

Abstract
G protein-coupled receptors (GPCRs) are targeted by about a third of clinically used drugs. Many GPCRs couple to more than one type of heterotrimeric G proteins, become phosphorylated by any of several different GRKs, and then bind one or more types of arrestin. Thus, classical therapeutically active drugs simultaneously initiate several branches of signaling, some of which are beneficial, whereas others result in unwanted on-target side effects. The development of novel compounds to selectively channel the signaling into the desired direction has the potential to become a breakthrough in health care. However, there are natural and technological hurdles that must be overcome. The fact that most GPCRs are subject to homologous desensitization, where the active receptor couples to G proteins, is phosphorylated by GRKs, and then binds arrestins, suggest that in most cases the GPCR conformations that facilitate their interactions with these three classes of binding partners significantly overlap. Thus, while partner-specific conformations might exist, they are likely low-probability states. GPCRs are inherently flexible, which suggests that complete bias is highly unlikely to be feasible: in the conformational ensemble induced by any ligand, there would be some conformations facilitating receptor coupling to unwanted partners. Things are further complicated by the fact that virtually every cell expresses numerous G proteins, several GRK subtypes, and two non-visual arrestins with distinct signaling capabilities. Finally, novel screening methods for measuring ligand bias must be devised, as the existing methods are not specifically for one particular branch of signaling.
1. Introduction
The classical paradigm of G protein-coupled receptor (GPCR) signaling posited that GPCRs communicate with the cell via heterotrimeric G proteins, and their signaling is terminated by receptor phosphorylation followed by arrestin binding (reviewed in (Carman & Benovic, 1998)). However, in the last two decades, numerous studies have suggested that receptor-bound arrestins initiate a second round of signaling, which is quite different from the G protein-mediated one (reviewed in (E. V. Gurevich & V. V. Gurevich, 2006; Peterson & Luttrell, 2017)). This phenomenon suggested the idea that GPCR signaling can be biased towards either G proteins or arrestins by using ligands that promote receptor interaction with one type of signal transducer but not with the other. Although the first reported examples of bias of a ligand or a GPCR described differential activation of different G protein subtypes (Eason, Jacinto, & Liggett, 1994; Spengler, et al., 1993), in recent years discussions of GPCR signaling bias have mostly focused on the G protein-arrestin dichotomy (DeWire & Violin, 2011; Violin & Lefkowitz, 2007; Whalen, Rajagopal, & Lefkowitz, 2011; Wisler, Xiao, Thomsen, & Lefkowitz, 2014). In many cases, it has been proposed that one signaling branch is responsible for the therapeutic effects of drugs, while the other mediates unwanted side effects. Therefore, the idea that biased drugs will be therapeutically superior to the traditional unbiased ones has become popular (extensively reviewed in (DeWire & Violin, 2011; Khoury, Clément, & Laporte, 2014; Luttrell, Maudsley, & Bohn, 2015; Rankovic, Brust, & Bohn, 2016; Whalen, et al., 2011; Wisler, et al., 2014)). For a number of pathological conditions, drugs that might specifically direct GPCR signaling to G protein- or arrestin-mediated pathways have been identified, and their beneficial effects have been demonstrated (reviewed in (Whalen, et al., 2011)). Here we consider the issue of GPCR signaling bias from a structural and mechanistic perspective, discussing the possibilities opened by this approach and its inherent limitations. We do not attempt to provide a comprehensive review of all reported cases where signaling bias has been documented, as this has already been done in the excellent reviews cited above.
2. Conformational heterogeneity of GPCRs
The classical view of GPCRs considered these receptors as on-off switches that oscillate between two conformations, active and inactive. Agonists were believed to shift the equilibrium towards the active conformation, inverse agonists – towards the inactive one, thereby suppressing constitutive activity – whereas neutral antagonists occupy the ligand binding site without affecting the conformational equilibrium. The classical extended ternary complex model of GPCR signaling (Samama, Cotecchia, Costa, & Lefkowitz, 1993) was based on this binary on-off concept (Fig. 1, classical paradigm). The idea that receptor signaling can be biased towards a particular G protein subtype or a particular arrestin implies that there must be several “active” GPCR conformational states, some of which are conducive to coupling to one signal transducer, while others facilitate the coupling to another (Fig. 1, biased signaling paradigm). Indeed, biophysical studies of the β2-adrenergic receptor (β2AR) (Manglik, et al., 2015) and rhodopsin (Van Eps, et al., 2017) have shown that the receptor exists in an equilibrium of several conformations in the inactive state, and activation (agonist in the case of β2AR or light in the case of rhodopsin) shifts this equilibrium, but conformational heterogeneity persists. Even the binding of the cognate G protein or G protein-mimicking nanobody in combination with receptor activation does not collapse this equilibrium to a single conformation. The documented conformational flexibility of activated GPCRs (Manglik, et al., 2015; Van Eps, et al., 2017) is consistent with the idea that the receptor can exist in several “active” states, which is a prerequisite for signaling selectivity: some of these states might be more conducive to coupling to a particular G protein, whereas others could lead to coupling to a different G protein or to arrestin. It has also been shown that the binding of different agonists induces distinct conformational equilibria, suggesting that agonist-specific conformations in an active GPCR can translate into signaling specificity (Fig. 1, biased signaling paradigm; also see (Wisler, et al., 2014) and references therein).
Fig. 1. Classical paradigm of GPCR activation and biased signaling.
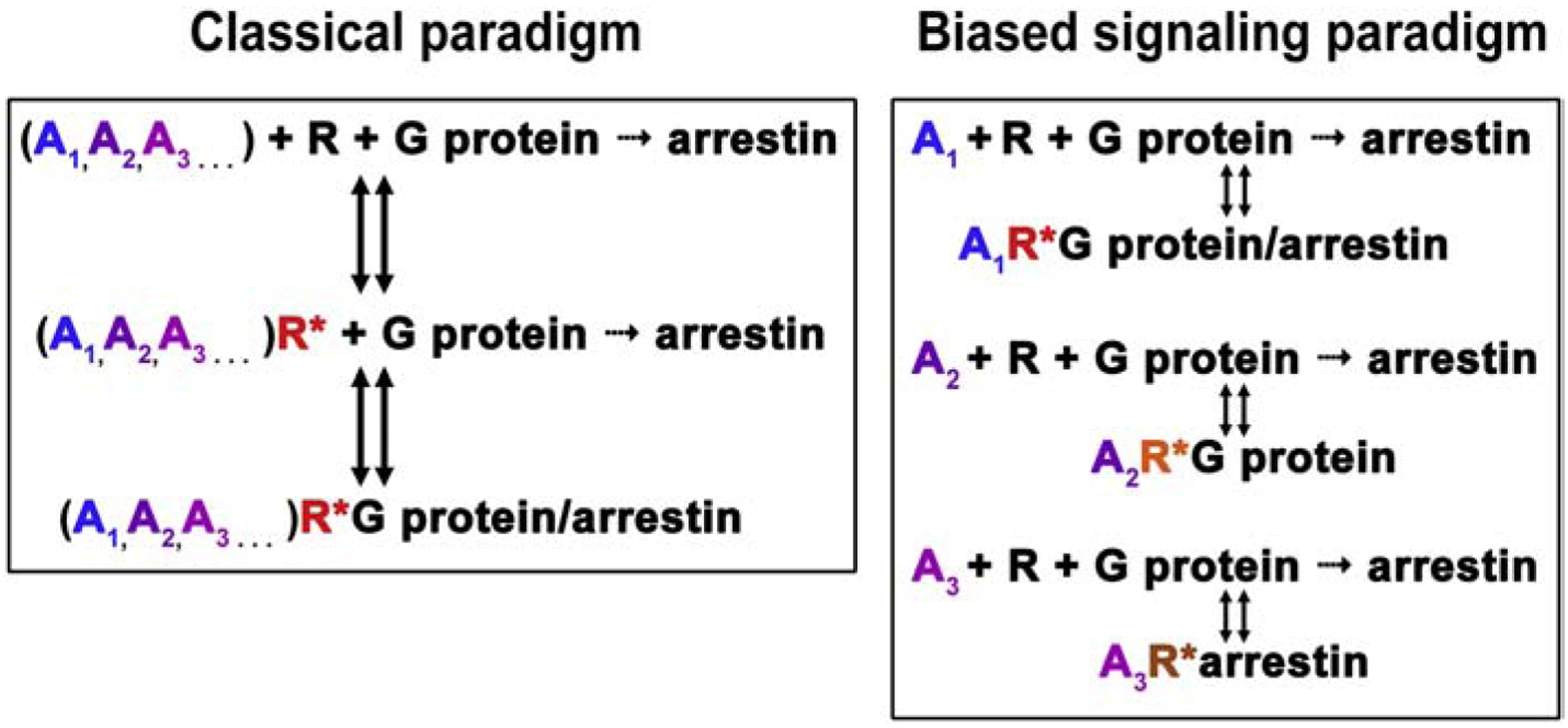
Classical extended ternary complex model (Samama, et al., 1993) of GPCR activation was based on the assumption that receptors have only two conformations, active (R*) and inactive (R). Receptors activated by any agonist (shown as differently colored A1, A2, A3) were thought to couple to G proteins, become phosphorylated by GRKs, and then bind arrestins. Arrestin binding terminated G protein-mediated signaling (Carman & Benovic, 1998). The paradigm of biased signaling is based on the idea that different agonists induce distinct active receptor conformations (shown as differently colored R*), some of which are equally good for G proteins and arrestins, whereas others differentially affect receptor interactions with these signaling partners, demonstrating bias towards G proteins or arrestins.
Several biophysical studies provide indirect evidence that distinct conformations of GPCRs correspond to preferential coupling to G proteins and arrestins. A study using fluorescently labeled purified Arg-vasopressin receptor type 2 and different agonists revealed that ligand-induced changes in the fluorescence of the label placed in transmembrane helix 6 (TM6) correlated with the ability of the ligand to activate G protein: the native unbiased agonist vasopressin and the synthetic G protein-biased agonist MCF14 induced changes in fluorescence, whereas the arrestin-biased agonist SR121463 did not (Rahmeh, et al., 2012). In contrast, the arrestin-biased agonist changed the fluorescence of the labels attached to TM7 and to cytoplasmic α-helix 8, but not the labels attached to TM6 (Rahmeh, et al., 2012). However, the receptor in this study was reconstituted into neutral amphipol, not into its native environment of the lipid bilayer. Similarly, a 19F-NMR-based study of purified β2AR suggested that G protein-biased agonists affect the position of TM6 in the active receptor, arrestin-biased agonists preferentially affect the position of TM7, and unbiased agonists affect both (Liu, Horst, Katritch, Stevens, & Wüthrich, 2012). However, the β2AR used in the latter study was modified by the deletion of the C-terminus (containing all the phosphorylation sites necessary for high-affinity arrestin binding (Fredericks, Pitcher, & Lefkowitz, 1996; Seibold, et al., 2000)), removal of part of the third intracellular loop, and introduction of the stabilizing mutation E122W (Liu, et al., 2012). In addition, the receptor in these experiments was in detergent (a mix of dodecyl maltoside and cholesterol hemisuccinate), rather than in the lipid bilayer (Liu, et al., 2012). It has been shown that arrestins do not interact with GPCRs in detergent micelles (Q. Chen, et al., 2015; Zhuang, et al., 2013), and that the conformational equilibrium of rhodopsin, a prototypical GPCR, in micelles and in lipid bilayer-containing nanodiscs is quite different (Van Eps, et al., 2017). Although it remains to be elucidated whether the findings are relevant for unmodified GPCRs in their native lipid environment, these data are suggestive.
