

Abstract
Introduction:
Troponin (TNN)-encoded cardiac troponins (Tn) are critical for sensing calcium and triggering myofilament contraction. TNN variants are associated with development of cardiomyopathy; however, recent advances in genetic analysis have identified rare population variants. It is unclear how certain variants are associated with disease while others are tolerated.
Objective:
To compare probands with TNNT2, TNNI3, and TNNC1 variants and utilize high-resolution variant comparison mapping of pathologic and rare population variants to identify loci associated with disease pathogenesis.
Methods:
Cardiomyopathy-associated TNN variants were identified in the literature and topology mapping conducted. Clinical features were compiled and compared. Rare population variants were obtained from the gnomAD database. Signal-to-noise (S:N) normalized pathologic variant frequency against population variant frequency. Abstract review of clinical phenotypes was applied to “significant” hotspots.
Results:
Probands were compiled (N=70 studies, 224 probands) as were rare variants (N=125748 exomes; 15708 genomes, MAF <0.001). TNNC1-positive probands demonstrated the youngest age of presentation (20.0 years; P=0.016 vs TNNT2; P=0.004 vs TNNI3) and the highest death, transplant, or ventricular fibrillation events (P=0.093 vs TNNT2; P=0.024 vs TNNI3; Kaplan Meir: P=0.025). S:N analysis yielded hot-spots of diagnostic significance within the tropomyosin-binding domains, α-helix 1, and the N-Terminus in TNNT2 with increased sudden cardiac death and ventricular fibrillation (P=0.004). The inhibitory region and C-Terminus in TNNI3 exhibited increased restrictive cardiomyopathy (P=0.008). HCM and RCM models tended to show increase calcium sensitivity and DCM decreased sensitivity (P<0.001). DCM and HCM studies typically showed no differences in Hill coefficient but were decreased in RCM models (P<0.001). All CM models showed mostly no changes to Fmax (P=0.239).
Conclusion:
TNNC1-positive probands had younger ages of diagnosis and poorer clinical outcomes. Mapping of TNN variants identified locations in TNNT2 and TNNI3 associated with heightened pathogenicity, RCM diagnosis, and increased risk of sudden death.
Keywords: Cardiomyopathy, signal-to-noise analysis, troponin, TNNT2, TNNI3, TNNC1, Troponin T, Troponin I, Troponin C
INTRODUCTION
Cardiomyopathies are a heterogeneous group of primary diseases of the heart muscle, that can predispose to heart failure and cardiovascular death, with risk of sudden cardiac death (SCD) at first presentation [1]. Although many classifications exist, the American Heart Association classifies primary cardiomyopathies by genetic, mixed genetic, and non-genetic variants. Structural genetic primary cardiomyopathies include hypertrophic cardiomyopathy (HCM), left ventricular non-compaction cardiomyopathy (LVNC), and arrhythmogenic right ventricular cardiomyopathy (ARVC). Mixed structural cardiomyopathies, or those with both genetic and non-genetic etiologies, include dilated cardiomyopathy (DCM) and restrictive cardiomyopathy (RCM) [2].
The etiology of the majority cardiomyopathies, independent of the clinical presentation, is believed to be genetic. Genetic variants resulting in molecular defects in the components of the contractile myofilaments represent a major cause of cardiomyopathies [3, 4]. The cardiac troponin molecular complex (Tn) is essential for the regulation of striated muscle contraction and is located along the sarcomere thin filament [5, 6]. Disruption of any of these processes likely leads to cardiac dysfunction and cardiomyopathy.
While variants in TNN-encoding genes are clear causes of cardiomyopathy, there is marked clinical heterogeneity. Genotype-phenotype correlations to explain this heterogeneity have been limited by small numbers of probands and a lack of comprehensive knowledge of population variants. Recent genetic studies have identified previously reported pathologic variants in large population studies as well as rare variants of unknown disease risk [7, 8].
It is unclear how certain TNN variants are associated with disease while others are physiologically tolerated. To address this, we set out to 1) create a compendium of cardiomyopathy (CM)-associated variants and population-associated variants, and 2) identify prognostic characteristics associated with genotype.
METHODS
Nomenclature
Nomenclature in this study is detailed in Supplemental Materials.
Compendium of cardiomyopathy-associated TNN variants
To compile pathologic TNN variants, PubMed and the Human Gene Mutation database were queried through June 2018 for studies identifying patients with cardiomyopathy and confirmed TNN variants [9]. Inclusion criteria for the studies included 1) study of individuals with primary cardiomyopathic disease, 2) comprehensive genetic analysis of the coding exons of TNNT2, TNNI3, or TNNC1, 3) availability of individual variants and their amino acid location. A variant from these studies was included in our analysis if it was 1) hosted by a proband, 2) described in the sentinel study reporting the proband, and 3) was denoted as pathogenic or likely pathogenic by ACMG criteria. A variant was excluded if 1) non-proband cases hosted the variant, 2) the variant was available and denoted as “benign” or “likely benign” by ClinVar or was denoted as a variant other than pathogenic or likely pathogenic by ACMG criteria, further described in our Supplemental Materials [10]. The latter exclusion criterion was applied to control for recent changes in the assessment of variant pathogenicity. Given the evolution of pathogenicity among some variants over time, as well as heterogeneity among individual studies included, we manually applied current ACMG criteria to re-assess the pathogenicity of all variants, independent of any previously assigned classifications [11]. Further inclusion criteria for S:N analysis considered whether the study was cohort-based. Variants were excluded from S:N analysis if they were sense, synonymous, intronic, UTR, or intergenic. Variants were abstracted and standardized based on consensus primary sequence TNNT2 (NP_001001430), TNNI3 (NP_000354), and TNNC1 (NP_003271) (Ensembl, release 93). Studies testing functional consequences of variants using animal models and in vitro assays were also compiled in a separate database [12].
Compendium of rare population-based TNN variants
To compile rare population variants in Tn, variants from the Genome Aggregation Database (gnomAD) were compiled [13]. These were utilized as “control” or “reference” alleles. Given the observation of likely pathogenic variants found among gnomAD cases, we determined the maximum credible minor allele frequency (MAF) of gnomAD-identified pathogenic variants and set an inclusion threshold of MAF <0.001. This MAF threshold was set to include any pathologic variants observed in gnomAD and has been validated for both heritable arrhythmias and cardiomyopathies [14–16].
Amino acid-level signal-to-noise analysis and structure modeling
Variant mapping and amino acid-level signal-to-noise (S:N) analysis was done as previously described and is fully detailed in the Supplemental Materials [17]. Following exclusion of TNN homozygous and compound and double heterozygote probands, variants were mapped along the protein topology for each protein and signal-to-noise calculations involved normalization of variant frequency against gnomAD variant frequency at each amino acid position. For each TNN gene, variant hotspots considered significant by S:N analysis were determined (Supplemental Methods). Probands with variants within each hotspot were identified and grouped by hotspot locations. These hotspots were further grouped based on functional domains. Clinical features of probands were compared amongst hotspots at each functional domain. A Bonferroni corrected threshold was set to determine clinically significant hotspots with outcome relevance.
Comparison Statistics
Fully detailed in Supplemental Materials.
