

Abstract
All drugs usually have side effects, which endanger the health of patients. To identify potential side effects of drugs, biological and pharmacological experiments are done but are expensive and time-consuming. So, computation-based methods have been developed to accurately and quickly predict side effects. To predict potential associations between drugs and side effects, we propose a novel method called the Triple Matrix Factorization- (TMF-) based model. TMF is built by the biprojection matrix and latent feature of kernels, which is based on Low Rank Approximation (LRA). LRA could construct a lower rank matrix to approximate the original matrix, which not only retains the characteristics of the original matrix but also reduces the storage space and computational complexity of the data. To fuse multivariate information, multiple kernel matrices are constructed and integrated via Kernel Target Alignment-based Multiple Kernel Learning (KTA-MKL) in drug and side effect space, respectively. Compared with other methods, our model achieves better performance on three benchmark datasets. The values of the Area Under the Precision-Recall curve (AUPR) are 0.677, 0.685, and 0.680 on three datasets, respectively.
1. Introduction
Drug treatment of patients' diseases may be accompanied by side effects, endangering the life and health of patients. Therefore, how to quickly and accurately find potential drug side effect information becomes an important step in the drug development process. The traditional methods to detect the side effects of drugs are usually biological and pharmacological experiments. These approaches often take a long time and huge capital investment. So, it is necessary to accurately and quickly predict the potential side effects of drugs through computation-based methods [1]. Most computation-based methods for predicting drug side effects used Machine Learning (ML) classification models to predict side effect categories by extracted features from the biochemical characteristics of drugs. ML has been widely used in the field of computational biology, containing potential disease-associated microRNAs [2, 3] or circRNAs [4], O-GlcNAcylation sites [5], prediction of DNA or RNA methylcytosine sites [6, 7], protein function identification [8–12], protein remote homology [13], analyzing microbiology [14], electron transport proteins [15], drug-target interactions [16], drug-side effect association [17, 18], protein-protein interactions [19, 20], and lncRNA-miRNA interactions [21].
Pauwels and Stoven develop a predictive model of drug-side effect association by Ordinary Canonical Correlation Analysis (OCCA) and Sparse Canonical Correlation Analysis (SCCA) [1, 22]. The input feature of OCCA and SCCA is extracted from chemical structures of drugs. Cheng and Wang proposed the Phenotypic Network Inference Model (PNIM) [23] to detect new potential drug-side effect associations. Mizutani and Stoven [24] utilized cooccurrence of drug profiles and protein interaction profiles to predict side effects. The Support Vector Machine (SVM) was used to build Adverse Drug Reaction (ADR) prediction, which is based on chemical structures, biological properties of drugs, and phenotypic characteristics [25]. Zhang et al. [26–28] built an ensemble method, which was based on the Integrated Neighborhood-Based Method (INBM) and Restricted Boltzmann Machine-Based Method (RBMBM). Matrix Factorization- (MF-) based methods have been widely used for link prediction in bipartite networks of systems biology. To predict drug-target interactions, Neighborhood Regularized Logistic Matrix Factorization (NRLMF) [29], Collaborative Matrix Factorization (CMF) [30], and Graph Regularized Matrix Factorization (GRMF) [31] were developed via the MF theory.
In our study, we develop a Triple Matrix Factorization- (TMF-) based model to identify the associations of drug and side effect. TMF employs the biprojection matrix and two latent feature matrices (from drug and side effect space) to estimate the strength of new drug-side effect associations. Latent feature matrices are built via Low Rank Approximation (LRA), which could construct a lower rank matrix to approximate the original matrix. To improve the performance of prediction, Kernel Target Alignment-based Multiple Kernel Learning (KTA-MKL) is used to integrate multiple kernel matrices in drug and side effect space, respectively. Our method can fuse multivariate information (multiple kernels) and obtain new associations through matrix projection. Compared with other existing methods, our model obtains better performance on three benchmark datasets.
2. Method
2.1. Problem Description
The dataset of drug-side effect association can be regarded as a bipartite network, which has n drugs and m side effects. The relationships of drug and side effect can be represented as a n × m adjacent matrix Y ∈ Rn×m. D = {d1, d2, ⋯, dn} and S = {s1, s2, ⋯, sm} are the drug and side effect sets, respectively. Yi,j = 1 denotes that drug di and side effect sj are related; otherwise, it is 0. The associations between drugs and side effect terms are shown in Figure 1. The solid lines link the known drug-side effect associations. The hollow circles and filled squares are drugs and side effects, respectively. The prediction of new associations is a recommender task.