3. Structures of GPCR complexes with signal transducers
The first solved structure of a GPCR bound to a cognate G protein (β2AR complex with Gs (Rasmussen, et al., 2011)) was followed by several GPCR-Gs structures in which the receptor-G protein interfaces looked remarkably similar (Carpenter, Nehmé, Warne, Leslie, & Tate, 2016; Liang, et al., 2017; Y. Zhang, et al., 2017). These structures confirmed the activation-dependent outward movement of the cytoplasmic ends of the transmembrane helices V and VI earlier described in rhodopsin (Farrens, Altenbach, Yang, Hubbell, & Khorana, 1996). Since all these structures involved the Gs, complexes of GPCRs with other G protein subtypes were necessary to test whether the receptor conformations conducive to coupling to a particular type of G protein are different. Rhodopsin has a somewhat different conformation in the structure of its complex with visual arrestin-1a, with a much smaller movement of the transmembrane helices (Y. Kang, et al., 2015; Zhou, et al., 2017). This was interpreted by some as an arrestin-specific conformation of the receptor; however, this interpretation relied on comparing two different types of receptors: Gs-coupled receptors, such as β2AR, and rhodopsin, which couples to transducin, a member of the Gi subfamily. Indeed, subsequent structures of rhodopsin in complex with Gi (Y. Kang, et al., 2018), as well as the μ-opioid receptor-Gi complex (Koehl, et al., 2018), revealed that the smaller outward movement of the transmembrane helices V and VI is a common feature of active Gi-coupled receptors. In fact, the structures of rhodopsin in complex with arrestin-1 and Gi are virtually identical (He, et al., 2017). So far, there are no structures of GPCRs in complex with the other major types of G proteins, Gq/11 or G12/13, so we do not know whether these G proteins prefer receptor conformations different from those preferred by Gs or Gi. Most importantly, many GPCRs couple to more than one type of G protein (Gesty-Palmer, et al., 2006; Violin & Lefkowitz, 2007). To test whether the selection of a partner depends on a particular conformation, we need to compare the structures of the same receptor in complex with different G proteins and arrestins, which is not possible with currently available data.
Arrestins preferentially bind active phosphorylated GPCRs (reviewed in (V. V. Gurevich & Gurevich, 2004)), so receptor phosphorylation by G protein-coupled receptor kinases (GRKs; see (E. V. Gurevich, Tesmer, Mushegian, & Gurevich, 2012) for review) is a prerequisite for arrestin binding to most GPCRs. If the receptor has a mutation precluding its phosphorylation by GRKs, it does not engage arrestins (Choi, et al., 2018). Thus, in order to make an arrestin-preferring conformation of a GPCR actually bind arrestin, in most cases the receptor must assume a conformation conducive to its phosphorylation by GRKs. GRKs are activated by physical interaction with the active receptor (C. Y. Chen, Dion, Kim, & Benovic, 1993; Palczewski, Buczylko, Kaplan, Polans, & Crabb, 1991) and engage the same cavity between the transmembrane α-helices (He, et al., 2017; Komolov, et al., 2017), which opens upon receptor activation (Farrens, et al., 1996), that is engaged by G proteins (Carpenter, et al., 2016; Y. Kang, et al., 2018; Koehl, et al., 2018; Liang, et al., 2017; Rasmussen, et al., 2011; Van Eps, et al., 2018; Y. Zhang, et al., 2017), as well as arrestins (Huang, et al., 2020; Y. Kang, et al., 2015; Staus, et al., 2020; Yin, et al., 2019; Zhou, et al., 2017). Generally, it is conceivable that a distinct set of conformations (e.g., affecting the extent of the opening of the inter-helical cavity in a GPCR), might be specific for GRKs, and that these conformations do not necessarily overlap with those favored by G proteins or arrestins.
Overall, the structures of GPCR complexes with G proteins, arrestins, and GRKs show that all three classes of proteins that preferentially bind active GPCRs use at least one common “clue”: engage the cavity that emerges between the cytoplasmic tips of the GPCR transmembrane α-helices (V. V. Gurevich & Gurevich, 2018c), which is a hallmark of GPCR activation (Farrens, et al., 1996; Rasmussen, et al., 2011). This is consistent with the experimentally observed competition of these proteins for active GPCRs (Krupnick, Gurevich, & Benovic, 1997; Pan, Gurevich, & Gurevich, 2003; Wilden, 1995), but does not necessarily mean that these three classes of GPCR binding partners prefer exactly the same conformation(s) of active receptors.
4. What does the mechanism of GPCR desensitization tell us about biased signaling?
Let’s recall the classical paradigm of two-step GPCR desensitization (Carman & Benovic, 1998). It posits that active GPCRs are phosphorylated by GRKs, and then active phosphorylated receptors bind arrestins. The original concept assumed that the same active conformation of the receptor activates G proteins, is recognized and phosphorylated by GRKs, and then binds arrestins (Fig. 1, classical paradigm). Since GRK/arrestin-mediated desensitization has been demonstrated by numerous labs working with many different GPCRs, we cannot doubt its existence. The concept of biased signaling implies that the GPCR conformations that are preferred by G proteins and arrestins are distinct (Fig. 1, biased signaling paradigm). Thus, the concept of homologous desensitization must be modified to accommodate this idea.
During the time that the receptor remains active, it encounters and sequentially activates numerous G protein molecules, ensuring signal amplification at this step. The greatest amplification is achieved in rod photoreceptors, where light-activated rhodopsin activates a molecule of the visual G protein transducin (which belongs to the Gi subfamily) every 2–3 milliseconds (Heck & Hofmann, 2001). Thus, activated rhodopsin must exist in the conformation(s) capable of G protein coupling most of the time. This requirement might be less strict in the case of non-visual GPCRs, where the rate of G protein activation is much lower. However, this slower G protein activation is partly determined by a much lower concentration of the cognate G proteins, as the concentration of transducin in the rhodopsin-containing rod outer segment is orders of magnitude higher than the concentration of any other G protein in any “normal” cell. Rods contain ~1 transducin molecule per 10 rhodopsin molecules (Hamm & Bownds, 1986), which translates into ~200 μM transducin in the rhodopsin-containing rod outer segment in the dark.
The active receptor binds GRK, thereby activating it, and is phosphorylated by the bound GRK (C. Y. Chen, et al., 1993; Palczewski, et al., 1991). Several phosphates must be attached to the receptor to make it a good binding partner for an arrestin (Azevedo, et al., 2015; V. V. Gurevich, et al., 1995; Mendez, et al., 2000; S.A. Vishnivetskiy, et al., 2007). Thus, GRKs could be processive, attaching several phosphates to the GPCR in one “sitting”, which would imply high-affinity binding of a GRK to a particular conformation of the active GPCR. Alternatively, agonist-activated GPCR might “sample” GRK-favoring conformations often enough to bind GRKs more than once within a fairly short time span, which is several seconds in the case of β2AR (Pitcher, Lohse, Codina, Caron, & Lefkowitz, 1992) and is likely to be similar for other non-visual GPCRs. In the case of rhodopsin, the time required for its sufficient phosphorylation to promote arrestin binding appears to be significantly shorter, about 30–40 milliseconds (Gross & Burns, 2010).
Arrestins bind to the active phosphorylated form of their cognate receptors, blocking G protein access to a GPCR, thereby terminating G protein-mediated signaling. Arrestin binding must also be fairly rapid, within a few dozen milliseconds of activation in the case of rhodopsin (Gross & Burns, 2010). Again, this means that the receptor samples arrestin-favoring conformations pretty often.
A recent study suggested that receptor phosphorylation facilitates its transition into arrestin-favoring conformation(s) (Shiraishi, et al., 2018). However, this study was performed with β2AR, which binds arrestins only transiently (Barak, Ferguson, Zhang, & Caron, 1997). Unlike many GPCRs, β2AR does not have the complete “phosphorylation code” (three precisely spaced phosphates) necessary for high-affinity arrestin binding (Zhou, et al., 2017). While the phosphates that “attract” arrestins can be localized in the receptor C-terminus (as in many GPCRs, such as β2AR (Fredericks, et al., 1996; Seibold, et al., 2000), vasopressin receptor (Shukla, et al., 2013), or Y2 neuropeptide Y receptor (Walther, et al., 2010)), they can be in other receptor elements, such as the third (Pals-Rylaarsdam, et al., 1997; Pals-Rylaarsdam & Hosey, 1997) or the second (Celver, Lowe, Kovoor, Gurevich, & Chavkin, 2001; J. Lowe, J. Celver, V. V. Gurevich, & C. Chavkin, 2002) cytoplasmic loop. While the phosphates attached to β2AR by non-GRK protein kinases do not attract arrestins (Hausdorff, et al., 1989), in some receptors they appear to do so (Tóth, et al., 2018), further increasing the variability of the receptor elements carrying the phosphates that attract arrestins. It is hard to imagine how phosphorylation in these different regions can push the receptor into the same arrestin-preferring set of conformations, as was proposed for β2AR (Shiraishi, et al., 2018). Moreover, different GRKs appear to phosphorylate the same receptor at different sites (Nobles, et al., 2011), yet arrestins bind all of these differentially phosphorylated GPCRs, albeit with distinct functional consequences (Kim, et al., 2005; Ren, et al., 2005).
Conceptually, the model of biased G protein- or arrestin-mediated GPCR signaling is not easily reconcilable with the well-established mechanism of GRK- and arrestin-dependent homologous desensitization. Evolution honed most GPCRs to work with unbiased native ligands and undergo efficient homologous desensitization. The simplest way to reconcile existing data with the idea of biased signaling is to assume that the GPCR conformations preferred by G proteins, GRKs, and arrestins significantly overlap. This does not necessarily mean that conformations that do not couple to G proteins, yet couple to GRKs, arrestins, or any combination of these partners, do not exist. It just means that conformations truly specific for a particular signal transducer likely represent a minor fraction of the conformational ensemble of active GPCRs (Fig. 2).
Fig. 2. Conformational landscape of active GPCRs.
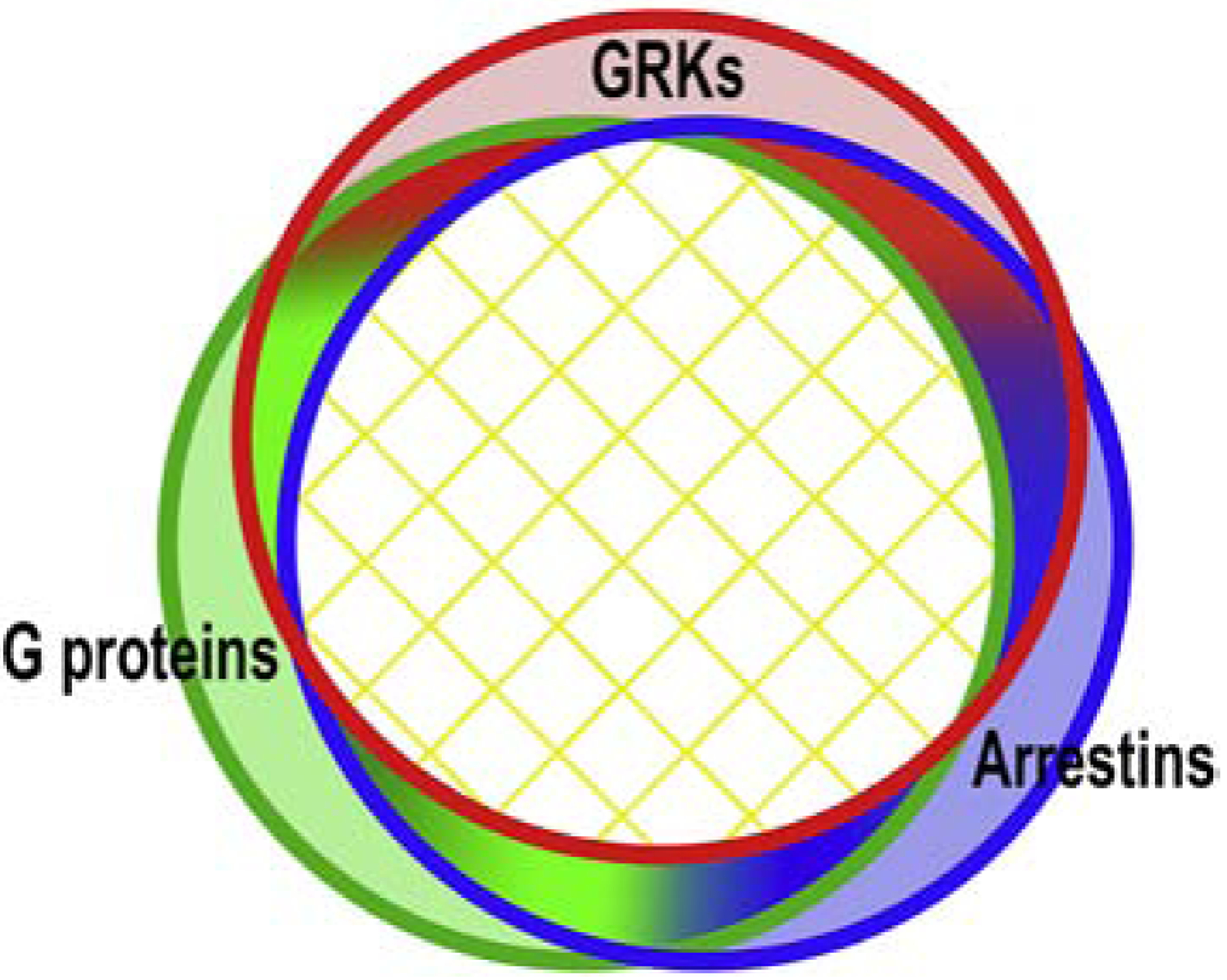
Active GPCRs exist in an equilibrium of multiple conformations. The great majority of active GPCR conformations are conducive to their coupling to G proteins, GRKs, and arrestins, i.e., are not biased toward a particular partner. Available data do not exclude the existence of GPCR conformations preferentially coupling to one class of these signal transducers. However, these “signaling-specific” conformations likely constitute a small fraction of the total. This simplistic diagram suggests that there might be conformations conducive to coupling to two out of three classes of these GPCR-binding proteins (gradient-filled areas). Conformations “good” for G proteins and arrestins but not for GRKs, while conceivable, do not make much biological sense: in most cases arrestin binding requires prior receptor phosphorylation. Similarly, conformations conductive to interactions with G proteins and GRKs but not arrestins are unlikely, since receptor phosphorylation by a GRK would favor arrestin binding. However, GPCRs likely do not stay in a single conformation for a long time, but oscillate among many, so the receptor might arrive at one of these G protein- + arrestin-preferring conformations after sampling the conformations “good” for GRKs, where it would be phosphorylated.
