

Abstract
Photoredox catalysis using proton-coupled electron transfer (PCET) has emerged as a powerful method for bond transformations. We previously employed traditional chemical oxidants to achieve multiple-site concerted proton-electron transfer (MS-CPET) activation of a C–H bond in a proof-of-concept fluorenyl-benzoate substrate. As described here, photoredox oxidation of the fluorenyl-benzoate follows the same rate constant vs driving force trend determined for thermal MS-CPET. Analogous photoredox catalysis enables C–H activation and H/D exchange in a number of additional substrates with favorably positioned bases. Mechanistic studies support our hypothesis that MS-CPET is a viable pathway for bond activation for substrates in which the C–H bond is weak, while stepwise carboxylate oxidation and hydrogen atom transfer likely predominates for stronger C–H bonds.
Graphical Abstract
Introduction
Selective activation and functionalization of C–H bonds under mild conditions remains a primary challenge in synthetic chemistry.1 Photoredox catalysis has emerged as a method for performing difficult bond transformations by harnessing the redox power of photocatalyst electronic excited states.2 Recent photoredox strategies have selectively activated strong X-H bonds by multiple-site concerted proton-electron transfer (MS-CPET).3 In this mechanism, electron transfer (ET) with a photocatalyst occurs simultaneously with proton transfer to a separate, basic site. For polar X-H bonds, hydrogen bonding of the base assists the concerted reaction (Scheme 1A). In contrast, nonpolar C–H bonds have typically been activated by stepwise heteroatom oxidation and then hydrogen atom transfer (HAT; Scheme 1B).3a,4
Scheme 1.
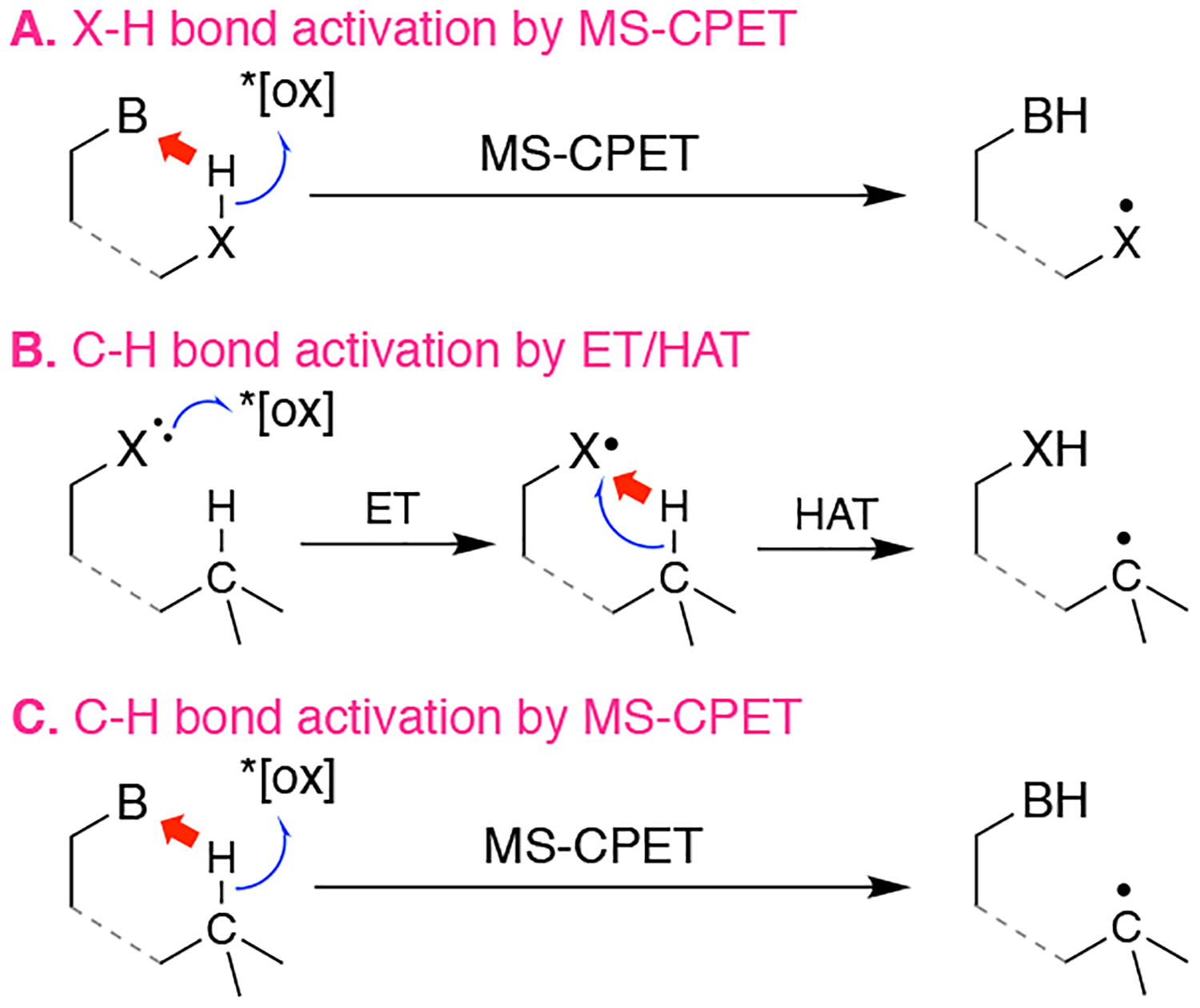
Photoredox PCET mechanisms for X-H and C–H bond activation. B represents a generic base, X is N or O. This work involves case C.
Recently, C–H bond activation by MS-CPET was reported using traditional chemical (“thermal”) oxidants and a proof-of concept substrate (1) containing a favorably positioned carboxylate base (Figure 1A).5 For 1, initial intermolecular ET is concerted with intramolecular proton transfer (PT) to the carboxylate base. Conversion of radical intermediate 1· to the observed lactone product (1-lac) involves additional loss of 1e−/1H+. The substrate scope of such reactions is restricted by incompatibility of carboxylate bases with many thermal oxidants such as ferrocenium cations.5a The direct reaction of an oxidant [electron poor] with a base [electron rich] is a known limitation of MS-CPET reactivity.6
Figure 1. A).
