

Abstract
Purpose:
We studied galactose supplementation in SLC35A2-congenital disorder of glycosylation (SLC35A2-CDG), caused by mono-allelic pathogenic variants in SLC35A2 (Xp11.23), encoding the ER and Golgi UDP-galactose transporter. Patients present with epileptic encephalopathy, developmental disability, growth deficiency, and dysmorphism.
Methods:
Ten patients with SLC35A2-CDG were supplemented with oral D-galactose for 18 weeks in escalating doses up to 1.5 g/kg/day. Outcome was assessed using the Nijmegen Pediatric CDG Rating Scale (NPCRS, ten patients) and by glycomics (eight patients).
Results:
SLC35A2-CDG patients demonstrated improvements in overall NPCRS (P=0.008), the current clinical assessment (P=0.007) and the system specific involvement (P=0.042) domains. Improvements were primarily in growth and development with five patients resuming developmental progress, that included postural control, response to stimuli, chewing and swallowing amelioration. Additionally, there were improvements in gastrointestinal symptoms and epilepsy. One patient in our study did not show any clinical improvement.
Galactose supplementation improved patients’ glycosylation with decreased ratios of incompletely formed to fully formed glycans (M-gal/di-sialo, P=0.012 and mono-sialo/di-sialo, P=0.017) and increased levels of a fully galactosylated N-glycan (P=0.05).
Conclusion:
Oral D-galactose supplementation results in clinical and biochemical improvement in SLC35A2-CDG. Galactose supplementation may partially overcome the Golgi UDP-galactose deficiency and improves galactosylation. Oral galactose is well-tolerated and shows promise as dietary therapy.
Keywords: SLC35A2-CDG, galactose, Nijmegen Pediatric CDG Rating Scale (NCPRS), glycan, glycosylation
Introduction
SLC35A2-CDG (MIM#300896), previously known as CDG-IIm, is a multisystem disease caused by mono-allelic pathogenic variants in SLC35A2 (Xp11.23). SLC35A2 encodes a transporter that imports UDP-galactose into the Golgi and the endoplasmic reticulum and is thus important for the formation of N- and O-linked glycans, glycosaminoglycans and glycosphingolipids. Currently 62 SLC35A2-CDG patients have been described 1–7, making up a sizeable percentage (7%) of the patient’s with CDG-II 7. SLC35A2-CDG presents predominantly with neurological features and has been described as an early-onset epileptic encephalopathy 1. Diagnosis of SLC35A2-CDG is challenging 7. About 95 % of patients have been diagnosed through next-generation sequencing 3.
Many patients with SLC35A2-CDG show severe epilepsy including hypsarrhythmia and West syndrome. They can also have severe global developmental delay and muscular hypotonia without seizures. Brain MRI can show cerebellar atrophy, thinning and shortening of the corpus callosum, delayed myelination, white matter hyperintensities, cerebral atrophy and dilatation of the lateral ventricles. Ocular findings of SLC35A2-CDG include cortical visual impairment, retinitis pigmentosa, nystagmus and strabismus. There is failure to thrive due to gastrointestinal disease leading to poor nutritional state and an impairment of the GH/IGF axis. Other affected organs include the liver (dysfunction, hepatomegaly), spleen (splenomegaly), kidney (nephrotic syndrome), and skeleton (rhizomelic shortening of limbs, craniosynostosis or scoliosis). Patients also have decreased coagulation factors, and there is a type 2 pattern on sialotransferrin isoelectric focusing which can spontaneously improve. 1–7
SLC35A2-CDG is an X-linked dominant disease (Xp11.13) affecting more females than males. In females, skewed X-inactivation can sometimes be observed. Variable degrees of mutant protein expression can modulate disease severity 4. In males, mosaicism has also been reported and most likely some degree of allelic expression is a prerequisite for life 7. Moreover, brain somatic mutations have been held responsible for isolated epilepsy and focal cortical dysplasia 8,9.
Sialotransferrin isoelectric focusing or mass spectroscopy, the commonly used diagnostic modalities in N-linked protein glycosylation disorders, are normal in the majority of patients(76%) 3. Moreover, within the same patient the profile can either improve or worsen over a few years (frequently normalizing by the age of 1-3 years). Interestingly, galactose intake could also have a role in modulation of the transferrin profile in early life 7.
