [Google Scholar]
- 28.Neshani A, Kakhki RK, Sankian M, Zare H, Chichaklu AH, Sayyadi M, et al. Modified genome comparison method: a new approach for identification of specific targets in molecular diagnostic tests using Mycobacterium tuberculosis complex as an example. BMC infectious diseases. 2018;18(1):517. doi: 10.1186/s12879-018-3417-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shin SH, Jhun BW, Kim S-Y, Choe J, Jeon K, Huh HJ, et al. Nontuberculous mycobacterial lung diseases caused by mixed infection with Mycobacterium avium complex and Mycobacterium abscessus complex. . Antimicrob Agents Chemother. 2018;62(10):e01105–18. doi: 10.1128/AAC.01105-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Irandoost M, Ghanbari MZ, Sakhaee F, Vaziri F, Jamnani FR, Siadat SD. High rates of Mycobacterium fortuitum isolation in respiratory samples from Iranian patients with suspected tuberculosis: is it clinically important? J Med Microbiol. 2018;67(9):1243–8. doi: 10.1099/jmm.0.000814. [DOI] [PubMed] [Google Scholar]