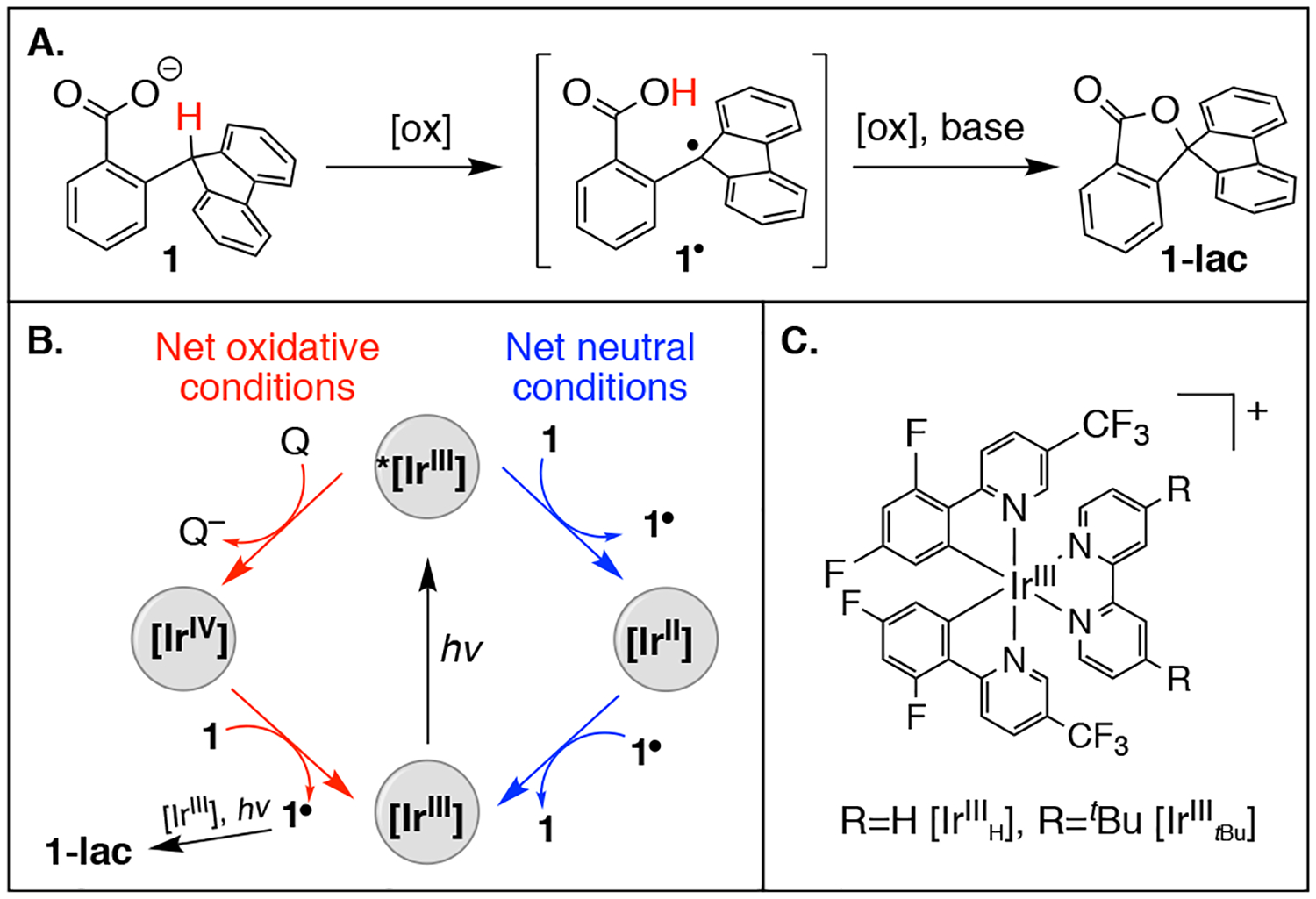
Oxidation of substrate 1. B) Photoredox cycle showing the flash-quench pathway for oxidation of 1 under oxidative conditions (red), and transient C–H activation of 1 by *[IrIII] under redox neutral conditions (blue). Q represents the Co(acac)3 quencher for oxidative photoredox. C) Ir photocatalysts in this work.
We elected to explore photocatalysis as a path to mitigate incompatibilities and enable further investigation of the fundamental aspects governing MS-CPET reactivity at C–H bonds. During the preparation of this manuscript, the Knowles and Alexanian groups reported a termolecular MS-CPET system in which the photocatalyst and phosphate base form a noncova-lent adduct that is capable of activating sp3 C–H bonds.7 We report here a photoredox system that oxidizes 1 and related substrates, achieves non-oxidative base-directed C–H activation on additional substrates, and investigates the competition between MS-CPET (Scheme 1C) and stepwise ET/HAT (Scheme 1B) mechanisms.
Results and Discussion
The 2-electron oxidation of 1 to 1-lac proceeds under oxidative photoredox conditions (Figures 1A and 1B). A 410 nm LED was used to irradiate a 3–5 mM solution of 1 (generated in situ by deprotonation of the carboxylic acid with 0.8 eq tetrabutylammonium acetate, TBAOAc), with 10 mol% iridium photocatalyst [IrIIIH] or [IrIIItBu],8 and ~1.2 eq of Co(acac)3 terminal oxidant, in deoxygenated acetonitrile. After 2–4 hours of irradiation, up to 90°% formation of the 1-lac final product was observed (1H NMR), with respect to equivalents of base and terminal oxidant.9 Importantly, omitting light, photocatalyst, terminal oxidant, or added base results in no or only trace 1-lac.10
A variety of factors influence the efficacy of this photoredox transform ation (see Supporting Information [SI] for additional discussion). Notably, using TBAOAc for in situ deprotonation enhances long-term photocatalyst stability, compared to tetrabutylammonium hydroxide (TBAOH) or DBU (1,8-diazabicyclo[5.4.0]undec-7-ene). We presume that the stoichiometric am ount of acetic acid formed during in situ deprotonation reduces the nucleophilicity of the 1 carboxylate, attenuating degradation of the photocatalysts as a result of nucleophilic attack on the ligands. However, both Ir catalysts degrade slowly under these basic conditions over many hours, as evidenced by red-shifting of the UV-visible absorption and emission spectra (see SI).11
Photo-oxidant stability is also enhanced when using < 1 equivalent of base, and adding protic solvents (e.g., MeOH). However, additions > 5°% v/v of MeOH significantly slow or halt the conversion of 1 to 1-lac, consistent with studies employing thermal oxidants.5a No additional products are formed under these more protic conditions. The selection of a term inal oxidant is somewhat limiting, as many weak oxidants react directly with 1 (e.g., N-bromo succinimide, methyl viologen), and other reagents common to photoredox (e.g., aryl diazonium salts) form reactive radicals that could interfere with the desired mechanistic analysis. Co(acac)3 meets the experimental limitations for our purposes,2a but its competitive absorbance throughout the visible region renders it less than optimal.