RESULTS
Creation of troponin variant database and meta-analysis
To identify all pathologic variants and rare population variants, we created a compendium of cardiomyopathy-associated variants and gnomAD-based population variants (Figure 1A). A comprehensive review of the literature identified 224 probands hosting variants in TNN from 70 studies. A total of 54 distinct variants were identified in TNNT2, 53 in TNNI3, and 16 in TNNC1 (Supplemental Table 1). Individual proband data for TNNT2, TNNI3, and TNNC1 are detailed in Supplemental Tables 2, 3, and 4, respectively. Three hundred and five rare population variants were collected from the gnomAD database v2.1.1 (N=125748 exomes; 15708 genomes, MAF <0.001).
Figure 1: A.
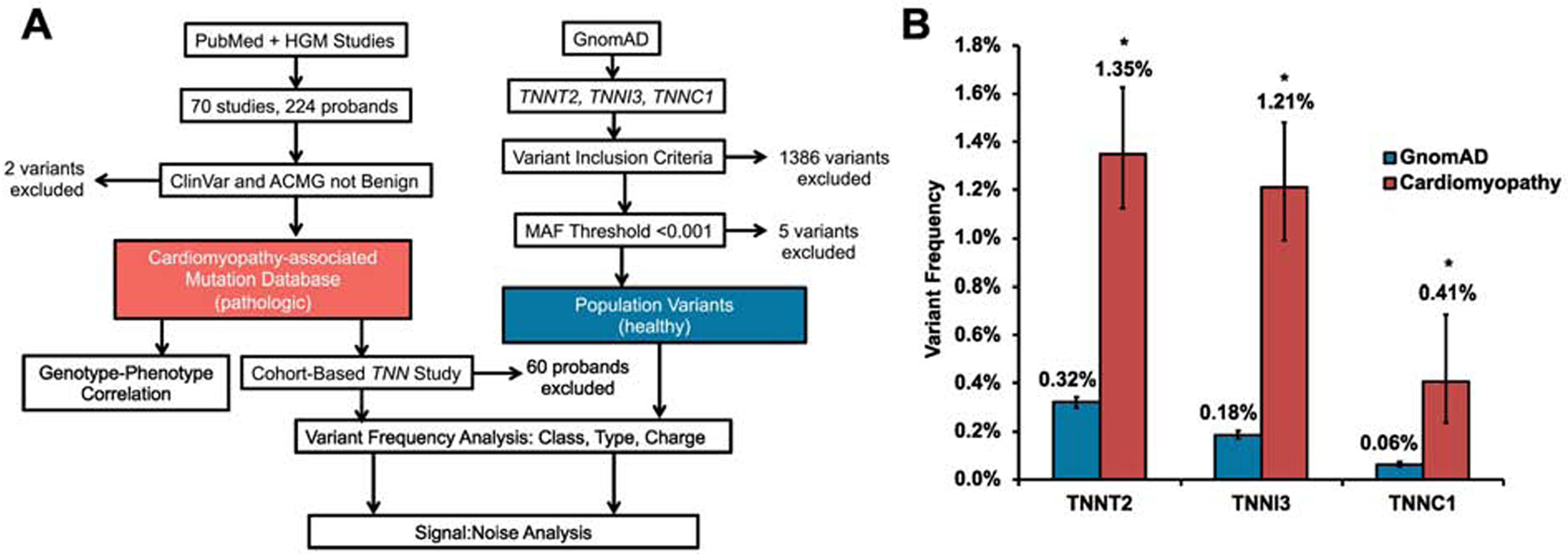
Study methodology and inclusion and exclusion criteria. B. Variant frequencies in variants and the gnomAD database. *P=0.0001
Frequency of pathologic troponin variants is higher among individuals with cardiomyopathy than rare population variants
While variants in TNN are classically believed to be rare among individuals with CM, CM-associated pathologic frequency was significantly higher in all individual TNN genes compared to all gnomAD variants, with frequencies of 1.35% (1.12–1.63), 1.21% (0.99–1.48), and 0.41% (0.24–0.69) in TNNT2, TNNI3, and TNNC1, respectively, compared to population cohorts of 0.32% (0.30–0.34), 0.18% (0.17–0.20), and 0.06% (0.05–0.07). Comparisons among all frequencies were significant (P<0.001). Variant frequencies are summarized in Figure 1B. Variant frequencies in TNNT2 were 0.83% (0.50–1.36) for DCM, 1.33% (1.07–1.67) for HCM, and 4.17% (0.74–20.24) for RCM. In TNNI3, variant frequencies were 0.57% (0.34–0.98) for DCM, 1.35% (1.07–1.71) for HCM, and 19.44% (9.75–35.03) for RCM. In TNNC1, variant frequencies were 0.61% (0.28–1.34) for DCM, 0.24% (0.10–0.57) for HCM, and 1.83% (0.62–5.24) for LVNC. Variant frequencies were consistently lower in DCM and were higher in rarer cardiomyopathy subtypes such as RCM and LVNC across all genes. S:N for each gene was 4.22, 6.72, and 6.83 for TNNT2, TNNI3, and TNNC1, respectively. These results indicate that, while rare, pathologic variants in TNN genes are more prevalent in clinical cases than rare population variants, and suggest these variants have meaningful diagnostic signal-to-noise relevance if there is a clinical diagnosis of CM.
TNNC1-variant positive probands have a worse prognosis compared to TNNT2 or TNNI3
To determine if the TNN gene mutated was correlated with clinical outcomes, we compared severity of cardiomyopathy and mortality among the three genes, looking specifically at heterozygous probands. TNNC1 probands were diagnosed within Almost 40% of the first decade of life (Figure 2A). TNNC1 probands were diagnosed at an earlier age with a mean age of 20.00±5.26 years, as compared to 33.70±1.99 years in TNNT2 and 39.30±2.49 years in TNNI3 (P=0.016 and P=0.004, respectively; Figure 2B). Fatal and potentially fatal events including death, transplant, and documented ventricular fibrillation (VF) affected 53.80% (29.14–76.79) of TNNC1 probands as compared to 26.50% (18.80–36.04) of TNNT2 probands and 22.40% (14.80–32.29) of TNNI3 probands (P=0.093 and P=0.024, respectively). Survival was compared and is plotted in Figure 2C (global P=0.025; TNNC1 vs TNNI3: P=0.008; TNNC1 vs TNNT2: P=0.019). TNNC1-positive probands were more likely to have LVNC, with 23.10% (8.18–50.25) having LVNC as compared to 1.10% (0.19–5.84) in TNNI3 and 1.00% (0.18–5.45) in TNNT2 probands (P=0.001 and P=0.010, respectively; Figure 2D). Conversely, 15.00% (9.18–23.70) of TNNI3 probands had RCM, as compared to 2.00% (0.55–7.00) of TNNT2 and 0.00% of TNNC1 probands (P=0.003 and P=0.001, respectively). Combined TNNT2, TNNI3, and TNNC1 proband clinical and demographic data are detailed in Supplemental Table 5. Data analysis prior to exclusion of compound and double heterozygote and homozygote probands are detailed in Supplemental Table 6. Taken together, TNNC1-positive probands demonstrated a more malignant phenotype compared to their TNNT2 and TNNI3 counterparts.
Figure 2: A.