Figure 1.
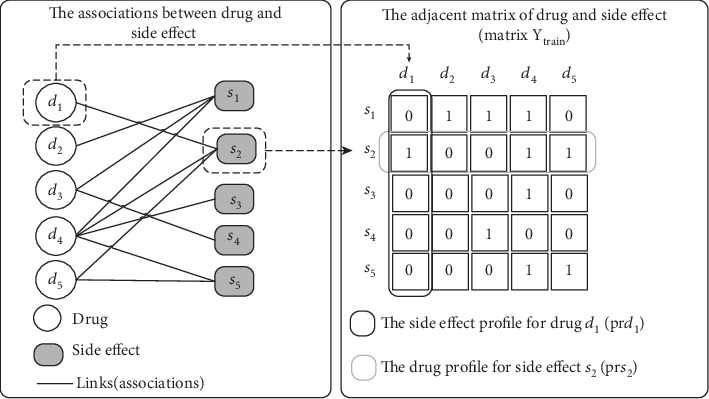
The schematic diagram of associations between drugs and side effects.
2.2. Drug Kernels and Side Effect Kernels
To predict the associations of drugs and side effects, we need to construct the relationship between drugs (or side effects). In this study, we build different kernels (similarity matrices) to describe the relationships of drugs (or side effects). In drug space, the fingerprint of 881 chemical substructures is employed to encode the drug chemical structure, which is shown in Figure 2. The fingerprint represents whether some substructures are present (1) or absent (0). What is more, the known links between drugs and side effect terms (a side effect profile for a specific drug) are also used to represent the information of the subjacent network, which is shown on the right side of Figure 1. In side effect space, the drug profile for a side effect also represents the subjacent network of side effects.
Figure 2.
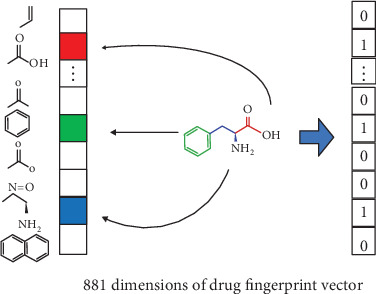
An example of the fingerprint vector.
There are four different types of measure functions, including Gaussian Interaction Profile (GIP) kernel [32–35], COsine Similarity (COS) [26], Correlation coefficient (Corr) [26], and Mutual Information (MI) [36, 37], which are employed to calculate the similarity between the drugs (or side effects).
For drug di and dk, the GIP kernel is defined as follows:
| (1) |
where γ is the bandwidth of the Gaussian kernel. γ is set as 1 in our study. prdi and prdk are the side effect profile of drug di and dk, respectively.
The COS is defined as follows:
| (2) |
The Corr kernel is calculated as follows:
| (3) |
In order to describe the degree of correlation between two random variables, we further use Mutual Information (MI) to measure the similarity between the two random variables:
| (4) |
where f(u) (f(v)) denotes the observed frequency of value u (v) in profile prdi (prdk). f(u, v) is the observed relative frequency. Similarly, the kernels of the fingerprint (drug space: KGIP−chem,d, KCOS−chem,d, KCorr−chem,d, and KMI−chem,d) and drug profile of side effects (side effect space: KGIP−link,s, KCOS−link,s, KCorr−link,s, and KMI−link,s) can be constructed via the above functions. The drug space has 8 kernels, and the side effect space has 4 kernels, which are listed in Table 1.
Table 1.
Summary of kernels for two feature spaces.