5. Is there G protein-independent arrestin-mediated signaling?
The ERK pathway is the second, after c-Src, signaling mechanism identified as activated via arrestin-dependent scaffolding (Luttrell, et al., 2001). It is arguably the best studied arrestin-dependent signaling pathway and often serves as a classic example of arrestin-mediated signaling. ERK is activated by a variety of mechanisms, some of which are GPCR-dependent (Luttrell, 2003), while others involve growth factor receptors (Garrington & Johnson, 1999; McKay & Morrison, 2007), death receptors (Sabio & Davis, 2014), and integrins (Stupack & Cheresh, 2002). ERK activation by many GPCR agonists peaks within a few minutes and then persists for up to 30–60 min. Ever since the demonstration that rapid ERK activation via some GPCRs is mediated by G proteins, whereas the later phase is arrestin-dependent (Ahn, Shenoy, Wei, & Lefkowitz, 2004; Gesty-Palmer, et al., 2006; Shenoy, et al., 2006), persistent ERK activation has been widely used as a readout for arrestin-mediated signaling. Yet it turned out that prolonged ERK signaling in response to GPCR activation can also be G protein-dependent (e.g., (Alvarez-Curto, et al., 2016; Luo, Busillo, & Benovic, 2008)). Thus, the timing of ERK activation cannot be used to discriminate between the mechanisms that depend on G proteins or arrestins.
Some early studies using siRNA knockdown led to the conclusion that arrestin signaling via ERK is G protein-independent (Shenoy, et al., 2006). The main advantage of protein knockdown is that it’s transient; therefore, the selection of atypical cells adjusted to the absence of the targeted protein, which might represent only a small fraction of the total cell population, does not occur during the course of the experiment. However, knockdown using siRNA or similar tools targeting messenger RNA has the disadvantage of being incomplete, so that functions that require a small fraction of the normal complement of a particular protein in the cell remain unaffected by the knockdown even when the protein in question plays a critical role. Therefore, any conclusion based on this approach that a protein is not required for a specific function remains doubtful. Several recent studies using CRISPR/Cas9 knockout, which eliminates targeted proteins with 100% efficiency, have challenged the idea that ERK activation is G protein independent. It was shown that fatty acid receptor FFA4 (Alvarez-Curto, et al., 2016) and numerous other GPCRs, including β2AR and the angiotensin II receptor – two GPCRs that are often used to demonstrate arrestin-mediated signaling (Grundmann, et al., 2018), do not activate ERK1/2 in the absence of functional G proteins. Furthermore, β2AR, which is often used as a model ligand-sensitive GPCR, has been shown to activate ERK1/2 in cells lacking both non-visual arrestins (Grundmann, et al., 2018; O’Hayre, et al., 2017), suggesting that arrestins are dispensable for the GPCR-driven ERK activation. These data led to questioning of the whole concept of G protein-independent arrestin-mediated signaling (Gutkind & Kostenis, 2018). However, the CRISPR/Cas9 approach also has a disadvantage: because this technique is genetic, it could lead to inadvertent selection of atypical cells that can survive in the absence of the targeted protein. In line with this notion, it was recently demonstrated that three lines of HEK293 cells that underwent CRISPR/Cas9-mediated knockout of the two non-visual arrestins independently in three different labs are quite different (Luttrell, et al., 2018). In particular, in one line arrestin-2/3 knockout resulted in more than two-fold increase in β2AR-stimulated ERK1/2 activation relative to the parental line where both arrestins were present, whereas in the other two it did not significantly change ERK1/2 activation stimulated by β2AR (Luttrell, et al., 2018). As far as GPCR signaling is concerned, the binding of arrestins to the receptor has dual function: it suppresses G protein coupling, while creating at the same time the arrestin-receptor complex that serves as a scaffold for certain branches of signaling. Thus, the most parsimonious mechanistic explanation of the observed differences is that in one cell line G protein-mediated ERK1/2 activation predominated, so that enhancing G protein signaling by arrestin elimination increased ERK1/2 activation. In contrast, it appears that in the other two lines both G protein signaling and arrestin-dependent scaffolding contributed to the ERK1/2 activation, so that simultaneous increase of one and abrogation of the other by arrestin knockout balanced each other, yielding no significant change.
It has traditionally been considered that arrestin has to interact with a GPCR in order to transition into an “active” conformation capable of assembling the signaling proteins such as the ERK cascade (Fig. 3A). Recently, the structural analysis of arrestin activation by GPCRs demonstrated that arrestins could be activated by transient interaction with active GPCRs and become competent to activate ERK after dissociation from the receptors, which are clustered in clathrin-coated endosomes (Kelsie Eichel, et al., 2018; K. Eichel, Jullié, & von Zastrow, 2016). Taking these data into account, Gutkind and Kostenis (Gutkind & Kostenis, 2018) proposed that arrestins do not activate ERK independently of G proteins but act as rheostats regulating the parameters of the ERK activity via receptor desensitization/internalization and scaffolding of signaling components. However, it remains unclear how the requirement for G protein activation could fit into this model from the mechanistic standpoint, for neither desensitization/internalization processes nor arrestin-mediated scaffolding require active G proteins.
Fig. 3. GPCR signaling.
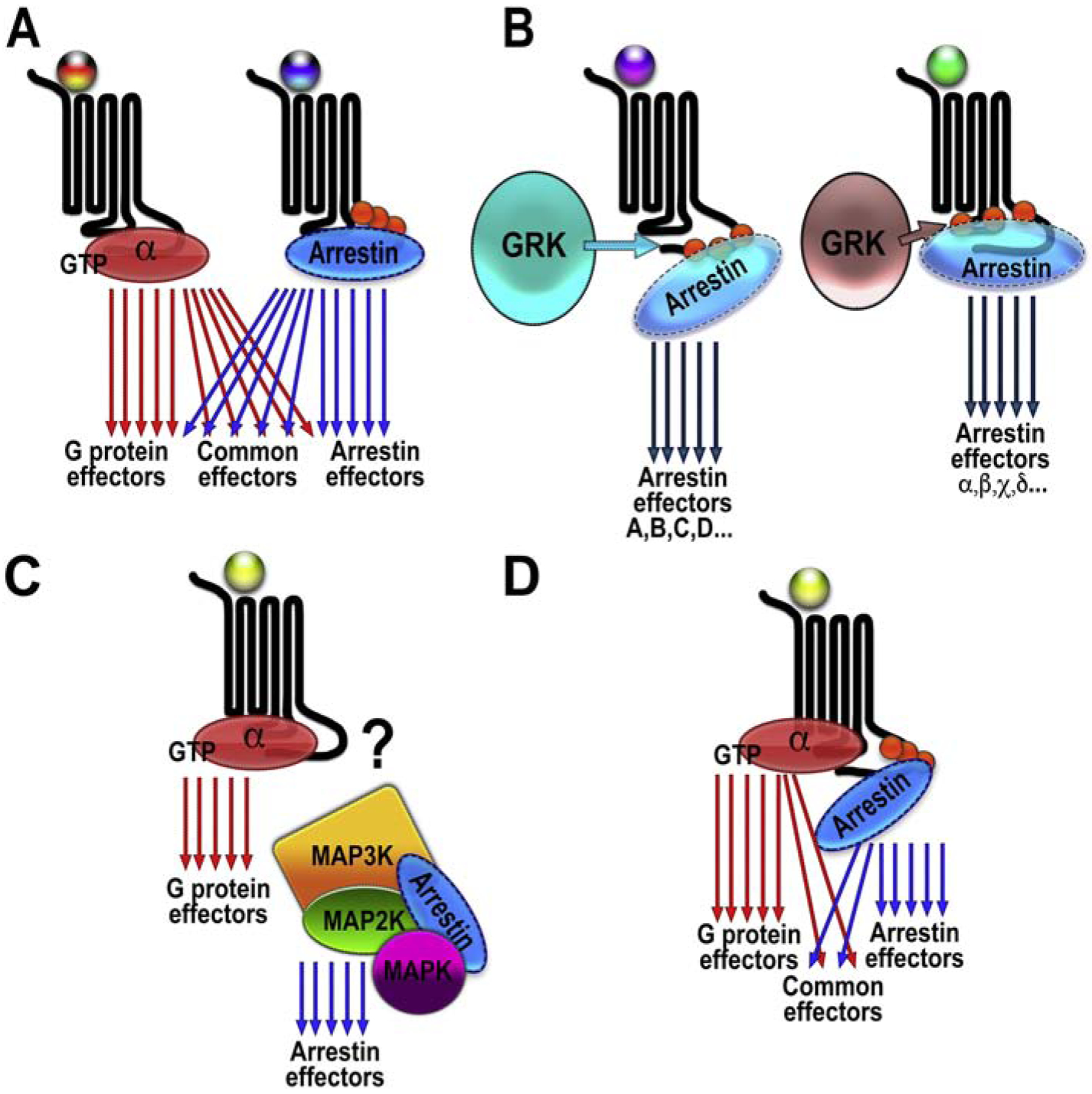
A. G protein-biased ligand (tri-colored ball with red in the middle) facilitates the activation of numerous effectors. Arrestin-biased ligand (tri-colored ball with blue in the middle) also facilitates the activation of many effectors, some of which are the same as those activated by G protein-mediated signaling (common effectors). B. Different ligands (shown as balls of different colors) might determine which particular GRK phosphorylates the receptor. Different GRKs generate distinct patterns of receptor-attached phosphates (barcodes) which might pre-determine the direction of arrestin-mediated signaling. Two non-overlapping sets of effectors are shown. C. Arrestin-3-mediated scaffolding of ASK1-MKK4/7-JNK3 cascade does not require GPCR input. Scaffolding would only be productive when the upstream-most MAP3K is active. Whether active GPCRs play any role in the regulation of signaling by complexes scaffolded by free arrestins (like the JNK cascade shown here) remains to be determined. D. An unbiased ligand (yellow ball) can promote the formation of the super-complex, where G protein and arrestin interact with the receptor simultaneously, with the G protein engaging the inter-helical cavity and arrestin binding to the phosphorylated receptor C-terminus. This would promote simultaneous activation of the G protein- and arrestin-dependent effectors.
It has been suggested that G protein activity might play a role in the activation of the upstream-most kinases in the MAP kinase cascade – MAP3Ks – such as Raf (V. V. Gurevich & Gurevich, 2018b). These kinases are activated via a set of complex mechanisms, and this is unlikely to be accomplished by a simple arrestin-based scaffolding. In fact, arrestin-dependent activation of MAP3Ks has never been demonstrated experimentally. It appears much more likely that these kinases are scaffolded by arrestins in the active form and have to be pre-activated via some arrestin-independent pathways, including those involving G proteins. In the absence of other activating mechanisms (which tend to be excluded by the standard experimental procedures), the activity of the entire cascade becomes dependent on the G protein activation, which may not necessarily be the case under more natural circumstances. Today, the role of G proteins in arrestin-mediated ERK activation remains controversial. There is currently no data on whether and how G protein activity affects other branches of signaling regulated by arrestins. However, if G protein activation by GPCRs proves to be indispensable for arrestin-dependent regulation of at least some signaling pathways, the necessity of both G protein and arrestin activity is hard to reconcile with the idea of G protein- or arrestin-biased signaling.
