

Summary
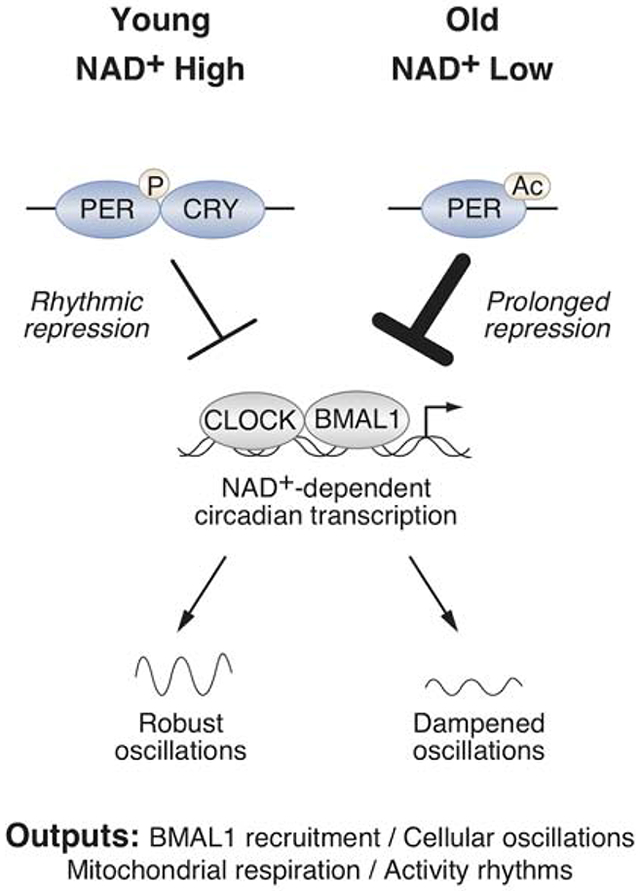
Disrupted sleep-wake and molecular circadian rhythms are a feature of aging associated with metabolic disease and reduced levels of NAD+, yet whether changes in nucleotide metabolism control circadian behavioral and genomic rhythms remains unknown. Here we reveal that supplementation with the NAD+ precursor nicotinamide riboside (NR) markedly reprograms metabolic and stress-response pathways that decline with aging through inhibition of the clock repressor PER2. NR enhances BMAL1 chromatin binding genome-wide through PER2K680 deacetylation, which in turn primes PER2 phosphorylation within a domain that controls nuclear transport and stability and which is mutated in human advanced sleep phase syndrome. In old mice, dampened BMAL1 chromatin binding, transcriptional oscillations, mitochondrial respiration rhythms, and late evening activity are restored by NAD+ repletion to youthful levels with NR. These results reveal effects of NAD+ on metabolism and the circadian system with aging through the spatiotemporal control of the molecular clock.
Graphical Abstract
Introduction
All mammals possess an internal circadian clock that couples sleep-wake and fasting-feeding cycles with the light-dark cycle (Bass and Lazar, 2016; Rey and Reddy, 2013). The core clock is a cell-autonomous transcription-translation feedback loop comprised of heterodimeric activators (CLOCK/BMAL1) that transcribe their own repressors (PERs/CRYs/REV-ERB) in a cycle that repeats itself every ~24 hrs. CLOCK/BMAL1 transcriptional activity during the light period is enabled through sequestration of nascent PER and CRY repressor proteins in the cytoplasm. During the dark period, PER and CRY translocate into the nucleus and associate into a macromolecular complex that inhibits CLOCK/BMAL1 and recruits regulators of rhythmic gene oscillation (Aryal et al., 2017; Hardin et al., 1990; Kume et al., 1999; Meyer et al., 2006; Yagita et al., 2002). Ubiquitin-mediated degradation of PER and CRY ends the repressive phase and permits a new cycle to begin (Eide et al., 2005). While this core clock cycle persists in the absence of external cues, the clock is also responsive to changes in environmental conditions through mechanisms that have not been fully elucidated (Bargiello and Young, 1984; Bargiello et al., 1984; Gallego and Virshup, 2007; Konopka and Benzer, 1971; Lowrey et al., 2000; Meng et al., 2008; Reddy et al., 1984; Toh et al., 2001; Zehring et al., 1984).
Mounting evidence has indicated that the circadian clock is intimately linked with metabolic homeostasis. In mice, genetic disruption of molecular clock leads to diet-induced obesity and metabolic disorders (Hirota et al., 2012; Marcheva et al., 2010; Paschos et al., 2012; Turek et al., 2005). Dysregulation of circadian rhythms in humans is also associated with obesity and increased risk for metabolic syndrome (Bass and Lazar, 2016; Rey and Reddy, 2013). The connection between clock disruption and metabolic disease can also be understood based on the observation that ~10% of the mammalian transcriptome is expressed rhythmically with enrichment in pathways involved in intermediary metabolism (Fang et al., 2014; Koike et al., 2012; Mure et al., 2018; Storch et al., 2002; Zhang et al., 2014). Conversely, changes in nutrient and high fat feeding lead to alterations in rhythms of locomotor activity, clock gene expression, and bioenergetics (Atger et al., 2015; Eckel-Mahan et al., 2013; Guan et al., 2018; Kohsaka et al., 2007; Sato et al., 2017; Tognini et al., 2017). However, how nutrients impact circadian function remains poorly understood.
Several lines of evidence indicate that decline in circadian processes is a hallmark of aging . Aging leads to increased daytime napping and a shift towards earlier hours of waking and sleep-onset in humans (Buysse et al., 1992; Ohayon et al., 2004; Roenneberg et al., 2007; Walch et al., 2016), as well as increased risk for developing obesity, diabetes, and metabolic syndrome (Folsom et al., 1993; Ford et al., 2002; Morley, 2008). At the molecular level, aging is associated with dampened oscillation of clock gene expression (Rosenberg et al., 1991; Sato et al., 2017; Solanas et al., 2017; Yamazaki et al., 2002), and genetic abrogation of Bmal1 in mice leads to a spectrum of premature aging disorders and reduces lifespan (Kondratov et al., 2006). Despite the link between aging and disrupted circadian rhythms, little is known regarding the underlying molecular mechanisms that mediate these phenotypes.
A clue to the mechanism underlying a decline in circadian rhythms and metabolism with age stems from the observation that the longevity-associated deacetylase SIRT1 interacts with the core clock complex, though how SIRT1 impacts the clock remain incompletely understood (Asher et al., 2008; Chang and Guarente, 2013; Nakahata et al., 2008; 2009; Ramsey et al., 2009). SIRT1 activity requires the sentinel metabolite nicotinamide adenine dinucleotide (NAD+) (Imai et al., 2000), which declines with age (Braidy et al., 2011; Camacho-Pereira et al., 2016; Massudi et al., 2012; Yoshino et al., 2011). Boosting NAD+ in rodents ameliorates age-associated insulin resistance, protects against noise-induced hearing loss, central brain injury, heart failure, diabetic neuropathy, enhances postpartum functions and extends lifespan (Brown et al., 2014; Cantó et al., 2012; Diguet et al., 2018; Ear et al., 2019; Massudi et al., 2012; Trammell et al., 2016; Vaur et al., 2017; Yoshino et al., 2011; Zhang et al., 2016), though whether these NAD+-dependent benefits improve circadian programming is unknown. Based on the observation that calorie restriction helps maintain hepatic circadian gene expression and the NAD+ metabolome (Sato et al., 2017), we hypothesized that reversing the decline in NAD+ with age would restore clock activity. Here we examine the effect of pharmacologic and genetic manipulations of NAD+ in the control of genome-wide circadian transcriptional rhythms and identify NAD+ as a mediator of PER2 nuclear extrusion and BMAL1 chromatin binding activity. We find that pharmacologic elevation of NAD+ failed to reprogram transcription in Bmal1 knockout mice, emphasizing a requirement for the molecular clock in NAD+ action. Moreover, provision of nicotinamide riboside (NR), an NAD+ precursor (Bieganowski and Brenner, 2004), in drinking water of old mice countered the decline in nighttime locomotor activity rhythms and enhanced molecular oscillations in mitochondrial respiration and transcription, revealing a metabolic feedback pathway to the clock important in health with aging.
Results
Supplementation with NR promotes global remodeling in the rhythmic expression of stress and metabolic gene networks through HSF1
To examine whether NAD+ affects genome-wide circadian transcription, we provided NR (~400 mg/kg/day) (Bieganowski and Brenner, 2004) in drinking water ad libitum to 4 month old mice for a period of 4 months, which resulted in a 4-5-fold increase in NAD+ levels in liver, soleus muscle, and hypothalamus without changing nicotinamide (Figures 1A, S1A–B). At the age of 8 months, livers were harvested for RNA-sequencing (RNA-seq) across six time points from ad lib-fed animals housed in 12:12 light:dark (LD) conditions. Analysis of rhythmic transcription using eJTK_Cycle revealed that 3,588 hepatic transcripts oscillated in animals provided water only, while NR supplementation reprogrammed ~50% of the oscillating hepatic transcriptome (Figure 1A). We identified three major categories of genes with altered diurnal expression following NR supplementation (Figure 1A), including those exhibiting (i) a loss (825 genes), (ii) a gain (753 genes), or (iii) a phase-shift in oscillation (512 genes) (FDR-adjusted p-values <0.1 or >0.9 for oscillating or non-oscillating genes, respectively) (Figure 1A–B, Table S1). We found that NR supplementation exerted distinct effects on gene expression rhythms at different phases of the light-dark cycle. Of the genes that lost oscillation following NR supplementation, 538 (65%) exhibited a peak in expression in either the early light (ZT2-ZT8) or the early dark (ZT16-ZT18) period in the H2O condition (Figure 1C). Interestingly, NR supplementation caused de novo oscillation of 505 (67%) of the gain genes within the same early light and early dark periods (Figure 1C). The early dark period also contained the largest group of phase-shifted genes; 120 (23%) genes newly peaked at ZT18 following NR supplementation, of which 79% phase-advanced from a peak at ZT0 in the H2O condition (Figure 1C). Gene ontology and promoter transcription factor (TF) motif analysis of genes that lost oscillations following NR revealed enrichment in pathways associated with translation, insulin signaling, and ketogenesis and in motifs that were bound by E26 transformation-specific (ETS) and FOXO (Forkhead) family TFs (Figure 2A). Transcripts that gained oscillation encompassed those involved in RNA splicing and nucleotide metabolism and were enriched with homeobox (HOX) and signal transducer and activator of transcription (STAT) motifs (Figure 2A). Phase-shifted genes were primarily involved in the spliceosome and enriched in the heat shock factor (HSF) motif (Figure 2A). We note that analyses in Figures 1–2 are also presented in Figure S1C using FDR-adjusted p-values of 0.05 and 0.95 for oscillating and non-oscillating genes, respectively, to identify genes for promoter motif and gene ontology analyses. Here, we reach the same conclusions as in Figures 1–2 with fewer genes (Figure S1C). These results implicate a broad range of collaborative TFs in mediating reprogramming of circadian transcription of stress and metabolic gene networks in liver following NAD+ elevation with NR.
Figure 1. Supplementation with the NAD+ precursor nicotinamide riboside elevates tissue NAD+ levels and promotes global remodeling of hepatic circadian transcriptome.
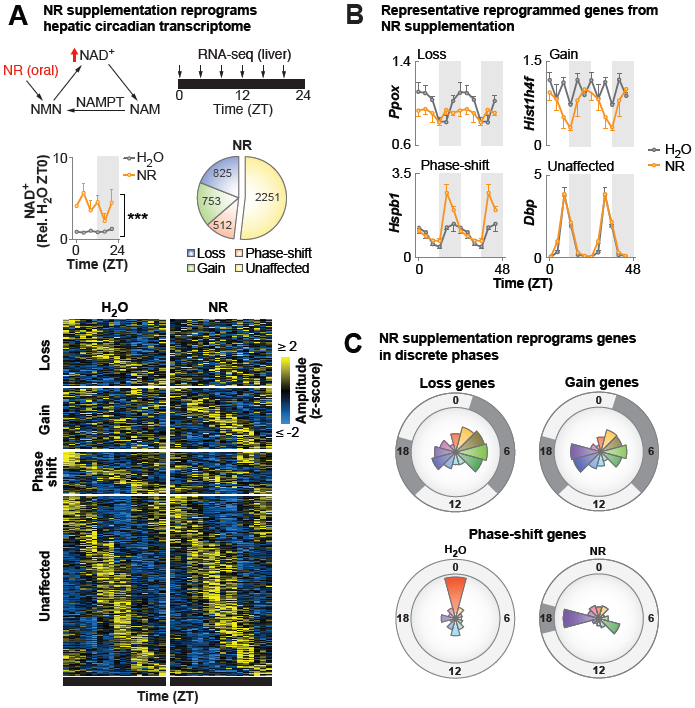
(A) Model depicting the NAD+ salvage pathway and nicotinamide riboside (NR) conversion to NAD+. Hepatic NAD+ quantified by HPLC in WT mice provided NR-supplemented or regular water for 4 months (***p≤0.001, ANOVA) (n=3/timepoint/group). eJTK_Cycle analyses following RNA-seq every 4 hrs across the 24 hr day identified transcripts that lost, gained, or phase-shifted >4 hrs oscillations following NR (FDR-adjusted p-value <0.1 and >0.9 for cycling and non-cycling genes, respectively). (B) Normalized counts (DESeq2) plotted for representative transcripts of genes that lost, gained, or phase-shifted (≥4 hrs) oscillations, or were unaffected, following NR. Data are double-plotted for better visualization. (C) For each NR-reprogrammed group, radial histograms show the number of genes whose oscillations peak within each 2-hr window, with the radius corresponding to 150 genes. Dark gray shading indicates phases with the largest number of affected genes. See also Figure S1 and Table S1.