| Chemical fingerprint (drug space) | Side effect profiles (drug space) | Drug profiles (side effect space) | |
|---|---|---|---|
| GIP | K GIP−chem,d | K GIP−link,d | K GIP−link,s |
| COS | K COS−chem,d | K COS−link,d | K COS−link,s |
| Corr | K Corr−chem,d | K Corr−link,d | K Corr−link,s |
| MI | K MI−chem,d | K MI−link,d | K MI−link,s |
2.3. Kernel Target Alignment-Based Multiple Kernel Learning
In our study, the kernel sets for drug space Kd = {K1,d, K2,d, ⋯, Kkd,d} and side effect space Ks = {K1,s, K2,s, ⋯, Kks,s} are combined via multiple kernel learning, respectively. kd and ks are the number of kernels in drug and side effect space, respectively. A heuristic approach of Kernel Target Alignment-based Multiple Kernel Learning (KTA-MKL) [38, 39] is employed to calculate the weights of each kernel. The optimal kernels of Kd∗ and Ks∗ can be obtained as follows:
| (5) |
where βd = {β1,d, β2,d, ⋯, βkd,d} and βs = {β1,s, β2,s, ⋯, βks,s} are the weights of kernels in drug and side effect space, respectively. KTA-MKL estimates the weight of each kernel by COsine Similarity of matrices (drug space):
| (6) |
where denotes the Frobenius norm. 〈P, Q〉F = Trace(PTQ) is the Frobenius inner product. The value of kernel alignment can describe similarity of two kernels. KTA-MKL estimates the value between the ideal kernel matrix and the drug kernel (or side effect kernel) as follows:
| (7) |
where Kideal,d = YtrainYtrainT ∈ Rn×n and Kideal,s = YtrainTYtrain ∈ Rm×m are the ideal kernels of drug and side effect, respectively, which are built via the training label (known associations).
2.4. Triple Matrix Factorization-Based Model
Inspired by MF [29–31, 40], the similarity between drugs (or side effects) can be approximated by the inner product of two drug (or side effect) features as follows:
| (8) |
where A and B are the matrices of Low Rank Approximation and rd and rs are the dimensions of the latent feature space in drug and side effect space, respectively. The objective function of TMF is defined as follows:
| (9) |
where Θ ∈ Rrd×rs is the biprojection matrix. λ is the regularization coefficient of Θ. In our study, λ is set as 1.
Let ∂J/∂Θ = 0, so we can obtain functions as follows:
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
where Equation (14) is a Sylvester equation. The final prediction can be constructed by
| (15) |
The overview of our proposed method is shown in Figure 3 and Algorithm 1.
Figure 3.
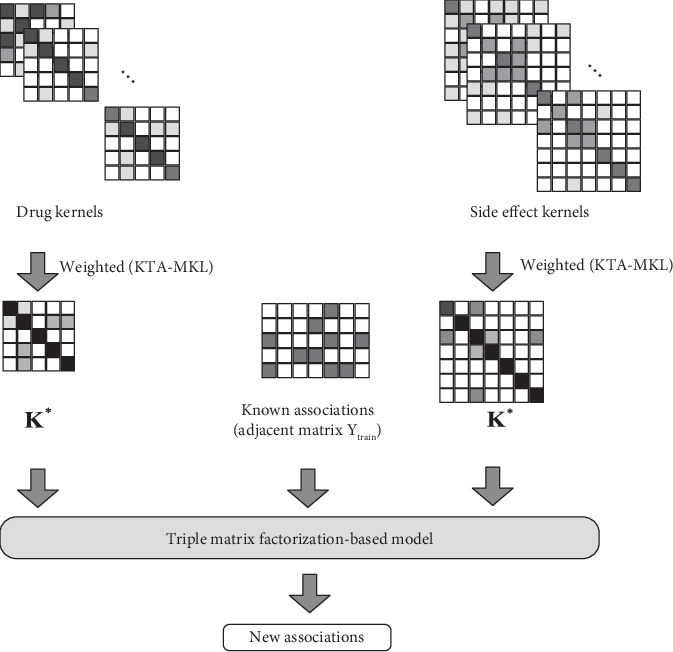
Overview of our method.
Algorithm 1.

Algorithm of our method.
3. Result
In this section, we employed benchmark dataset to evaluate our approach and compared it with other existing methods.
3.1. Datasets
In order to test the performance of our model, three types of datasets are employed in our study. They are Pauwels's dataset, Liu's dataset, and Mizutani's dataset, which are collected from the DrugBank [41], SIDe Effect Resource (SIDER) [42], KEGG DRUG [43], and PubChemCompound [44, 45]. Table 2 lists benchmark datasets of this study.
Table 2.
Three benchmark datasets.
| Datasets | Drugs | Side effects | Associations |
|---|---|---|---|
| Pauwels's dataset | 888 | 1385 | 61,102 |
| Mizutani's dataset | 658 | 1339 | 49,051 |
| Liu's dataset | 832 | 1385 | 59,205 |
3.2. Evaluation Measurements
The training adjacent matrix can be obtained via randomly setting known associations as 0. In this study, we use 5-fold Cross-Validation (5-CV) and 5-fold local Cross-Validation (5 local CV) to test our method. 5-CV randomly sets known associations as 0 in the whole matrix. 5 local CV is employed to evaluate the prediction of new drugs, which do not have any side effect information. 5 local CV sets some rows of the adjacent matrix as 0 to test related drugs. The Area Under the Precision-Recall curve (AUPR) and Area Under the receiver operating Characteristic curve (AUC) are utilized to evaluate the performance of prediction.