To probe the identity of the active oxidant during these reactions, Stern-Volmer quenching experiments were employed. The excited state *[IrIII] reacts rapidly with Co(acac)3 with kq ≅ 108 M−1is−1 (Figure 1B, top red arrow).10 This forms [IrIV], which can oxidize 1. The direct reaction of *[IrIII] with 1 is slower, (kq ≅ 106 M−1s−1, see below). Nearly equimolar Co(acac)3 and substrate 1 are employed in the reaction mixture, and [IrIV] persists in solution longer than *[IrIII], so we expect [IrIV] to be the predominant oxidant under oxidative photoredox conditions (Figure 1B, red pathway).
C–H bond activation was also probed under net-redox neutral photoredox conditions, in the absence of Co(acac)3 term inal oxidant. No formation of 1-lac is observed under these conditions. However, 1 can be transiently oxidized to 1· by *[IrIII] (Figure 1B, blue pathway). Formation of reduced photosensitizer ([IrII]) is observed upon quenching with 1, by comparison of transient spectra to that of electrochemically-reduced photosensitizer, and to [IrII] photochemically produced in the presence of an established reductive quencher, tritolylamine.10 This confirms electron transfer from 1 to *IrIII. Stern-Volmer analysis indicates a rate constant for luminescence quenching of *[IrIIItBu] by 1 of approximately kq = 4(±2) × 106 M−1s−1 (comparing luminescence lifetimes at varying concentration of 1). There is significant batch-to-batch variability in the rate constant vs concentration dependence12 (also see SI), possibly due to small impurities (e.g., water hydrogen-bonding to the carboxylate in 1) that could affect the observed reactivity.
Additional evidence for transient C–H activation under net redox-neutral conditions was obtained from deuterium incorporation studies (Scheme 2). The fluorenyl C–H of 1 in solution does not exchange with methanol-OD within 24 hours, in the absence of photocatalyst and light.5a However, irradiating a solution of 1-h (denoting isotopic substitution at the fluorenyl carbon) and *[IrIII] with ~2°% v/v d4-methanol in d3-MeCN led to 80°% conversion to 1-d (1H NMR and HR-ESI-MS). Enrichment is envisioned to occur because the carboxylic acid proton of intermediate 1· exchanges with ~0.5 M CD3 OD to give 1·-CO2 D competitively with its reduction to 1-d by the quenching product [IrII] (≤ 150 μM) under these net redox neutral conditions. Therefore, deuterium incorporation can be used as a marker for transient C–H activation by *[IrIII].
Scheme 2.
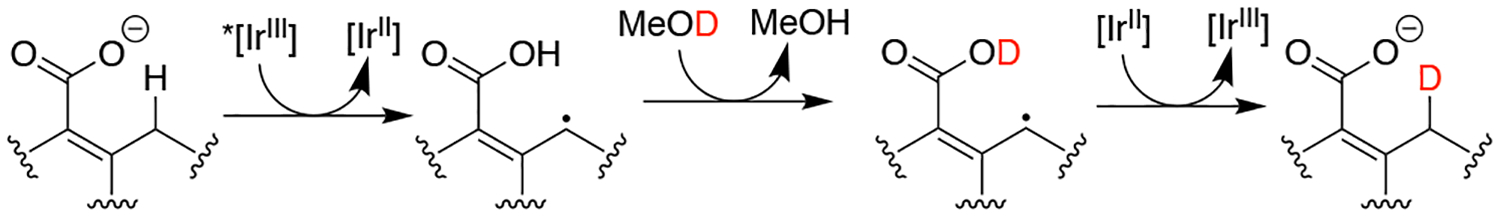
Deuterium incorporation under net-redox neutral photoredox conditions is a marker for C–H activation.
This H/D exchange methodology has allowed us to explore photoredox activation of C–H bonds in other substrates with a positioned base (Table 1), including many that were unreactive with thermal oxidants (5–10). Deuterium incorporation was observed for substrates 1-10 (NMR and/or HR-ESI-MS). In addition, irradiating 1-6 under oxidative photoredox conditions - with Co(acac)3 - showed some formation of the oxidized lactone product (1H NMR and/or HR-ESI-MS, denoted “+ox” in Table 1).
Table 1.
Deuterium incorporationa and lactone form ationb for various substrates under photoredox conditions.
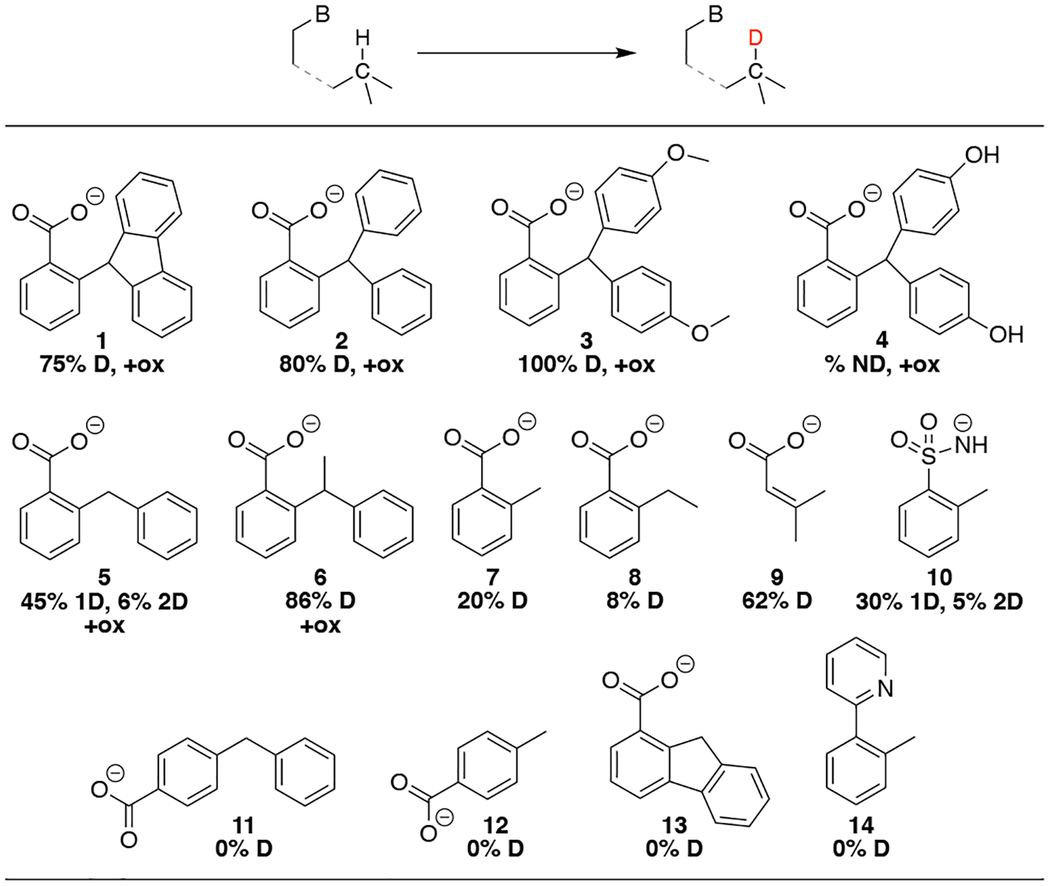
|
D incorporation under net redox-neutral conditions (~3 mM substrate acid, 0.8 eq TBAOAc, 10 mol % [IrIII], 2% d4-t-MeOD (v/v) in MeCN, irradiated overnight, 410 nm.) % D determined by HRESI-MS. “2D” indicates double deuteration. % D not determined for 4 due to low solubility in MeCN.