Figure 2. NR drives rhythmic expression of stress and metabolic gene networks through the recruitment of HSF1.
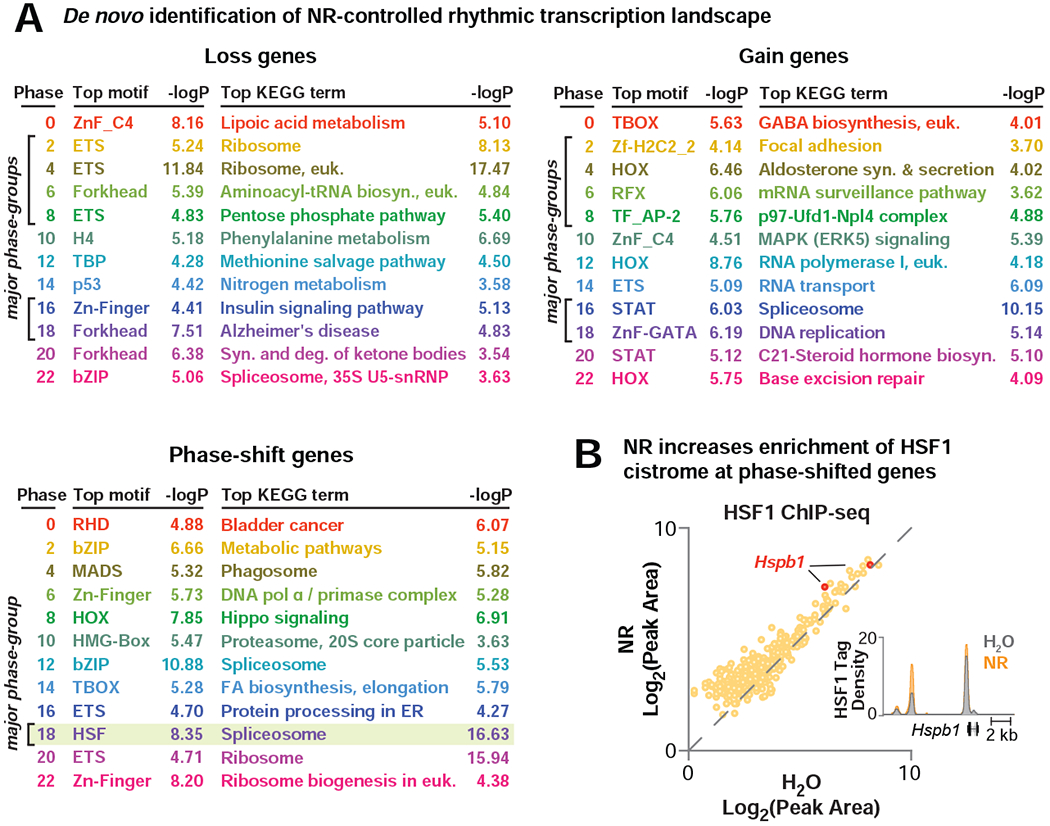
(A) The top enriched promoter-proximal TF motifs (± 2 kb of the transcription start site, JASPAR motif set) and KEGG ontology terms are shown for each NR-reprogrammed group. Each phase is uniquely colored and corresponds to colors in radial histograms in Figure 1C; phases with the largest number of affected genes are indicated by brackets. (B) ChIP-seq for HSF1 in liver (ZT8) following 2 months of NR-supplementation compared to H2O. Scatter plot depicts average log-transformed tag densities for HSF1 peaks that annotate to NR phase-shifted genes (n=2). UCSC genome browser image of average HSF1 tag density at the Hspb1 locus in liver during H2O or NR supplementation. Maximum track height is indicated on the y-axis, and gene orientation is indicated. See also Figure S1.
The identification of HSF1 as an NAD+-responsive factor highly enriched in phase-shifted genes prompted us to test whether HSF1 chromatin binding was induced following NR treatment. We next performed HSF1 ChIP-sequencing in liver of NR-treated mice at ZT8 and found that NR induced a ~27% increase in HSF1 chromatin binding proximal to genes phase-shifted by NR (p=7.8E-9), which included HSF1 targets such as Hspb1 (Figure 2B) (Mahat et al., 2016). The increase in HSF1 binding occurred independent of changes in body temperature (Figure S1D). Together, these results indicate that HSF1 participates in the phase-shifting response to NR.
NAD+ is synthesized de novo from tryptophan and from three vitamin precursors made available by digestion of the NAD+ metabolomes in foods (Bogan and Brenner, 2008). Because NAD+-consuming enzymes degrade NAD+, inhibition of the nicotinamide salvage pathway by Nampt inactivation in combination with fasting is a powerful way to depress NAD+-dependent processes (Yang and Sauve, 2016). We therefore next tested whether depletion of NAD+ also impacts circadian transcriptional rhythms by performing RNA-seq in 18-hr fasted liver-specific Nampt KO mice (L-Nampt−/−), which have a 30-60% reduction in hepatic NAD+ (Figure 3A). We observed reprogramming of ~50% of the oscillating transcripts in the liver of fasted L-Nampt−/− mice relative to the fasted control (Figure 3A, Table S2). Importantly, L-Nampt−/− mice displayed reduced expression of genes that were elevated by NR, and increased expression of those that were down-regulated by NR (binomial test, p=6.387E-11) (Figure 3B). Together with the finding that NR supplementation reprogrammed circadian transcription of stress and metabolic gene networks (Figure 1A), these data indicate that NAD+ is necessary and sufficient for circadian transcriptional regulation.
Figure 3. Abrogation of NAD+ biosynthesis induces genome-wide disruption of the hepatic circadian transcriptome.
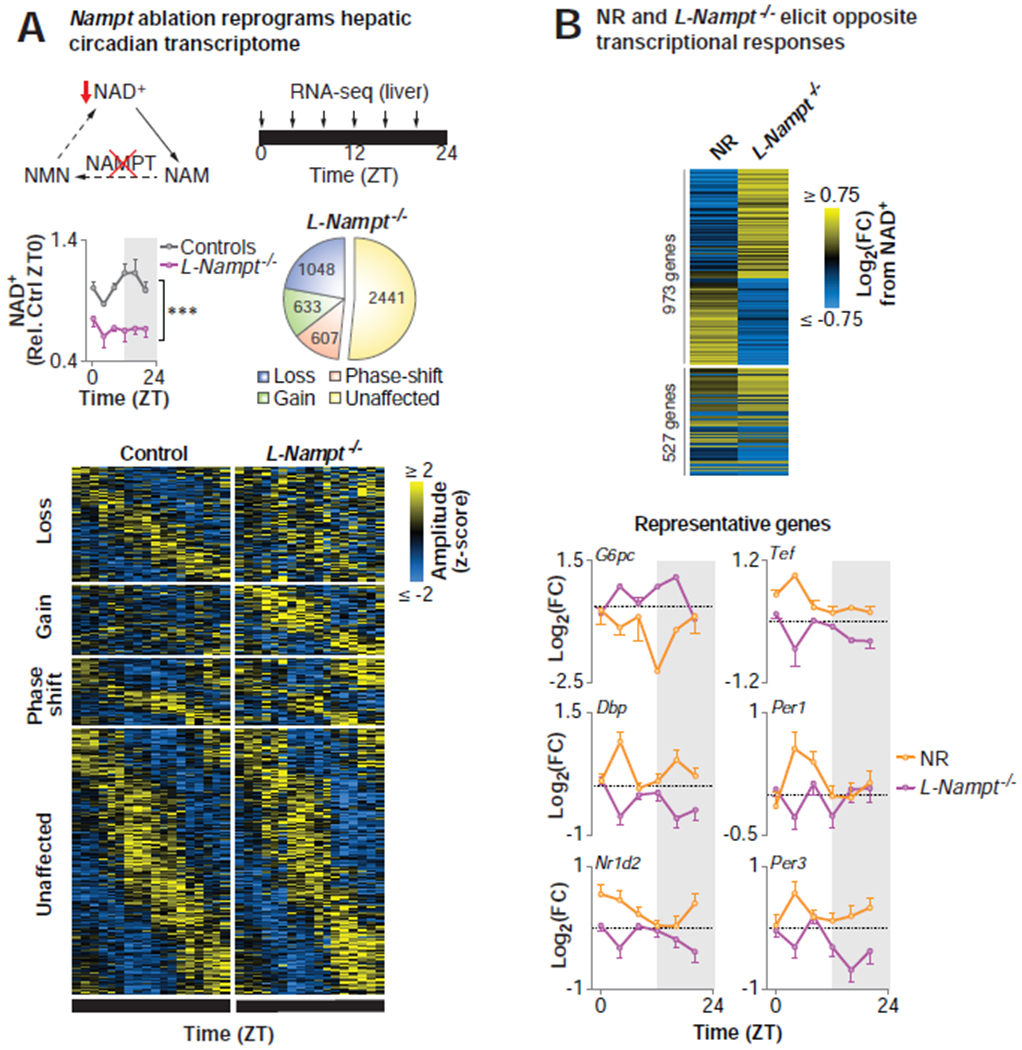
(A) Model depicting the NAD+ salvage pathway, highlighting the contribution of NAMPT to NAD+ biosynthesis. Hepatic NAD+ quantified by HPLC in 4-6 mo old liver-specific Nampt knockout (L-Nampt−/−) and control (Alb-Cre and Namptfx/fx) mice following an 18-hr pre-collection fast (***p<0.001, ANOVA) (n=2-6/timepoint/group). eJTK_Cycle analyses following RNA-seq every 4 hrs across the 24 hr day identified transcripts that lost, gained, or phase-shifted (≥4 hrs) oscillations following Nampt ablation (FDR-adjusted p-value <0.1 and >0.9 for cycling and non-cycling genes, respectively) (n=2-6/timepoint/group). (B) Heat map depicting the maximal diurnal differential transcriptional responses to NR supplementation and NAD+ depletion in L-Nampt−/− mice for the 1500 most up- and down-regulated genes. Log-transformed fold change at each timepoint from NR relative to H2O (orange) and L-Nampt−/− relative to control (purple) for representative genes (n=2-6/timepoint/group). See also Figure S1 and Table S2.
Requirement for BMAL1 in chromatin remodeling and HSF1 recruitment following pharmacologic NAD+ elevation
Given that increased NAD+ by NR supplementation altered the landscape of rhythmic transcription in liver (Figure 1A), we next sought to examine whether BMAL1 was necessary for modulation of rhythmic transcription by NAD+ (Figure 4A). We compared 4-month old wild-type (WT) and Bmal1 knockout (Bmal1−/−) mice following administration of the NAD+ precursor nicotinamide mononucleotide (NMN) (Figure S1E). NMN administration allowed us to study the effect of elevation of NAD+ at the zenith of BMAL1 DNA binding, which corresponds with the lowest point of NAD+ oscillations (ZT6) (Koike et al., 2012). In WT mice, NMN supplementation led to differential expression of 299 genes (FDR-adjusted p-value <0.1), including increased expression of the clock repressors Per1 and Cry2 (Figure 4B, Table S3), indicating that NAD+ exerts transcriptional control in part through induction of the clock activators CLOCK/BMAL1. Surprisingly, ~95% of the genes differentially expressed in WT liver were no longer differentially expressed in the Bmal1 mutants, suggesting a primary role for BMAL1 in regulating gene expression in response to NAD+ (Figure 4B, Table S3). We next tested whether BMAL1 chromatin binding was affected by NMN supplementation. Indeed, ChIP-seq for BMAL1 showed a ~25% increase in binding density following NMN at ZT6 (p=9.5E-47), including at canonical BMAL1 target genes Per1 and Cry2 (Figure 4C, Table S3). To determine whether BMAL1 was also necessary for the induction of HSF1 during NR supplementation, we performed HSF1 ChIP-seq in both WT and Bmal1−/− mice supplemented with NR for 2 months at ZT8. Strikingly, we observed that the increase in HSF1 chromatin binding at NR-phase-shifted genes was abrogated in Bmal1 mutants (Figure 4D). Given previous evidence that BMAL1 modulates chromatin structure in liver (Menet et al., 2014), we reasoned that induction of BMAL1 activity from increased NAD+ may enhance chromatin accessibility. Indeed, the assay for transposase accessible chromatin-sequencing (ATAC-seq) revealed decreased nucleosome-free signal at HSF1 binding sites at phase-shifted genes in Bmal1−/− mice, suggesting a role for BMAL1 in establishing HSF1-accessible chromatin binding sites (Figure 4E). Together, these findings show that NAD+ drives circadian transcription by stabilizing BMAL1 chromatin binding and facilitating the recruitment of collaborative TFs such as HSF1.
Figure 4. Requirement for BMAL1 in transcriptional reprogramming and HSF1 recruitment following pharmacologic NAD+ elevation.