Currently, two other CDG types (PGM1-CDG and TMEM165-CDG) that are similarly notable for an undergalactosylation of the N-glycans, have shown good responses to galactose supplementation 10,11. Moreover, Dörre et al. treated one patient with SLC35A2-CDG with galactose 1g/kg of body weight divided in five equal doses after demonstrating its success in improving glycosylation in CHO-Lec8 cell expressing the mutant allele 4. This led to a marked improvement in sialotransferrin isoelectric focusing with near-normalization after 6 months 4. However, another group could not demonstrate the beneficial effect of galactose on patient fibroblasts in vitro 2.
We performed a treatment trial with galactose in patients with SLC35A2-CDG.
Patients and Methods
Patients
Patients with known pathogenic variants were recruited from the Mayo Clinic, Rochester, USA; Children’s Hospital of Philadelphia, Philadelphia, USA; Texas Children’s Hospital, Houston, Texas, USA; University of Catania, Catania, Italy; Tartu University Hospital, Tartu, Estonia; Children’s & Women’s Health Centre of British Columbia, Vancouver, Canada and Copenhagen University Hospital, Copenhagen, Denmark.
The protocol was approved by the institutional review board (Tulane University Hayward Genetics Center #IRB 517339-4) and registered on https://clinicaltrials.gov/ct2/show/NCT02955264.
Methods
Glycan analysis by mass spectrometry
From 8 patients (patients 1-3, 5-7 and 9-10) samples were collected before and after the galactose trial for semi-quantitative N-glycan mass spectrometry analysis (ESI-QTOF) 12.
Briefly, heparinized plasma was combined with an internal control (sialylglycopeptide) and digested in buffered RapiGest SFTM solution. N-glycans were cleaved with PNGase FTM, reacted with RapiFluor-MS, and isolated on a HILIC column. Mass spectrometric analysis was performed on the Waters’ Synapt G2 Si QTOF as previously described.
Normal values were derived from a previously obtained cohort of 31 healthy controls.
All samples were analyzed at the Children’s Hospital of Philadelphia, Philadelphia, USA.
Galactose supplementation
Participants in this non-blinded observational pilot study received oral galactose supplementation over 18 weeks. D-Galactose intake was increased in a stepwise fashion over the study period as follows (weeks 0-6: 0.5 g/kg per day, weeks 7-12: 1.0 g/kg per day, weeks 13-18: 1.5 g/kg per day), to minimize gastrointestinal and metabolic side effects. Participants were instructed to continue their regular diet in combination with daily oral galactose supplementation in a single morning dose. The maximum daily dose of galactose any patient received was 50 g (this amount is within recommended daily intake). Prior studies investigating focal segmental glomerulosclerosis have demonstrated this amount of galactose to be safe and tolerable by patients 11,13.
At the start and the end of galactose therapy, a clinical and metabolic baseline was established: at each time point in the study, patients underwent a clinical examination including an assessment for changes and side effects or concerns regarding the galactose supplementation. Urinary galactitol levels were measured as a safety parameter.
The clinical characteristics and disease progression score of the patients were documented and followed by the Nijmegen CDG rating scale 14. This score collects information on I: current function (current history on vision, hearing, communication, feeding, and mobility); II: system specific involvement (history in the last 6 months on seizures, encephalopathy, bleeding diathesis or coagulation defect, gastrointestinal, endocrine, respiratory, cardiovascular, renal, liver function, and blood anomalies) and III: the current clinical assessment (growth, development, vision, strabismus and eye movement, myopathy, ataxia, pyramidal and extrapyramidal symptoms, and neuropathy). Higher scores are associated with worse clinical disease 15. These scores were calculated at each investigation site for each patient by the attending physician (authors RB, KÕ, RS, SG, EM, AE).
Statistics
SPSS 22 for windows (SPSS Inc., Chicago, IL, USA) was used for statistical analysis. All results are expressed as the means ± standard deviation. For differences between two continuous variables in two groups, the Wilcoxon signed rank test was used. A p-value < 0.05 was considered statistically significant. No adjustment for multiple testing was performed for the Glycan analysis as these are dependent variables.