6. Diversity of arrestin and GRK subtypes
Another issue routinely ignored in the discussion of bias is the existence of arrestin isoforms: all vertebrates, including mammals, have two non-visual arrestins (bony fish that underwent an extra whole genome duplication have three) (E. V. Gurevich & V. V. Gurevich, 2006; Indrischek, Prohaska, Gurevich, Gurevich, & Stadler, 2017). While they are highly homologous (Attramadal, et al., 1992; Indrischek, et al., 2017; Sterne-Marr, et al., 1993), the fact that vertebrate evolution kept them for millions of years suggests that they are not identical. In fact, several functional differences have been reported. Arrestin-3 (a.k.a. β-arrestin2) has higher affinity for many GPCRs (Barak, et al., 1997; Santini, Penn, Gagnon, Benovic, & Keen, 2000), although there are GPCRs (Pera, et al., 2015; D. L. Zhang, et al., 2018) and other membrane proteins (Lipsky, et al., 2008) that prefer arrestin-2 (a.k.a. β-arrestin1). Arrestin-3 has higher affinity for clathrin (Goodman, et al., 1996) and, in contrast to the closely related arrestin-2, facilitates JNK3 activation in cells (McDonald, et al., 2000; Miller, et al., 2001; Perry, et al., 2019; Seo, Tsakem, Breitman, & Gurevich, 2011; Song, Coffa, Fu, & Gurevich, 2009; Zhan, Kaoud, Dalby, & Gurevich, 2011). Interestingly, arrestin-2 also binds many of the kinases of the ASK1-MKK4/7-JNK3 cascade (Seo, et al., 2011; Song, Raman, Gurevich, Vishnivetskiy, & Gurevich, 2006; Zhan, et al., 2011), so it can serve as a dominant-negative subtype in this pathway, similar to the arrestin-3 mutant deficient in JNK3 activation (Breitman, et al., 2012). In fact, a dominant-negative effect of arrestin-2 on arrestin-3-mediated signaling has been reported (Ahn, Wei, Garrison, & Lefkowitz, 2004). Arrestin-2 was selectively implicated in the activation of Rho GTPase (Barnes, et al., 2005), apparently by suppressing the GTPase-activating function of ARHGAP21 (Anthony, et al., 2011). In some cases, both non-visual arrestins are needed for full response, which also suggests that their functions are non-redundant (Ge, Shenoy, Lefkowitz, & DeFea, 2004).
While the interactions of both arrestin-2 and −3 with numerous trafficking and signaling proteins are similar, some binding partners are subtype-specific (Xiao, et al., 2007). Thus, a generic “arrestin bias” does not really describe a biological function: a specific bias toward arrestin-2 or −3 needs to be determined. The consequences of GPCRs recruiting one arrestin subtype or the other can be as different as the consequences of the recruitment of distinct G protein subfamilies. Moreover, both arrestin-2 and −3 have at least two splice variants (Sterne-Marr, et al., 1993). An element that is present in the longer isoform of arrestin-2 but is spliced out in the shorter one has been shown to participate in clathrin binding (D. S. Kang, et al., 2009).
For the great majority of GPCRs, arrestins bind with high affinity only to phosphorylated receptors (reviewed in (V. V. Gurevich & Gurevich, 2004)). Thus, before the question of arrestin binding arises, GRKs must do their part. Most cells express at least four different GRK subtypes, and many GRKs also have splice variants (E. V. Gurevich, et al., 2012), so that the number of distinct GRK forms in every cell is quite large. Even though GPCR phosphorylation by all GRKs results in arrestin recruitment (Kim, et al., 2005; Ren, et al., 2005; Zidar, Violin, Whalen, & Lefkowitz, 2009), bias toward a particular GRK subtype could also exist and have functional significance (Fig. 3B). Unfortunately, GPCR phosphorylation, which is a prerequisite for arrestin binding, was not investigated in most studies of biased signaling. Although there is direct (Nobles, et al., 2011) and indirect (Kim, et al., 2005; Ren, et al., 2005) evidence that different GRKs target different phosphorylation sites in the same GPCR, the level of receptor phosphorylation and the sites targeted were not determined in most studies. While arrestins require three receptor-attached phosphates (some might be substituted by negatively charged residues) (Galliera, et al., 2004; Mendez, et al., 2000; Mukherjee, et al., 2002; S.A. Vishnivetskiy, et al., 2007; Zhou, et al., 2017) for high-affinity binding, most GPCRs have a lot more potential GRK phosphorylation sites. Some sites appear to be more important for arrestin recruitment than others (Azevedo, et al., 2015), which is reflected in different rates of receptor internalization when certain sites are missing or remain unphosphorylated (Lau, et al., 2011).
The idea that different patterns of phosphorylation might result in distinct signaling outcomes was suggested in 2008 (Tobin, Butcher, & Kong, 2008). Indeed, the consequences of GPCR phosphorylation by different GRKs could be strikingly different (Kim, et al., 2005; Ren, et al., 2005; Zidar, et al., 2009). In the case of β2AR, it was experimentally demonstrated that GRK2 and GRK6 phosphorylate different sites, and their phosphorylation differentially affects arrestin-dependent signaling (Choi, et al., 2018; Nobles, et al., 2011). Based on these data, it was proposed that GPCR phosphorylation by different GRKs establishes patterns (termed “barcodes”), each leading to arrestin recruitment in different conformations and, consequently, to distinct downstream signaling events (barcode hypothesis) (Nobles, et al., 2011) (Fig. 3B). The different conformations assumed by arrestins upon binding to differentially phosphorylated receptors is the simplest mechanistic explanation of the available data. This idea was first suggested in 2006 (V. V. Gurevich & E. V. Gurevich, 2006). Recent structures of the arrestin-receptor complex, in which arrestins were found in different “poses” relative to the receptor they bind (Huang, et al., 2020; Y. Kang, et al., 2015; Staus, et al., 2020; Yin, et al., 2019), support this notion, although in these cases the issue of the effect of differential phosphorylation of the same receptor on the mode of arrestin binding was not addressed. Existing data suggest that the nature of the GRK that phosphorylates the receptor and the resulting phosphorylation pattern are crucial for the control of the G protein signaling, as well as of the signaling bias (Choi, et al., 2018; Kim, et al., 2005; Ren, et al., 2005). This important aspect is routinely missing from the discussion of biased signaling, which is presented as a G protein vs arrestin dichotomy, while the critical role of the GRKs is often ignored. Yet in view of the existing data, however fragmentary, the bias toward a particular GRK could be the key bias that predetermines the direction of arrestin-mediated signaling.
7. How typical is the ERK pathway for arrestin-dependent signaling?
Arrestins are known to regulate a large and extremely diverse set of signaling pathways (Peterson & Luttrell, 2017). It is hardly conceivable that all these pathways depend in a similar fashion on the arrestin interactions with GPCRs. However, the dependence of a particular signaling function of arrestins on their interactions with GPCRs is rarely studied or discussed. Even though both ERK1/2 and JNK1/2/3 are MAP kinases activated by three-tiered MAP kinase cascades, the modes of activation of these two types of MAP kinases by arrestins differ in an important way. The activation of ERK1/2 sends a pro-proliferative, pro-survival signal. So far, all studies agree that arrestin-mediated ERK activation requires the formation of the arrestin-receptor complex (Breitman, et al., 2012; Coffa, Breitman, Hanson, et al., 2011; Coffa, Breitman, Spiller, & Gurevich, 2011; Luttrell, et al., 2001). Moreover, ERK binding to arrestins in the absence of GPCR activation cannot be detected without cross-linking, whereas upon receptor activation it is easily detectable by co-immunoprecipitation (Coffa, Breitman, Hanson, et al., 2011; Luttrell, et al., 2001; Song, et al., 2009), suggesting that the affinity of ERK1/2 for free non-visual arrestins in the cytoplasm is much lower than its affinity for the GPCR-associated arrestins or for arrestins “activated” by their previous transient interactions with active GPCRs (Kelsie Eichel, et al., 2018; K. Eichel, et al., 2016; Latorraca, et al., 2018). Importantly, arrestins are considered “activated” when they assume a conformation resembling that in the complex with a GPCR. Thus, while receptor-bound or activated arrestins certainly serve as scaffolds for the cRaf-MEK1-ERK1/2 cascades, as originally proposed (Luttrell, et al., 2001), free “inactive” arrestins cannot fulfill this function.
JNK family kinases are generally anti-proliferative; they facilitate differentiation and sometimes apoptosis. JNKs are activated by death receptors (Sabio & Davis, 2014), stressors (Fujino, et al., 2007), and other inputs. Arrestin-3, but not the closely related arrestin-2, facilitates JNK3 activation via the ASK1-MKK4/7-JNK3 cascade (McDonald, et al., 2000). Although arrestin-dependent JNK3 phosphorylation was first demonstrated to occur in response to GPCR activation (McDonald, et al., 2000), soon thereafter it was shown not to require receptor action (Miller, et al., 2001; Seo, et al., 2011) (Fig. 3C). Moreover, an arrestin-3 mutant with a 7-residue deletion in the inter-domain hinge, which precludes its binding to GPCRs (Hanson, et al., 2007; S. A. Vishnivetskiy, Hirsch, Velez, Gurevich, & Gurevich, 2002), was found to facilitate JNK3 activation as efficiently as wild type arrestin-3 (Breitman, et al., 2012; Song, et al., 2009). In contrast, the arrestin-3–3A mutant, which binds GPCRs more avidly than wild type, fails to activate JNK3 (Breitman, et al., 2012). In arrestin-3–3A, the mutations result in disengagement of the C-tail, which is considered the main conformational requirement for arrestin “activation” upon binding to a GPCR (Kelsie Eichel, et al., 2018; Latorraca, et al., 2018). Although arrestin-dependent activation of the ERK cascade seems to require arrestin to assume a conformation similar to the receptor-bound one, the structural requirements for arrestin-dependent activation of the JNK pathway are apparently different.
Arrestin-3 has also been shown to promote the activation of at least some isoforms of the ubiquitously expressed JNK1 and JNK2 (Kook, et al., 2014). The phosphorylation of purified JNK1, JNK2, and JNK3 in vitro by purified MKK4 and MKK7 is facilitated by purified arrestin-3 in the absence of receptors (Kook, et al., 2014; Zhan, et al., 2011), indicating that GPCRs are not required for this process (Fig. 3C). Interestingly, a 25-residue arrestin-3-derived peptide, but not its homolog derived from arrestin-2, was found to facilitate JNK3 activation both in vitro and in cells (Zhan, et al., 2016). This peptide does not have most of the receptor-binding elements of arrestin-3, so these data demonstrate yet again that GPCRs are not required even in cells, where GPCRs are always present. While there is experimental evidence that G proteins might be necessary for arrestin-dependent ERK activation by some GPCRs (Alvarez-Curto, et al., 2016; Grundmann, et al., 2018), there is no such evidence in the case of JNKs, although there is no direct evidence to the contrary. On that score, the jury is still out (V. V. Gurevich & Gurevich, 2018a, 2018b). Direct experimental testing of the G protein dependence of JNK activation is needed.
Thus, in discussing the signaling function of arrestins, we should keep in mind the differences in arrestin-dependent activation of different signaling pathways. First, such activation may or may not require arrestin binding to a GPCR; second, it may or may not require G protein activity. Although arrestins have been reported to interact with dozens of signaling proteins (Xiao, et al., 2007), presumably regulating a variety of signaling pathways, in most cases it has not been determined whether arrestin binding to a GPCR and/or G protein activation is required for a particular downstream outcome. It is conceivable that GPCR-independent arrestin functions play a role in the action of various drugs, particularly in long-term adaptations to chronic drug use. However, this issue remains to be investigated experimentally. Obviously, neither unbiased nor biased GPCR ligands are likely to turn on GPCR-independent arrestin signaling (Fig. 3C).