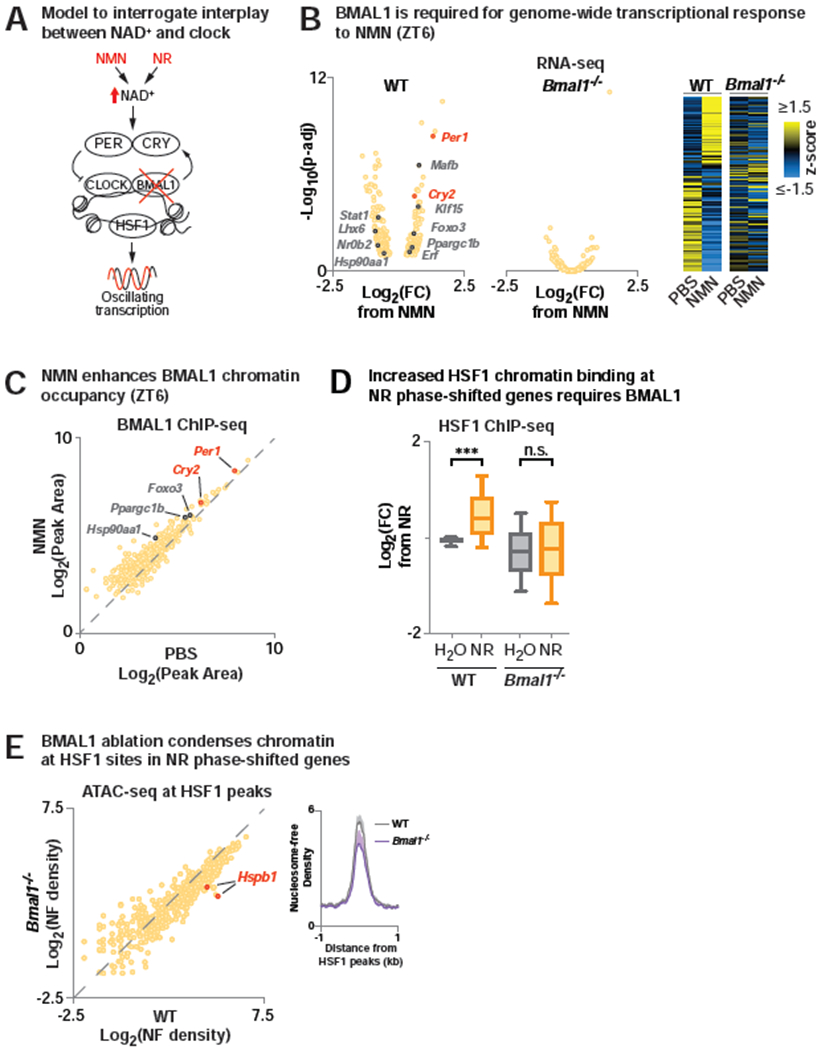
(A) Model to interrogate the role of BMAL1 in NAD+-dependent transcriptional oscillations. (B) RNA-seq in liver (ZT6) in WT and Bmal1−/− mice injected with NMN or PBS at ZT2. (Left) Log-transformed fold-change (NMN/PBS) and FDR-adjusted p-values shown for genes differentially expressed by NMN in WT liver. (Right) Heatmap depicting differentially expressed genes from NMN in WT and Bmal1−/− liver. RNA-seq data is transformed by row z-score across treatments and genotypes and sorted by fold change from NMN in WT (FDR-adjusted p-value <0.1) (n=2-3). (C) BMAL1 ChIP-seq in liver (ZT6) following NMN or PBS injection at ZT2. Scatter plot of average log-transformed tag-densities for BMAL1 peaks that annotate to genes differentially expressed by NMN in WT liver (n=2). (D) ChIP-seq for HSF1 in WT and Bmal1−/− liver at ZT8 following 2 months of NR supplementation compared to H2O. Log-transformed fold-change in average HSF1 tag-density from H2O-treated WT mice for peaks that annotate to NR phase-shifted genes (n=2) (*** p < 0.001). (E) ATAC-seq in liver (ZT8) of WT and Bmal1−/− mice. Average nucleosome free reads are quantified at HSF1 peaks that annotate to NR phase-shifted genes. (n=2-4). See also Figure S1 and Table S3.
NAD+ and SIRT1 regulate nuclear localization of the clock repressor complex
NAD+ is an obligate substrate of the major nuclear class III deacetylase SIRT1 (Imai et al., 2000) which is also found in complex with CLOCK/BMAL1 (Asher et al., 2008; Nakahata et al., 2008); yet the role of SIRT1 in genome-wide transcription during NR supplementation has not been determined. To evaluate whether NAD+-induced genome-wide reprogramming requires SIRT1, we performed RNA- and BMAL1 ChIP-seq following elevation of NAD+ through the acute administration of NMN in liver-specific Sirt1 KO (L-Sirt1−/−) and control mice (Figure 5A). NMN was injected at ZT2 and tissues collected at ZT6 in order to elevate NAD+ at the time of day when it is lowest (Figure 3A). We detected 277 genes that were significantly differentially expressed by NMN in the control mice (FDR-adjusted p-value <0.1), with 140 genes up-regulated (Log2FC>0, red dots) and 137 genes down-regulated (Log2FC<0, blue dots) by NMN (Figure 5A). In L-Sirt1−/− liver, 233 (84%) of the NMN-responsive genes were no longer significantly differentially expressed and 260 (94%) had a smaller absolute fold change in expression from NMN (binomial test, p=2.2E-16) (Figure 5A), indicating that SIRT1 is necessary for the response to NAD+. Similarly, loss of SIRT1 prevented induction of BMAL1 chromatin binding in response to NMN (Figure 5A, Table S3) (p<2.15E-140). Together, these data indicate that NAD+ mediates global transcription through both SIRT1 and BMAL1.
Figure 5. Pharmacologic and genetic manipulations of NAD+ and SIRT1 mediate nuclear localization of the clock repressor complex through PER2 deacetylation to control transcription.
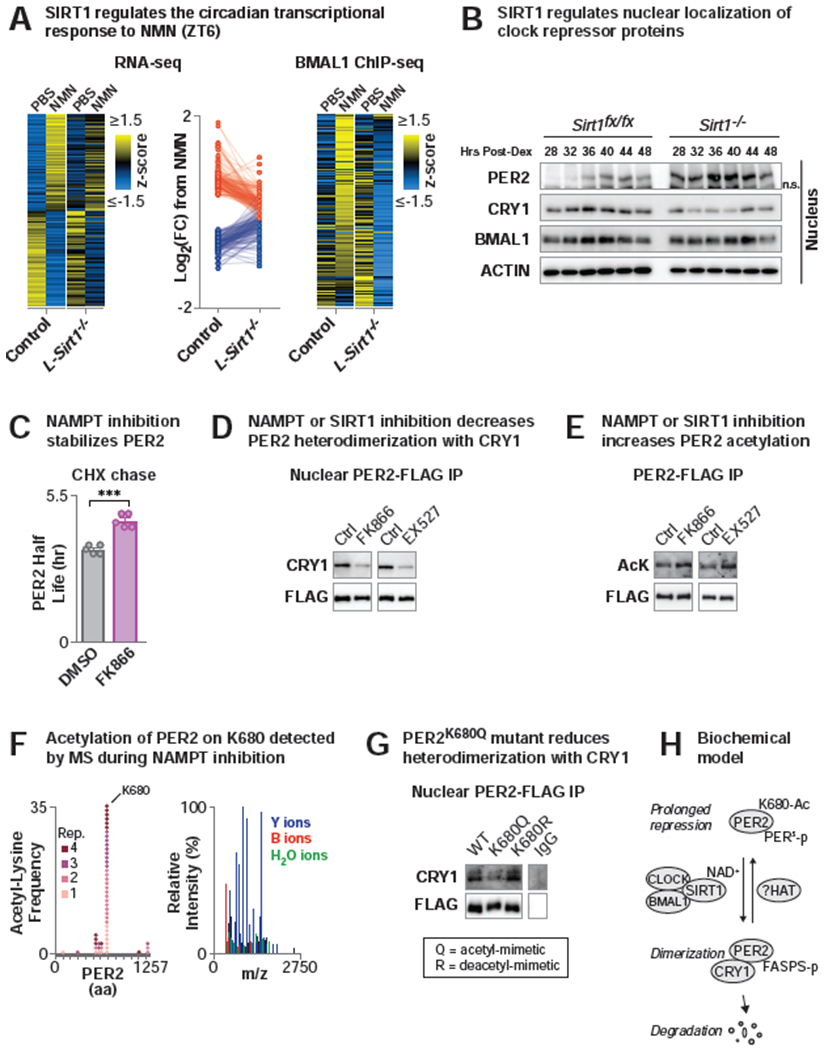
(A) (Left and middle) RNA-seq in liver (ZT6) in control (Sirt1fx/fx) and liver-specific Sirt1 knockout (L-Sirt1−/−) (Alb-Cre;Sirt−/−) mice following NMN or PBS injection at ZT2. (Left) Data for heatmap was transformed by row z-score across treatments and genotypes and sorted by fold change from NMN in control (n=3-4). (Middle) Log-transformed fold-change (NMN/PBS) in gene expression in control and L-Sirt1−/− mice shown for genes differentially expressed by NMN in control liver (FDR-adjusted p-value <0.1). Red and blue dots indicate genes that are induced or repressed, respectively, by NMN in control liver (n=3-4). (Right) BMAL1 ChIP-seq in control and L-Sirt1−/− liver (ZT6) following NMN or PBS injection at ZT2. Heatmap depicts row z-score transformed average tag-densities for BMAL1 peaks that annotate to genes differentially expressed by NMN in WT liver (n=2). (B) Clock repressor protein levels in nuclear fractions from dexamethasone-synchronized Sirt1fx/fx and Sirt1−/− MEFs. “n.s.” indicates non-specific band. (C) PER2 half-life (hr) following cycloheximide (CHX) addition to DMSO- or FK866-treated MEFs expressing the PER2::LUCIFERASE protein from the endogenous Per2 locus. (***p<0.01). (D) CRY1 western blotting following PER2-FLAG immunoprecipitation from nuclei of HEK293 cells treated with either FK866 (10 nM, 24 hr) or EX527 (1μM, 24 hr). (E) Acetyl-lysine and FLAG Western blotting of immunoprecipitated FLAG-PER2 following FK866 or EX527 treatment in HEK293 cells. (F) IP-LC-MS/MS analysis of PER2 immunoprecipitated from FK866-treated HEK293 cells. Frequency of unique spectra containing acetyl-lysine modifications shown across 4 replicates and representative spectra for PER2 acetyl K680. (G) Western blotting of CRY1 following PER2-FLAG immunoprecipitation from nuclei of HEK293 cells expressing WT, acetyl-mimetic (K680Q), or de-acetyl-mimetic (K680R) PER2 mutants. The IgG sample was run on the same gel as the other samples shown. (H) Biochemical model demonstrating mechanism by which NAD+ alleviates prolonged repression of BMAL1 by nuclear PER2 monomer: SIRT1 uses NAD+ to deacetylate PER2 on lysine 680, driving dimerization with CRY1, PER2 phosphorylation at the FASPS site, and ultimately PER2 degradation. See also Figures S2 and S3 and Tables S3 and S4.
While the transcriptional co-activator PGC1α is activated by SIRT1 and has been reported to induce Bmal1 RNA expression (Foteinou et al., 2018; Liu et al., 2007; Rodgers et al., 2005), we did not observe changes in Bmal1 RNA with either NMN or in liver-specific Sirt1 knockout animals (Table S3); however, there were marked increases in the RNA expression of the clock repressors Per1 and Cry2 in response to NAD+ (Figures 1A, 4B, Table S3). These observations suggest that NAD+ influences BMAL1 transcriptional activity rather than expression of Bmal1 through PGC1α. SIRT1 inactivation has also been reported to increase H3K9/14 acetylation at the BMAL1-binding site at the Dbp locus (Nakahata et al., 2008). However, ChIP-sequencing for histone acetyl marks that are SIRT1-dependent (H3K9Ac) and SIRT1-independent (H3K27Ac) following NR supplementation revealed increased acetylation of both marks at the Dbp locus and genome-wide (Figure S2A). Specifically, NR increased the average H3K9Ac tag density for all peaks identified genome-wide by 31% (p<3.7E-264) and specifically increased tag density for peaks at the Dbp locus by 29% (p<0.00043) (Figure S2A). Similarly, NR increased the average H3K27Ac tag-density at all peaks identified across the genome by 26% (p<1.4E-139) and at peaks at the Dbp locus by 22% (p<0.0015) (Figure S2A). Together, increased histone acetylation and SIRT1-dependent and independent sites following NR supplementation suggests that these effects are not directly dependent on SIRT1. Lastly, we were unable to observe differences in BMAL1 acetylation using an anti-acetyl-BMAL1 antibody (Millipore AB15396) (data not shown).