Results
Clinical findings
Ten patients (two males, 2-2.6y and eight females 0.5-26y) were enrolled in this study. The most common clinical features in this cohort of patients were developmental delay, short stature, epilepsy, and hypotonia. The NPCRS severity score varied widely from 8-40 (the lower score depicts the better clinical state, see table 1). The average patient (total NCPRS score 28.7 ± 9.7) had moderate visual impairment (not fully corrected with glasses or inattention to large objects in the visual field). They could not communicate effectively with either parents or strangers and were attending special school or nursery. Many had global developmental delay without clear developmental progress and were fully reliant on parents with no contribution to self-care. Most patients were wheelchair dependent. Four of the patients had seizures. Many of the patients had mild feeding problems (such as choking, emesis, and anorexia resulting in decreased intake) and gastrointestinal issues such as vomiting or diarrhea 1-3 times/week. On average, patients’ growth and weight was less than the second centile but were growing parallel to it. Elevated serum transaminases without liver failure were seen in five patients.
Table 1:
Characteristics of included patients.
| Patient number | Gender, age | Genotoypea | Abnl TIEF | Initial NCPRS score | Final NCPRS score | Previous reported patient |
|---|---|---|---|---|---|---|
| 1 | F,4y | c.389A>G (p.Tyr130Cys) | Normal | 39 | 32 | Patient 9 in 7; CDG-0469 in 3 |
| 2 | M, 2.6y | c.670C>T (p.Leu224Phe) | Abnormal | 8 | 5 | Patient 1 in 7 |
| 3 | F, 27y | c.991G>A, (p.Val331Ile) | Normal | 21 | 17 | CDG-0173 in 3 |
| 4 | F, 10y | c.815 G>A (p.Trp272Ter) | Normal | 39 | 33 | |
| 5 | F, 2.6y | c.211G>A (p.Val71Met) | Normal | 22 | 19 | CDG-187 in 3 |
| 6 | F, 11y | c.935C>A (p.Ser312Tyr) | Normal | 40 | 38 | CDG-0164 in 3 |
| 7 | F, 0.5y | c.523C>T (p.Leu175Phe) | Abnormal | 24 | 17 | CDG-0308 in 3 |
| 8 | F, 5y | c.466_468delTCC (p.Ser156del) | Normal | 32 | 32 | Patient 15 in 7 |
| 9 | F, 1.3y | c.923C>T (p.Ser308Phe) | Abnormal | 27 | 25 | 19 |
| 10 | M, 1y | c.944T>C (p.Leu315Pro) | Abnormal | 35 | 30 | CDG-0460 in 7 |
reference transcript NM_001042498.2
Glycosylation abnormalities
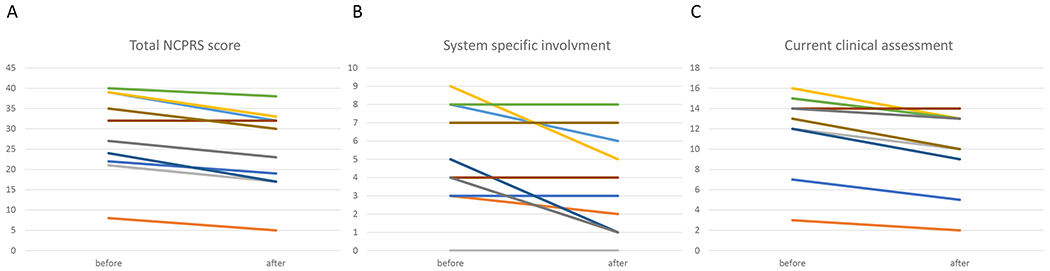
Only four out of the ten patients had a type 2 pattern on transferrin isoelectric focusing. These were all patients younger than 3 years of age. Pre-treatment samples from 8 patients (patients 1-3, 5-7 and 9-10) were available for a recently described quantitative N-glycan analysis with demonstrated increased clinical sensitivity and specificity12. All 8 patients demonstrated pre-treatment abnormalities in galactosylation with the quantitative N-glycan assay. In SLC35A2-CDG, galactosylation is likely affected due to defective transport of UDP-Gal into the Golgi resulting in decreased substrate for the β1,4-galactosyltransferase (B4GALT)3. This enzyme acts on Hex3HexNac4 (asialo, agalacto biantennary glycans) (A-Gal 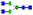 ) and adds a galactose residue to form Hex4HexNAc4 (monogalactosylated, asialo, biantennary) (MGAS
) and adds a galactose residue to form Hex4HexNAc4 (monogalactosylated, asialo, biantennary) (MGAS  ), which is elongated to Neu5Ac1Hex4HexNAc4 (monosialo, monogalactosylated, biantennary) (M-gal
), which is elongated to Neu5Ac1Hex4HexNAc4 (monosialo, monogalactosylated, biantennary) (M-gal  ) and also adds a second galactose residue to form Neu5Ac1Hex5HexNAc4 (M-Sialo, monosialo, biantennary
) and also adds a second galactose residue to form Neu5Ac1Hex5HexNAc4 (M-Sialo, monosialo, biantennary  ) that is then elongated to the mature glycan Neu5Ac2Hex5HexNAc4 (Disialo biantennary) (Di-sialo
) that is then elongated to the mature glycan Neu5Ac2Hex5HexNAc4 (Disialo biantennary) (Di-sialo  ). All SLC35A2-CDG patients had abnormalities of at least one of these glycans: increased A-gal (4/8 patients), increased MGAS (5/8), increased M-gal (6/8) or decreased Di-sialo (2/8).