Oxidative photoredox of 1-6 (~6 mM substrate acid, 0.9 eq TBAOAc, 10 mol % [IrIII], 1.2 eq Co(acac)3), MeCN, irradiated overnight, 410 nm) gave corresponding lactone product by NMR and/or HR-ESI-MS, (“+ox”). No lactone product observed for 7. Substrates 8-14 not screened for formation of lactones.
The substrates in Table 1 span a range of C–H bond strengths from ~74 kcal/mol (1)5a to ~87 kcal/mol (ortho-toluate, 7),13 and include an example with an internal sulfon-amide base (10). Importantly, no evidence of deuterium incorporation is seen for para-carboxylate substrates 11 and 12, indicating the importance of base position for reactivity. There is also no observed deuteration for 14, which has a weaker pyridine base.14,15 Time course NMR studies with substrates 1, phenyl o-toluate (5) and o-toluate (7) indicate that the majority of deuteration occurs within the first 20–30 minutes.10 This is a rapid, simple, and regioselective method for isotope incorporation that is complementary to a recently reported photoredox strategy for amine-directed deuterium and tritium incorporation.16
Preliminary studies explored functionalization of the carbon radical intermediates generated under redox neutral conditions. Methylvinylketone (MVK) and TEMPO have been employed by others as traps to achieve photoredox-mediated CC and C–O bond formations.3a,4b,17 Acetonitrile solutions of substrate 7 (toluate) with 10 mol % [IrIIItBu] and 2 eq of the trap were irradiated at 410 nm for 16 hrs. HR-ESI-MS spectra of the resulting mixtures showed anions corresponding to TEMPO–CH2C6H4CO2− and CH3C(O)(CH2)3C6H4CO2−.10
The H/D exchange and trapping results show that photoredox C–H activation occurs for a variety of benzylic substrates with internal carboxylates. Previously reported rate constants for thermal oxidations of 1 correlate with the E1/2 of the ferricenium and triarylaminium oxidants used.5a,5c The approximate rate constant for photoredox C–H bond cleavage in 1 by *[IrIIItBu] fits this same correlation (Figure 2), supporting an MS-CPET mechanism.18
Figure 2.
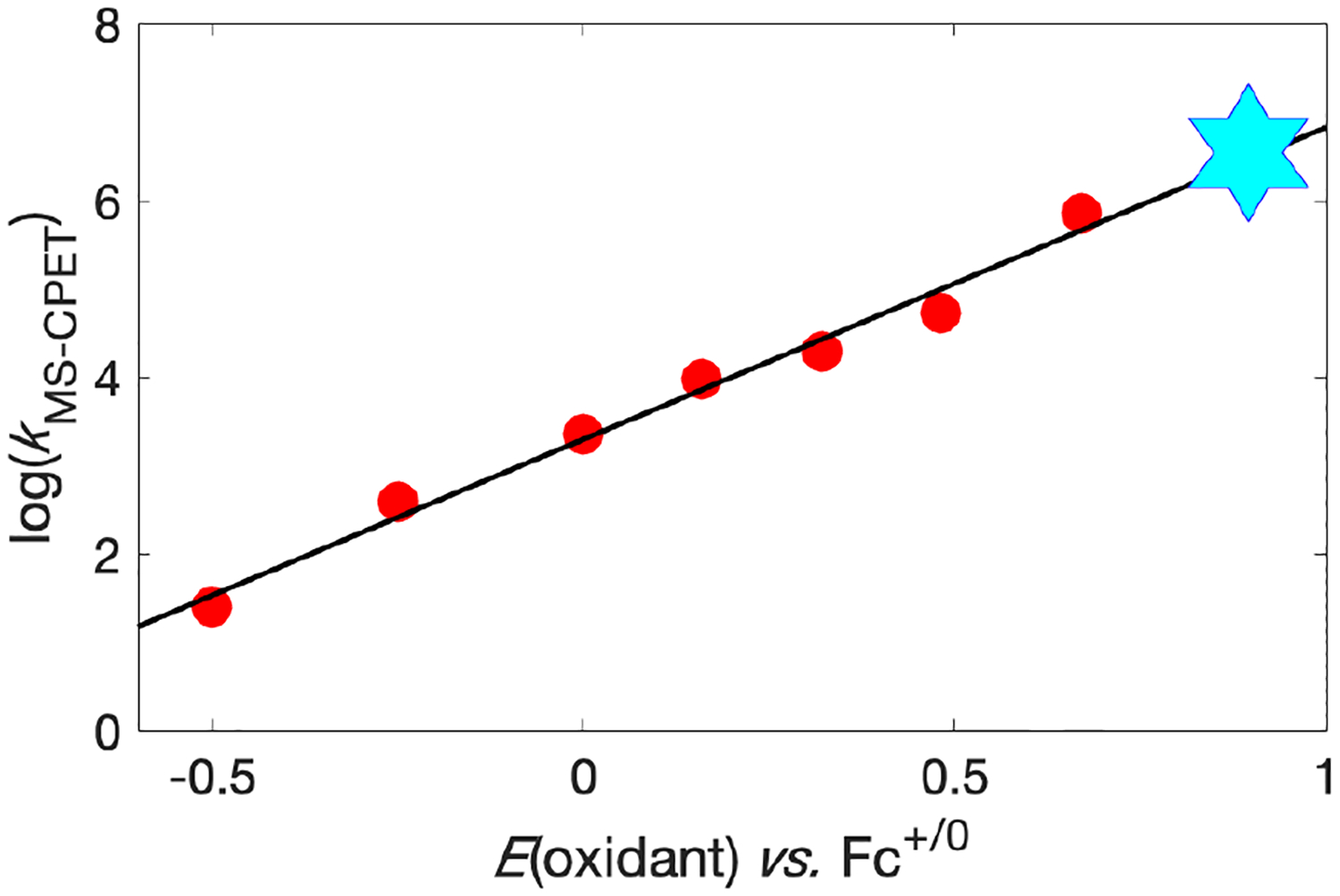
Plot of rate constant vs. oxidant strength for oxidation of 1. Red circles and trendline are thermal oxidations;5a cyan star is oxidation by *[IrIIItBu] (this work). Star size encompasses uncertainty in KMS–CPET and E(oxidant).