SIRT1 has been reported to promote the deacetylation and degradation of PER2 (Asher et al., 2008), although the molecular mechanism whereby PER2 acetylation affects PER2 stability remain unknown. Given that NAD+ did not appear to directly affect transcription of Bmal1 or acetylation of histones, we sought to examine whether NAD+ regulates SIRT1-dependent recruitment of BMAL1 to chromatin through control of the PER/CRY repressor complex. During the light period, cytoplasmic sequestration of clock repressors PER and CRY enables sustained CLOCK/BMAL1 activation of transcription. During the dark period, PER and CRY proteins translocate into the nucleus where they inhibit CLOCK/BMAL1 (Aryal et al., 2017; Hardin et al., 1990; Koike et al., 2012; Kume et al., 1999; Meyer et al., 2006; Michael et al., 2017; Yagita et al., 2002; Ye et al., 2014). Phosphorylation and ubiquitin-mediated degradation of PER and CRY then inhibits the repressive phase (Eide et al., 2005). Since PER and CRY proteins undergo nuclear transport, association, and stability (Gallego and Virshup, 2007; Meng et al., 2008), we first sought to test whether NAD+/SIRT1 impacts CRY and PER subcellular distribution across the day. Surprisingly, in Sirt1 knockout cells, we found markedly increased nuclear accumulation of PER2 compared to controls, whereas we observed reduced levels of nuclear PER1, CRY1, and CRY2 (Figures 5B, S2B). Further, PER2 exhibited increased stability upon cycloheximide chase following NAD+ depletion with the NAMPT inhibitor FK866 (Figures 5C). Finally, PER2-FLAG displayed reduced binding to CRY1 within the nucleus following either NAMPT inhibition with FK866 or SIRT1 inhibition with EX527 (Figure 5D). These findings indicate that NAD+-SIRT1 controls the spatiotemporal distribution, abundance, and oligomerization of core clock repressor proteins.
We next hypothesized that NAD+-SIRT1 may control changes in PER2 localization and stability through posttranslational modification (PTM) of PER2. Indeed, NAMPT and SIRT1 inhibition with FK866 and EX527 treatment, respectively, increased acetylation of FLAG-PER2 (Figure 5E), consistent with the prior report that loss of SIRT1 increased PER2 acetylation (Asher et al., 2008). To identify the specific acetylated residues on PER2, we subjected PER2 to LC-MS/MS under conditions of NAD+ depletion through FK866 treatment and identified hyperacetylation of PER2 at lysine 680 (Figure 5F, Table S4). Mutation of PER2K680 to the acetyl-mimetic PER2K680Q revealed decreased association with the repressor CRY1 within nucleus relative to WT and the deacetyl-mimetic PER2K680R (Figure 5G), consistent with a role for NAD+ and SIRT1 in the association of PER2 with CRY1 (Figure 5D). Moreover, the primary amino acid sequence proximate to PER2K680 is similar to that of Drosophila PER and is known to mediate intermolecular complex formation with CRY1 (Hennig et al., 2009; Yildiz et al., 2005). These data indicate that acetylation modulates the nucleocytoplasmic transport of the PER2-CRY1 complex.
In addition to acetylation of PER2K680, we observed regulation of PER2 phosphorylation following NAMPT inhibition with FK866. Indeed, acetylation of PER2K680 occurs near the orthologous sites of impaired phosphorylation in human (PER2S659) and Drosophila (PER2S693) variants that cause circadian and sleep disruption (Bargiello and Young, 1984; Bargiello et al., 1984; Reddy et al., 1984; Toh et al., 2001; Top et al., 2018; Zehring et al., 1984). We identified numerous sites of PER2 phosphorylation in NAD+-depleted conditions (Figure S3A–B), including at PER2S693, which is orthologous to the PERS site in Drosophila that regulates its stability and activity (Garbe et al., 2013; Toh et al., 2001; Vanselow et al., 2006) and near the β-TRCP binding domain, which is necessary for ubiquitin-mediated degradation of PER2 (Figure S3A–B) (Zhou et al., 2015). We next sought to test whether acetylation of PER2K680 is critical in priming phosphorylation of PER2S659, which is mutated in familial advanced sleep-phase syndrome (FASPS) in humans (Toh et al., 2001). Immunoprecipitation of the acetyl-mimetic mutant PER2K680Q and immunoblotting for phosphorylation at PER2S659 revealed decreased phosphorylation at this site relative to WT PER2 and the deacetyl-mimetic PER2K680R mutant (Figure S3C). Thus, NAD+/SIRT1 controls PER2 acetylation and coordinates PER2 phosphorylation through a pathway similar to that involved in human sleep/wake regulation (Figure 5H).
NR enhances circadian transcription and evening locomotor activity in old mice
Aging is associated with decreased circadian amplitude and robustness in both mice and humans (Bass and Lazar, 2016; Campisi et al., 2019; Rey and Reddy, 2013). Further, Bmal1 mutant mice display premature aging and have low NAD+ levels (Kondratov et al., 2006; Ramsey et al., 2009). NAD+ levels decline with age, and provision of both NR and NMN have been shown to ameliorate age-associated insulin resistance, protect against noise-induced hearing loss, central brain injury, heart failure, diabetic neuropathy, enhance postpartum functions and extend lifespan in rodents (Brown et al., 2014; Cantó et al., 2012; Diguet et al., 2018; Ear et al., 2019; Massudi et al., 2012; Trammell et al., 2016; Vaur et al., 2017; Yoshino et al., 2011; Zhang et al., 2016). We therefore sought to test the hypothesis that restoration of NAD+ levels in old mice might counteract the age-related decline in circadian rhythms (Figure 6A). We chose to examine “young” mice at 10 months of age and “old” mice at 22 months of age because NAD+ declines in mice older than 12 months of age and there is no change in weight between mice at these ages (Figure S4A) (Braidy et al., 2011; Camacho-Pereira et al., 2016; Turturro et al., 1999). We first compared levels of the NAD+-SIRT1 target clock protein PER2 in young and old mice, in addition to determining the genome-wide binding of BMAL1 in liver from the same populations of young and old mice (Figure 6B–C). We observed an increase in PER2 in old mice, consistent with enhanced repression (Figure 6B). Additionally, old mice displayed reduced amplitude of BMAL1 binding across the day with significantly reduced binding during the light period (Figures 6C, S4B, Table S5). Remarkably, NR supplementation for 6 months in old mice (Figure S4A) led to a dramatic reduction in PER2 (Figure 6B) and a corresponding induction in BMAL1 DNA binding (p=1.4E-237), particularly at the zenith of BMAL1 activity (ZT8) (Koike et al., 2012) (Figures 6C, S4B, Table S5). Gene ontology analysis of genes proximal to BMAL1 peaks in NR- but not H2O-treated old mice revealed enrichment in pathways associated with metabolism and longevity and in genes with protein structures containing the NAD+-binding Rossman-fold (Figure S4C). Of note, the age-dependent decline in BMAL1 chromatin occupancy was independent of changes in total BMAL1 protein levels (Figure 6B). Together, our findings indicate that decreased NAD+ in old mice causes increased PER2 that diminishes circadian rhythms.
Figure 6. NR enhances circadian transcription and evening locomotor activity in old mice.
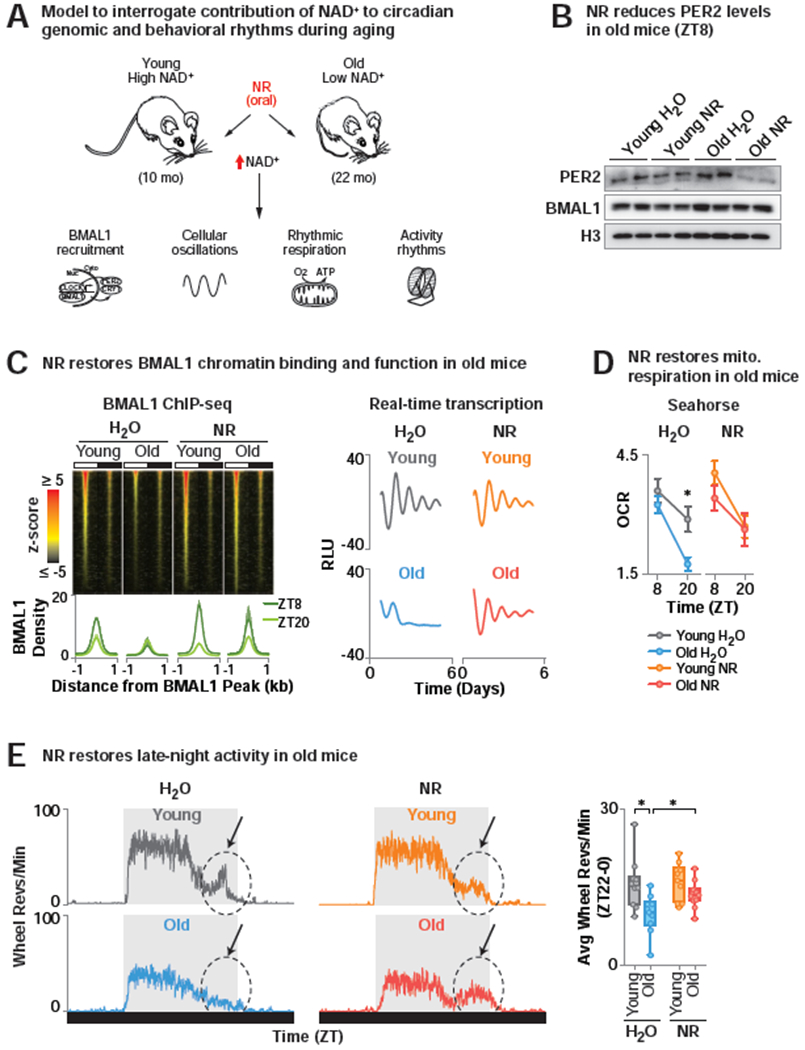
(A) Model to interrogate the contribution of NAD+ to circadian genomic and behavioral rhythmic phenotypes during aging. (B) PER2 and BMAL1 levels in liver (ZT8) of 10 month (“young”) and 22 month (“old”) WT mice treated either with NR-supplemented or regular drinking water for 6 months. (C) (Top) BMAL1 ChIP-seq in liver (ZT8 and ZT20). Z-score normalized average tag-densities for genomic regions corresponding to the center of each BMAL1 peak identified in old mice treated with NR ± 1 kb and sorted by size (n=2) (Bottom) Average BMAL1 tag-density and standard error for both replicates within each group. (right) Average background-subtracted PER2::LUC oscillations from soleus of young and old water- and NR-treated mice transgenic for the Per2::Luciferase reporter monitored ex vivo in a lumicycle (n=3). (D) Oxygen consumption rates (OCR) of mitochondria isolated from young and old, water- and NR-treated liver (ZT8 and ZT20) by Seahorse Bioanalyzer. FCCP-treated respiration with palmitate relative to basal (*p<0.05) (n=4-5). (E) (Left) Representative average 24 hr profile of ad lib wheel running activity of young and old water- and NR-treated WT mice. Arrow denotes ‘late-night activity’. (Right) Quantification of average wheel revolutions/min in the late night (ZT22-0) (n=8-10) (*p<0.05). See also Figure S4 and Table S5.
Since NAD+ regulates transcription by activating BMAL1 in young mice, we hypothesized that age-associated decline in BMAL1 chromatin binding and the correspondingly low NAD+ was associated with alterations in circadian transcription. To test this, we first measured BMAL1 transcriptional activity using live-cell luminometry recording in old (22 mo) genetic reporter mice expressing the Per2::Luciferase(Luc) transcriptional reporter compared to young (10 mo) mice and under NAD+ repletion with NR (Yoo et al., 2005) (Figure 6C). We observed a 90% decrease in amplitude of PER2-LUC oscillations in soleus of old mice (p<0.03) that was restored in old NR-treated mice to within 40% of the amplitude in young mice (Figure 6C). To next determine whether decreased NAD+ with age altered circadian transcriptional reprogramming by collaborative TFs, we performed ChIP-seq for HSF1 (Figures 2, 4D–E). Similar to the reduced genome-wide DNA binding by BMAL1 in old mice, HSF1 also displayed reduced chromatin binding during the daytime in old mice which was restored following NR supplementation (Figure S4D). Aging is also associated with decline in mitochondrial function (Petersen et al., 2003), a clock-controlled process (Peek et al., 2013; Turek et al., 2005). Indeed, “metabolism” is a top GO term associated with decreased BMAL1 chromatin binding in old age (Figure S4C). Thus, we analyzed respiration in isolated mitochondria harvested from liver of young and old mice provided chronic supplementation with NR. Intriguingly, we observed reduced oxygen consumption in old mice in the presence of palmitic acid specifically at ZT20, which was rescued by NR supplementation (Figure 6D). Thus, restoration of NAD+ levels in old age with NR impacts BMAL1 chromatin binding, stress-responsive transcription and a circadian decline in fat oxidation.
In addition to cell and molecular signatures of reduced circadian function, a behavioral hallmark of aging in humans is disruption of circadian sleep-wake rhythms especially in the evening (Buysse et al., 1992; Ohayon et al., 2004; Roenneberg et al., 2007; Walch et al., 2016). We therefore next assessed the impact of aging and NAD+ elevation by NR supplementation on 24-hr rest-activity patterns of old mice. While old mice with low NAD+ displayed significantly diminished late-night activity between ZT22 and ZT0 (a period that corresponds to human late afternoon), NR supplementation restored this locomotor activity (denoted by arrow in Figure 6E). Similarly, animals deficient in NAD+ (CAGCreER; Namptfx/+) displayed reduced late-night locomotor activity (Figure S4E). Thus, countering the decline in NAD+ in old age leads to both restoration of youthful patterns of transcription and circadian physical activity.