8. Can changes in the kinetics of G protein activation be confused with arrestin-mediated signaling?
In many cases when ligand bias is discussed, arrestin-mediated signaling is proposed as an alternative to the G protein-dependent signaling. However, it is critical to analyze what was experimentally demonstrated in each study, rather than what was implied or assumed. Let us consider some of the most recent examples. Accessory protein MRAP2 was shown to bias ghrelin receptor GHSR1a towards G proteins (Rouault, et al., 2020). In this study, G protein-mediated signaling was measured directly, whereas only arrestin recruitment to the receptor was experimentally determined. The activation of RhoA was used as a readout of arrestin-mediated signaling in this study (Rouault, et al., 2020). Yet it is well established that RhoA can also be activated via G protein-mediated signaling (reviewed in (Siehler, 2009)). Thus, this readout can be used only when its dependence on arrestins has been established (e.g., by demonstrating the absence of this signaling in arrestin-2/3 knockout cells), which was not done in this case. In a recent study of the role of GRKs in the bias of β2AR ligands, arrestin recruitment and receptor internalization were measured, but no measurements of any signaling that might involve arrestins were performed (Choi, et al., 2018). In another study of glucagon-like peptide receptor 1, ERK activation was used as a readout of arrestin-mediated signaling (Lei, et al., 2018). As discussed above, this is a highly questionable readout, as G proteins often play a key role in GPCR-dependent ERK activation (Alvarez-Curto, et al., 2016; Grundmann, et al., 2018). Moreover, receptor-dependent ERK activation was observed in the case of β2AR in arrestin-2/3 knockout cells (O’Hayre, et al., 2017). The study by Lei et al. did not demonstrate the requirement for, or even the involvement of, arrestins in the ERK activation observed in these experiments (Lei, et al., 2018). These studies are representative: in dozens of previous studies, only arrestin recruitment and/or GPCR internalization was directly measured to demonstrate bias. If any signaling readout was used, its dependence on arrestins, as opposed to G proteins, was assumed rather than experimentally demonstrated.
We must keep in mind that differences in arrestin binding to GPCRs and in the kinetics of receptor internalization inevitably affect the time course of G protein activation, which can yield different biological outcomes observed in vivo (e.g., like those described in (Gesty-Palmer & Luttrell, 2011)). The phosphorylation of some GPCRs by GRKs (Arshavsky, Dizhoor, Shestakova, & Philippov, 1985; Benovic, et al., 1987), as well as β2AR phosphorylation by PKA (Benovic, et al., 1985; Daaka, Luttrell, & Lefkowitz, 1997; Martin, Whalen, Zamah, Pierce, & Lefkowitz, 2004), has been shown to directly affect G protein coupling. Moreover, GRK2 and GRK3 bind the free βγ-subunit of G proteins (Koch, Inglese, Stone, & Lefkowitz, 1993; Tesmer, Kawano, Shankaranarayanan, Kozasa, & Tesmer, 2005), thereby directly modulating the G protein-mediated signaling (Raveh, Cooper, Guy-David, & Reuveny, 2010). Thus, in many cases we don’t even need to invoke arrestin-mediated signaling to explain bias. The asymmetrical enhancement or reduction in the GRK/arrestin function and the resulting changes in the canonical GPCR signaling via G proteins would suffice. As far as the signaling role of arrestins goes, at the moment we have many potential suspects (both non-visual arrestins interact with a wide variety of signaling proteins (Xiao, et al., 2007)), but none of these signaling mechanisms are strictly arrestin-dependent. Furthermore, there is no reason to suppose that even if arrestin-mediated signaling is the cause of the observed bias, it is the same signaling in all cases. Thus, if arrestin-mediated signaling is suspected to have a therapeutically beneficial effect or, conversely, to drive a pathological process, it is critically important to identify the specific branch of signaling in question. Unless and until this is accomplished, we cannot simply assume that in every case of a ligand or receptor bias, seen as the differential propensity to recruit arrestins or to internalize the receptor, arrestin-mediated signaling is involved. To summarize: ligand bias can be quite real and therapeutically beneficial simply due to changes in the dynamics of G protein activation, even if no arrestin-mediated signaling is involved.
9. The promise and limitations of biased GPCR ligands
Most GPCRs couple to more than one type of G protein and also bind one or more arrestin variant upon phosphorylation by several distinct GRKs. In quite a few cases, some branches of GPCR signaling were shown to be therapeutically beneficial, whereas others caused unwanted on-target side effects. Thus, the development of biased ligands that can initiate certain branches of signaling, but not others, has an obvious value for therapy (reviewed in (Gesty-Palmer & Luttrell, 2011; Luttrell, et al., 2015; Violin & Lefkowitz, 2007; Whalen, et al., 2011; Wisler, et al., 2014)). As discussed above, it is most often assumed that the bias stems from a ligand directing the GPCR signaling towards either G protein- or arrestin-dependent signaling.
If GPCRs initiated two independent non-overlapping branches of signaling, one via G proteins and the other via arrestins, functional screening of GPCR ligands, both orthosteric and allosteric, for potential bias would be relatively straightforward. Several recent studies suggested ingenious ways to determine ligand bias based on screening data (Costa-Neto, Parreiras-E-Silva, & Bouvier, 2016; Kenakin & Christopoulos, 2013a, 2013b; Kenakin, Watson, Muniz-Medina, Christopoulos, & Novick, 2012; Luttrell, et al., 2015), so this is not the major hurdle today. This is, however, a rather artificial situation, and in all cases arrestin recruitment/receptor internalization is measured in lieu of arrestin-dependent signaling. It is acknowledged that translating the findings of ligand bias from such screens to the behavior of these ligands in cultured cells, let alone in animals, is problematic (Kenakin & Christopoulos, 2013b). The main difficulty is that the field lacks a readout of signaling specific for arrestins in general, let alone for any particular arrestin species. While ERK activation has been used for this purpose, it is clearly inadequate (Alvarez-Curto, et al., 2016; Grundmann, et al., 2018; O’Hayre, et al., 2017). It is not even evident that there could be such a readout, since the signaling cascades activated by G proteins and arrestins significantly overlap. The ERK signaling, for example, is readily activated by GPCRs independently of arrestins (O’Hayre, et al., 2017). Moreover, under physiological conditions, GPCRs are certainly not the main drivers of MAPK activation: growth factor and death receptors, integrins, and stressors are responsible for MAPK activation in most cases. While in cell culture one can make MAPK activation dependent on GPCRs by eliminating other inputs (serum starvation silences growth factor and death receptors; plating cells on poly-D-lysine, which binds but does not activate integrins, excludes their role, etc.), this cannot be done in patients, where biased drugs are meant to function. So far, no viable unambiguous readout for arrestin-dependent signaling has emerged even for screening purposes. This is not surprising considering the multitude of signaling pathways activated by both G proteins and arrestins, as well as the considerable overlap between these two sets (Fig. 3A).
Recent studies have revealed yet another potential difficulty of exploiting G protein vs arrestin bias. Experiments with a chimeric GPCR having the seven transmembrane helices of β2AR and the C-terminus of V2 vasopressin receptor revealed that a single activated receptor can bind both G protein and arrestin simultaneously (Nguyen, et al., 2019; Thomsen, et al., 2016) (Fig. 3D). The arrestin molecule in this complex only interacts with the phosphorylated receptor C-terminus, rather than with both the phosphorylated elements and the cavity between the TM helices, as in the crystal structure of arrestin-1 complex with rhodopsin (Y. Kang, et al., 2015; Zhou, et al., 2017) and arrestin-2 complex with non-visual GPCRs (Huang, et al., 2020; Staus, et al., 2020; Yin, et al., 2019). Nevertheless, arrestin in this complex appears to be signaling-competent (Cahill, et al., 2017). Thus, this complex seems to be able to signal in both directions at the same time (Fig. 3D). This suggests that even ligands specifically inducing a biased subset of conformations of the 7TM GPCR core conducive to G protein coupling cannot prevent arrestin-mediated signaling, if the receptor is phosphorylated by GRKs.
Another impediment to efficient biased signaling is that GPCRs are inherently flexible, always existing in an equilibrium of multiple conformations. It is highly unlikely that any ligand can shift the equilibrium to such an extent as to preclude receptor coupling to one of the partners, while significantly facilitating the coupling to another. Thus, achieving complete bias with any ligand structure does not seem feasible. While there is no doubt that we can design mutant GPCRs that specifically couple to G proteins or arrestins (Choi, et al., 2018; Hu, et al., 2016; Nakajima & Wess, 2012), we cannot replace native GPCRs in patients with these mutants. Still, it appears entirely possible that, in complex with certain compounds, some GPCRs can assume conformations conducive to G protein activation but not to GRK phosphorylation and/or arrestin binding, or vice versa. Indeed, evolution has actually used this tool: if a GPCR has multiple endogenous ligands, these ligands can show signaling bias. For example, chemokine receptors can bind several ligands, and these ligands have differential ability to trigger G protein activation and arrestin recruitment (Amarandi, Hjortø, Rosenkilde, & Karlshøj, 2016; Zweemer, Toraskar, Heitman, & Ijzerman, 2014). Another example of GPCRs with multiple endogenous ligands that also show ligand bias are μ-opioid and somatostatin receptors (Thompson, Canals, & Poole, 2014). Some GPCRs are naturally “biased” towards G proteins, as their structure slows down (e.g., in the case of μ-opioid receptor (J. D. Lowe, J. P. Celver, V. V. Gurevich, & C. Chavkin, 2002)) or totally precludes (e.g., in the case of β3-adrenergic receptor (Liggett, Freedman, Schwinn, & Lefkowitz, 1993)) GRK- and arrestin-dependent homologous desensitization.
From the therapeutic standpoint, even partial bias has potential value. For example, the ligands that change the probability of type 1 parathyroid hormone receptor coupling to Gs, Gq/11, or arrestins have profoundly different biological effects in vivo than unbiased agonists (Gesty-Palmer & Luttrell, 2011). Even partial reduction of the receptor’s propensity to get phosphorylated and bind arrestins would greatly increase G protein-mediated signaling and change its time course relative to that induced by unbiased agonists, as well as reduce the effects of the arrestin-receptor complex (Luttrell, et al., 2015). The opposite bias can reduce (although likely not eliminate) G protein signaling and potentially enhance the signaling by the arrestin-receptor complex (Violin & Lefkowitz, 2007). Thus, biased agonists, as well as biased allosteric modulators of GPCRs, should be developed and explored, even though 100% bias is unlikely to be possible.
The screening of drugs has to be significantly modified, though. In most cases screening methods measure G protein activation. To this end, chimeric G proteins with modified C-terminus of the α-subunit are used to channel the signaling of any GPCR to the Gq/11 pathway; consequent increase in intracellular calcium can easily be measured in high-throughput format using calcium-sensitive fluorescent dies (Coward, Chan, Wada, Humphries, & Conklin, 1999). While this method has certainly contributed to drug discovery efforts, its drawback is that the kinetics of the return of the calcium signal back to baseline does not depend on receptor desensitization (i.e., the rate of phosphorylation and arrestin binding) as much as on the GPCR-independent cellular mechanisms ensuring calcium homeostasis. Thus, an agonist that favors G proteins over GRKs/arrestins would produce essentially the same signal as an unbiased agonist. The screening for specific arrestin-mediated signaling that cannot be attributed to G proteins will only be possible when unambiguous readouts are established. The arrestin recruitment and/or GPCR internalization is not an adequate indicator of the involvement, or lack thereof, of arrestin-mediated signaling. Similarly, the changes in the activity of ubiquitous signaling pathways responsive to a large variety of regulatory influences cannot simply be assumed to be arrestin-dependent unless proven so. “Zero functional G” cells (Alvarez-Curto, et al., 2016; Grundmann, et al., 2018) can be used to screen for arrestin-mediated signaling, but their drawback is the potential selection of atypical cells that can survive without G proteins. These cells might not faithfully recapitulate signaling mechanisms existing in “normal” cells, i.e., those that are meant to be targeted in patients, which have the full complement of G proteins.