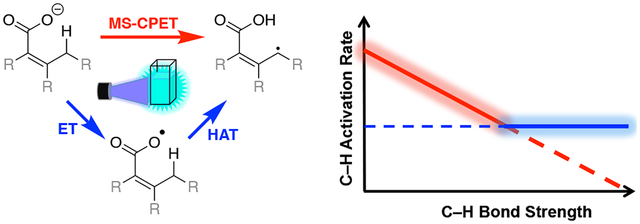
To explore whether [IrIV] and/or *[IrIII] can directly oxidize some carboxylates to carboxyl radicals, we estimated the reduction potentials from cyclic voltammetry and optical estimates of the E0–0 energies (Table 2, Supporting Information Section 4). These values show that the ground state IrIV species are capable of oxidizing carboxylates by outer-sphere electron transfer, and very likely the excited states as well. This is consistent with Glorius’ report that *[IrIIItBu] oxidizes benzoate to PhCO2·, which then activate a variety of aliphatic C–H bonds by intermolecular HAT (Scheme 1B).4a Our transient absorption experiments show the formation of reduced photosensitizer ([IrII]) in the presence of benzoate, further supporting this possibility. This stepwise mechanism may be expected to outcompete a concerted one (MS-CPET) for substrates with stronger C–H bonds, in which MS-CPET is expected to be slower (conceptually illustrated in Figure 3).
Table 2.
Measured and estimated reduction potentials vs. ferrocene+/0 (Fc+/0) in MeCN.a
| Redox Couple | E1/2 | References |
|---|---|---|
| E1/2(IrIVH/IrIIIH) | ~1.38 V | estimated herea |
| E1/2(IrIVtBu/IrIIItBu) | ~1.35 V | estimated herea |
| E1/2(*IrIIIH/IrIIH) | ~1.05 V | estimated hereb |
| E1/2(*IrIItBu/IrIItBu) | ~0.95 V | estimated hereb |
| E1/2(RCO2•/RCO2−) | ~0.9 V | 3a,4a,19 |
Quasi-reversible waves by cyclic voltammetry; see Supporting Information, Section 4.1.
See discussion of the optical E0–0 energies in Supporting Information, Section 4.2.
Figure 3.
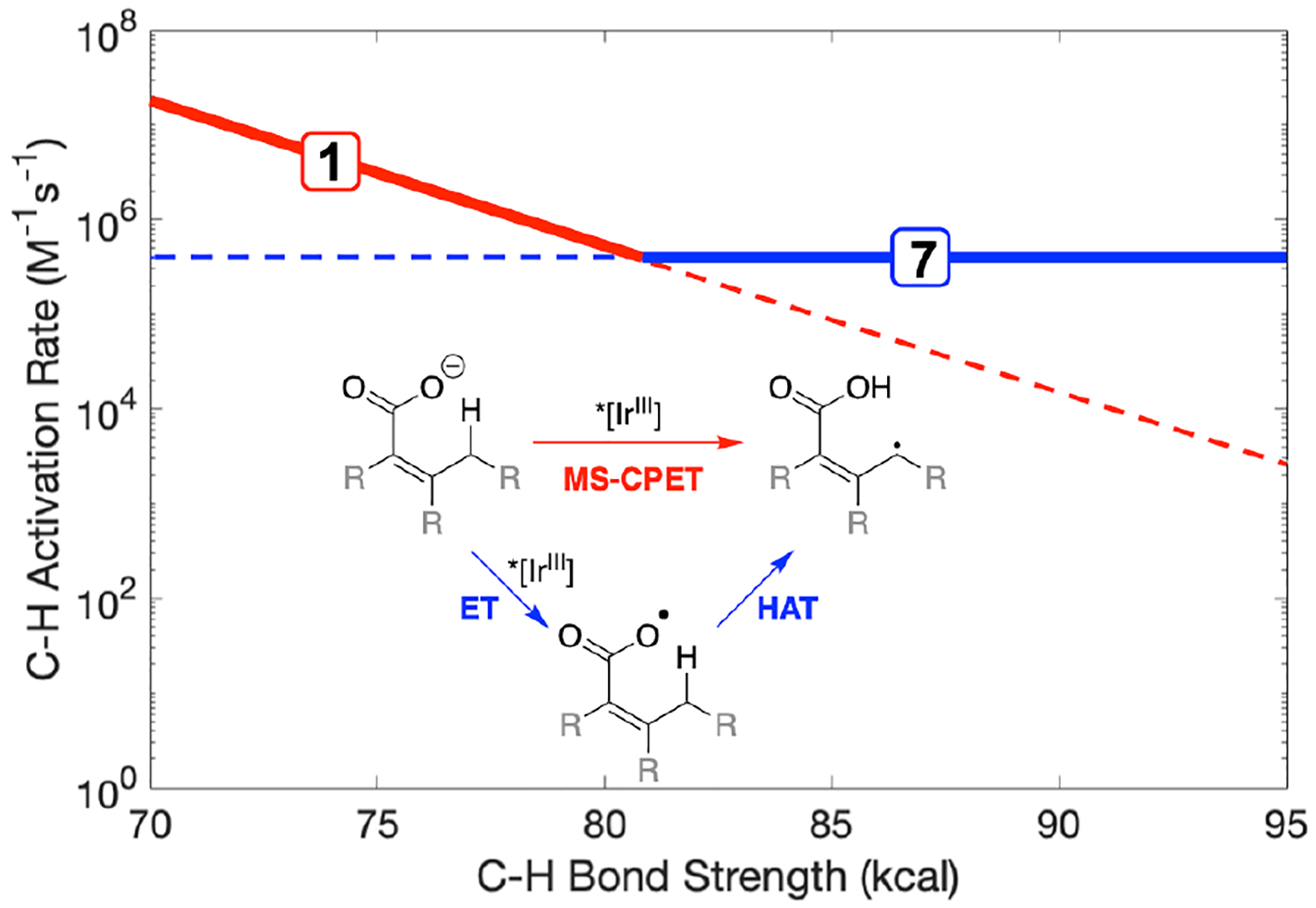
Conceptual transition between concerted and stepwise C–H activation mechanisms with a single oxidant. Boxed numbers represent measured kq for substrates 1 and 7. The blue line (ET/HAT), representing measured kq for benzoate, is independent of C–H bond strength. The red line (MS-CPET) represents the k vs. driving force relationship for thermal oxidations of substrate 1.5a,5b Inset: schematic of MS-CPET (red) and ET/HAT (blue) mechanisms.