Discussion
NAD+ coordination of transcription and behavior through the molecular clock
Here we elucidated the mechanism of transcriptional remodeling by NAD+ and defined a role for the core molecular clock in this process. Studies in yeast were the first to identify NAD+ as a cofactor for histone-modifying enzymes involved in the response to caloric restriction, and subsequent work implicated an intersection between the NAD+ pathway and the core clock. Further, in both aging and metabolic stress, there is decline in NAD+, yet how changes in NAD+ might modulate activity of core clock transcription factors has remained incompletely known. Given that pharmacologic NAD+ elevation improves healthspan (Brown et al., 2014; Cantó et al., 2012; Ear et al., 2019; Massudi et al., 2012; Trammell et al., 2016; Vaur et al., 2017; Yoshino et al., 2011; Zhang et al., 2016), the potential remains that some of the benefits of NAD+ may involve circadian reprogramming. Results presented here implicate a role for NAD+ in remodeling the circadian transcriptome through deacetylation of the core clock repressor PER2, which in turn promotes the activity of CLOCK/BMAL1. Our studies clarify the mechanisms underlying SIRT1-mediated regulation of the clock, for which discrepant mechanisms were previously proposed (Asher et al., 2008; Nakahata et al., 2008). Our identification of acetylation on PER2K680 by mass spectrometry and implication of this site in nuclear PER2/CRY1 heterodimer formation and CK1-dependent phosphorylation supports findings that proposed a role for SIRT1 in destabilizing PER2 (Asher et al., 2008). Further, our discovery that NR increases acetylation of both H3K9 and H3K27 genome-wide and at the Dbp locus suggests that changes in histone acetylation may be an indirect effect of SIRT1 activity.
Time of day as a variable in the metabolic control of epigenetics
Increasing evidence suggests that metabolic cues provide feedback to regulate epigenetic enzymes important in gene expression. For example, metabolites produced through the TCA cycle, such as alpha-ketoglutarate and 2-hydroxyglutarate, are modulators of the jumonji family of demethylases (Dang et al., 2009; Losman et al., 2013; Tsukada et al., 2006). However, how NAD+ affects global transcription has remained an open question. Our observation that NR supplementation exerts broad effects on rhythmic gene transcription in liver and our identification of NAD+ as a regulator of SIRT1-mediated deacetylation of PER2 establishes a mechanism by which NAD+ regulates the clock. It will be interesting to further probe the effects of NAD+ on clock repressor complex formation and nuclear-cytoplasmic shuttling. Supplementation with NR revealed a collaboration with HSF1 in a phase-specific set of remodeled genes, consistent with transcriptomic evidence that HSF1 induces transcription of metabolic genes (Mahat et al., 2016). Additionally, recent genomic analyses in liver following high-fat feeding suggest that macronutrients reprogram transcription through the recruitment of collaborative TFs (SREBP/PPARα) (Guan et al., 2018). In this regard, core clock proteins have been shown to shape the chromatin landscape through direct effects on nucleosome remodeling (Menet et al., 2014), through long-range chromatin interactions (Aguilar-Arnal et al., 2013; Kim et al., 2018; Mermet et al., 2018; Sobel et al., 2017), and through recruitment of regulatory cofactors (Duong et al., 2011). One possibility is that local changes in chromatin accessibility may arise in response to tethering of SIRT1 to the CLOCK/BMAL1/PER2 complex. Such interactions may affect phase-specific recruitment of collaborative TFs, many of which are SIRT1 targets (Purushotham et al., 2009; Walker et al., 2010; Westerheide et al., 2009). Conversely, reciprocal regulation of clock oscillations by temperature may involve similar TF interactions (Buhr et al., 2010; Reinke et al., 2008; Saini et al., 2012). It is tempting to speculate that cells undergoing shifts in NAD+ metabolism may exhibit alterations in rhythmic transcription, such as in immune cells with activation and genotoxic stress.
Epigenetic regulation of circadian behavioral and metabolic cycles in aging and disease
Disruption of circadian patterns of sleep/wake activity, as occurs in shiftwork, neurodegenerative disorders, and aging, has been tied to metabolic disorders. Our results show that NAD+ acts through SIRT1 to deacetylate PER2 and regulate phosphorylation of a CK1 site on PER2 that is mutated in human familial advanced sleep phase (Toh et al., 2001). Whether NAD+ repletion might enhance metabolism through the alignment of feeding and sleep time remains to be established. One therapeutic implication may be that targeting SIRT1 activity via NAD+ (Ondracek et al., 2017) may ameliorate age-related decline in behavior and metabolism . At the cellular level, modulating circadian function through targeting PER2K680 may provide a therapeutic avenue in disease contexts including sleep loss associated with neurodegenerative and shiftwork disorders. Enhancing the amplitude of daily rhythms in oxidative metabolism may be one mechanism by which NAD+ repletion improves resistance to metabolic stress with age.
STAR METHODS
Resource Availability
Lead Contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joseph Bass (j-bass@northwestern.edu).
Materials Availability
No new materials or reagents were generated in this study.
Data and Code Availability
RNA-seq, ChIP-seq, and ATAC-seq data sets have been deposited in NCBI’s Gene Expression Omnibus database (GSE133989).
Experimental Model and Subject Details
Mice
All animal procedures were in accordance with guidelines of the Institutional Animal Care and Use Committee, and all mice were housed at 23-25°C in the Center for Comparative Medicine at Northwestern University and maintained under 12hr light:12hr dark (LD) cycles with ad lib access to regular chow and water unless otherwise indicated. Mice were maintained on Teklad chow 7912 containing 100 mg niacin / kg diet for the duration of this study in order to provide vitamin B3 in excess of daily nutritional requirements of mice (15 mg niacin / kg diet) (National Research Council (US) Subcommittee on Laboratory Animal Nutrition, 1995). 4 and 16 month old wild-type C57BL/6NCrl male mice were obtained from Charles River or the NIA colony at Charles River, respectively. Bmal1−/− and Per2::Luciferase mice were a gift from J. Takahashi (UT Southwestern). Nampfx/fx and Sirt1fx/fx mice were a gift from S. Imai (Washington University in St. Louis). Alb-Cre and CAGCRE-ER mice were purchased from Jackson Laboratories. Mice crossed to CAGCRE-ER received 3 tamoxifen (Sigma) injections (200 μg/g body weight i.p. in corn oil) administered every other day.
Cell Lines
Per2::Luciferase MEFs were a gift from J. Takahashi (UT Southwestern). Sirt1fx/fx MEFs were isolated from pregnant mice at embryonic day 14 or 15 following trypsin digestion as previously (Peek et al., 2013). MEFs were immortalized by lentivirus transduction of shRNAs targeting p19ARF (Addgene 14090, Northwestern Skin Disease Research Center). Recombination of LoxP sites was achieved through 3 treatments with adenovirus expressing Cre or empty vector (Vector Biolabs, MOI 500). MEFs were synchronized with dexamethasone (100 nM, 30 min) and collected at indicated timepoints. pCMV-SPORT2-PER2 expressing plasmid (Addgene 16204) was subjected to site-directed mutagenesis according to manufacturer instructions (NEB) and transiently transfected into low-passage HEK293 cells (Clontech).
Methods Details
Pharmacologic NAD+ supplementation
Freshly prepared, sterile-filtered nicotinamide mononucleotide (NMN, Oriental Yeast Company) was injected (i.p. 500 mg/kg in PBS) 4 hours prior to sample collection. Nicotinamide riboside (NR, Chromadex) was prepared to 3.2 g/L in drinking water, sterile filtered, and provided ad lib to mice in autoclaved, light-protected water bottles for 4 months to examine the impact of NR supplementation on the hepatic circadian transcriptome (Cantó et al., 2012). For HSF1 ChIPsequencing studies, NR was provided for 2 months since Canto et al demonstrated that similar metabolic phenotypes were apparent even after only 1 month of treatment (Cantó et al., 2012). NR was replaced every 3 days after lights off to prevent bacterial growth and minimize behavioral disruptions. Water consumption rates were monitored for 1 month and were not different between H2O and NR-supplemented mice. Mice drank ~3.75 mL water per day, achieving a dose of ~400 mg/kg in a 30 g mouse.
Nucleotide measurements
NAD+ was quantified as previously described (Peek et al., 2013). Briefly, cut and weighed tissue was homogenized in perchloric acid (Sigma) in the TissueLyzer (Qiagen). Following neutralization with K2CO3, supernatant was diluted 1:1 in mobile phase and analyzed by HPLC (Shimadzu) on a Supelco LC-18 column (Sigma), UV-Vis detector at 260 nm. Nicotinamide was quantified as previously described (Trammell and Brenner, 2013) by buffered, boiled ethanol extraction followed by liquid-chromatography mass-spectrometry by NRomics at the University of Iowa.
RNA-sequencing and analysis
RNA was isolated from 10-20mg of liver tissue using trizol reagent and purified on the Zymo Direct-Zol RNA miniprep kit. RNA quality was assessed by Bioanalyzer (Agilent). RNAs with a RNA Integrity Number (RIN) greater than 8 were used for library prep. Libraries were constructed from 250 ng of RNA using the NEBNext RNA Ultra Directional library prep kit from NEB. Average library size and concentration were determined by Bioanalyzer and qPCR (NEBNext Library Quant kit), respectively, prior to pooling. Pooled libraries were quantified by qPCR and loaded onto a NextSeq 500/550 v2 high output flow cell from Illumina (FC-404-2002) and 75bp single-end sequencing was performed on a NextSeq 500 sequencer. Libraries were sequenced to an average depth of ~15M or ~40M aligned reads for rhythmic or differential analyses, respectively. Using standard parameters, sequences were aligned to the mm10 genome with STAR (v2.5.2) and assigned to Ensembl features (GRCm38.vM12) with subread:featureCounts (v1.5.1).
Analysis of differential expression at each timepoint was performed with standard parameters in DESeq2 (v1.24.0) after removal of genes with 0 read counts. FDR-adjusted p-value cutoff for significance was set to 0.1. DESeq2-normalized counts were used for analysis of rhythmic expression with eJTK_Cycle (v3.1.R). Period lengths were allowed between 20 and 28 hours to account for sampling frequency (4 hours). Adjusted p-values of 0.1 and 0.9 were chosen for ‘rhythmic’ and ‘non-rhythmic’ genes respectively. Genes were classified as “unaffected” if they were similarly rhythmic in both conditions, as “gain” if they were non-rhythmic in the control condition and rhythmic in the experimental condition, as “loss” if they were rhythmic in the control condition and non-rhythmic in the experimental condition, and “phase-shift” if they were rhythmic in both conditions but were annotated with peak phases that were greater than 4 hours different. Heatmaps were sorted by peak phase for the control condition for loss, unaffected, and phase-shift reprogramming groups, while they were sorted by peak phase of the experimental condition for the gain reprogramming group. Gene ontology and promoter motif analysis were performed on genes from each reprogramming group that were sub-divided by phase. Gene ontology analysis of the KEGG term database was performed with the HOMER findGO command. Enrichment of known motifs +/− 2 kb of the TSS were determined using the HOMER findMotif command. The JASPAR vertebrate DNA binding motif database was used as the known motif set with a threshold set to 6.5.
ChIP-sequencing and analysis
Liver was collected at the indicated time points and processed for ChIP-seq as previously published (Perelis et al., 2015). Briefly, liver was fixed in disuccinimidyl glutarate (2mM in PBS + 1% DMSO, 30 min) and formaldehyde (1% in PBS, 10 min) prior to aliquoting and freezing at −80°C. For BMAL1 (antibody produced in Bass Lab as in (Perelis et al., 2015), Millipore ABE2599) and HSF1 (Cell Signaling 4356) ChIP-Seq, nucleii from a quarter of a liver were isolated in buffer (150 mM NaCl, 5 mM EDTA pH8, 50 mM Tris-HCl pH8, 0.35% NP-40) in the presence of protease inhibitors by needle, then sheared in 1% SDS buffer in a sonicator (diagenode) for 6 cycles 30on/30off at 4°C. An aliquot for input samples was taken after shearing. Chromatin was diluted 1:10 in dilution buffer (.01% SDS, 1.1% Triton X-100, 167 mM NaCl, 1.2 mM EDTA pH8, 1.67 mM Tris-HCl pH8), and immunoprecipitations with 15 μg of antibody were performed overnight at 4°C, rotating. Secondary-conjugated, BSA-blocked paramagnetic beads were used to pull down protein/DNA complexes. Chromatin was de-crosslinked and purified by Qiagen MinElute column prior to library prep with NEBNext Ultra II library prep kit. Inputs were quantified by Qubit Fluorometer (Invitrogen) for each liver, and 2 ng of equimolar pools of the replicates per condition were processed into libraries as above. Libraries were size-selected to 200-600 bp on the Sage Pippin Prep prior to PCR amplification. Libraries were processed and pooled for sequencing as above. Libraries were sequenced to an average depth of ~15M aligned reads.
Bowtie2 (v2.2.4) was used to align sequencing data to the mm10 genome with standard parameters. For each ChIP, peaks that enrich over the appropriate input were called using the HOMER findPeaks command with settings -style factor, -size 275, -fragLength 250. For scatter plots, heatmaps, box and whisker plots, tag-density for individual peaks for each replicate as described was quantified by HOMER annotatePeaks with setting -size given, and averaged. For histograms, the average tag-density across numerous peaks for each replicate was determined with annotatePeaks with the settings -hist 5, size −2000. For gene ontology analyses, sam files for both replicates were merged (samtools1.2 merge command) and peaks that enrich over the input were identified as above and compared across groups with HOMER mergePeaks. Gene ontology analysis of KEGG and Interpro databases was performed with HOMER findGO command using non-redundant genes annotated from identified peaks. For tornado plots, tag-density +/− 1kb from the center of the peak as described was generated from merged sam files using the HOMER annotatePeaks command with settings -ghist 25, and -size 2000. Tornado plots were z-score normalized and sorted from max-min signal according to old, NR-treated mice at ZT8.