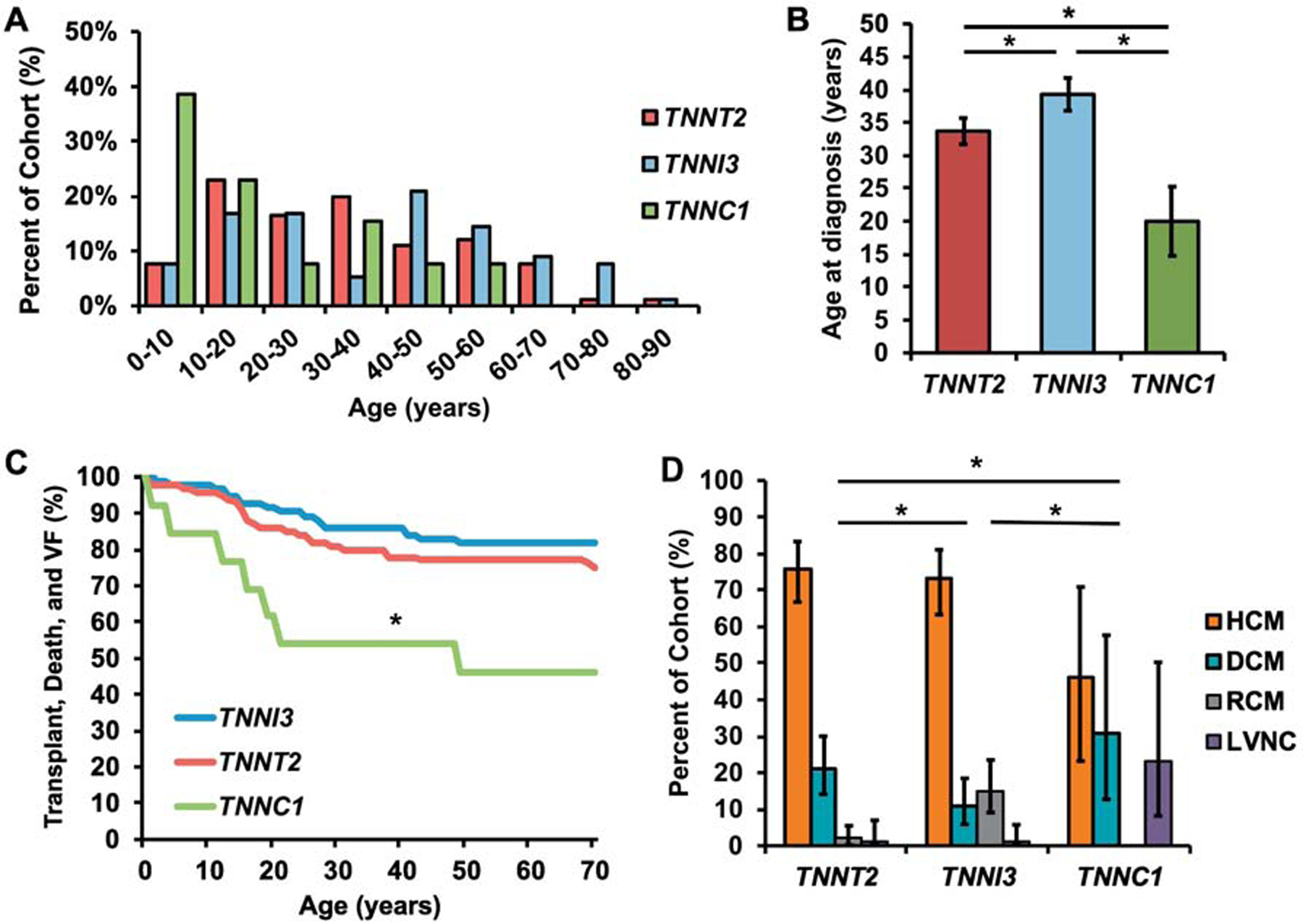
Histogram exhibiting distribution of ages of diagnoses in TNNT2 (red), TNNI3 (blue), and TNNC1 (green) probands. B. Histogram exhibiting average proband age in TNNT2, TNNI3, and TNNC1. C. Survival curve for TNNT2 (red), TNNI3 (blue), and TNNC1 (green) probands; * TNNC1 vs TNNI3: P=0.008; TNNC1 vs TNNT2: P=0.019. D. Histogram exhibiting the distribution of hypertrophic cardiomyopathy (HCM; orange), dilated cardiomyopathy (DCM; blue), restrictive cardiomyopathy (RCM; grey), and left ventricular noncompaction cardiomyopathy (LVNC; purple) in TNNT2, TNNI3, and TNNC1 probands. *indicates significance
Higher troponin variant zygosity conveys an earlier age of diagnosis and a negative prognosis
Given recent evidence that variant burden has prognostic relevance in cardiomyopathic disease, we next explored the impact of variant zygosity and compound and double heterozygosity on prognosis.[18, 19] Overall, 91.97% (87.66–94.86) of all TNN-positive probands were heterozygous and 1.34% (0.46–3.86) were homozygous for pathologic variants across all TNN genes, 2.23% (0.96–5.12) carried compound heterozygous variants, and 4.46% (2.44–8.02) were double heterozygote. Initially, we grouped all probands with homozygous variation and compound and double heterozygosity and compared them to those with heterozygous variants. Of clinical variables explored, age of diagnosis was 16.0 years (SEM: 3.50) in our higher variant burden group compared to heterozygous probands who had a mean age of 35.1 years (SEM: 1.54; P<0.001). Other variables were similar in both groups (P>0.05).
Although proband numbers were small when looking at each group separately, we next sought to address differences in clinical variables based on type of variant burden. We found that the majority of homozygous probands were transplanted or died, although only 3 probands were available (Supplemental Table 7). Death or transplant is higher among homozygous probands than those with heterozygous variants where only 8.20% (5.09–12.85) were transplanted and 24.50% (19.00–30.96) were either transplanted or died. Compound heterozygous probands were found to be younger at time of diagnosis? than their single variant counterparts as well (8.20±2.58 years vs 35.1±1.54 years; Figure 3A; Supplemental Table 8). Double heterozygote probands also had younger ages of presentation (18.40±5.83 years vs 35.1±1.54 years) compared to those with single variants (Figure 3B; Supplemental Table 9). Overall, homozygosity, double heterozygosity, and compound heterozygosity were rare yet correlated with worse outcomes among all TNN genes.
Figure 3: A.