We have probed the mechanistic possibilities of stepwise ET/HAT vs. MS-CPET (Scheme 1B vs. C) by comparing *[IrIIItBu] luminescence quenching by different substrates. Benzoate (which can only undergo direct ET) quenches the *[IrIIItBu] luminescence with an observed rate constant that is very sensitive to conditions. For benzoate formed in situ from benzoic acid and TBAOAc, kq = 4× 105 M−1s−1. In contrast, kq for TBA-benzoate in dry acetonitrile (no acetic acid as potential hydrogen bond partner) is approximately two orders of magnitude faster20 (4× 107 M−1s−1 at low concentrations, in reasonable agreement with other reports).4a Assuming that all luminescence quenching by benzoate/HOAc is due to ET, the rate constant of kq = 4× 105 M−1s−1 gives an approximate bound at which ET/HAT becomes competitive with MS-CPET under these acetic acid-tuned conditions (Figure 3). For substrate 7 (toluate), which has one of the strongest C–H bonds in Table 1, the kq = 4× 105 M−1s−1, suggests that ET/HAT is a likely mechanism for C–H activation.
The substrates in Table 1 span the range of bond strengths between that of 1 and 7, and are expected to span the transition between mechanisms. Trityl substrates 2, 3, and 4 have similar C–H bond strengths to 1 and probably follow an MS-CPET mechanism, while phenyl toluate (5) may undergo predominantly ET/HAT, or a combination of both mechanisms. Limited solubility of these substrates (< 20 mM) has precluded Stern-Volmer analysis. Similarly, the available data do not allow drawing mechanistic conclusions for the reduction of the fluorenyl radical, the last part of the isotope exchange pathway (Scheme 2, Supporting Information 6.2).
Conclusion
The results presented above demonstrate that photoredox can be used as a mild, versatile method for base-directed C–H activation, deuterium incorporation, and functionalization. Photoredox conditions can replace thermal oxidants to accomplish MS-CPET at the fluorenyl C–H bond in 1, opening new avenues to probe fundamental aspects of this concerted mechanism. For many substrates, two mechanistic pathways are possible under the highly oxidizing conditions generated during photoredox. MS-CPET is likely in competition with the two-step pathway of rate-limiting ET followed by rapid HAT, which has been reported in other studies.4a The stepwise ET/HAT path is favored by a more easily oxidized carboxylate and by stronger C–H bonds, since it is independent of the C–H bond strength. MS-CPET may be favored when the C–H bond is weak, and the proton acceptor (typically carboxylate here) is basic and difficult to oxidize. We hope these insights will facilitate further development of strategies for activating C–H bonds, and deeper explorations into the fundamental aspects of MS-CPET processes at carbon.
Experimental
Materials
Substrates 1 (including both isotopic variants 1-h and 1-d), 2, S15 and 6 were synthesized as described previously.5a,21 Substrate 3 was synthesized as described below. The remaining chemicals were obtained from commercial sources and used without further purification unless otherwise noted. Co(acac)3, and substrates 10 and S20 were obtained from Alfa Aesar; substrate S16 was obtained from Alfa chemicals; substrates 4, 5, 8, and 14 were obtained from Santa Cruz Bio-technology; substrate 13 was obtained from Acros Organics; substrate 7 was obtained from Tokyo Chemical Industry; tetrabutylammonium acetate (TBAOAc) was obtained from Sigma Aldrich and dried in a vacuum oven; benzoic acid, 1,3,5-trimethoxybenzene (TMB), tetrabutylammonium hydroxide (TBAOH, 1M solution in methanol), tetrabutylammonium benzoate (TBAOBz), [IrIIIH] and [IrIIItBu], and substrates 9, 12, S17, S18, S19, S21, S22, S23 were obtained from Sigma Aldrich. Phthalic anhydride was obtained from Sigma Aldrich and recrystallized from chloroform before use. Substrate 11 was obtained from Trans World Chemicals. Anaerobic acetonitrile (Burdick Jackson low water) was sparged with argon, and was plumbed directly into the glovebox and used without additional purification/drying. Deuterated solvents were obtained from Cambridge Isotope Lab, deoxygenated by freeze-pump-thaw, and stored in an N2-filled glovebox.
Synthesis
2-(bis(4-m ethoxyphenyl)m ethyl)benzoic acid (3).
Phenolphthalein (1.0 g, 3.1 mmol, 1 eq), K2 CO3 (1.3 g, 9.3 mmol, 3 eq), and acetone (20 mL) were stirred at reflux for 10 minutes to ensure deprotonation. Then, iodomethane (3.1 mL, 6.2 mmol, 2 eq) was added. The reaction was quenched after 22 hrs with 20 mL 2 M NaOH. The aqueous layer was re-acidified with 1 M HCl until a white precipitate formed. The aqueous layer was extracted with Et2O (3 × 40 mL), dried over Na2SO4, and concentrated. The crude product was purified on a silica column using pure EtOAc to yield the triply methylated methyl 2-(bis(4-methoxyphenyl)methyl)benzoate. The methyl ester (3.1 mmol) was deprotected by refluxing in KOHaq (1.7 g in 50 mL H2O) for 5 hours. Then the reaction mixture was cooled to room temperature and acidified with ~40 mL 1 M HCl until a white precipitate formed. The aqueous layer was extracted with Et2O (2 × 40 mL) and dried over Na2SO4. The resulting white solid was recrystallized from MeOH; yield 110 mg, 10.1%. 1H NMR (400 MHz, CD3 CN) δ (ppm): 7.85 (d, 1H), 7.45 (t, 1H), 7.31 (t, 1H), 7.05 (d, 1H), 6.95 (d, 4H), 6.84 (d, 4H), 6.49 (s, 1H), 3.75 (s, 6H). 13C{1H} NMR (100 MHz, CD3 CN) δ (ppm): 169.2, 159.1 146.4, 137.3, 132.6, 131.6, 131.5, 131.4, 131.2, 127.2, 114.5, 55.8, 51.2. HR-MS (negative ion mode, for the carboxylate anion): m/z 347.1319 (theoretical: 347.1284).