ATAC-sequencing and analysis
100 mg of fresh liver was dounced in ice-cold PBS, passed through a 40 uM filter, and 50,000 cells were processed as described previously (Corces et al., 2017). Briefly, samples were resuspended in ATAC-RSB (10 mM Tris-HCl pH 7.4, 10 mM NaCl, and 3 mM MgCl2 in water) containing 0.1% IGEPAL, 0.1% Tween-20, and 0.01% digitonin and incubated on ice for 3 min prior to 20x dilution in ATAC-RSB containing 0.01% Tween-20. Nuclear pellets were subjected to tagmentation at 37°C for 30 min in Nextera TD buffer (Illumina) containing 33% PBS, 0.01% digitonin, 0.1% Tween-20, and 2.5 units Nextera transposase enzyme (Illumina). Following transposition reaction, DNA was purified using MinElute columns (Qiagen), subjected to 9 amplification cycles, size-selected by Sage Pippin Prep for fragments between 100-1000 bp, and sequenced by 42 bp paired-end sequencing to an average depth of ~50M aligned reads per sample. Nucleosome-free and nucleosome-bound fragments were segregated based on size distribution using the R Package ATACSeqQ (Ou et al., 2018). The HOMER findPeaks command with settings -size 150 and -fragLength 41 was used to find nucleosome free regions/peaks. Scatterplots and histograms were made as described in the ChIP-sequencing method above.
Western blotting
Transfections occurred 24 hours after seeding and media changed at 48 hours after seeding. Cells were collected at indicated time points or 72 hours after seeding. FK866 (10 nM) (Sigma) or EX527 (1μM) (Sigma) was diluted in DMSO and supplemented to cells without changing media 24 hours before collection. For subcellular localization experiments, cytoplasmic fractions were isolated from the supernatant following treatment with CER1/CER2 reagents according to manufacturer instructions (Thermo, NE-PER kit). Nuclear fractions were isolated by resuspending the nuclear pellet in 5 volumes RIPA buffer and shearing (Diagenode) 3 cycles at 30sec on/30sec off. Whole cell lysates were isolated by treating cell pellets with RIPA as above. All lysis buffers contained 1X protease inhibitors (Roche), 1X phosphatase inhibitors (Roche), and 1μM Trichostatin A (Sigma) and 20mM nicotinamide (Sigma) to inhibit deacetylases. Anti-FLAG-PER2 immunoprecipitations were carried out by incubating 0.25 mg protein lysate with 30 μL of 50% slurry of anti-FLAG conjugated paramagnetic beads (Sigma) in PBS + 5% Tween-20 in protein lo-bind tubes (Eppendorf) overnight at 4°C, rotating. Western blots for H3 (CST 9715), β-ACTIN (CST 4970), GAPDH (CST 5174), BMAL1 (Santa Cruz sc-48790), p-PER2-S659 (Thermo PA5-38901), PER2 (gift from C. Lee (Florida State University)), PER1 (Abcam ab136451), CRY2 (Alpha Diagnostic CRY21-A), CRY1 (Bethyl A302-614A), and FLAG-HRP (Sigma). HRP-conjugated secondary antibodies were from Sigma and Rockland (TrueBlot).
Ex vivo PER2::LUC measurements
Soleus was excised from transgenic mice expressing the full-length PER2 fused to LUCIFERASE (PER2::LUC) and cultured ex vivo in a lumicycle (Actimetrics) as previously described (Yoo et al., 2005). Briefly, the full-length soleus muscle was spread flat on a 0.2 μm filter (Millipore) exposed to luciferin-containing media (Dulbecco’s modified Eagle’s medium (DMEM; Gibco, 1.2 ml) containing sodium bicarbonate (352.5 μg/ml), 10 mM HEPES (Gibco), 2 mM L-glutamine, 2% B-27 serum-free supplement (Invitrogen), penicillin (25 U/ml), streptomycin (Gibco, 20 μg/ml), and 0.1 mM luciferin sodium salt (Biosynth AG)) on the basal side. Dishes were sealed with vacuum grease and a round cover slip and maintained in a lumicycle at 37°C. Amplitude was determined by calculating the half-difference in height of the background-subtracted maxima and minima on the 3rd phase of oscillation (~3 days after start of recordings).
PER2::LUC stability
MEFs expressing full-length PER2 fused to LUCIFERASE (PER2::LUC) were tested for PER2 stability in a lumicycle as described previously (Zhou et al., 2015). Briefly, MEFs were synchronized with dexamethasone, then incubated in a lumicycle (Actimetrics) in media containing luciferin as above. 17 hours post-shock, FK866 (30 nM) or DMSO was added to the dish without changing the media. 22 hours post-shock, cycloheximide (40 μg/mL) was added to the dish without changing the media. Background-subtracted luminescence readings were normalized to signal at 21.5 hours post-shock. Half-life was calculated as the time following cycloheximide addition in which the luminescence signal was decreased by greater than 50%.
LC-MS/MS analysis of post-translational modifications
FLAG-tagged PER2 was overexpressed in HEK293 cells treated with FK866 (Sigma) or DMSO and immunoprecipitated from whole-cell lysate as above. Immunoprecipitate was gel-purified and a ~1 cm x 0.5 cm piece was cut from the gel at the location that corresponds to the predominant FLAG-PER2 band (between 150 kDa and 160 kDa) and transferred to protein lo-bind tubes (Eppendorf). Gel pieces were washed with ammonium bicarbonate (Ambic, Fisher) and acetonitrile (Fisher), reduced with dithiothreitol (Promega), washed with Ambic/iodoacetamide (Sigma), then subjected to in-gel proteolysis with 300 ng trypsin (Promega) at 30°C overnight, and washed with Ambic and formic acid (ThermoFisher). Samples were then submitted to the Northwestern Proteomics Center of Excellence where peptides were analyzed by LC-MS/MS using a using a Dionex UltiMate 3000 Rapid Separation nanoLC and a Q Exactive™ HF Hybrid Quadrupole-Orbitrap™ Mass Spectrometer (Thermo Fisher Scientific Inc, San Jose, CA). Approximately 1 μg of peptide samples was loaded onto the trap column, which was 150 μm x 3 cm in-house packed with 3 um C18 beads. The analytical column was a 75 um x 10.5 cm PicoChip column packed with 3 um C18 beads (New Objective, Inc. Woburn, MA). The flow rate was kept at 300nL/min. Solvent A was 0.1% FA in water and Solvent B was 0.1% FA in ACN. The peptide was separated on a 120-min analytical gradient from 5% ACN/0.1% FA to 40% ACN/0.1% FA. The mass spectrometer was operated in data-dependent mode. The source voltage was 2.10 kV and the capillary temperature was 320 degrees C. MS1 scans were acquired from 300-2000m/z at 60,000 resolving power and automatic gain control (AGC) set to 3x106. The top 15 most abundant precursor ions in each MS1 scan were selected for fragmentation. Precursors were selected with an isolation width of 2 Da and fragmented by Higher-energy collisional dissociation (HCD) at 30% normalized collision energy in the HCD cell. Previously selected ions were dynamically excluded from re-selection for 20 seconds. The MS2 AGC was set to 1x105. All samples were run in duplicates. Proteins were identified from the tandem mass spectra extracted by Xcalibur version 4.0. MS/MS spectra were searched against the SwissProt Homo Sapiens database contain mouse proteins of interest using Mascot search engine (Matrix Science, London, UK; version 2.5.1). All searches included carbamidomethyl cysteine as a fixed modification and oxidized Met, phosphorylated Ser Thr and Tyr, deamidated Asn and Gln, acetylated N-term, and acetylation on Lys as variable modifications. Three missed tryptic cleavages were allowed. The MS1 precursor mass tolerance was set to 10 ppm and the MS2 tolerance was set to 0.05 Da. A 1% false discovery rate cutoff was applied at the peptide level. Only proteins with a minimum of two peptides above the cutoff were considered for further study. The search result was visualized by Scaffold (version 4.8.3. Proteome Software, INC., Portland, OR). Acetylation of PER2K680 was manually validated.
Behavioral analysis
Locomotor activity was analyzed in young and old mice treated with NR or H2O or in CAGCreER; Namptfx/fx mice and controls before and after tamoxifen treatment. Mice were singly housed in standard mouse cages equipped with running wheels in standard 12:12 LD conditions. Average 24-hour wheel running activity from each mouse was determined from a 14-day period for each mouse using ClockLab software (Actimetrics). The average activity between ZT22 and ZT0 was quantified.
Live animal body temperature monitoring
Mice were implanted with a IPTT-300 temperature transponder into the subcutaneous dorsal region. Following one week of recovery, body temperature was assessed every 2 hrs for 24 hrs using a hand-held DAS-8007-IUS reader from outside the cage, enabling remote body temperature measurements with minimal disruption to the animals (Bio Medic Data Systems). Mice had ad lib access to food and water for the duration of the experiment.
Oxygen consumption rates
Liver mitochondrial respiratory rhythms were measured as previously described (Peek et al., 2013). Briefly, mitochondria were isolated from fresh liver in mitochondrial isolation buffer (70 mM sucrose, 210 mM mannitol, 5 mM HEPES, 1 mM EGTA, and 0.5% (w/v) fatty acid-free BSA (pH 7.2)) prior to resuspension in 1xMAS buffer (70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1.0 mM EGTA, and 0.2% (w/v) fatty acid-free BSA (pH 7.2). 10 μg of mitochondria in 1xMAS buffer containing 80 μM palmitoyl-carnitine was loaded into each well of a 96-well Seahorse bioanalyzer (Agilent) and centrifuged for 20 min in 2000g at 4°C to adhere mitochondria. Oxygen consumption rates were taken basally and following each sequential injection of 4mM ADP, 10μM oligomycin, 10μM FCCP, and 10μM antimycin A (Sigma). FCCP-treated, “maximal” OCR was normalized to basal readings to control for collection/plate differences and quantified.
Quantitation and statistical analysis
Statistical analysis was performed with unpaired two-tailed Student’s t test except where otherwise noted in results, figure legends, or methods. Behavioral analysis of CAGCRE=ER;Namptfx/fx mice before and after tamoxifen administration was performed with a paired two-tailed Student’s t test. Where appropriate, data are represented as mean +/− SEM. Differences were considered significant when p<0.05 except where otherwise noted in results, figure legends, or methods. Two-way ANOVA was performed to compare effects of NR-treatment or L-Nampt−/− on NAD+ and nicotinamide rhythms. P-value was reported for variation from the drug/genetic intervention.
Supplementary Material
Table S1. NR reprograms hepatic diurnal transcriptome. Related to Figure 1.
Table S2. Genetic depletion of NAMPT reprograms hepatic diurnal transcriptome. Related to Figure 3.
Table S3. Transcriptional response to pharmacologic elevation of NAD+ require both BMAL1 and SIRT1. Related to Figures 4–5.
Table S4. LC/MS-MS reveals PER2 acetylation at K680. Related to Figure 5.