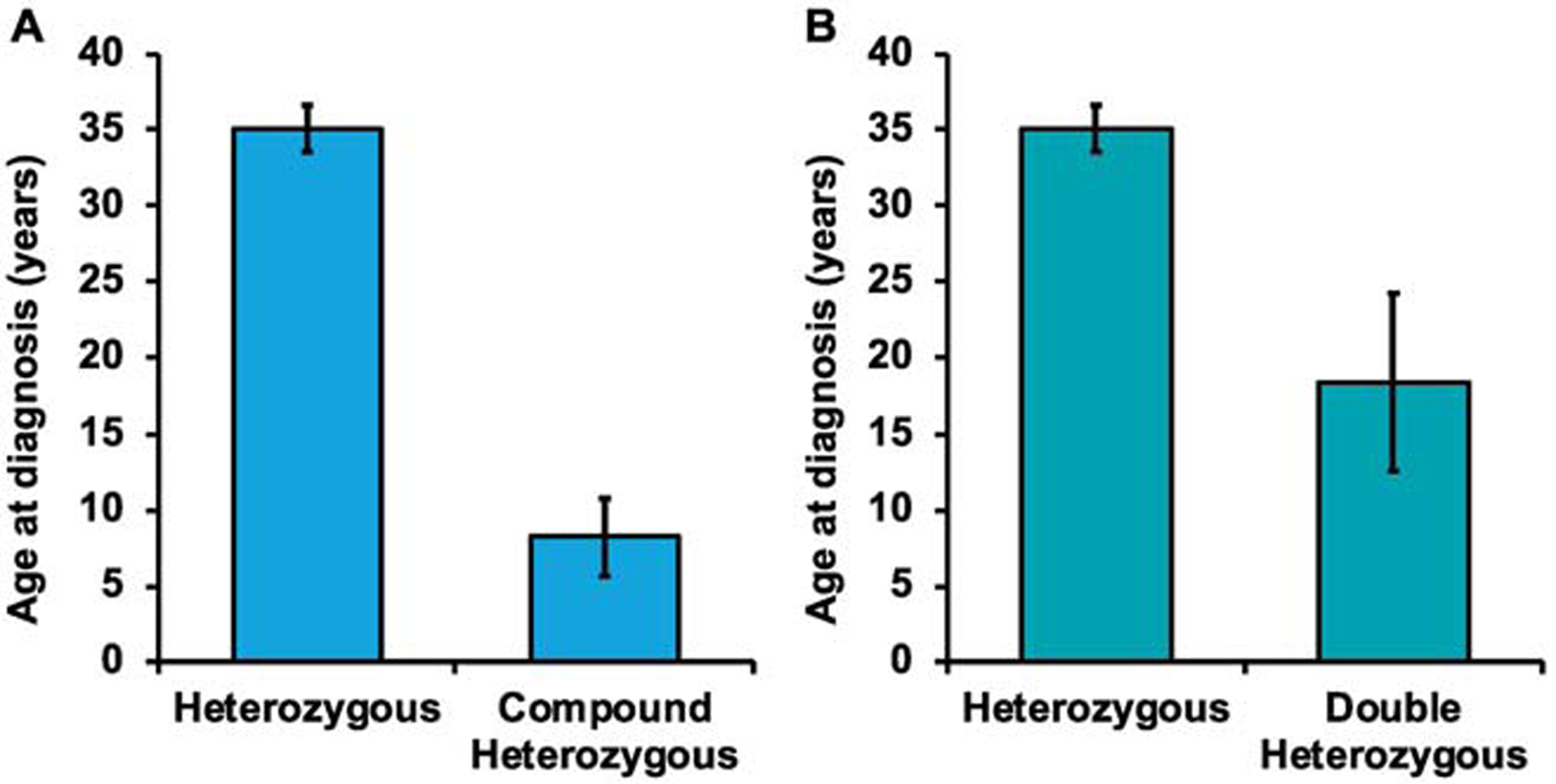
Histogram exhibiting distribution of ages of diagnoses in probands with compound heterozygosity and probands with heterozygosity. B. Histogram exhibiting distribution of ages of diagnoses in probands with double heterozygosity and probands with heterozygosity. *indicates significance
Amino acid-level signal-to-noise analysis identifies variant hotspots that drive prognostic associations
Given the recent observation that pathologic TNN variants have been observed within the gnomAD database, we sought to integrate rare population variants to determine areas that may drive diagnostic and prognostic influence of the TNN CM-associated variants. Amino acid-level S:N has recently been shown as a method to normalize pathologic variation against the rare population variation to identify pathologic hotspots [17]. In this way, we sought to identify areas of pathologic and prognostic relevance distinct from rare population variation. Based on pre-analysis significance of double and compound heterozygote and homozygote probands, only heterozygote probands were included in this analysis.
Hotspots of diagnostic relevance were isolated in TNNT2 within the N-terminus, tropomyosin binding domains, and α-helix 1 (Figure 4). Probands hosting variants at these locations demonstrated significant differences in sudden cardiac death/ventricular fibrillation (SCD/VF; P=0.004) and diagnosis (P<0.001), with SCD/death/transplant (P=0.053) and age (P=0.052) trending towards significance. Hotspot 3 (aa85–95; S:N= 310) proved to be highly diagnostically relevant, with 26.3% SCD/VF and 31.6% SCD/VF/death, with all probands having HCM. Hotspots 2 (aa73–80; SN: 31) and 15 (aa277–281, SN: 61) exhibited significantly higher SCD/VF. Hotspot 11 (aa204–206; S:N: 61) exhibited 100% transplant and a young age of diagnosis at 8.3y, although only two probands are identified at this location. Hot-spot 12 (aa209–211, SN: 17) also had a higher amount of SCD/death/transplant. Five different hotspots of clinical significance were isolated within vital functional domains. S:N hotspots are listed in Supplemental Table 10 and hotspots grouped by functional domains are listed in Supplemental Table 11.
Figure 4:
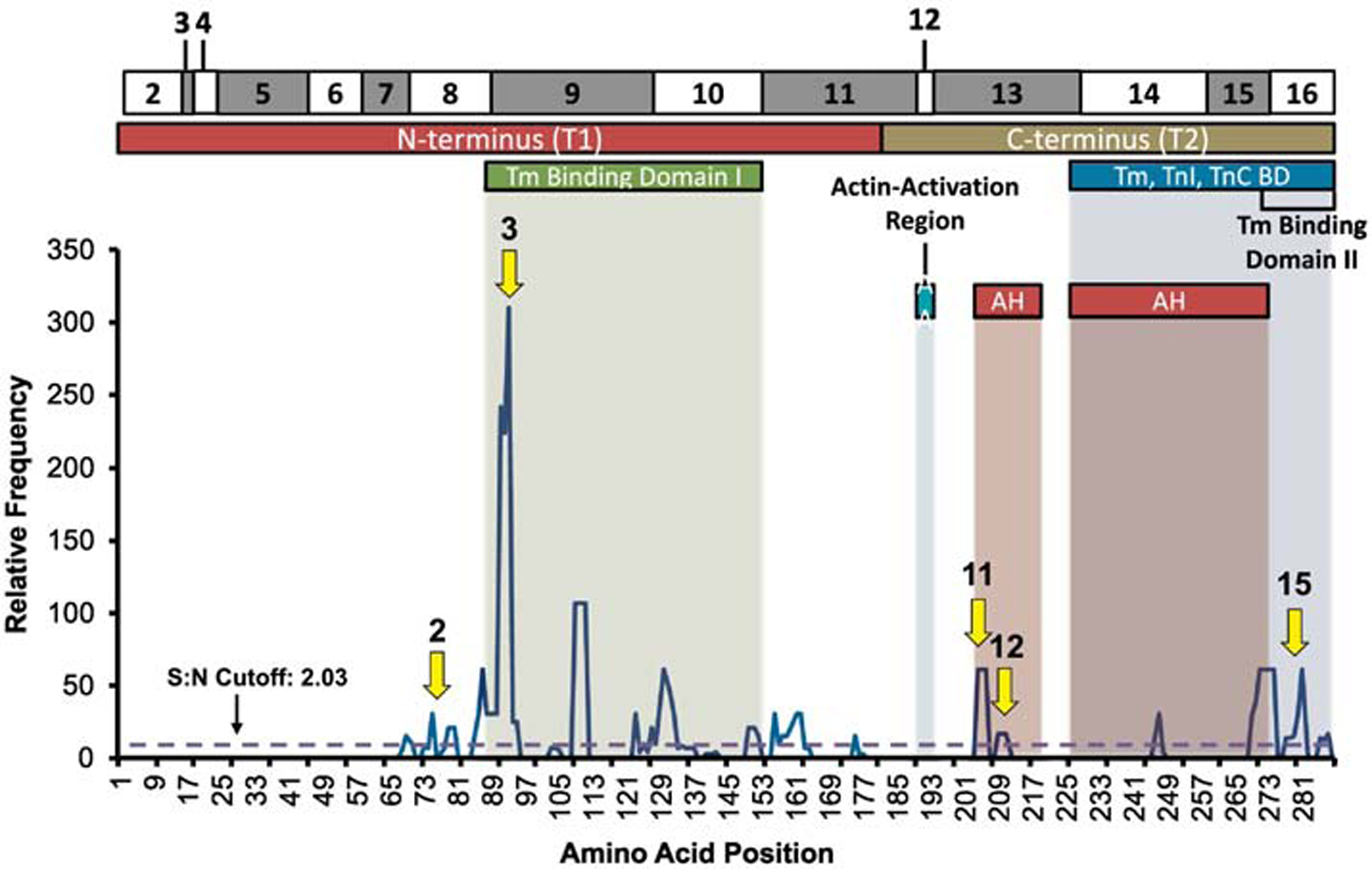
Signal-to-noise ratio analysis of TNNT2 probands mapped against TnT amino acid location and domain: N-Terminus, tropomyosin (Tm) binding domains, actin-activation region, the Tm, TnI, and TnC binding domain, and alpha helix (AH) 1 and 2. Arrows point to significant hotspots with outcome relevance.