Photoredox Reactions
Photoredox reaction mixtures were prepared in a nitrogen-filled glovebox using deoxygenated, deuterated solvent, and placed in sealed J-Young NMR tubes. Blue/violet light irradiation was applied between 45 minutes and 16 hours (depending on the experiment), using a 410 nm LED apparatus assembled in house. Lamp components were purchased from Mouser Electronics: 410 nm LED (e.g., LZ1–10UB00–01U8), heat sink, focusing lens, and associated lamp components. The distance between lamp and J-Young tube was typically 15–20 cm. No effort was made to cool the samples during irradiation, and no filters were used. More details for these and other experiments are given in the Supporting Information.
For net oxidative photoredox reactions, samples typically included 5 mM substrate, 0.8–0.9 eq TBAOAc as in situ base, 6 mM Co(acac)3 as terminal oxidant, and 75–150 μM Ir photocatalyst in d3-acetonitrile. For net neutral photoredox (H/D exchange) conditions, samples typically included 3 mM substrate, ~0.8 eq TBAOAc, and ~150 μM Ir photocatalyst in d3-acetonitrile with 2°% (v/v) d4-methanol. Deuterium incorporation was first assessed by 1H NMR spectroscopy, then the sample was diluted to 1 mM in acetonitrile, and a High Resolution Mass Spectrum (HR-MS) was collected. 1H NMR spectra were collected in the Yale Chemical and Biophysical Instrumentation Center on either a 400 MHz or 500 MHz Agilent NMR spectrometer. HR-MS was performed with a Thermo Scientific Orbitrap Velos Pro Mass Spectrometer. HR-MS data were recorded in anion mode with a resolution (FWHM) of at least 30,000 at 400 m/z and averaged over at least 100 spectra. The spray needle was held at 2.5kV with an injection rate of 2.5μL/min, source temperature 40°C, capillary temperature 225°C and sheath gas flow rate of 8 a.u. Instrument parameters were held constant for all MS experiments. Isotope patterns were modeled by hand with natural abundance isotopic distribution. Linear combinations of isotope patterns for substrates containing 1, 2, or 3 deuterium that best matched the experimental data were used to determine % D incorporation.
Electrochemistry
Cyclic voltammograms were collected to determine the ground state potentials of the two photocatalysts, [IrH] and [IrtBu]. The experiments were performed in a nitrogen-filled glovebox, in acetonitrile with 0.1M TBAPF6 electrolyte. The three electrode setup consisted of a glassy carbon working electrode, platinum counter electrode, and pseudo reference electrode with a silver wire encased in a fritted compartment containing 100 mM TBAPF6. After collection of the voltammograms for the photocatalysts, ferrocene was added as an internal standard and voltammograms were corrected to the Fc+/0 potential. This addition of ferrocene caused the IrIV/III couple to become less reversible.
Luminescence and Transient Absorption
For luminescence and transient absorption experiments, samples were prepared in a nitrogen-filled glovebox using deoxygenated acetonitrile, placed in quartz cuvettes with inner path lengths of 2 mm × 1cm quartz cuvettes equipped with airtight Kontes stoppers. Samples typically contained 100–300 μM photocatalyst and 0.5–16 mM substrate. UV-visible absorption spectra were acquired on an Agilent 8453 UV-visible spectrophotometer. Time Resolved and Steady State luminescence measurements were conducted on a TCSPC TD_Fluor Horiba Fluorolog 3 Time Domain Fluorimeter (time resolved studies used a pulsed 390 nm LED) in the Yale Chemical and Biophysical Instrumentation Center. Time resolved m easurements were acquired until a maximum intensity of approximately 2000 counts. For Stern-Volmer Luminescence experiments specifically, samples contained 80–100 μM iridium photocatalyst [IrtBu] and a wide range (0–200 mM) of substrate concentrations in acetonitrile unless otherwise noted. Kinetics traces were fit to a monoexponential decay using MATLAB software, and rate constants were determined from the slope of the best fit line for rate vs. concentration of substrate plots. For Transient Absorption experiments, excitation was performed at 430 nm with a Spectra-Physics Nd/YAG laser (Quanta-Ray) and OPO (basiScan). Probe light was supplied by an Edinburgh Instruments Xe900 xenon lamp directed at a 90° angle to the excitation beam. Multiple-wavelength transient absorption changes were monitored with an Andor iStar300 series CCD camera. Single-wavelength kinetics (luminescence and transient absorption) were monitored at 530 nm using an LP920-K UV-Vis detector. Samples were excited along the long path length and probed at 90 degrees (through the short path length dimension). Data were analyzed, fit, and plotted using MATLAB software. References19 and20 are cited in the SI.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge NIH 2R01GM50422 for funding this work. FSM thanks the Air Force Office of Scientific Research (AFOSR) under grants FA9550-17-1-0267 (DURIP) and FA9550-18-1-0213. We thank Prof. Mark A. Johnson for helpful discussions, Brian Koronkiewicz for assistance with transient absorption experiments, and Scott Coste for help with a final experiment and final manuscript preparation.