10. Conclusions
Biased GPCR ligands that preferentially direct signaling to particular G proteins, GRKs, and/or arrestins hold promise for better therapeutic outcomes. However, the bias is unlikely to be absolute: other (unwanted) signaling pathways would likely be activated along with the preferred one. Moreover, in the quest to reduce or eliminate the unwanted signaling, the preferred pathway is also likely to be impaired. A huge technical hurdle in the development of biased GPCR-targeting drugs is the issue of screening. Methods sensitive to the rates of desensitization of G protein signaling and specific arrestin-dependent readouts that are proven not to involve G proteins are necessary. Most cells express several GRK subtypes and two non-visual arrestins with distinct signaling capabilities. Most GRKs and both non-visual arrestins have splice variants, which are also functionally distinct. Thus, generic arrestin or GRK bias is an oversimplification: in reality it matters which GRK phosphorylates a GPCR and which arrestin subtype is recruited as a result. Matters are further complicated by the fact that arrestins and GRKs participate in various signaling pathways independently of GPCR inputs. Thus, while the development of biased GPCR ligands is a worthy goal, we need to proceed down this path with our eyes open, keeping in mind the complexities of bias, the limitations of existing technologies, and possible alternative interpretations of observed differences in biological outcomes.
Acknowledgements
This project was supported by NIH grants RO1 EY011500, R35 GM122491, and Cornelius Vanderbilt Endowed Chair (Vanderbilt University) (VVG), as well as NIH grants RO1 NS065868 and RO1 DA030103 (EVG).
Abbreviations:
- GPCR
G protein-coupled receptor
- GRK
G protein-coupled receptor kinase
- β2AR
β2-adrenergic receptor
- TM
transmembrane helix
- NMR
nuclear magnetic resonance
- EPR
electron paramagnetic resonance
- DEER
double electron-electron resonance (a pulsed EPR technique)
- ERK
extracellular signal-regulated kinase
- cRaf, MEK, MKK
mitogen-activated protein kinase kinase
- JNK
c-Jun N-terminal kinase
- ASK
apoptosis signal-regulating kinase
- MAP kinases
mitogen-activated protein kinases
- CRISPR
clustered regularly interspaced short palindromic repeats
- Cas-9
CRISPR-associated protein-9 nuclease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
We use the systematic names of arrestin proteins, where the number after the dash indicates the order of cloning: arrestin-1 (historic names S-antigen, 48 kDa protein, visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin).
Conflict of interest
The authors declare no conflict of interest.
References
- Ahn S, Shenoy SK, Wei H, & Lefkowitz RJ (2004). Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem, 279, 35518–35525. [DOI] [PubMed] [Google Scholar]
- Ahn S, Wei H, Garrison TR, & Lefkowitz RJ (2004). Reciprocal regulation of angiotensin receptor-activated extracellular signal-regulated kinases by beta-arrestins 1 and 2. J Biol Chem, 279, 7807–7811. [DOI] [PubMed] [Google Scholar]
- Alvarez-Curto E, Inoue A, Jenkins L, Raihan SZ, Prihandoko R, Tobin AB, & Milligan G (2016). Targeted Elimination of G Proteins and Arrestins Defines Their Specific Contributions to Both Intensity and Duration of G Protein-coupled Receptor Signaling. J Biol Chem, 291, 27147–27159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarandi R-M, Hjortø GM, Rosenkilde MM, & Karlshøj S (2016). Chapter Eight - Probing Biased Signaling in Chemokine Receptors In Handel TM (Ed.), Methods in Enzymology (Vol. 570, pp. 155–186): Academic Press. [DOI] [PubMed] [Google Scholar]
- Anthony DF, Sin YY, Vadrevu S, Advant N, Day JP, Byrne AM, Lynch MJ, Milligan G, Houslay MD, & Baillie GS (2011). β-Arrestin 1 inhibits the GTPase-activating protein function of ARHGAP21, promoting activation of RhoA following angiotensin II type 1A receptor stimulation. Mol Cell Biol, 31, 1066–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arshavsky VY, Dizhoor AM, Shestakova IK, & Philippov P (1985). The effect of rhodopsin phosphorylation on the light-dependent activation of phosphodiesterase from bovine rod outer segments. FEBS Lett, 181, 264–266. [DOI] [PubMed] [Google Scholar]
- Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, Snyder SH, Caron MG, & Lefkowitz RJ (1992). Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J Biol Chem, 267, 17882–17890. [PubMed] [Google Scholar]
- Azevedo AW, Doan T, Moaven H, Sokal I, Baameur F, Vishnivetskiy SA, Homan KT, Tesmer JJ, Gurevich VV, Chen J, & Rieke F (2015). C-terminal threonines and serines play distinct roles in the desensitization of rhodopsin, a G protein-coupled receptor. Elife, 4, doi: 10.7554/eLife.05981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak LS, Ferguson SS, Zhang J, & Caron MG (1997). A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J Biol Chem, 272, 27497–27500. [DOI] [PubMed] [Google Scholar]
- Barnes WG, Reiter E, Violin JD, Ren XR, Milligan G, & Lefkowitz RJ (2005). beta-Arrestin 1 and Galphaq/11 coordinately activate RhoA and stress fiber formation following receptor stimulation. J Biol Chem, 280, 8041–8050. [DOI] [PubMed] [Google Scholar]
- Benovic JL, Kühn H, Weyand I, Codina J, Caron MG, & Lefkowitz RJ (1987). Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein). Proc Nat Acad Sci USA, 84, 8879–8882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benovic JL, Pike LJ, Cerione RA, Staniszewski C, Yoshimasa T, Codina J, Caron MG, & Lefkowitz RJ (1985). Phosphorylation of the mammalian beta-adrenergic receptor by cyclic AMP-dependent protein kinase. Regulation of the rate of receptor phosphorylation and dephosphorylation by agonist occupancy and effects on coupling of the receptor to the stimulatory guanine nucleotide regulatory protein. J Biol Chem, 260, 7094–7101. [PubMed] [Google Scholar]
- Breitman M, Kook S, Gimenez LE, Lizama BN, Palazzo MC, Gurevich EV, & Gurevich VV (2012). Silent scaffolds: inhibition of c-Jun N-terminal kinase 3 activity in the cell by a dominant-negative arrestin-3 mutant. J Biol Chem, 287, 19653–19664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill TJ 3rd, Thomsen AR, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, Huang LY, Kahsai AW, Bassoni DL, Gavino BJ, Lamerdin JE, Triest S, Shukla AK, Berger B, Little J 4th, Antar A, Blanc A, Qu CX, Chen X, Kawakami K, Inoue A, Aoki J, Steyaert J, Sun JP, Bouvier M, Skiniotis G, & Lefkowitz RJ (2017). Distinct conformations of GPCR-β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc Natl Acad Sci U S A, 114, 2562–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV, & Benovic JL (1998). G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol, 8, 335–344. [DOI] [PubMed] [Google Scholar]
- Carpenter B, Nehmé R, Warne T, Leslie AG, & Tate CG (2016). Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature, 536, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celver J, Lowe J, Kovoor A, Gurevich VV, & Chavkin C (2001). Threonine 180 is requred for G protein-coupled receptor kinase 3 and b-arrestin mediated desensitization of the m-opioid receptor in Xenopus oocytes. J Biol Chem, 276, 4894–4900. [DOI] [PubMed] [Google Scholar]
- Chen CY, Dion SB, Kim CM, & Benovic JL (1993). Beta-adrenergic receptor kinase. Agonist-dependent receptor binding promotes kinase activation. J Biol Chem, 268, 7825–7831. [PubMed] [Google Scholar]
- Chen Q, Vishnivetskiy SA, Zhuang T, Cho MK, Thaker TM, Sanders CR, Gurevich VV, & Iverson TM (2015). The rhodopsin-arrestin-1 interaction in bicelles. Methods Mol Biol, 1271, 77–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Staus DP, Wingler LM, Ahn S, Pani B, Capel WD, & Lefkowitz RJ (2018). G protein-coupled receptor kinases (GRKs) orchestrate biased agonism at the β2-adrenergic receptor. Sci Signal, 11, 544. [DOI] [PubMed] [Google Scholar]
- Coffa S, Breitman M, Hanson SM, Callaway K, Kook S, Dalby KN, & Gurevich VV (2011). The Effect of Arrestin Conformation on the Recruitment of c-Raf1, MEK1, and ERK1/2 Activation. PLoS One, 6, e28723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffa S, Breitman M, Spiller BW, & Gurevich VV (2011). A single mutation in arrestin-2 prevents ERK1/2 activation by reducing c-Raf1 binding. Biochemistry, 50, 6951–6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Neto CM, Parreiras-E-Silva LT, & Bouvier M (2016). A Pluridimensional View of Biased Agonism. Mol Pharmacol, 90, 587–595. [DOI] [PubMed] [Google Scholar]
- Coward P, Chan SD, Wada HG, Humphries GM, & Conklin BR (1999). Chimeric G proteins allow a high-throughput signaling assay of Gi-coupled receptors. Anal Biochem, 270, 242–248. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, & Lefkowitz RJ (1997). Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature, 390, 88–91. [DOI] [PubMed] [Google Scholar]
- DeWire SM, & Violin JD (2011). Biased ligands for better cardiovascular drugs: dissecting G-protein-coupled receptor pharmacology. Circ Res, 109, 205–216. [DOI] [PubMed] [Google Scholar]
- Eason MG, Jacinto MT, & Liggett SB (1994). Contribution of ligand structure to activation of alpha 2-adrenergic receptor subtype coupling to Gs. Mol Pharmacol, 45, 696–702. [PubMed] [Google Scholar]
- Eichel K, Jullié D, Barsi-Rhyne B, Latorraca NR, Masureel M, Sibarita J-B, Dror RO, & von Zastrow M (2018). Catalytic activation of β-arrestin by GPCRs. Nature, 557, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichel K, Jullié D, & von Zastrow M (2016). β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nature Cell Biology, 18, 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrens DL, Altenbach C, Yang K, Hubbell WL, & Khorana HG (1996). Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science, 274, 768–770. [DOI] [PubMed] [Google Scholar]
- Fredericks ZL, Pitcher JA, & Lefkowitz RJ (1996). Identification of the G protein-coupled receptor kinase phosphorylation sites in the human beta2-adrenergic receptor. J Biol Chem, 271, 13796–13803. [DOI] [PubMed] [Google Scholar]
- Fujino G, Noguchi T, Matsuzawa A, Yamauchi S, Saitoh M, Takeda K, & Ichijo H (2007). Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Mol Cell Biol, 27, 8152–8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galliera E, Jala VR, Trent JO, Bonecchi R, Signorelli P, Lefkowitz RJ, Mantovani A, Locati M, & Haribabu B (2004). beta-Arrestin-dependent constitutive internalization of the human chemokine decoy receptor D6. J Biol Chem, 279, 25590–25597. [DOI] [PubMed] [Google Scholar]
- Garrington TP, & Johnson GL (1999). Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol, 11, 211–218. [DOI] [PubMed] [Google Scholar]
- Ge L, Shenoy SK, Lefkowitz RJ, & DeFea K (2004). Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both beta-arrestin-1 and −2. J Biol Chem, 279, 55419–55424. [DOI] [PubMed] [Google Scholar]
- Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, & Lefkowitz RJ (2006). Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem, 281, 10856–10864. [DOI] [PubMed] [Google Scholar]
- Gesty-Palmer D, & Luttrell LM (2011). Refining efficacy: exploiting functional selectivity for drug discovery. Adv Pharmacol, 62, 79–107. [DOI] [PubMed] [Google Scholar]
- Goodman OB Jr., Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, & Benovic JL (1996). Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature, 383, 447–450. [DOI] [PubMed] [Google Scholar]