TNNI3 proband hotspots were similarly compared. Most hotspots and probands clustered within the inhibitory region, switch peptide region, and the C-terminus (Figure 5). Hotspots of diagnostic relevance had higher rates of heart failure (P=0.030), as well as less common cardiomyopathy subtypes such as RCM (P=0.008). Hotspots 9 (aa140–146; S:N= 58), 12 (aa161–167; S:N= 96), 13 (aa169–187; S:N=225), and 14 (aa195–210; S:N=80) had, respectively, 60.0%, 90.0%, 100.0%, and 50.0% of probands who have heart failure. Hotspots 9 and 13 both have an increased amount of RCM with 13.3% and 9.5%, respectively. S:N hotspots are listed in Supplemental Table 12 and hotspots grouped by functional domains are listed in Supplemental Table 13.
Figure 5:
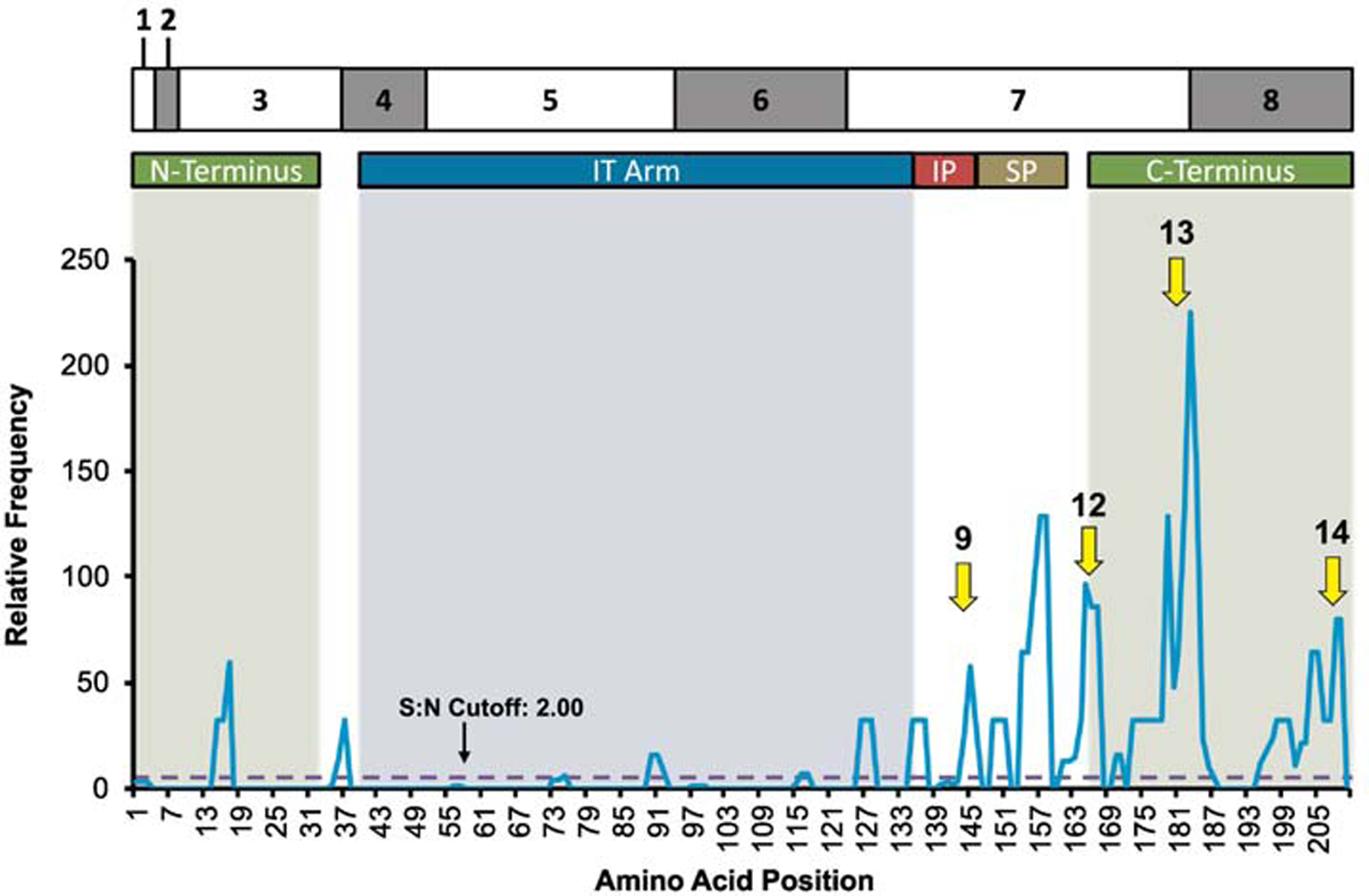
Signal-to-noise ratio analysis of TNNI3 probands mapped against TnI amino acid location and domains: N-terminal extension, I-T arm, inhibitory region (IR), switch-peptide domain (SP), and C-terminus/mobile domain. Arrows point to significant hotspots with outcome relevance.
TNNC1 proband hotspots were compared similarly. However, no hotspots of diagnostic significance were identified (Supplemental Figure 1). This is likely due to the small number of probands and/or high pathogenicity of all hotspots. S:N hotspots are listed in Supplemental Table 14.
Analysis of inheritance and variant alterations
We sought to analyze the effect of inheritance and polarity and charge changes on phenotype. This is detailed in the Supplemental results.
Functional Consequences of TnT, TnI, and TnC Variants
Functional characteristics of pathologic variants were compiled from 96 different studies. Testing of biochemical/biophysical properties of thin filaments and the troponin macromolecular complex were variable amongst most studies, although common themes were seen. We compared results of common studies including calcium sensitivity, Fmax, and Hill coefficient. Statistical differences between CM models were exhibited in pCa50, determined by concentration of Ca2+ at which the force is half of the maximal value and a marker of calcium sensitivity, and in Hill coefficient, a measure of cooperative action/cooperativity. For pCa50, 68.0% of HCM models (83/122 models) and 76.9% of RCM models (20/26 models) demonstrated an increased pCa50, while 52.5% of DCM models (21/40 models) showed decreased pCa50 (P<0.001). Conversely, 51.9% of HCM models (28/54 models) and 92.9% of DCM models (26/28 models) showed no change in the Hill coefficient while 60.0% of RCM models (9/15 models) showed decreases in Hill coefficient (P<0.001). No difference was found in Fmax, a measure of maximal force, among cardiomyopathy types. Although data was scare, HCM models tended to show increased maximal velocity of shortening, where changes were exhibited, and DCM decreased maximal velocity of shortening, were changes were exhibited. HCM and RCM models tended to show increase calcium sensitivity and DCM decreased sensitivity. This is fully detailed in Supplemental Table 16. Taken together, these findings suggest variants associated with specific types of cardiomyopathy impart similar biophysical effects.