3.3. Selecting Optimal Parameters
In this section, we use the grid search method to get the optimal rd and rs. We test different values of and from 100 to the max value with the step of 100. The results of the grid search method are shown in Figure 4 (on Mizutani's dataset by 5-CV). rd = 700 and rs = 800 are the best parameters (AUPR) on Mizutani's dataset. In Figure 4, the lower value of AURP and AUC is blue, and the higher value is yellow. On the other two datasets, we use the same parameters of rd and rs.
Figure 4.

The AUC and AUPR values (under different rd and rs).
3.4. Performance of Different Kernels
We evaluate the performance of multiple kernels and single kernel on three datasets. The results of prediction are listed in Table 3 and Figure 5. Obviously, the kernels of KMI−link,d and KMI−link,s have better performance on Pauwels's dataset (AUPR: 0.6557, AUC: 0.9079), Mizutani's dataset (AUPR: 0.6615, AUC: 0.9369), and Liu's dataset (AUPR: 0.6587, AUC: 0.9408). In addition, the KTA-MKL model achieves the best results on Pauwels's dataset (AUPR: 0.6765, AUC: 0.9434), Mizutani's dataset (AUPR: 0.6847, AUC: 0.9409), and Liu's dataset (AUPR: 0.6801, AUC: 0.9426), respectively. KTA-MKL could combine kernels from different sources via the heuristic method, which is better mean weighted.
Table 3.
The performance of different kernels via 5-fold Cross-Validation.
| Models | Pauwels's dataset | Mizutani's dataset | Liu's dataset | |||
|---|---|---|---|---|---|---|
| AUPR | AUC | AUPR | AUC | AUPR | AUC | |
| K GIP−chem,d & KGIP−link,sa | 0.4420 | 0.8950 | 0.4735 | 0.9148 | 0.4718 | 0.9145 |
| K COS−chem,d & KCOS−link,sa | 0.4892 | 0.8994 | 0.5343 | 0.9070 | 0.5224 | 0.9067 |
| K Corr−chem,d & KCorr−link,sa | 0.4994 | 0.8981 | 0.5217 | 0.9005 | 0.5143 | 0.9026 |
| K MI−chem,d & KMI−link,sa | 0.4978 | 0.9079 | 0.5591 | 0.9214 | 0.5529 | 0.9238 |
| K GIP−link,d & KGIP−link,sb | 0.6254 | 0.9300 | 0.6623 | 0.9376 | 0.6574 | 0.9398 |
| K COS−link,d & KCOS−link,sb | 0.5861 | 0.9035 | 0.6324 | 0.9090 | 0.6252 | 0.9087 |
| K Corr−link,d & KCorr−link,sb | 0.5833 | 0.8999 | 0.6123 | 0.9014 | 0.6047 | 0.9013 |
| K MI−link,d & KMI−link,sb | 0.6557 | 0.9428 | 0.6615 | 0.9369 | 0.6587 | 0.9408 |
| Mean weightedc | 0.6598 | 0.9353 | 0.6724 | 0.9280 | 0.6651 | 0.9285 |
| KTA-MKLc | 0.6765 | 0.9434 | 0.6847 | 0.9409 | 0.6801 | 0.9426 |
aThe TMF uses the drug fingerprint and drug profile for side effects. bThe TMF uses the side effect profile for drugs and drug profile for side effects. cThe TMF uses the drug fingerprint, side effect profile for drugs, and drug profile for side effects.
Figure 5.