- Gross OP, & Burns ME (2010). Control of rhodopsin’s active lifetime by arrestin-1 expression in mammalian rods. J Neurosci, 30, 3450–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmann M, Merten N, Malfacini D, Inoue A, Preis P, Simon K, Rüttiger N, Ziegler N, Benkel T, Schmitt NK, Ishida S, Müller I, Reher R, Kawakami K, Inoue A, Rick U, Kühl T, Imhof D, Aoki J, König GM, Hoffmann C, Gomeza J, Wess J, & Kostenis E (2018). Lack of beta-arrestin signaling in the absence of active G proteins. Nat Commun, 9, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, & Gurevich VV (2006). Arrestins are ubiquitous regulators of cellular signaling pathways. Genome Biol, 7, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Tesmer JJ, Mushegian A, & Gurevich VV (2012). G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther, 133, 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Dion SB, Onorato JJ, Ptasienski J, Kim CM, Sterne-Marr R, Hosey MM, & Benovic JL (1995). Arrestin interaction with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, b2-adrenergic, and m2 muscarinic cholinergic receptors. J Biol Chem, 270, 720–731. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, & Gurevich EV (2004). The molecular acrobatics of arrestin activation. Trends Pharmacol Sci, 25, 105–111. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, & Gurevich EV (2006). The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharm Ther, 110, 465–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, & Gurevich EV (2018a). Arrestin-mediated signaling: Is there a controversy? World J Biol Chem, 9, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, & Gurevich EV (2018b). Arrestins and G proteins in cellular signaling: The coin has two sides. Sci Signal, 11, eaav1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, & Gurevich EV (2018c). GPCRs and Signal Transducers: Interaction Stoichiometry. Trends Pharmacol Sci, 39, 672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutkind JS, & Kostenis E (2018). Arrestins as rheostats of GPCR signalling. Nat Rev Mol Cell Biol, 19, 615–616. [DOI] [PubMed] [Google Scholar]
- Hamm HE, & Bownds MD (1986). Protein complement of rod outer segments of frog retina. Biochemistry, 25, 4512–4523. [DOI] [PubMed] [Google Scholar]
- Hanson SM, Cleghorn WM, Francis DJ, Vishnivetskiy SA, Raman D, Song X, Nair KS, Slepak VZ, Klug CS, & Gurevich VV (2007). Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J Mol Biol, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausdorff WP, Bouvier M, O’Dowd BF, Irons GP, Caron MG, & Lefkowitz RJ (1989). Phosphorylation sites on two domains of the beta 2-adrenergic receptor are involved in distinct pathways of receptor desensitization. J Biol Chem, 264, 12657–12665. [PubMed] [Google Scholar]
- He Y, Gao X, Goswami D, Hou L, Pal K, Yin Y, Zhao G, Ernst OP, Griffin P, Melcher K, & Xu HE (2017). Molecular assembly of rhodopsin with G protein-coupled receptor kinases. Cell Res, 27, 728–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck M, & Hofmann KP (2001). Maximal rate and nucleotide dependence of rhodopsin catalyzed transducin activation: initial rate analysis based on a double displacement mechanism. J Biol Chem, 276, 10000–10009. [DOI] [PubMed] [Google Scholar]
- Hu J, Stern M, Gimenez LE, Wanka L, Zhu L, Rossi M, Meister J, Inoue A, Beck-Sickinger AG, Gurevich VV, & Wess J (2016). A G Protein-biased Designer G Protein-coupled Receptor Useful for Studying the Physiological Relevance of Gq/11-dependent Signaling Pathways. J Biol Chem, 291, 7809–7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Masureel M, Qianhui Q, Janetzko J, Inoue A, Kato HE, Robertson MJ, Nguyen KC, Glenn JS, Skiniotis G, & Kobilka BK (2020). Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indrischek H, Prohaska SJ, Gurevich VV, Gurevich EV, & Stadler PF (2017). Uncovering missing pieces: duplication and deletion history of arrestins in deuterostomes. BMC Evol Biol, 17, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DS, Kern RC, Puthenveedu MA, von Zastrow M, Williams JC, & Benovic JL (2009). Structure of an arrestin2-clathrin complex reveals a novel clathrin binding domain that modulates receptor trafficking. J Biol Chem, 284, 29860–29872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Kuybeda O, de Waal PW, Mukherjee S, Van Eps N, Dutka P, Zhou XE, Bartesaghi A, Erramilli S, Morizumi T, Gu X, Yin Y, Liu P, Jiang Y, Meng X, Zhao G, Melcher K, Ernst OP, Kossiakoff AA, Subramaniam S, & Xu HE (2018). Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature, 558, 553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, Xu Q, de Waal PW, Ke J, Tan MHE, Zhang C, Moeller A, West GM, Van Eps N, Caro LN, Vishnivetskiy SA, Lee RJ, Suino-Powell KM, Gu X, Pal K, Ma J, Zhi X, Boutet S, Williams GJ, Messerschmidt M, Gati C, Zatsepin NA, Wang D, James D, Basu S, Roy-Chowdhuty S, Conrad S, Coe J, Liu H, Lisova S, Kupitz C, Grotjohann I, Fromme R, Jiang Y, Tan M, Yang H, Li J, Wang M, Zheng Z, Li D, Zhao Y, Standfuss J, Diederichs K, Dong Y, Potter CS, Carragher B, Caffrey M, Jiang H, Chapman HN, Spence JCH, Fromme P, Weierstall U, Ernst OP, Katritch V, Gurevich VV, Griffin PR, Hubbell WL, Stevens RC, Cherezov V, Melcher K, & Xu HE (2015). Crystal structure of rhodopsin bound to arrestin determined by femtosecond X-ray laser. Nature, 523, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, & Christopoulos A (2013a). Measurements of ligand bias and functional affinity. Nat Rev Drug Discov, 12, 483. [DOI] [PubMed] [Google Scholar]
- Kenakin T, & Christopoulos A (2013b). Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov, 12, 205–216. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, & Novick S (2012). A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci, 3, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury E, Clément S, & Laporte SA (2014). Allosteric and biased g protein-coupled receptor signaling regulation: potentials for new therapeutics. Front Endocrinol (Lausanne), 5, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, & Lefkowitz RJ (2005). Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Nat Acad Sci USA, 102, 1442–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch WJ, Inglese J, Stone WC, & Lefkowitz RJ (1993). The binding site for the beta gamma subunits of heterotrimeric G proteins on the beta-adrenergic receptor kinase. J Biol Chem, 268, 8256–8260. [PubMed] [Google Scholar]
- Koehl A, Hu H, Maeda S, Zhang Y, Qu Q, Paggi JM, Latorraca NR, Hilger D, Dawson R, Matile H, Schertler GFX, Granier S, Weis WI, Dror RO, Manglik A, Skiniotis G, & Kobilka BK (2018). Structure of the μ-opioid receptor-Gi protein complex. Nature, 558, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komolov KE, Du Y, Duc NM, Betz RM, Rodrigues JPGLM, Leib RD, Patra D, Skiniotis G, Adams CM, Dror RO, Chung KY, Kobilka BK, & Benovic JL (2017). Structural and Functional Analysis of a β2-Adrenergic Receptor Complex with GRK5. Cell, 169, 407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kook S, Zhan X, Kaoud TS, Dalby KN, Gurevich VV, & Gurevich EV (2014). Arrestin-3 binds JNK1 and JNK2 and facilitates the activation of these ubiquitous JNK isoforms in cells via scaffolding. J Biol Chem, 288, 37332–37242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupnick JG, Gurevich VV, & Benovic JL (1997). Mechanism of quenching of phototransduction. Binding competition between arrestin and transducin for phosphorhodopsin. J Biol Chem, 272, 18125–18131. [DOI] [PubMed] [Google Scholar]
- Latorraca NR, Wang JK, Bauer B, Townshend RJL, Hollingsworth SA, Olivieri JE, Xu HE, Sommer ME, & Dror RO (2018). Molecular mechanism of GPCR-mediated arrestin activation. Nature, 557, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau EK, Trester-Zedlitz M, Trinidad JC, Kotowski SJ, Krutchinsky AN, Burlingame AL, & von Zastrow M (2011). Quantitative encoding of the effect of a partial agonist on individual opioid receptors by multisite phosphorylation and threshold detection. Sci Signal, 4, ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei S, Clydesdale L, Dai A, Cai X, Feng Y, Yang D, Liang YL, Koole C, Zhao P, Coudrat T, Christopoulos A, Wang MW, Wootten D, & Sexton PM (2018). Two distinct domains of the glucagon-like peptide-1 receptor control peptide-mediated biased agonism. J Biol Chem, 293, 9370–9387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang YL, Khoshouei M, Radjainia M, Zhang Y, Glukhova A, Tarrasch J, Thal DM, Furness SGB, Christopoulos G, Coudrat T, Danev R, Baumeister W, Miller LJ, Christopoulos A, Kobilka BK, Wootten D, Skiniotis G, & Sexton PM (2017). Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature, 546, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggett SB, Freedman NJ, Schwinn DA, & Lefkowitz RJ (1993). Structural basis for receptor subtype-specific regulation revealed by a chimeric beta 3/beta 2-adrenergic receptor. Proc Natl Acad Sci U S A, 90, 3665–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsky R, Potts EM, Tarzami ST, Puckerin AA, Stocks J, Schecter AD, Sobie EA, Akar FG, & Diversé-Pierluissi MA (2008). beta-Adrenergic receptor activation induces internalization of cardiac Cav1.2 channel complexes through a beta-arrestin 1-mediated pathway. J Biol Chem, 283, 17221–17226. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Liu JJ, Horst R, Katritch V, Stevens RC, & Wüthrich K (2012). Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science, 335, 1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J, Celver J, Gurevich VV, & Chavkin C (2002). mu-opioid receptors desensitize less rapidly than d-opioid receptors due to less efficient activation of arrestin. J Biol Chem, 277, 15729–15735. [DOI] [PubMed] [Google Scholar]
- Lowe JD, Celver JP, Gurevich VV, & Chavkin C (2002). mu-Opioid receptors desensitize less rapidly than delta-opioid receptors due to less efficient activation of arrestin. J Biol Chem, 277, 15729–15735. [DOI] [PubMed] [Google Scholar]
- Luo J, Busillo JM, & Benovic JL (2008). M3 muscarinic acetylcholine receptor-mediated signaling is regulated by distinct mechanisms. Mol Pharmacol, 74, 338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM (2003). ‘Location, location, location’: activation and targeting of MAP kinases by G protein-coupled receptors. J Mol Endocrinol, 30, 117–126. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Maudsley S, & Bohn LM (2015). Fulfilling the Promise of “Biased” G Protein-Coupled Receptor Agonism. Mol Pharmacol, 88, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, & Lefkowitz RJ (2001). Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci U S A, 98, 2449–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM, Wang J, Plouffe B, Smith JS, Yamani L, Kaur S, Jean-Charles P-Y, Gauthier C, Lee M-H, Pani B, Kim J, Ahn S, Rajagopal S, Reiter E, Bouvier M, Shenoy SK, Laporte SA, Rockman HA, & Lefkowitz RJ (2018). Manifold roles of beta-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci Signal, 11, eaat7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, Prosser RS, & Kobilka BK (2015). Structural Insights into the Dynamic Process of β2-Adrenergic Receptor Signaling. Cell, 161, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin NP, Whalen EJ, Zamah MA, Pierce KL, & Lefkowitz RJ (2004). PKA-mediated phosphorylation of the beta1-adrenergic receptor promotes Gs/Gi switching. Cell Signal, 16, 1397–1403. [DOI] [PubMed] [Google Scholar]
- McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, & Lefkowitz RJ (2000). Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science, 290, 1574–1577. [DOI] [PubMed] [Google Scholar]
- McKay MM, & Morrison DK (2007). Integrating signals from RTKs to ERK/MAPK. Oncogene, 26, 3113–3121. [DOI] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Roca A, Lem J, Wu LW, Simon MI, Baylor DA, & Chen J (2000). Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron, 28, 153–164. [DOI] [PubMed] [Google Scholar]
- Miller WE, McDonald PH, Cai SF, Field ME, Davis RJ, & Lefkowitz RJ (2001). Identification of a motif in the carboxyl terminus of beta -arrestin2 responsible for activation of JNK3. J Biol Chem, 276, 27770–27777. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Gurevich VV, Preninger A, Hamm HE, Bader M-F, Fazleabas AT, Birnbaumer L, & Hunzicker-Dunn M (2002). Aspartic acid 564 in the third cytoplasmic loop of luteinizing hormone/choriogonadotropin receptor is crucial for phosphorylation-independent interaction with arrestin2. J Biol Chem, 277, 17916–17927. [DOI] [PubMed] [Google Scholar]
- Nakajima K, & Wess J (2012). Design and functional characterization of a novel, arrestin-biased designer G protein-coupled receptor. Mol Pharmacol, 82, 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AH, Thomsen ARB, Cahill TJ 3rd, Huang R, Huang LY, Marcink T, Clarke OB, Heissel S, Masoudi A, Ben-Hail D, Samaan F, Dandey VP, Tan YZ, Hong C, Mahoney JP, Triest S, Little J 4th, Chen X, Sunahara R, Steyaert J, Molina H, Yu Z, des Georges A, & Lefkowitz RJ (2019). Structure of an endosomal signaling GPCR G protein-β-arrestin megacomplex. Nat Struct Mol Biol, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, Shenoy SK, Gygi SP, & Lefkowitz RJ (2011). Distinct Phosphorylation Sites on the {beta}2-Adrenergic Receptor Establish a Barcode That Encodes Differential Functions of {beta}-Arrestin. Sci Signal, 4(ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hayre M, Eichel K, Avino S, Zhao X, Steffen DJ, Feng X, Kawakami K, Aoki J, Messer K, Sunahara R, Inoue A, von Zastrow M, & Gutkind JS (2017). Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signaling to ERK. Sci Signal, 10, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Buczylko J, Kaplan MW, Polans AS, & Crabb JW (1991). Mechanism of rhodopsin kinase activation. J Biol Chem, 266, 12949–12955. [PubMed] [Google Scholar]