). All SLC35A2-CDG patients had abnormalities of at least one of these glycans: increased A-gal (4/8 patients), increased MGAS (5/8), increased M-gal (6/8) or decreased Di-sialo (2/8).
Clinical effect of treatment
After 18 weeks of galactose treatment, mean total NPCRS score improved from 28.7 ± 9.7 to 24.6 ± 9.6 (P = 0.008) (See figure 1a and supplementary table 1). There was a statistically significant improvement in the system specific involvement 5.1 ± 2.7 to 3.6 ± 2.6 (P = 0.042) (See figure 1b) and the current clinical assessment 12.1 ± 3.8 to 10.2 ± 3.7 (P = 0.007) domains (See figure 1c). There was an improvement in growth (P = 0.023) with all patients following normal growth trajectory. All but two patients made developmental progress (P = 0.008) that included postural control, amelioration in response to visual and auditory stimuli, chewing and swallowing. Of the four patients that initially had epilepsy with >5 generalized tonic-clonic seizures/month or >20 absence or myoclonic seizures/month, 2 improved and had no further epileptic episodes. Notably, one patient reduced the number of antiepileptic drugs from >3 to one. However, due to the small number, this change was not statistically significant. One patient in our study (patient 8) did not show any clinical improvement.
Figure 1.
a Total score in Nijmegen pediatric CDG rating scale (before and after galactose supplementation)
Mean total NPCRS score improved from 28.7 ± 9.7 to 24.6 ± 9.6 (P = 0.008)
b System specific involvement in NCPRS (before and after galactose supplementation)
System specific involvement improved from 5.1 ± 2.7 to 3.6 ± 2.6 (P = 0.042)
c Current clinical assessment in NCPRS (before and after galactose supplementation)
Current clinical assessment improved from 12.1 ± 3.8 to 10.2 ± 3.7 (P = 0.007)
No serious adverse effects of the treatment were encountered.
Glycosylation changes after treatment
With galactose supplementation, there was a significant increase in total normally formed Di-sialo glycans from 23.60 ± 9.61 to 27.91 ± 9.57 (nl range 23.19-42.50; P = 0.050). Moreover, there was a decrease in the M-Gal/Di-Sialo ratio, from 0.82 ± 1.25 to 0.45 ± 0.64 (normal 0.02-0.07, P = 0.012) (See figure 2a), as galactosylation was improved. Furthermore, there was an improvement in the next synthetic step leading to a decrease in the M-Sialo/Di-Sialo ratio, from 0.52 ± 0.10 to 0.42 ± 0.09 (normal 0.20 – 0.52, P = 0.017) (See figure 2b).
Figure 2. Glycomics before and after galactose supplementation.
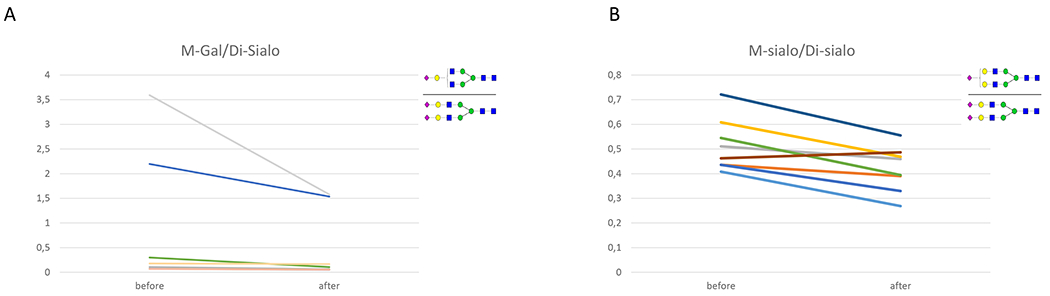
a: A decrease in the mono-Gal/Di sialo upon treatment from 0.74 ± 1.27 to 0.45 ± 0.68 (p=0.011)
b: A decrease in mono-sialic/disialic upon treatment from 0.51 ± 0.1 to 0.42 ± 0.09 (p=0.017)
Looking specifically at the underglycosylated glycans, there was a decrease of A-Gal, MGAS and M-Gal in all patients who had levels outside the reference range at the start of the trial (See table 2).