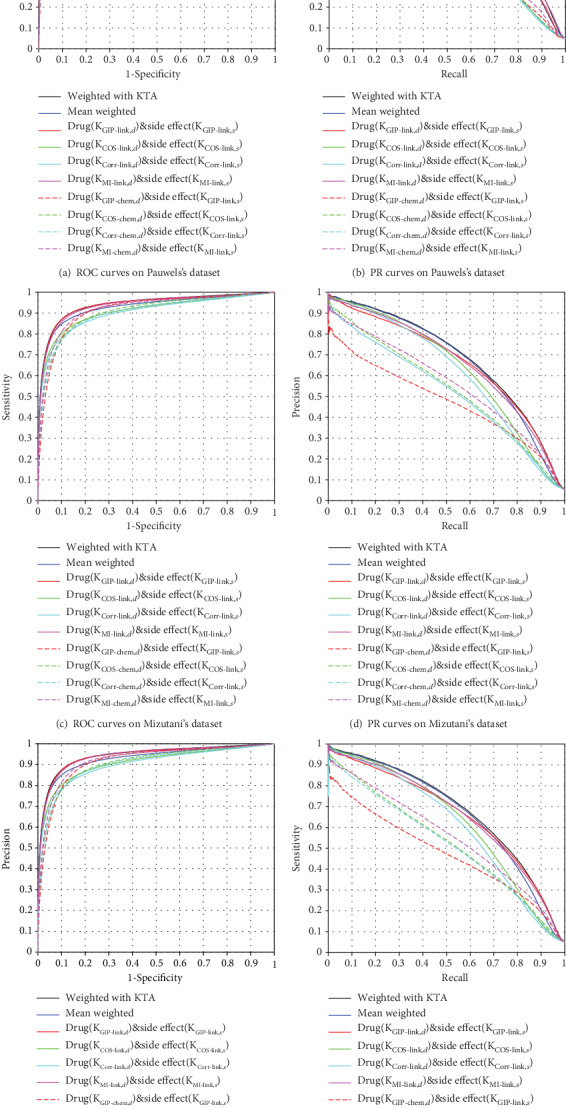
The ROC and PR curves of different models (single kernel and multiple kernels).
In Table 4, we list the weight of each kernel on three datasets. We can find that the weights of KMI−link,d and KMI−link,s are the highest than other kernels. At the same time, their performance is also the best. KTA-MKL could reduce bias of kernels by the low weights.
Table 4.
The kernel weights on three datasets.
| Kernel | Pauwels's dataset | Mizutani's dataset | Liu's dataset |
|---|---|---|---|
| K GIP−chem,d | 0.1159 | 0.1168 | 0.1167 |
| K COS−chem,d | 0.1224 | 0.1226 | 0.1226 |
| K Corr−chem,d | 0.1200 | 0.1203 | 0.1203 |
| K MI−chem,d | 0.1113 | 0.1122 | 0.1116 |
| K GIP−link,d | 0.0596 | 0.0621 | 0.0613 |
| K COS−link,d | 0.1538 | 0.1533 | 0.1528 |
| K Corr−link,d | 0.1507 | 0.1498 | 0.1497 |
| K MI−link,d | 0.1664 | 0.1628 | 0.1650 |
| K GIP−link,s | 0.0151 | 0.0173 | 0.0152 |
| K COS−link,s | 0.3286 | 0.3374 | 0.3380 |
| K Corr−link,s | 0.2909 | 0.2865 | 0.2855 |
| K MI−link,s | 0.3654 | 0.3588 | 0.3613 |
3.5. Comparison with Existing Methods
To evaluate the performance of the TMF model, we compare it with other methods. The results are listed in Table 5. Obviously, our method (TMF) achieves the best results on Pauwels's dataset (AUPR: 0.677), Mizutani's dataset (AUPR: 0.685), and Liu's dataset (AUPR: 0.680). Zhang et al.'s work (ensemble model) [26] obtained the good performance of AUPRs (0.660, 0.666, and 0.661). The best AUCs (0.954, 0.950, and 0.953) are achieved by Neighborhood Regularized Logistic Matrix Factorization (NRLMF) [29], which is also based on Matrix Factorization (MF). The results of other MF-based models, including Collaborative Matrix Factorization (CMF) [30] and Graph Regularized Matrix Factorization (GRMF) [31], are competitive. Local and Global Consistency (LGC) [18] is our previous work. LGC obtains the second best results of AUPR (0.668, 0.673, and 0.670) on three datasets, respectively.
Table 5.