Footnotes
Supporting Information
Additional experimental procedures and analytical methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- 1. (a).Hartwig JF Evolution of C–H Bond Functionalization from Methane to Methodology. J. Am. Chem. Soc 2016, 138, 2–24; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shul’pin BG New Trends in Oxidative Functionalization of Carbon-Hydrogen Bonds: A Review. Catalysts 2016, 6, 50; [Google Scholar]; (c) Crabtree RH Alkane C–H Activation and Functionalization with Homogeneous Transition Metal Catalysts: A Century of Progress-a New Millennium in Prospect. J. Chem. Soc., Dalton Trans 2001, 2437–2450; [Google Scholar]; (d) Liao K; Negretti S; Musaev DG; Bacsa J; Davies HML Site-Selective and Stereoselective Functionalization of Unactivated C–H Bonds. Nature 2016, 533, 230–234. [DOI] [PubMed] [Google Scholar]
- 2. (a).Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schultz DM; Yoon TP Solar Synthesis: Prospects in Visible Light Photocatalysis. Science 2014, 343, 1239176; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nicewicz DA; Nguyen TM Recent Applications of Organic Dyes as Photoredox Catalysts in Organic Synthesis. ACS Catalysis 2014, 4, 355–360; [Google Scholar]; (d) Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. (a).Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Catalytic Alkylation of Remote C–H Bonds Enabled by Proton-Coupled Electron Transfer. Nature 2016, 539, 268–271; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yayla HG; Wang H; Tarantino KT; Orbe HS; Knowles RR Catalytic Ring-Opening of Cyclic Alcohols Enabled by PCET Activation of Strong O-H Bonds. J. Am. Chem. Soc 2016, 138, 10794–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. (a).Mukherjee S; Maji B; Tlahuext-Aca A; Glorius F Visible-Light-Promoted Activation of Unactivated C(sp3)–H Bonds and Their Selective Trifluoromethylthiolation. J. Am. Chem. Soc 2016, 138, 16200–16203; [DOI] [PubMed] [Google Scholar]; (b) Jeffrey JL; Terrett JA; MacMillan DWC O-H Hydrogen Bonding Promotes H-Atom Transfer from α C–H Bonds for C-Alkylation of Alcohols. Science 2015, 349, 1532–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. (a).Markle TF; Darcy JW; Mayer JM A New Strategy to Efficiently Cleave and Form C–H Bonds Using Proton-Coupled Electron Transfer. Sci. Adv 2018, 4, eaat5776; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Darcy JW; Kolmar SS; Mayer JM Transition State Asymmetry in C–H Bond Cleavage by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2019, 141, 10777–10787; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sayfutyarova ER; Goldsmith ZK; Hammes-Schiffer S Theoretical Study of C–H Bond Cleavage Via Concerted Proton-Coupled Electron Transfer in Fluorenyl-Benzoates. J. Am. Chem. Soc 2018, 140, 15641–15645; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sayfutyarova ER; Lam Y-C; Hammes-Schiffer S Strategies for Enhancing the Rate Constant of C–H Bond Cleavage by Concerted Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2019, 141, 15183–15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waidmann CR; Miller AJM; Ng C-WA; Scheuermann ML; Porter TR; Tronic TA; Mayer JM Using Combinations of Oxidants and Bases as PCET Reactants: Thermochemical and Practical Considerations. Energy Environ. Sci 2012, 5, 7771–7780. [Google Scholar]
- 7.Morton CM; Zhu Q; Ripberger H; Troian-Gautier L; Toa ZSD; Knowles RR; Alexanian EJ C–H Alkylation Via Multisite-Proton-Coupled Electron Transfer of an Aliphatic C–H Bond. J. Am. Chem. Soc 2019, 141, 13253–13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The two photocatalysts have very similar photophysical properties, also see SI. [IrH] is a slightly more potent oxidant (~100 mV), and [IrtBu] is slightly more robust in the presence of carboxylates. Oxidative photoredox studies were performed using [IrH] for historical reasons. Stern Volmer quenching experiments intentionally used [IrtBu] due to its slightly enhanced stability. The two catalysts were used interchangeably in the redox-neutral deuterium exchange studies, and appeared to be equally effective.
- 9.The oxidation of 1 to 1-lac is a 2e−, 2H+ reaction, but only one equivalent of base and terminal oxidant were used. This was due to the dark color of Co(acac)3 oxidant (which absorbed much of the incident light) and instability of the Ir photocatalyst in the presence of excess TBAOAc base.
- 10.See SI.
- 11. (a).These shifts in photocatalyst absorption and emission spectrum only occur after irradiation. In the absence of irradiation, the addition of carboxylate-containing substrates to MeCN solutions of [IrIII] do not result in shifting of the photocatalyst absorption or emission spectra, indicating the absence of non-covalent photosensitizer-substrate complexes. Such complexes has been implicated by such shifting in CH2Cl2 solutions.7,11bFarney EP; Chapman SJ; Swords WB; Torelli MD; Hamers RJ; Yoon TP Discovery and Elucidation of Counteranion Dependence in Photoredox Catalysis J. Am. Chew. Soc 2019,141, 15, 6385–6391
- 12.Previously reported reactions of 1 with thermal oxidants showed KIEs of 2–4. The batch-to-batch variability of 1-H is on this same order, precluding conclusive determination of a KIE for photoinitiated reactions
- 13.Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Cliem. Rev 2010,110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.In acetonitrile solution, the pKas of acetic acid (22) and benzoic acid (24) are significantly higher than that of pyridine (13) - see SI for further discussion and references.
- 15. (a).Kutt A; Leito I; Kaljurand I; Sooväli L; Vlasov VM; Yagupolskii LM; Koppel IA A Comprehensive Self-Consistent Spectrophotometric Acidity Scale of Neutral Brønsted Acids in Acetonitrile. J. Org. Chem 2006, 71, 2829–2838; [DOI] [PubMed] [Google Scholar]; (b) Lõkov M; Tshepelevitsh S; Heering A: Plieger PG; Vianello R; Leito I On the Basicity of Conjugated Nitrogen Heterocycles in Different Media. Eur. J. Org. Chem 2017, 2017,4475–4489. [Google Scholar]
- 16.Loh YY; Nagao K; Hoover AJ; Hesk D; Rivera NR; Colletti SL; Davies IW: MacMillan DWC Photoredox-Catalyzed Deuteration and Tritiation of Pharmaceutical Compounds. Science 2017, 555, 1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gentry EC; Rono LJ; Hale ME; Matsuura R; Knowles RR Enantioselective Synthesis of Pyrroloindolines Via Noncovalent Stabilization of Indole Radical Cations and Applications to the Synthesis of Alkaloid Natural Products. J. Am. Chem. Soc 2018,140, 3394–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The reduction reaction that re-forms the C–H or C–D bond could similarly be MS-CPET, or could proceed through a stepwise mechanism. See SI for discussion.; the data available do not allow a confident assignment. See SI Section 6.2 for discussion.
- 19.Galicia M; Gonzalez FJ Electrochemical Oxidation of Tetrabutylammonium Salts of Aliphatic Carboxylic Acids in Acetonitrile. J. Electrochem. Soc 2002, 149, D46–D50. [Google Scholar]
- 20.The relationship between observed quenching rate and benzoate concentration is non-linear over a large range of concentrations. This is an indication of side reactions.
- 21.DeZutter CB; Horner JH; Newcomb M Rate Constants for 1, 5-and 1, 6-Hydrogen Atom Transfer Reactions of Mono-, Di-, and Tri-Aryl-Substituted Donors, Models for Hydrogen Atom Transfers in Polyunsaturated Fatty Acid Radicals. J. Phys. Chem. A 2008,112, 1891–1896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.