Table 2:
Glycan findings on ESI-QTOF before and after treatment
| Glycan structure | Before galactose | After galactose | P-value |
|---|---|---|---|
| 13C6-Neu5Ac2Hex5HexNac4 (IS) | 116553.33 ± 57108.29 | 142085 ± 55888.93 | N.S. |
| Hex5HexNAc2 (Man 5) | 3.13 ± 0.72 | 2.83 ± 0.74 | N.S. |
| Hex3HexNAc4 (Asialo, agalacto, biantennary) (A-gal) | 3.81 ± 6 | 3.18 ± 5.04 | N.S. |
| Hex6HexNAc2 (Man 6) | 3 ± 0.66 | 2.72 ± 0.59 | N.S. |
| Fuc1Hex3HexNAc4 (Asialo, agalacto, biantennary fucosylated) | 6.99 ± 2.28 | 6.67 ± 2.11 | N.S. |
| Hex4HexNAc4 (Monogalactosylated, asialo, biantennary) | 1.25 ± 0.81 | 1.06 ± 0.6 | N.S. |
| Neu5Ac1Fuc1Hex3HexNAc3 (Fucosylated Mono-antennary glycan) | 0.12 ± 0.04 | 0.09 ± 0.04 | 0.027 |
| Hex7HexNAc2 (Man 7) | 0.98 ± 0.29 | 0.83 ± 0.16 | N.S. |
| Neu5Ac1Hex4HexNAc3 (Monosialo,galactosylated,mono N-acetyl glucosamine biantennary) (M-GlcNAc) | 3.15 ± 1.04 | 3.04 ± 0.99 | N.S. |
| Hex8HexNAc2 (Man8) | 1.09 ± 0.35 | 0.94 ± 0.17 | N.S. |
| Neu5Ac1Hex4HexNAc4 (Monosialo, monogalactosylated, mono N-acetylglucosamine biantennary)(M-gal) | 7.34 ± 7.99 | 6.83 ± 7.96 | 0.011 |
| Fuc1Hex5HexNAc4 | 5.01 ± 1.8 | 4.5 ± 1.23 | N.S. |
| Hex9HexNAc2 (Man 9) | 0.85 ± 0.27 | 0.76 ± 0.17 | N.S. |
| Neu5Ac1Fuc1Hex4HexNAc4 (Monogalactosylated fucosylated biantennary) | 1.88 ± 0.4 | 1.83 ± 0.55 | N.S. |
| Neu5Ac1Hex5HexNAc4 (monosialo, biantennary) (M-sialo) | 11.44 ± 4.07 | 11.09 ± 3.16 | N.S. |
| Neu5Ac2Hex5HexNAc4 (Disialo biantennary) (Di-sialo) | 24.01 ± 9.68 | 27.91 ± 10.23 | 0.049 |
| Neu5Ac2Hex6HexNAc5 (Disialo triantennary) | 0.51 ± 0.26 | 0.61 ± 0.31 | N.S. |
| Neu5Ac2Fuc1Hex6HexNAc5 (Disialo triantennary fucosylated) | 0.2 ± 0.11 | 0.22 ± 0.11 | N.S. |
| Neu5Ac3Hex6HexNAc5 (Trisialo triantennary) (Tri-sialo) | 0.78 ± 0.36 | 1.09 ± 0.58 | 0.042 |
| Neu5Ac3Fuc1Hex6HexNAc5 (Trisialo triantennary fucosylated) | 0.24 ± 0.15 | 0.3 ± 0.15 | N.S. |
| NeuAc3Hex7HexNAc6 (Trisialo tetra-antennary) | 0.06 ± 0.06 | 0.07 ± 0.07 | N.S. |
| Total Glycan Signal | 3004.08 ± 653.6 | 2478.09 ± 323.51 | N.S. |
| Selected ratios | |||
| Man5/9 | 4.11 ± 1.49 | 3.98 ± 1.82 | N.S. |
| Calculated Di-sialo Concentration | 0.48 ± 0.43 | 0.44 ± 0.31 | N.S. |
| M-gal/Di-SA | 0.74 ± 1.27 | 0.45 ± 0.68 | 0.011 |
| M-sial/di-sial | 0.51 ± 0.1 | 0.42 ± 0.09 | 0.017 |
There was no apparent correlation between age, sex, clinical severity, or mutation type with clinical or biochemical response.