Comparison to existing methods via 5-fold Cross-Validation.
| Datasets | Methods | AUPR | AUC |
|---|---|---|---|
| Pauwels | Pauwels's methoda | 0.389 ± N/A | 0.897 ± N/A |
| Liu's methoda | 0.345 ± N/A | 0.920 ± N/A | |
| Cheng's methoda | 0.588 ± N/A | 0.922 ± N/A | |
| RBMBMa [26] | 0.612 ± N/A | 0.941 ± N/A | |
| INBMa [26] | 0.641 ± N/A | 0.934 ± N/A | |
| Ensemble modela [26] | 0.660 ± N/A | 0.949 ± N/A | |
| CMFb | 0.646 ± 0.007 | 0.939 ± 0.005 | |
| GRMFb | 0.643 ± 0.006 | 0.937 ± 0.005 | |
| NRLMFb | 0.654 ± 0.005 | 0.954 ± 0.005 | |
| LGCb | 0.668 ± 0.008 | 0.952 ± 0.007 | |
| Our method | 0.677 ± 0.004 | 0.943 ± 0.003 | |
|
| |||
| Mizutani | Mizutani's methoda | 0.412 ± N/A | 0.890 ± N/A |
| Liu's methoda | 0.366 ± N/A | 0.918 ± N/A | |
| Cheng's methoda | 0.599 ± N/A | 0.923 ± N/A | |
| RBMBMa [26] | 0.619 ± N/A | 0.939 ± N/A | |
| INBMa [26] | 0.646 ± N/A | 0.932 ± N/A | |
| Ensemble modela [26] | 0.666 ± N/A | 0.946 ± N/A | |
| CMFb | 0.645 ± 0.005 | 0.938 ± 0.006 | |
| GRMFb | 0.646 ± 0.007 | 0.937 ± 0.007 | |
| NRLMFb | 0.660 ± 0.006 | 0.950 ± 0.005 | |
| LGCb | 0.673 ± 0.007 | 0.948 ± 0.007 | |
| Our method | 0.685 ± 0.006 | 0.941 ± 0.008 | |
|
| |||
| Liu | Liu's methoda | 0.278 ± N/A | 0.907 ± N/A |
| Cheng's methoda | 0.592 ± N/A | 0.922 ± N/A | |
| RBMBMa [26] | 0.616 ± N/A | 0.941 ± N/A | |
| INBMa [26] | 0.641 ± N/A | 0.934 ± N/A | |
| Ensemble modela [26] | 0.661 ± N/A | 0.948 ± N/A | |
| CMFb | 0.649 ± 0.006 | 0.938 ± 0.005 | |
| GRMFb | 0.650 ± 0.007 | 0.938 ± 0.008 | |
| NRLMFb | 0.656 ± 0.005 | 0.953 ± 0.006 | |
| LGCb | 0.670 ± 0.008 | 0.951 ± 0.007 | |
| Our method | 0.680 ± 0.005 | 0.943 ± 0.006 | |
3.6. Local CV and Case Study
In some cases, certain drugs are new and have no information of side effects. The 5 local CV is employed to test the performance of the side effect prediction for new drugs. In this section, we also compare TMF with other MF-based models, including NRLMF, CMF, and GRMF. The results are listed in Table 6 and Figure 6.
Table 6.
Comparison with MF-based models via 5-fold local Cross-Validation.
| Datasets | Methods | AUPR | AUC |
|---|---|---|---|
| Pauwels | CMF∗ | 0.382 ± 0.006 | 0.894 ± 0.004 |
| GRMF∗ | 0.358 ± 0.008 | 0.883 ± 0.005 | |
| NRLMF∗ | 0.374 ± 0.007 | 0.886 ± 0.004 | |
| Our method | 0.392 ± 0.008 | 0.889 ± 0.004 | |
|
| |||
| Mizutani | CMF∗ | 0.395 ± 0.005 | 0.889 ± 0.004 |
| GRMF∗ | 0.392 ± 0.008 | 0.890 ± 0.006 | |
| NRLMF∗ | 0.390 ± 0.006 | 0.882 ± 0.005 | |
| Our method | 0.399 ± 0.013 | 0.886 ± 0.003 | |
|
| |||
| Liu | CMF∗ | 0.393 ± 0.007 | 0.894 ± 0.005 |
| GRMF∗ | 0.379 ± 0.008 | 0.895 ± 0.006 | |
| NRLMF∗ | 0.398 ± 0.006 | 0.897 ± 0.004 | |
| Our method | 0.401 ± 0.015 | 0.891 ± 0.004 | |
∗Results are derived from [18].
Figure 6.