DISCUSSION
Variant Investigation in Cardiomyopathy
In light of the rapid progression of precision or personalized medicine, efforts to utilize an individual’s specific genetic abnormality to predict outcomes and guide therapy are underway. Thus, understanding both the functional and prognostic implications of rare variation is critical to the development of personalized medicine.[20] Recent studies have highlighted that specific genetic variation that is associated with the development of cardiomyopathy, in particular HCM, may guide rational drug design and the development of novel pharmacotherapies [21]. For instance, chronic administration of the small molecule MYK-461 has been shown to reduce signs of HCM and reduce profibrotic gene expression in mice with heterozygous variants in the myosin heavy chain [22]. Further, establishing the diagnostic and prognostic impact of known pathologic variation on sarcomeric genes has formed the foundation for potential gene therapies. For example, CRISPR-Cas9 targeting of a GAGT deletion in MYBPC3 in human embryos and expression of functional cMYBPC in MYBPC3 knockout mice with use of AAV therapy [23, 24]. Overall, our findings that specific genetic variants were associated with specific prognostic outcomes reinforces the role of genetic testing in personalized care of cardiomyopathies.
Genetic investigations have revealed a large number of variants in Tn subunits linked to CM. Prognostic utility of genetic variants and their impact on phenotype has been controversial. Early studies have found that myofilament-positive HCM patients had more severe cardiac dysfunction, higher likelihood of progressive heart failure, and higher rates of cardiovascular death [25, 26]. More recently, it has become clear in larger cohort studies that sarcomere positive cardiomyopathy is associated with a poorer prognosis compared to their sarcomere negative counterparts, with higher endpoints of cardiovascular death, progression to end-stage heart failure, and non-fatal stroke [27, 28]. At the individual gene level, some early correlations have been identified such as heightened pathogenicity and SCD in TNNT2 at residue 92 and heightened variant rates in codon 145 in TNNI3 causing RCM or HCM [26, 29]. However, these multigenerational family studies have been difficult to replicate in large population studies. Nonetheless, significantly more probands identified in the literature will aid in determining gene-level prognostic correlations. With the advancement of genetic testing and newly discovered variants, we set out to determine TNN gene-specific prognostic associations.
Our compiled data demonstrated that TNNC1 probands had the youngest age of diagnosis and the highest rates of potentially fatal events. TNNT2 and TNNI3 probands had similar rates of death, SCD, and documented VF, although TNNI3 probands had a propensity for RCM. Although not statistically significant, more than 40% of TNNC1 probands lacked a family history as compared to lower proportions in TNNT2 and TNNI3. De novo variants play an important role in severe, early-onset diseases and are significant in sporadic schizophrenia, neurodevelopmental disorders, and congenital heart disease [30–33]. The combination of an earlier disease onset and increased negative outcomes in TNNC1 probands could explain the increased amount of de novo variants. The probable explanation is survival to transmission of the mutant allele was less likely given its lethality. This would further be supported by the scarcity of TNNC1 probands identified in the literature (7.6% of all TNN probands identified). Further, although outside of our study’s set time period, recently identified TNNC1 probands have exhibited similar findings in our study, including young ages of diagnosis, death, and negative family histories [34, 35]. Comparison of survival between each of the TNN genes demonstrated a significantly worse prognosis in individuals hosting likely pathologic variants in TNNC1 compared to TNNT2 and TNNI3.
Variant Burden
Variant burden is an important factor that affects clinical variability in variant-positive cardiomyopathy probands. Compound heterozygosity, double heterozygosity, and homozygosity are linked with more severe phenotypes, both in humans and animal models. Although less common than single variants, compound heterozygotes make up around 5% of HCM probands [36]. Presence of multiple variants in HCM patients has been linked to earlier onset of disease, a more severe phenotype, and a higher prevalence of major cardiac events [36–39]. Similar findings of increased SCD and arrhythmic events were found in ARVC associated with compound heterozygosity [40, 41]. Homozygosity in the HCM cohort is also associated with rates of SCD, and more significant hypertrophy [42]. Our analysis earlier onset of disease, higher cardiac further amplifies these findings. Compound heterozygotes and double heterozygotes, regardless of variant class, were associated with a younger age of diagnosis while homozygosity was associated with a higher rate of transplant and death, where over 50% of homozygous probands were transplanted and all probands were transplanted or died.
Insights with Signal-to-Noise Analysis
Pathologic variants within the TNN genes are frequently linked to significant functional changes. Recently, studies have exhibited that some pathologic variants are found within the general population [7, 8, 43, 44]. Our primary goal was to address recent observations of pathologic variants in population-based data by S:N analysis.
In TnT, distinct locations with poor prognosis were found within the N-terminus, the tropomyosin binding domains, and α-helix 1. TNNT2 variants in or near the N-tail T1 domain (aa79–170) can directly interact with overlapping tropomyosin (Tm) [45]. Variants between 92 and 110 were found to impair tropomyosin-dependent functions of TnT and alter secondary structure in the regions of Ca2+-dependent interaction between TnI and TnC [45, 46]. Hotspot 3 (aa85–95), located in this area, exhibited the highest S:N ratio in our analysis and was a highly pathogenic phenotype. The C-terminal region of TnT (i.e. TnT2 digest fragment) is known to interact with other Tn subunits and also binds the tropomyosin molecule; the last 14 amino acid residues are critical for stability of the inactive state [47, 48]. Interestingly, variant hotspots were located within the C-terminus of TnT, one within the first α-helix and another towards the C-terminal end, within 14 amino acids of the terminal end. In TnI, several hotspots were located within the C-terminus. The mobile domain (aa168–210 or aa163–210 as suggested by Yamada et al.) is vital in the interaction with actin and N-TnC and has been shown to stabilize interactions between TnI and actin in the relaxed state of thin filament [49–51]. Even deletion of the last three residues in the C-terminus is associated with significant functional compromise [52].
The most recent findings regarding TNN variants are in the TNNC1 gene. Prognosis among most probands was significantly poor and lack of comparatively pathogenic hotspots is likely secondary to this. Interdomain communication within TnC may mean that single variants could affect function of multiple domains [53, 54]. However, an area of significance where variants are detrimental to protein function include the N-helix, an important modulator of Ca2+ binding affinity and myofilament regulation [55–58].
Although hotspot domain location may explain pathogenicity, Tn domain function and variant alterations are still unclear. Residue changes have functional consequences inherent in their location, but variants can also affect covalent, post-translational modifications and Tn proteins may have non-canonical roles [59, 60]. Further, our functional studies exhibited some common biochemical property alterations common in each cardiomyopathy subtype. This included heightened calcium sensitivity in HCM and RCM compared to DCM, where a decrease was seen. Heightened calcium sensitivity is thought to contribute to hypercontractility and have been linked to development of arrhythmias [61, 62]. RCM also exhibited decreased cooperative action of myofilaments, a process thought to have a diminishing effect on force development [63]. Regardless, functional consequences remain highly variable amongst variants studied.