Table S5. NR restores rhythmic BMAL1 chromatin binding. Related to Figure 6.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-BMAL1 (ChIP-Seq) | J. Bass, Millipore | ABE2599 (Perelis et al., 2015) |
| Anti-BMAL1 (WB) | Santa Cruz | sc-48790 |
| Anti-HSF1 | Cell Signaling | 4356 |
| Anti-H3 | Cell Signaling | 9715 |
| Anti-H3K27Ac | Active Motif | 39133 |
| Anti-H3K9Ac | Active Motif | 39137 |
| Anti-β-ACTIN | Cell Signaling | 4970 |
| Anti-GAPDH | Cell Signaling | 5174 |
| Anti-p-PER2-S659 | Thermo Fisher | PA5-38901 |
| Anti-PER2 | C. Lee (Florida State University) | |
| Anti-PER1 | Abcam | ab136451 |
| Anti-CRY2 | Alpha Diagnostic | CRY21-A |
| Anti-CRY1 | Bethyl | A302-614A |
| Anti-FLAG-HRP | Sigma | F1804-50UG |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma | T5648-1G |
| Corn Oil | Sigma | C8267-500ML |
| Trypsin | Fisher Scientific | 25300054 |
| Dexamethasone | Sigma | D4902-100MG |
| Nicotinamide mononucleotide (NMN) | Oriental Yeast Company | 44500900-1g |
| PBS | Mediatech | 21-040-CM |
| Nicotinamide riboside (NR) | Chromadex | N001 |
| Perchloric acid | Sigma | 244252-100ML |
| K2CO3 | Sigma | 367877-10G |
| Trizol Reagent | Molecular Research Center | TR118 |
| Disuccinimidyl glutarate | Proteochem | C1104-1 gram |
| DMSO | Sigma | D8418-100ML |
| Formaldehyde | Polysciences | 18814 |
| Sodium chloride (NaCl) | Fischer Scientific | BP35-10 |
| Ethylenediaminetetraacetic acid (EDTA) | Lonza | 51234 |
| Trizma® hydrochloride solution | Millipore-Sigma | T2319-1L |
| NP-40 | Sigma | NP40s |
| SDS | Sigma | L3771-ikg |
| Triton X-100 | Sigma | X100-100ML |
| MgCl2 | Sigma | M8266 |
| IGEPAL | Sigma | I8896-50ML |
| Tween-20 | Sigma | P1754-500ML |
| Digitonin | Sigma | D141-100MG |
| Nextera transposase enzyme | Illumina | 15027865 |
| Nextera TD buffer | Illumina | 15027865 |
| FK866 | Sigma | F8557-25MG |
| EX527 | Tocris | 2780 |
| HEPES | Gibco | 15630080 |
| NE-PER kit | ThermoFisher | 78835 |
| B27 Supplement | Invitrogen | 17504-001 |
| Trichostatin A | Sigma | T8552-1MG |
| Nicotinamide | Sigma | N3501-100MG |
| Ammonium bicarbonate | ThermoFisher | A643500 |
| Acetonitrile | ThermoFisher | A955500 |
| Dithiothreitol | Thermo Fisher | R0861 |
| Ambic/iodoacetamide | Sigma | I1149-5G |
| Formic acid | ThermoFisher | P128905 |
| Dulbecco’s modified Eagle’s medium (DMEM) | Gibco | MT10013CV |
| Sodium bicarbonate | Sigma | S5761-500G |
| Fetal bovine serum (FBS) | Atlanta Biological | S11550 |
| Penicillin-Streptomycin | Gibco | 15140122 |
| Luciferin sodium salt | Biosynth AG | L8220 |
| Cycloheximide | Sigma | 239764-1GM |
| Protease inhibitor cocktail, Complete mini | Roche Applied Science | 11836170001 |
| Phosphatase inhibitor cocktail, phoSTOP | Roche Applied Science | 04906845001 |
| Anti-FLAG conjugated paramagnetic beads | Sigma | M8823-1ML |
| L-Glutamine | Thermo Fisher Scientific | 25030081 |
| Sucrose | Sigma | S7903-1KG |
| KH2PO4 | Sigma | P3786-100g |
| Palmitoyl-carnitine | Sigma | P1645-5MG |
| Oligomycin | Sigma | O4876-5MG |
| FCCP | Sigma | C2920-10MG |
| Antimycin A | Sigma | A8674-25MG |
| Mannitol | Sigma | M9546-250G |
| EGTA | Bostonbioproducts | BM-151 |
| Fatty acid-free BSA | Sigma | A7511-5G |
| Critical Commercial Assays | ||
| Zymo Direct-Zol RNA miniprep kit | Zymo Research | R2051 |
| NEBNext RNA Ultra Directional Library Preparation Kit | New England Biolabs | E7420S |
| NEBNext Library Quant Kit | New England Biolabs | E7630S |
| NEBNext Ultra II Library Prep Kit | New England Biolabs | E7645S |
| Q5 site directed mutagenesis kit | New England Biolabs | E0552S |
| MinElute columns | Qiagen | 28006 |
| Software and Algorithms | ||
| Bowtie (v2.2.4) | http://bowtie-bio.sourceforge.net/index.shtml | (Langmead et al., 2009) |
| subread:featureCounts (v1.5.1). | (Liao et al., 2014) | |
| STAR (v2.5.2) | (Dobin et al., 2013) | |
| DESeq2 (v1.24.0) | (Love et al., 2014) | |
| eJTK_Cycle (v3.1.R). | (Hutchison et al., 2015) | |
| ClockLab Data Collection and Analysis System (v6.0) | http://actimetrics.com | |
| R Package ATACSeqQC | (Ou et al., 2018) | |
| HOMER (v4.10) | http://homer.ucsd.edu/homer/index.html | (Heinz et al., 2010) |
| Xcalibur (v4.0) | ||
| Mascot search engine (v2.5.1). | Matrix Science, London, UK | |
| MS/MS spectra scaffold (v4.8.3) | Proteome Software, Portland, OR | |
| Deposited Data | ||
| RNA-seq, ChIP-seq, ATAC-seq | NCBI GEO | GSE133989 |
| Experimental Models (Cells) | ||
| Per2::Luciferase MEFs | J. Takahashi (UT Southwestern) | |
| Sirt1−/− MEFs | J. Bass Lab | Harvested from Sirt1fx/fx mice treated with 3 rounds Adeno-Cre (Vector Biolabs) |
| HEK293 cells | Clontech | |
| Experimental Models (Mice) | ||
| C57BL/6CR (4 mo, 16 mo) | Charles River, NIA Colony | |
| Bmal1−/− | J. Takahashi (UT Southwestern) | |
| Per2::Luciferase | J. Takahashi (UT Southwestern) | |
| Namptfx/fx | S. Imai (Washington University) | |
| Sirt1fx/fx | S. Imai (Washington University) | |
| Alb-Cre | Jackson Laboratories | |
| CAGCre-ER | Jackson Laboratories | |
| Recombinant DNA | ||
| shRNA targeting p19ARF | Addgene | 14090 |
| pCMV-SPORT2-PER2 | Addgene | 16204 |
Acknowledgments
We thank all members of the Bass, Barish, and Allada laboratories for helpful discussions, and Biliana Marcheva for help with the figures. Research support was from: the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grants R01DK090625, R01DK100814, and 1R01DK113011-01A1, and the Chicago Biomedical Consortium S-007 (to J.B.); the National Institute on Aging (NIA) grant P01AG011412 (to J.B. and C.B.); NIDDK grants 5K01DK105137-03 and 1R03DK116012-01 (to C.B.P.); the National Research Service Award (NRSA) grant F30DK116481 (to B.J.W.); the Swedish Research Council grant 2014-6888 and the Swedish Society for Medical Research (to J.C.); the National Institute of Neurological Disorders and Stroke (NINDS) grant R21NS099813 (to C.L.); the National Heart, Lung, and Blood Institute (NHLBI) grant R01HL147545 and the Roy J. Carver Trust (to C.B.); and the NSF-Simons Center for Quantitative Biology (Simons Foundation/SFARI 597491-RWC) and the National Science Foundation (1764421) (to R.B.). Proteomics services were performed by the Northwestern Proteomics Core Facility, supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center and the National Resource for Translational and Developmental Proteomics supported by P41 GM108569.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: C.B. is the inventor of intellectual property on the nutritional and therapeutic uses of NR. He serves as chief scientific advisor of ChromaDex, which licensed, developed, and commercialized NR technologies, and holds stock in ChromaDex.
References
- Aguilar-Arnal L, Hakim O, Patel VR, Baldi P, Hager GL, and Sassone-Corsi P (2013). Cycles in spatial and temporal chromosomal organization driven by the circadian clock. Nat. Struct. Mol. Biol 20, 1206–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryal RP, Kwak PB, Tamayo AG, Gebert M, Chiu P-L, Walz T, and Weitz CJ (2017). Macromolecular Assemblies of the Mammalian Circadian Clock. Mol. Cell 67, 770–782.e776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, and Schibler U (2008). SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134, 317–328. [DOI] [PubMed] [Google Scholar]
- Atger F, Gobet C, Marquis J, Martin E, Wang J, Weger B, Lefebvre G, Descombes P, Naef F, and Gachon F (2015). Circadian and feeding rhythms differentially affect rhythmic mRNA transcription and translation in mouse liver. Proc. Natl. Acad. Sci. U.S.a 112, E6579–E6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargiello TA, and Young MW (1984). Molecular genetics of a biological clock in Drosophila. Pnas 81, 2142–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargiello TA, Jackson FR, and Young MW (1984). Restoration of circadian behavioural rhythms by gene transfer in Drosophila. Nature 312, 752–754. [DOI] [PubMed] [Google Scholar]
- Bass J, and Lazar MA (2016). Circadian time signatures of fitness and disease. Science 354, 994–999. [DOI] [PubMed] [Google Scholar]
- Bieganowski P, and Brenner C (2004). Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 117, 495–502. [DOI] [PubMed] [Google Scholar]
- Bogan KL, and Brenner C (2008). Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr 28, 115–130. [DOI] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, and Grant R (2011). Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 6, e19194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Brown KD, Maqsood S, Huang J-Y, Pan Y, Harkcom W, Li W, Sauve A, Verdin E, and Jaffrey SR (2014). Activation of SIRT3 by the NAD+ precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metabolism 20, 1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhr ED, Yoo S-H, and Takahashi JS (2010). Temperature as a universal resetting cue for mammalian circadian oscillators. Science 330, 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buysse DJ, Browman KE, Monk TH, Reynolds CF, Fasiczka AL, and Kupfer DJ (1992). Napping and 24-hour sleep/wake patterns in healthy elderly and young adults. J Am Geriatr Soc 40, 779–786. [DOI] [PubMed] [Google Scholar]
- Camacho-Pereira J, Tarragó MG, Chini CCS, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, et al. (2016). CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metabolism 23, 1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, and Verdin E (2019). From discoveries in ageing research to therapeutics for healthy ageing. Nature 571, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, et al. (2012). The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metabolism 15, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H-C, and Guarente L (2013). SIRT1 Mediates Central Circadian Control in the SCN by a Mechanism that Decays with Aging. Cell 153, 1448–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diguet N, Trammell SAJ, Tannous C, Deloux R, Piquereau J, Mougenot N, Gouge A, Gressette M, Manoury B, Blanc J, et al. (2018). Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation 137, 2256–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong HA, Robles MS, Knutti D, and Weitz CJ (2011). A Molecular Mechanism for Circadian Clock Negative Feedback. Science 332, 1436–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ear PH, Chadda A, Gumusoglu SB, Schmidt MS, Vogeler S, Malicoat J, Kadel J, Moore MM, Migaud ME, Stevens HE, et al. (2019). Maternal Nicotinamide Riboside Enhances Postpartum Weight Loss, Juvenile Offspring Development, and Neurogenesis of Adult Offspring. Cell Rep 26, 969–983.e4. [DOI] [PubMed] [Google Scholar]
- Eckel-Mahan KL, Patel VR, de Mateo S, Orozco-Solis R, Ceglia NJ, Sahar S, Dilag-Penilla SA, Dyar KA, Baldi P, and Sassone-Corsi P (2013). Reprogramming of the circadian clock by nutritional challenge. Cell 155, 1464–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide EJ, Woolf MF, Kang H, Woolf P, Hurst W, Camacho F, Vielhaber EL, Giovanni A, and Virshup DM (2005). Control of Mammalian Circadian Rhythm by CKIε-Regulated Proteasome-Mediated PER2 Degradation. Mol. Cell. Biol. 25, 2795–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang B, Everett LJ, Jager J, Briggs E, Armour SM, Feng D, Roy A, Gerhart-Hines Z, Sun Z, and Lazar MA (2014). Circadian enhancers coordinate multiple phases of rhythmic gene transcription in vivo. Cell 159, 1140–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folsom AR, Kaye SA, Sellers TA, Hong CP, Cerhan JR, Potter JD, and Prineas RJ (1993). Body fat distribution and 5-year risk of death in older women. Jama 269, 483–487. [PubMed] [Google Scholar]
- Ford ES, Giles WH, and Dietz WH (2002). Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. Jama 287, 356–359. [DOI] [PubMed] [Google Scholar]
- Foteinou PT, Venkataraman A, Francey LJ, Anafi RC, Hogenesch JB, and Doyle FJ (2018). Computational and experimental insights into the circadian effects of SIRT1. Proc. Natl. Acad. Sci. U.S.a 374, 201803410–201803416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego M, and Virshup DM (2007). Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Bio 8, 139–148. [DOI] [PubMed] [Google Scholar]
- Garbe DS, Fang Y, Zheng X, Sowcik M, Anjum R, Gygi SP, and Sehgal A (2013). Cooperative interaction between phosphorylation sites on PERIOD maintains circadian period in Drosophila. PLoS Genet. 9, e1003749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan D, Xiong Y, Borck PC, Jang C, Doulias P-T, Papazyan R, Bin Fang, Jiang C, Zhang Y, Briggs ER, et al. (2018). Diet-Induced Circadian Enhancer Remodeling Synchronizes Opposing Hepatic Lipid Metabolic Processes. Cell 174, 831–842.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin PE, Hall JC, and Rosbash M (1990). Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature 343, 536–540. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig S, Strauss HM, Vanselow K, Yildiz O, Schulze S, Arens J, Kramer A, and Wolf E (2009). Structural and Functional Analyses of PAS Domain Interactions of the Clock Proteins Drosophila PERIOD and Mouse PERIOD2. PLoS Biol 7, e1000094–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T, Lee JW, St John PC, Sawa M, Iwaisako K, Noguchi T, Pongsawakul PY, Sonntag T, Welsh DK, Brenner DA, et al. (2012). Identification of Small Molecule Activators of Cryptochrome. Science 337, 1094–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison AL, Maienschein-Cline M, Chiang AH, Tabei SMA, Gudjonson H, Bahroos N, Allada R, and Dinner AR (2015). Improved statistical methods enable greater sensitivity in rhythm detection for genome-wide data. PLoS Comput. Biol. 11, e1004094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, and Guarente L (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800. [DOI] [PubMed] [Google Scholar]
- Kim YH, Marhon SA, Zhang Y, Steger DJ, Won K-J, and Lazar MA (2018). Rev-erba dynamically modulates chromatin looping to control circadian gene transcription. Science 359, 1274–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohsaka A, Laposky AD, Ramsey KM, Estrada C, Joshu C, Kobayashi Y, Turek FW, and Bass J (2007). High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metabolism 6, 414–421. [DOI] [PubMed] [Google Scholar]