- Pals-Rylaarsdam R, Gurevich VV, Lee KB, Ptasienski J, Benovic JL, & Hosey MM (1997). Internalization of the m2 muscarinic acetylcholine receptor: arrestin-independent and -dependent pathways. J Biol Chem, 272, 23682–23689. [DOI] [PubMed] [Google Scholar]
- Pals-Rylaarsdam R, & Hosey MM (1997). Two homologous phosphorylation domains differentially contribute to desensitization and internalization of the m2 muscarinic acetylcholine receptor. J Biol Chem, 272, 14152–14158. [DOI] [PubMed] [Google Scholar]
- Pan L, Gurevich EV, & Gurevich VV (2003). The nature of the arrestin x receptor complex determines the ultimate fate of the internalized receptor. J Biol Chem, 278, 11623–11632. [DOI] [PubMed] [Google Scholar]
- Pera T, Hegde A, Deshpande DA, Morgan SJ, Tiegs BC, Theriot BS, Choi YH, Walker JK, & Penn RB (2015). Specificity of arrestin subtypes in regulating airway smooth muscle G protein-coupled receptor signaling and function. FASEB J, 29, 4227–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry NA, Kaoud TS, Ortega OO, Kaya AI, Marcus DJ, Pleinis JM, Berndt S, Chen Q, Zhan X, Dalby KN, Lopez CF, Iverson TM, & Gurevich VV (2019). Arrestin-3 scaffolding of the JNK3 cascade suggests a mechanism for signal amplification. Proc Natl Acad Sci U S A, 116, 810–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson YK, & Luttrell LM (2017). The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol Rev, 69, 256–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher J, Lohse MJ, Codina J, Caron MG, & Lefkowitz RJ (1992). Desensitization of the isolated beta 2-adrenergic receptor by beta-adrenergic receptor kinase, cAMP-dependent protein kinase, and protein kinase C occurs via distinct molecular mechanisms. Biochemistry, 31, 3193–3197. [DOI] [PubMed] [Google Scholar]
- Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, Durroux T, Sharma KS, Durand G, Pucci B, Trinquet E, Zwier JM, Deupi X, Bron P, Banères JL, Mouillac B, & Granier S (2012). Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci U S A, 109, 6733–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankovic Z, Brust TF, & Bohn LM (2016). Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg Med Chem Lett, 26, 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, & Kobilka BK (2011). Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature, 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh A, Cooper A, Guy-David L, & Reuveny E (2010). Nonenzymatic rapid control of GIRK channel function by a G protein-coupled receptor kinase. Cell, 143, 750–760. [DOI] [PubMed] [Google Scholar]
- Ren XR, Reiter E, Ahn S, Kim J, Chen W, & Lefkowitz RJ (2005). Different G protein-coupled receptor kinases govern G protein and beta-arrestin mediated signaling of V2 vasopressin receptor. Proc Nat Acad Sci USA, 102, 1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouault AAJ, Rosselli-Murai LK, Hernandez CC, Gimenez LE, Tall GG, & Sebag JA (2020). The GPCR accessory protein MRAP2 regulates both biased signaling and constitutive activity of the ghrelin receptor GHSR1a. Sci Signal, 13, eaax4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, & Davis RJ (2014). TNF and MAP kinase signalling pathways. Semin Immunol, 26, 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, & Lefkowitz RJ (1993). A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem, 268, 4625–4636. [PubMed] [Google Scholar]
- Santini F, Penn RB, Gagnon AW, Benovic JL, & Keen JH (2000). Selective recruitment of arrestin-3 to clathrin coated pits upon stimulation of G protein-coupled receptors. J Cell Sci, 113, 2463–2470. [DOI] [PubMed] [Google Scholar]
- Seibold A, Williams B, Huang ZF, Friedman J, Moore RH, Knoll BJ, & Clark RB (2000). Localization of the sites mediating desensitization of the beta(2)-adrenergic receptor by the GRK pathway. Mol Pharmacol, 58, 1162–1173. [DOI] [PubMed] [Google Scholar]
- Seo J, Tsakem EL, Breitman M, & Gurevich VV (2011). Identification of arrestin-3-specific residues necessary for JNK3 activation. J Biol Chem, 286, 27894–27901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, & Lefkowitz RJ (2006). beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem, 281, 1261–1273. [DOI] [PubMed] [Google Scholar]
- Shiraishi Y, Natsume M, Kofuku Y, Imai S, Nakata K, Mizukoshi T, Ueda T, Iwaï H, & Shimada I (2018). Phosphorylation-induced conformation of β2-adrenoceptor related to arrestin recruitment revealed by NMR. Nat Commun, 9, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, Staus DP, Hilger D, Uysal S, Huang LY, Paduch M, Tripathi-Shukla P, Koide A, Koide S, Weis WI, Kossiakoff AA, Kobilka BK, & Lefkowitz RJ (2013). Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature, 497, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siehler S (2009). Regulation of RhoGEF proteins by G12/13-coupled receptors. Br J Pharmacol, 158, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Coffa S, Fu H, & Gurevich VV (2009). How does arrestin assemble MAPKs into a signaling complex? J Biol Chem, 284, 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Raman D, Gurevich EV, Vishnivetskiy SA, & Gurevich VV (2006). Visual and both non-visual arrestins in their “inactive” conformation bind JNK3 and Mdm2 and relocalize them from the nucleus to the cytoplasm. J Biol Chem, 281, 21491–21499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler D, Waeber C, Pantaloni C, Holsboer F, Bockaert J, Seeburg PH, & Journot L (1993). Differential signal transduction by five splice variants of the PACAP receptor. Nature, 365, 170–175. [DOI] [PubMed] [Google Scholar]
- Staus DP, Hu H, Robertson MJ, Kleinhenz ALW, Wingler LM, Capel WD, Latorraca NR, Lefkowitz RJ, & Skiniotis G (2020). Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterne-Marr R, Gurevich VV, Goldsmith P, Bodine RC, Sanders C, Donoso LA, & Benovic JL (1993). Polypeptide variants of beta-arrestin and arrestin3. J Biol Chem, 268, 15640–15648. [PubMed] [Google Scholar]
- Stupack DG, & Cheresh DA (2002). Get a ligand, get a life: integrins, signaling and cell survival. J Cell Sci, 115, 3729–3738. [DOI] [PubMed] [Google Scholar]
- Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T, & Tesmer JJ (2005). Snapshot of activated G proteins at the membrane: the Galphaq-GRK2-Gbetagamma complex. Science, 310, 1686–1690. [DOI] [PubMed] [Google Scholar]
- Thompson GL, Canals M, & Poole DP (2014). Biological redundancy of endogenous GPCR ligands in the gut and the potential for endogenous functional selectivity. Frontiers in Pharmacology, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen ARB, Plouffe B, Cahill III TJ, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, Huang L, Breton B, Heydenreich FM, Sunahara RK, Skiniotis G, Bouvier M, & Lefkowitz RJ (2016). GPCR-G Protein-β-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell, 166, 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin AB, Butcher AJ, & Kong KC (2008). Location, location, location…site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci, 29, 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth AD, Prokop S, Gyombolai P, Várnai P, Balla A, Gurevich VV, Hunyady L, & Turu G (2018). Heterologous phosphorylation-induced formation of a stability lock permits regulation of inactive receptors by β-arrestins. J Biol Chem, 293, 876–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eps N, Altenbach C, Caro LN, Latorraca NR, Hollingsworth SA, Dror RO, Ernst OP, & Hubbell WL (2018). Gi- and Gs-coupled GPCRs show different modes of G-protein binding. Proc Natl Acad Sci U S A, 115, 2383–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eps N, Caro LN, Morizumi T, Kusnetzow AK, Szczepek M, Hofmann KP, Bayburt TH, Sligar SG, Ernst OP, & Hubbell WL (2017). Conformational equilibria of light-activated rhodopsin in nanodiscs. Proc Natl Acad Sci U S A, 114, E3268–E3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, & Lefkowitz RJ (2007). Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci, 28, 416–422. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Hirsch JA, Velez M-G, Gurevich YV, & Gurevich VV (2002). Transition of arrestin in the active receptor-binding state requires an extended interdomain hinge. J Biol Chem, 277, 43961–43968. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Raman D, Wei J, Kennedy MJ, Hurley JB, & Gurevich VV (2007). Regulation of arrestin binding by rhodopsin phosphorylation level. J Biol Chem, 282, 32075–32083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther C, Nagel S, Gimenez LE, Morl K, Gurevich VV, & Beck-Sickinger AG (2010). Ligand-induced internalization and recycling of the human neuropeptide Y2 receptor is regulated by its carboxyl-terminal tail. J Biol Chem, 285, 41578–41590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen EJ, Rajagopal S, & Lefkowitz RJ (2011). Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol Med, 17, 126–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilden U (1995). Duration and amplitude of the light-induced cGMP hydrolysis in vertebrate photoreceptors are regulated by multiple phosphorylation of rhodopsin and by arrestin binding. Biochemistry, 34, 1446–1454. [DOI] [PubMed] [Google Scholar]
- Wisler JW, Xiao K, Thomsen AR, & Lefkowitz RJ (2014). Recent developments in biased agonism. Curr Opin Cell Biol, 27, 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, Yates JR, & Lefkowitz RJ (2007). Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci U S A, 104, 12011–12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W, Li Z, Jin M, Yin YL, de Waal PW, Pal K, Yin Y, Gao X, He Y, Gao J, Wang X, Zhang Y, Zhou H, Melcher K, Jiang Y, Cong Y, Zhou XE, Yu X, & Xu HE (2019). A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res, 29, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Kaoud TS, Dalby KN, & Gurevich VV (2011). Non-visual arrestins function as simple scaffolds assembling MKK4- JNK3α2 signaling complex. Biochemistry, 50, 10520–10529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Stoy H, Kaoud TS, Perry NA, Chen Q, Vucak G, Perez A, Els-Heindl S, Slagis JV, Iverson TM, Beck-Sickinger AG, Gurevich EV, Dalby KN, & Gurevich VV (2016). Peptide mini-scaffold facilitates JNK3 activation in cells. Sci Rep, 6, 21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DL, Sun YJ, Ma ML, Wang YJ, Lin H, Li RR, Liang ZL, Gao Y, Yang Z, He DF, Lin A, Mo H, Lu YJ, Li MJ, Kong W, Chung KY, Yi F, Li JY, Qin YY, Li J, Thomsen ARB, Kahsai AW, Chen ZJ, Xu ZG, Liu M, Li D, Yu X, & Sun JP (2018). Gq activity- and β-arrestin-1 scaffolding-mediated ADGRG2/CFTR coupling are required for male fertility. Elife, 7, pii: e33432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sun B, Feng D, Hu H, Chu M, Qu Q, Tarrasch JT, Li S, Kobilka TS, Kobilka BK, & Skiniotis G (2017). Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature, 546, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XE, He Y, de Waal PW, Gao X, Kang Y, Van Eps N, Yin Y, Pal K, Goswami D, White TA, Barty A, Latorraca NR, Chapman HN, Hubbell WL, Dror RO, Stevens RC, Cherezov V, Gurevich VV, Griffin PR, Ernst OP, Melcher K, & Xu HE (2017). Structural Identification of Phosphorylation Codes for Arrestin Recruitment by G protein-Coupled Receptors. Cell, 170, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang T, Chen Q, Cho M-K, Vishnivetskiy SA, Iverson TI, Gurevich VV, & Hubbell WL (2013). Involvement of Distinct Arrestin-1 Elements in Binding to Different Functional Forms of Rhodopsin. Proc Nat Acad Sci USA, 110, 942–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidar DA, Violin JD, Whalen EJ, & Lefkowitz RJ (2009). Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc Nat Acad Sci USA, 106, 9649–9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweemer AJM, Toraskar J, Heitman LH, & Ijzerman AP (2014). Bias in chemokine receptor signalling. Trends in Immunology, 35, 243–252. [DOI] [PubMed] [Google Scholar]