In conclusion, our data exhibited that TNNC1-positive probands carried the poorest prognosis. Mapping of TNN variants revealed distinct locations in TNNT2 and TNNI3 associated with high risk of variant pathogenicity, early presentation, RCM diagnosis, and increased risk of VF and SCD. Future investigations should focus on TNNC1 variants as more probands are discovered. Furthermore, probands with de novo variants, homozygosity, and compound heterozygotes warrant further prospective investigations.
Supplementary Material
CLINICAL PERSPECTIVES.
In light of population-based data showing variants, it has become unclear how certain variants are well tolerated and others pathogenic. S:N analysis is a viable tool to better analyze variant hotspots, while integrating rare population variants. S:N analysis can be used to identify pathologic hotspots in other genes that demonstrate rare variants found in ostensibly healthy individuals.
Highlights:
It is unclear how certain variants are well tolerated and others pathogenic.
TNNC1-positive probands carried the poorest prognosis.
Mapping of TNN revealed locations in TNNT2 and TNNI3 associated with pathogenicity.
De novo variants, homozygosity, and compound heterozygotes have poorer prognosis.
Acknowledgments
GRANT SUPPORT
JRP is supported by the NIH Grant R01-HL128683. MSP is supported by AHA SDG #16SDG29120002. UF CTSI is supported by the NIH UL1TR001427. APL is supported by the Pediatric and Congenital Electrophysiology Society Paul C. Gillette Award, pilot grant funding from the Baylor College of Medicine Department of Pediatrics and Duke University School of Medicine.
ABBREVIATIONS
- DCM
dilated cardiomyopathy
- HCM
hypertrophic cardiomyopathy
- LVNC
left ventricular non-compaction cardiomyopathy
- RCM
restrictive cardiomyopathy
- S:N
signal to noise
- SCD
sudden cardiac death
- Tn
cardiac troponin molecular complex
- TnC
Troponin C
- TnI
Troponin I
- TNN
Troponin Gene
- TnT
Troponin T
- VF
ventricular fibrillation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST:
No conflicts of interest to declare.
LIMITATIONS
Studies compiled were variable in data collection and were performed retrospectively. Comprehensive clinical data were not always available for probands and patients may have been lost to follow-up. Given that TNN variants are rare, we were likely underpowered to detect the prognostic impact of TNNC1 variants. Further, when calculating variant frequencies based on cardiomyopathy subtype, it is possible that minor cardiomyopathy variant frequencies are inflated due to limited patients screened in the literature for these genes. Further, homozygous, compound, and double heterozygous cases were small in number which limits our statistical analysis. Despite these limitations, this study represents the largest and most comprehensive study to date.
REFERENCES
- 1.Wexler R, Elton T, Pleister A, Feldman D. Cardiomyopathy: An Overview. Am. Fam. Physician 2009;79(9):778–84. [PMC free article] [PubMed] [Google Scholar]
- 2.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary Definitions and Classification of the Cardiomyopathies. Circulation. 2006;113(14):1807. [DOI] [PubMed] [Google Scholar]
- 3.Parvatiyar MS, Pinto JR, Dweck D, Potter JD. Cardiac Troponin Mutations and Restrictive Cardiomyopathy. J. Biomed. Biotechnol 2010;2010:350706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J. Mol. Cell. Cardiol 2010;48(5):882–92. [DOI] [PubMed] [Google Scholar]
- 5.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol. Rev 2000;80(2):853–924. [DOI] [PubMed] [Google Scholar]
- 6.de AMM, Parvatiyar MS, Yang W, de Oliveira GP, Pinto JR. The missing links within troponin. Arch. Biochem. Biophys 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave-Castillo LM, Wolfgeher D, McNally EM. Population-based variation in cardiomyopathy genes. Circ. Cardiovasc. Genet 2012;5(4):391–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andreasen C, Nielsen JB, Refsgaard L, Holst AG, Christensen AH, Andreasen L, et al. New population-based exome data are questioning the pathogenicity of previously cardiomyopathy-associated genetic variants. European journal of human genetics : EJHG. 2013;21(9):918–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat 2003;21(6):577–81. [DOI] [PubMed] [Google Scholar]
- 10.Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med 2015;17(5):405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res. 2018;46(D1):D754–D61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genetics in medicine : official journal of the American College of Medical Genetics. 2017;19(2):192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landstrom AP, Fernandez E, Rosenfeld JA, Yang Y, Dailey-Schwartz AL, Miyake CY, et al. Amino acid-level signal-to-noise analysis of incidentally identified variants in genes associated with long QT syndrome during pediatric whole exome sequencing reflects background genetic noise. Heart Rhythm. 2018;15(7):1042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landstrom AP, Dailey-Schwartz CY, et al. Interpreting AL, Rosenfeld JA, Yang Y, McLean MJ, Miyake Incidentally Identified Variants in Genes Associated With Catecholaminergic Polymorphic Ventricular Tachycardia in a Large Cohort of Clinical Whole-Exome Genetic Test Referrals. Circ Arrhythm Electrophysiol. 2017;10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Au - Jones EG, Au - Landstrom AP. Determining the Likelihood of Variant Pathogenicity Using Amino Acid-level Signal-to-Noise Analysis of Genetic Variation. JoVE. 2019(143):e58907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, et al. Myofilament Protein Gene Mutation Screening and Outcome of Patients With Hypertrophic Cardiomyopathy. Mayo Clin. Proc 2008;83(6):630–8. [DOI] [PubMed] [Google Scholar]
- 19.Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J. Med. Genet 2005;42(10):e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X Gene mutation-based and specific therapies in precision medicine. J. Cell. Mol. Med 2016;20(4):577–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dainis AM, Ashley EA. Cardiovascular Precision Medicine in the Genomics Era. JACC. Basic to translational science. 2018;3(2):313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science (New York, N.Y.). 2016;351(6273):617–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma H, Marti-Gutierrez N, Park S-W, Wu J, Lee Y, Suzuki K, et al. Correction of a pathogenic gene mutation in human embryos. Nature. 2017;548(7668):413–9. [DOI] [PubMed] [Google Scholar]
- 24.Mearini G, Stimpel D, Geertz B, Weinberger F, Krämer E, Schlossarek S, et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nature Communications. 2014;5(1):5515. [DOI] [PubMed] [Google Scholar]
- 25.Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin. Proc 2008;83(6):630–8. [DOI] [PubMed] [Google Scholar]
- 26.Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O’Donoghue A, et al. Mutations in the Genes for Cardiac Troponin T and α-Tropomyosin in Hypertrophic Cardiomyopathy. N. Engl. J. Med 1995;332(16):1058–65. [DOI] [PubMed] [Google Scholar]
- 27.Sedaghat-Hamedani F, Kayvanpour E, Tugrul OF, Lai A, Amr A, Haas J, et al. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: a meta-analysis on 7675 individuals. Clin. Res. Cardiol 2018;107(1):30–41. [DOI] [PubMed] [Google Scholar]
- 28.Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138(14):1387–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van den Wijngaard A, Volders P, Van Tintelen JP, Jongbloed JD, van den Berg MP, Lekanne Deprez RH, et al. Recurrent and founder mutations in the Netherlands: cardiac Troponin I (TNNI3) gene mutations as a cause of severe forms of hypertrophic and restrictive cardiomyopathy. Neth. Heart J 2011;19(7–8):344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Acuna-Hidalgo R, Veltman JA, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016;17:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu B, Roos JL, Levy S, van Rensburg EJ, Gogos JA, Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet 2008;40(7):880–5. [DOI] [PubMed] [Google Scholar]