As transferrin isoelectric focusing was only abnormal in the four youngest patients, there were too few patients to perform a subgroup analysis in the patients overtly demonstrating glycan abnormalities.
Discussion
This is the first pilot study of galactose therapy in SLC35A2-CDG. In this CDG there is deficient import of UDP-Gal into the ER or the Golgi where it is required by β1,4-galactosyltransferase to elongate the growing glycan chain. We have previously demonstrated in control fibroblasts that galactose feeding increases cytoplasmic UDP-gal 11. In vivo, our patients exhibit an increase in normally formed, mature glycans and a decrease of undergalactosylated glycans, which is mirrored in measurable clinical improvement.
Nutritional therapy is increasingly used in CDG. It has been demonstrated to be beneficial mainly in PGM1-CDG (treatment with galactose) and in MPI-CDG (treatment with mannose) 16. Interestingly in SLC35C1-CDG, a defect in the UDP-fucose transporter, fucose supplementation also has been shown beneficial in some patients 17. This suggests that increasing the Golgi-cytoplasmic gradient of the UDP-hexose can improve its import. However, this increased transport remains to be demonstrated in vitro.
It is tempting to speculate that additional uridine supplementation to increase UDP-Gal production could further ameliorate the clinical picture. In SLC35A2-CDG, an X-linked disorder, where mosaicism or skewed X-inactivation has been documented previously, it is very likely that some SLC35A2 function remains.
We have shown that galactose has a beneficial effect on the epilepsy phenotype in 2/4 patients. It has been recently shown in multiple studies that focal cortical dysplasia may be associated with somatic pathogenic variants in SLC35A2 8,9,18. There may thus possibly be a role for galactose therapy in epilepsy management of these patients.
Interestingly, patients’ transferrin glycosylation profile can improve spontaneously within the first 3 years of life, which is presumed to be related to hepatic selection of less affected hepatocytes. Nevertheless, we have demonstrated that subtle glycosylation changes detectable by plasma quantitative N-glycan analysis via ESI-QTOF remain in older patients. These are glycans derived from all plasma proteins with the non-liver protein immunoglobulin G as the major contributor. Galactose therapy also improves these analytes.
One of the limitations of our study is that transferrin glycosylation can spontaneously improve. This makes it difficult to assign a direct correlation between patient clinical improvement and glycosylation improvement. However, our results are suggestive because of the short study period. Spontaneous improvement of transferrin glycosylation occurs in the first years of life and is not accompanied by clinical improvement 7. Since SLC35A2-CDG has primarily a neurological presentation, it is likely that the most significant improvements in glycosylation are necessary in the brain, for which there is not a readily assayable source. Another limitation of our study is its unblinded, non-placebo controlled character and clinical improvement was scored by the treating physicians. Future assessment of galactose efficacy in SLC35A2-CDG should include a double-blind, randomized, placebo-controlled study. Since this is a very rare condition, a double-blind, cross-over control study design is necessary. Future studies could also look at the efficacy of adding uridine supplementation as a precursor of UDP-Gal on clinical and glycosylation improvements. Additionally, assessment of plasma N-glycans following discontinuation of galactose therapy could increase the certainty that improvement of glycosylation is really due to the galactose therapy.
Given pre-treatment abnormalities in all SLC35A2-CDG patients tested, our results suggest that quantitative N-glycan analysis of plasma proteins may be a useful diagnostic tool for SLC35A2-CDG. Because of the good safety profile of galactose supplementation and improvements seen in almost all patients, we suggest that galactose could be trialed in individual SLC35A2-CDG patients.