- Koike N, Yoo S-H, Huang H-C, Kumar V, Lee C, Kim T-K, and Takahashi JS (2012). Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, and Antoch MP (2006). Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 20, 1868–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka RJ, and Benzer S (1971). Clock mutants of Drosophila melanogaster. PNAS 68, 2112–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, and Reppert SM (1999). mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell 98, 193–205. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu C, Li S, Liu T, Borjigin J, and Lin JD (2007). Transcriptional coactivator PGC-1a integrates the mammalian clock and energy metabolism. Nature 447, 477–481. [DOI] [PubMed] [Google Scholar]
- Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, and Kaelin WG (2013). (R)-2-Hydroxyglutarate Is Sufficient to Promote Leukemogenesis and Its Effects Are Reversible. Science 339, 1621–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey PL, Shimomura K, Antoch MP, Yamazaki S, Zemenides PD, Ralph MR, Menaker M, and Takahashi JS (2000). Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science 288, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Salamanca HH, Duarte FM, Danko CG, and Lis JT (2016). Mammalian Heat Shock Response and Mechanisms Underlying Its Genome-wide Transcriptional Regulation. Mol. Cell 62, 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, et al. (2010). Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 466, 627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, and Guillemin GJ (2012). Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS ONE 7, e42357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menet JS, Pescatore S, and Rosbash M (2014). CLOCK:BMAL1 is a pioneer-like transcription factor. Genes Dev. 28, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q-J, Logunova L, Maywood ES, Gallego M, Lebiecki J, Brown TM, Sládek M, Semikhodskii AS, Glossop NRJ, Piggins HD, et al. (2008). Setting clock speed in mammals: the CK1 epsilon tau mutation in mice accelerates circadian pacemakers by selectively destabilizing PERIOD proteins. Neuron 58, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermet J, Yeung J, Hurni C, Mauvoisin D, Gustafson K, Jouffe C, Nicolas D, Emmenegger Y, Gobet C, Franken P, et al. (2018). Clock-dependent chromatin topology modulates circadian transcription and behavior. Genes Dev. 32, 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer P, Saez L, and Young MW (2006). PER-TIM interactions in living Drosophila cells: an interval timer for the circadian clock. Science 311, 226–229. [DOI] [PubMed] [Google Scholar]
- Michael AK, Fribourgh JL, Chelliah Y, Sandate CR, Hura GL, Schneidman-Duhovny D, Tripathi SM, Takahashi JS, and Partch CL (2017). Formation of a repressive complex in the mammalian circadian clock is mediated by the secondary pocket of CRY1. Proc. Natl. Acad. Sci. U.S.a 114, 1560–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley JE (2008). Diabetes and aging: epidemiologic overview. Clin. Geriatr. Med. 24, 395-405-v. [DOI] [PubMed] [Google Scholar]
- Mure LS, Le HD, Benegiamo G, Chang MW, Rios L, Jillani N, Ngotho M, Kariuki T, Dkhissi-Benyahya O, Cooper HM, et al. (2018). Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 22, eaao0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, Guarente LP, and Sassone-Corsi P (2008). The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 134, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y, Sahar S, Astarita G, Kaluzova M, and Sassone-Corsi P (2009). Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 324, 654–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council (US) Subcommittee on Laboratory Animal Nutrition (1995). Nutrient Requirements of Laboratory Animals: Fourth Revised Edition, 1995. [PubMed] [Google Scholar]
- Ohayon MM, Carskadon MA, Guilleminault C, and Vitiello MV (2004). Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep 27, 1255–1273. [DOI] [PubMed] [Google Scholar]
- Ondracek CR, Frappier V, Ringel AE, Wolberger C, and Guarente L (2017). Mutations that Allow SIR2 Orthologs to Function in a NAD(+)-Depleted Environment. Cell Rep 18, 2310–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou J, Liu H, Yu J, Kelliher MA, Castilla LH, Lawson ND, and Zhu LJ (2018). ATACseqQC: a Bioconductor package for post-alignment quality assessment of ATAC-seq data. BMC Genomics 19, 169–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschos GK, Ibrahim S, Song W-L, Kunieda T, Grant G, Reyes TM, Bradfield CA, Vaughan CH, Eiden M, Masoodi M, et al. (2012). Obesity in mice with adipocyte-specific deletion of clock component Arntl. Nat. Med 18, 1768–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek CB, Affinati AH, Ramsey KM, Kuo H-Y, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, et al. (2013). Circadian Clock NAD+ Cycle Drives Mitochondrial Oxidative Metabolism in Mice. Science 342, 1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelis M, Marcheva B, Ramsey KM, Schipma MJ, Hutchison AL, Taguchi A, Peek CB, Hong H, Huang W, Omura C, et al. (2015). Pancreatic β cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science 350, aac4250–aac4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, and Shulman GI (2003). Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300, 1140–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, and Li X (2009). Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metabolism 9, 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, et al. (2009). Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 324, 651–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P, Zehring WA, Wheeler DA, Pirrotta V, Hadfield C, Hall JC, and Rosbash M (1984). Molecular analysis of the period locus in Drosophila melanogaster and identification of a transcript involved in biological rhythms. Cell 38, 701–710. [DOI] [PubMed] [Google Scholar]
- Reinke H, Saini C, Fleury-Olela F, Dibner C, Benjamin IJ, and Schibler U (2008). Differential display of DNA-binding proteins reveals heat-shock factor 1 as a circadian transcription factor. Genes Dev. 22, 331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey G, and Reddy AB (2013). Connecting cellular metabolism to circadian clocks. Trends Cell Biol. 23, 234–241. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, and Puigserver P (2005). Nutrient control of glucose homeostasis through a complex of PGC-1|[alpha]| and SIRT1. Nature 434, 113–118. [DOI] [PubMed] [Google Scholar]
- Roenneberg T, Kuehnle T, Juda M, Kantermann T, Allebrandt K, Gordijn M, and Merrow M (2007). Epidemiology of the human circadian clock. Sleep Med Rev 11, 429–438. [DOI] [PubMed] [Google Scholar]
- Rosenberg RS, Zee PC, and Turek FW (1991). Phase response curves to light in young and old hamsters. Am. J. Physiol. 261, R491–R495. [DOI] [PubMed] [Google Scholar]
- Saini C, Morf J, Stratmann M, Gos P, and Schibler U (2012). Simulated body temperature rhythms reveal the phase-shifting behavior and plasticity of mammalian circadian oscillators. Genes Dev. 26, 567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Solanas G, Peixoto FO, Bee L, Symeonidi A, Schmidt MS, Brenner C, Masri S, Benitah SA, and Sassone-Corsi P (2017). Circadian Reprogramming in the Liver Identifies Metabolic Pathways of Aging. Cell 170, 664–677.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel JA, Krier I, Andersin T, Raghav S, Canella D, Gilardi F, Kalantzi AS, Rey G, Weger B, Gachon F, et al. (2017). Transcriptional regulatory logic of the diurnal cycle in the mouse liver. PLoS Biol 15, e2001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solanas G, Peixoto FO, Perdiguero E, Jardi M, Ruiz-Bonilla V, Datta D, Symeonidi A, Castellanos A, Welz P-S, Caballero JM, et al. (2017). Aged Stem Cells Reprogram Their Daily Rhythmic Functions to Adapt to Stress. Cell 170, 678–692.e20. [DOI] [PubMed] [Google Scholar]
- Storch K-F, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, and Weitz CJ (2002). Extensive and divergent circadian gene expression in liver and heart. Nature 417, 78–83. [DOI] [PubMed] [Google Scholar]
- Tognini P, Murakami M, Liu Y, Eckel-Mahan KL, Newman JC, Verdin E, Baldi P, and Sassone-Corsi P (2017). Distinct Circadian Signatures in Liver and Gut Clocks Revealed by Ketogenic Diet. Cell Metabolism 26, 523–538.e525. [DOI] [PubMed] [Google Scholar]
- Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, Ptacek LJ, and Fu YH (2001). An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science 291, 1040–1043. [DOI] [PubMed] [Google Scholar]
- Top D, O’Neil JL, Merz GE, Dusad K, Crane BR, and Young MW (2018). CK1/Doubletime activity delays transcription activation in the circadian clock. Elife 7, 2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trammell SAJ, Weidemann BJ, Chadda A, Yorek MS, Holmes A, Coppey LJ, Obrosov A, Kardon RH, Yorek MA, and Brenner C (2016). Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Scientific Reports 6, 26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trammell SA, and Brenner C (2013). TARGETED, LCMS-BASED METABOLOMICS FOR QUANTITATIVE MEASUREMENT OF NAD+ METABOLITES. Computational and Structural Biotechnology Journal 4, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y-I, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, and Zhang Y (2006). Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816. [DOI] [PubMed] [Google Scholar]
- Turek F, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen D, et al. (2005). Obesity and metabolic syndrome in circadian Clock mutant mice. Science 308, 1043–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turturro A, Witt WW, Lewis S, Hass BS, Lipman RD, and Hart RW (1999). Growth curves and survival characteristics of the animals used in the Biomarkers of Aging Program. J. Gerontol. a Biol. Sci. Med. Sci 54, B492–B501. [DOI] [PubMed] [Google Scholar]
- Vanselow K, Vanselow JT, Westermark PO, Reischl S, Maier B, Korte T, Herrmann A, Herzel H, Schlosser A, and Kramer A (2006). Differential effects of PER2 phosphorylation: molecular basis for the human familial advanced sleep phase syndrome (FASPS). Genes Dev. 20, 2660–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaur P, Brugg B, Mericskay M, Li Z, Schmidt MS, Vivien D, Orset C, Jacotot E, Brenner C, and Duplus E (2017). Nicotinamide riboside, a form of vitamin B3, protects against excitotoxicity-induced axonal degeneration. Faseb J. 31, 5440–5452. [DOI] [PubMed] [Google Scholar]
- Walch OJ, Cochran A, and Forger DB (2016). A global quantification of “normal” sleep schedules using smartphone data. Sci Adv 2, e1501705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AK, Yang F, Jiang K, Ji J-Y, Watts JL, Purushotham A, Boss O, Hirsch ML, Ribich S, Smith JJ, et al. (2010). Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 24, 1403–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide SD, Anckar J, Stevens SM, Sistonen L, and Morimoto RI (2009). Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323, 1063–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagita K, Tamanini F, Yasuda M, Hoeijmakers JHJ, van der Horst GTJ, and Okamura H (2002). Nucleocytoplasmic shuttling and mCRY-dependent inhibition of ubiquitylation of the mPER2 clock protein. Embo J. 21, 1301–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Straume M, Tei H, Sakaki Y, Menaker M, and Block GD (2002). Effects of aging on central and peripheral mammalian clocks. Pnas 99, 10801–10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, and Sauve AA (2016). NAD+metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 1864, 1787–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye R, Selby CP, Chiou Y-Y, Ozkan-Dagliyan I, Gaddameedhi S, and Sancar A (2014). Dual modes of CLOCK:BMAL1 inhibition mediated by Cryptochrome and Period proteins in the mammalian circadian clock. Genes Dev. 28, 1989–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz O, Doi M, Yujnovsky I, Cardone L, Berndt A, Hennig S, Schulze S, Urbanke C, Sassone-Corsi P, and Wolf E (2005). Crystal structure and interactions of the PAS repeat region of the Drosophila clock protein PERIOD. Mol. Cell 17, 69–82. [DOI] [PubMed] [Google Scholar]
- Yoo S-H, Ko CH, Lowrey PL, Buhr ED, Song E-J, Chang S, Yoo OJ, Yamazaki S, Lee C, and Takahashi JS (2005). A noncanonical E-box enhancer drives mouse Period2 circadian oscillations in vivo. Pnas 102, 2608–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, and Imai S-I (2011). Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metabolism 14, 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehring WA, Wheeler DA, Reddy P, KONOPKA RJ, Kyriacou CP, Rosbash M, and Hall JC (1984). P-element transformation with period locus DNA restores rhythmicity to mutant, arrhythmic Drosophila melanogaster. Cell 39, 369–376. [DOI] [PubMed] [Google Scholar]
- Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R, et al. (2016). NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. [DOI] [PubMed] [Google Scholar]
- Zhang R, Lahens NF, Ballance HI, Hughes ME, and Hogenesch JB (2014). A circadian gene expression atlas in mammals: implications for biology and medicine. Proc. Natl. Acad. Sci. U.S.a 111, 16219–16224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Kim JK, Eng GWL, Forger DB, and Virshup DM (2015). A Period2 Phosphoswitch Regulates and Temperature Compensates Circadian Period. Mol. Cell 60, 77–88. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. NR reprograms hepatic diurnal transcriptome. Related to Figure 1.
Table S2. Genetic depletion of NAMPT reprograms hepatic diurnal transcriptome. Related to Figure 3.
Table S3. Transcriptional response to pharmacologic elevation of NAD+ require both BMAL1 and SIRT1. Related to Figures 4–5.
Table S4. LC/MS-MS reveals PER2 acetylation at K680. Related to Figure 5.
Table S5. NR restores rhythmic BMAL1 chromatin binding. Related to Figure 6.
Data Availability Statement
RNA-seq, ChIP-seq, and ATAC-seq data sets have been deposited in NCBI’s Gene Expression Omnibus database (GSE133989).