- 32.Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, et al. De novo mutations in Congenital Heart Disease with Neurodevelopmental and Other Birth Defects. Science (New York, N.Y.). 2015;350(6265):1262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Priest JR, Osoegawa K, Mohammed N, Nanda V, Kundu R, Schultz K, et al. De Novo and Rare Variants at Multiple Loci Support the Oligogenic Origins of Atrioventricular Septal Heart Defects. PLoS Genet. 2016;12(4):e1005963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Landim-Vieira M, Johnston JR, Ji W, Mis EK, Tijerino J, Spencer-Manzon M, et al. Familial Dilated Cardiomyopathy Associated With a Novel Combination of Compound Heterozygous TNNC1 Variants. Front. Physiol 2020;10:1612-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnston JR, Landim-Vieira M, Marques MA, de Oliveira GAP, Gonzalez-Martinez D, Moraes AH, et al. The intrinsically disordered C terminus of troponin T binds to troponin C to modulate myocardial force generation. J. Biol. Chem 2019;294(52):20054–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burns C, Bagnall RD, Lam L, Semsarian C, Ingles J. Multiple Gene Variants in Hypertrophic Cardiomyopathy in the Era of Next-Generation Sequencing. Circ. Cardiovasc. Genet 2017;10(4). [DOI] [PubMed] [Google Scholar]
- 37.Richard P, Isnard R, Carrier L, Dubourg O, Donatien Y, Mathieu B, et al. Double heterozygosity for mutations in the beta-myosin heavy chain and in the cardiac myosin binding protein C genes in a family with hypertrophic cardiomyopathy. J. Med. Genet 1999;36(7):542–5. [PMC free article] [PubMed] [Google Scholar]
- 38.Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J. Med. Genet 2005;42(10):e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, et al. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol 2004;44(9):1903–10. [DOI] [PubMed] [Google Scholar]
- 40.Rigato I, Bauce B, Rampazzo A, Zorzi A, Pilichou K, Mazzotti E, et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet 2013;6(6):533–42. [DOI] [PubMed] [Google Scholar]
- 41.Bauce B, Nava A, Beffagna G, Basso C, Lorenzon A, Smaniotto G, et al. Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2010;7(1):22–9. [DOI] [PubMed] [Google Scholar]
- 42.Kelly M, Semsarian C. Multiple Mutations in Genetic Cardiovascular Disease. Circ. Cardiovasc. Genet 2009;2(2):182–90. [DOI] [PubMed] [Google Scholar]
- 43.Hall CL, Sutanto H, Dalageorgou C, McKenna WJ, Syrris P, Futema M. Frequency of genetic variants associated with arrhythmogenic right ventricular cardiomyopathy in the genome aggregation database. European journal of human genetics : EJHG. 2018;26(9):1312–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nouhravesh N, Ahlberg G, Ghouse J, Andreasen C, Svendsen JH, Haunsø S, et al. Analyses of more than 60,000 exomes questions the role of numerous genes previously associated with dilated cardiomyopathy. Molecular genetics & genomic medicine. 2016;4(6):617–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manning EP, Tardiff JC, Schwartz SD. Molecular effects of familial hypertrophic cardiomyopathy-related mutations in the TNT1 domain of cTnT. J. Mol. Biol 2012;421(1):54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palm T, Graboski S, Hitchcock-DeGregori SE, Greenfield NJ. Disease-causing mutations in cardiac troponin T: identification of a critical tropomyosin-binding region. Biophys. J 2001;81(5):2827–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katrukha IA. Human cardiac troponin complex. Structure and functions. Biochemistry (Mosc.). 2013;78(13):1447–65. [DOI] [PubMed] [Google Scholar]
- 48.Franklin Andrew J, Baxley T, Kobayashi T, Chalovich Joseph M. The C-Terminus of Troponin T Is Essential for Maintaining the Inactive State of Regulated Actin. Biophys. J 2012;102(11):2536–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bohlooli Ghashghaee N, Li KL, Solaro RJ, Dong WJ. Role of the C-terminus mobile domain of cardiac troponin I in the regulation of thin filament activation in skinned papillary muscle strips. Arch. Biochem. Biophys 2018;648:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meyer NL, Chase PB. Role of cardiac troponin I carboxy terminal mobile domain and linker sequence in regulating cardiac contraction. Arch. Biochem. Biophys 2016;601:80–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamada Y, Namba K, Fujii T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nature Communications. 2020;11(1):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gilda JE, Xu Q, Martinez ME, Nguyen ST, Chase PB, Gomes AV. The functional significance of the last 5 residues of the C-terminus of cardiac troponin I. Arch. Biochem. Biophys 2016;601:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Veltri T, de Oliveira GAP, Bienkiewicz EA, Palhano FL, Marques MdA, Moraes AH, et al. Amide hydrogens reveal a temperature-dependent structural transition that enhances site-II Ca(2+)-binding affinity in a C-domain mutant of cardiac troponin C. Sci. Rep 2017;7(1):691-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marques MdA, Pinto JR, Moraes AH, Iqbal A, de Magalhães MTQ, Monteiro J, et al. Allosteric Transmission along a Loosely Structured Backbone Allows a Cardiac Troponin C Mutant to Function with Only One Ca(2+) Ion. The Journal of biological chemistry. 2017;292(6):2379–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith L, Greenfield NJ, Hitchcock-DeGregori SE. The effects of deletion of the amino-terminal helix on troponin C function and stability. J. Biol. Chem 1994;269(13):9857–63. [PubMed] [Google Scholar]
- 56.Smith L, Greenfield NJ, Hitchcock-DeGregori SE. Mutations in the N- and D-helices of the N-domain of troponin C affect the C-domain and regulatory function. Biophys. J 1999;76(1 Pt 1):400–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chandra M, da Silva EF, Sorenson MM, Ferro JA, Pearlstone JR, Nash BE, et al. The effects of N helix deletion and mutant F29W on the Ca2+ binding and functional properties of chicken skeletal muscle troponin. J. Biol. Chem 1994;269(21):14988–94. [PubMed] [Google Scholar]
- 58.Pinto JR, Parvatiyar MS, Jones MA, Liang J, Ackerman MJ, Potter JD. A Functional and Structural Study of Troponin C Mutations Related to Hypertrophic Cardiomyopathy. The Journal of Biological Chemistry. 2009;284(28):19090–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chase PB, Szczypinski MP, Soto EP. Nuclear tropomyosin and troponin in striated muscle: new roles in a new locale? J. Muscle Res. Cell Motil 2013;34(3–4):275–84. [DOI] [PubMed] [Google Scholar]
- 60.Johnston JR, Chase PB, Pinto JR. Troponin through the looking-glass: emerging roles beyond regulation of striated muscle contraction. Oncotarget. 2017;9(1):1461–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stücker S, Kresin N, Carrier L, Friedrich FW. Nebivolol Desensitizes Myofilaments of a Hypertrophic Cardiomyopathy Mouse Model. Front. Physiol 2017;8:558-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, et al. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J. Clin. Invest 2008;118(12):3893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gao WD, Pérez NG, Seidman CE, Seidman JG, Marbán E. Altered cardiac excitation-contraction coupling in mutant mice with familial hypertrophic cardiomyopathy. The Journal of clinical investigation. 1999;103(5):661–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.