Supplementary Material
Acknowledgements
Peter Witters is supported by the Clinical Research Foundation of University Hospitals Leuven, Leuven, Belgium. Andrew Edmondson is supported by National Institutes of Health grant T32 GM008638. Katrin Õunap is supported by Estonian Research Council grant PUT355 and PRG471. This study is partially supported by National Institutes of Health grant 1 U54 NS115198-01
Footnotes
Conflict of interests
There are no conflicts of interests for any of the authors.
References
- 1.Kodera H, Nakamura K, Osaka H, et al. De novo mutations in SLC35A2 encoding a UDP-galactose transporter cause early-onset epileptic encephalopathy. Hum Mutat. 2013;34:1708–1714. [DOI] [PubMed] [Google Scholar]
- 2.Ng BG, Buckingham KJ, Raymond K, et al. Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. Am J Hum Genet. 2013;92:632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng BG, Sosicka P, Agadi S, et al. SLC35A2-CDG: Functional characterization, expanded molecular, clinical, and biochemical phenotypes of 30 unreported Individuals. Hum Mutat. 2019;40:908–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dörre K, Olczak M, Wada Y, et al. A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): molecular basis, clinical phenotype, and therapeutic approach. J Inherit Metab Dis. 2015;38:931–940. [DOI] [PubMed] [Google Scholar]
- 5.Kimizu T, Takahashi Y, Oboshi T, et al. A case of early onset epileptic encephalopathy with de novo mutation in SLC35A2: Clinical features and treatment for epilepsy. Brain Dev. 2017;39:256–260. [DOI] [PubMed] [Google Scholar]
- 6.Westenfield K, Sarafoglou K, Speltz LC, et al. Mosaicism of the UDP-Galactose transporter SLC35A2 in a female causing a congenital disorder of glycosylation: a case report. BMC Med Genet. 2018;19:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vals M-A, Ashikov A, Ilves P, et al. Clinical, neuroradiological, and biochemical features of SLC35A2-CDG patients. J Inherit Metab Dis. 2019;42:553–564. [DOI] [PubMed] [Google Scholar]
- 8.Sim NS, Seo Y, Lim JS, et al. Brain somatic mutations in SLC35A2 cause intractable epilepsy with aberrant N-glycosylation. Neurol Genet. 2018;4:e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winawer MR, Griffin NG, Samanamud J, et al. Somatic SLC35A2 variants in the brain are associated with intractable neocortical epilepsy. Ann Neurol. 2018;83:1133–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morelle W, Potelle S, Witters P, et al. Galactose Supplementation in Patients With TMEM165-CDG Rescues the Glycosylation Defects. J Clin Endocrinol Metab. 2017;102:1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radenkovic S, Bird MJ, Emmerzaal TL, et al. The Metabolic Map into the Pathomechanism and Treatment of PGM1-CDG. Am J Hum Genet. 2019;104:835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Li X, Edmondson A, et al. Increased Clinical Sensitivity and Specificity of Plasma Protein N-Glycan Profiling for Diagnosing Congenital Disorders of Glycosylation by Use of Flow Injection-Electrospray Ionization-Quadrupole Time-of-Flight Mass Spectrometry. Clin Chem. 2019;65:653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Smet E, Rioux J-P, Ammann H, Déziel C, Quérin S. FSGS permeability factor-associated nephrotic syndrome: remission after oral galactose therapy. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc - Eur Ren Assoc. 2009;24:2938–2940. [DOI] [PubMed] [Google Scholar]
- 14.Achouitar S, Mohamed M, Gardeitchik T, et al. Nijmegen paediatric CDG rating scale: a novel tool to assess disease progression. J Inherit Metab Dis. 2011;34:923–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Witters P, Honzik T, Bauchart E, et al. Long-term follow-up in PMM2-CDG: are we ready to start treatment trials? Genet Med Off J Am Coll Med Genet. 2019;21:1181–1188. [DOI] [PubMed] [Google Scholar]
- 16.Witters P, Cassiman D, Morava E. Nutritional Therapies in Congenital Disorders of Glycosylation (CDG). Nutrients. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marquardt T, Lühn K, Srikrishna G, Freeze HH, Harms E, Vestweber D. Correction of leukocyte adhesion deficiency type II with oral fucose. Blood. 1999;94:3976–3985. [PubMed] [Google Scholar]
- 18.Baldassari S, Ribierre T, Marsan E, et al. Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol (Berl). Epub 2019. August 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yates TM, Suri M, Desurkar A, et al. SLC35A2-related congenital disorder of glycosylation: Defining the phenotype. Eur J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc. 2018;22:1095–1102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.