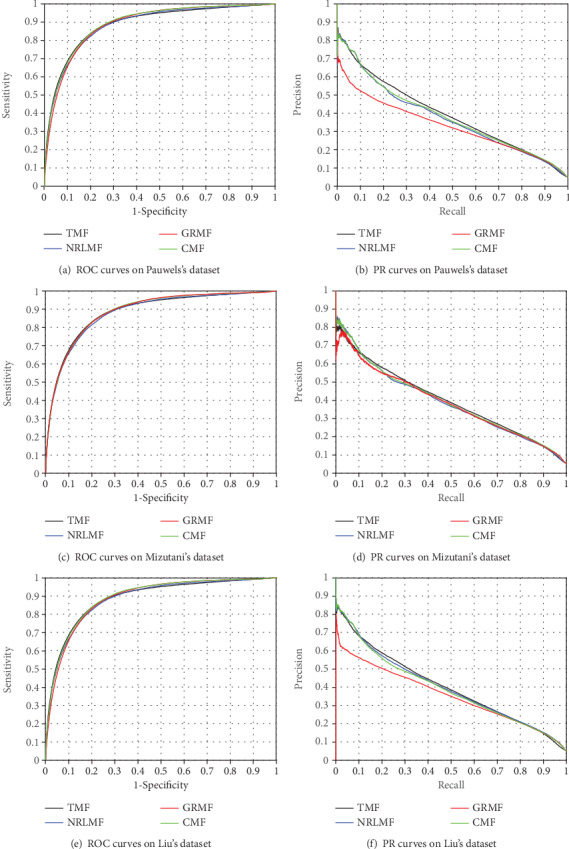
The ROC and PR curves of different methods via 5 local CV.
The proposed method (TMF) achieves the best results of AUPRs on Pauwels's dataset (AUPR: 0.392), Mizutani's dataset (AUPR: 0.399), and Liu's dataset (AUPR: 0.401). Other MF-based models also are still comparable with our results. NRLMF obtains AUPRs of 0.374, 0.390, and 0.398 on three datasets, respectively.
To predict the side effects of a new drug, our model calculates the strength of associations between the new drug and all existing side effects. The predictive strength scores of TMF will be ranked by descending order. The higher the value of the score, the higher the possibility of associations. In this section, we discuss two cases (drug caffeine and captopril on Mizutani's dataset) of top 10 associations predicted. The details are listed in Tables 7 and 8. Results are checked by the masked associations between drug caffeine (or captopril) and side effects.
Table 7.
Top 10 ranks of predictive side effects for drug caffeine.
| Side effect | Score | Ranks | Confirmed |
|---|---|---|---|
| Diarrhea | 0.3992 | 1 | Yes |
| Diabetic neuropathy | 0.3893 | 2 | Yes |
| Varicocele | 0.3844 | 3 | Yes |
| Gynecomastia | 0.3815 | 4 | Yes |
| Conjunctivitis | 0.3794 | 5 | Yes |
| Telangiectasia | 0.3737 | 6 | No |
| Lump | 0.3663 | 7 | Yes |
| Dyskinesia | 0.3638 | 8 | No |
| Palpitations | 0.3632 | 9 | No |
| Fecal incontinence | 0.3563 | 10 | Yes |
Table 8.
Top 10 ranks of predictive side effects for drug captopril.
| Side effect | Score | Ranks | Confirmed |
|---|---|---|---|
| Diarrhea | 0.4150 | 1 | No |
| Diabetic neuropathy | 0.4043 | 2 | Yes |
| Varicocele | 0.4004 | 3 | Yes |
| Conjunctivitis | 0.3973 | 4 | Yes |
| Gynecomastia | 0.3938 | 5 | Yes |
| Myoglobinuria | 0.3885 | 6 | No |
| Esophageal varices | 0.3854 | 7 | Yes |
| Lump | 0.3806 | 8 | Yes |
| Palpitations | 0.3770 | 9 | No |
| Eclampsia | 0.3674 | 10 | Yes |
3.7. Running Time
We evaluate the performance for predictive models of running time. The results of test are listed in Table 9. The running time of CMF is less than our method (TMF), LGC, GRMF, and NRLMF on Pauwels's dataset (910 seconds), Mizutani's dataset (757 seconds), and Liu's dataset (846 seconds). TMF costs 977, 873, and 929 seconds, which are less than the ensemble model [26].
Table 9.
The running time (seconds) via 5-fold Cross-Validation.
4. Conclusion and Discussion
In this study, we develop a Triple Matrix Factorization-based model to predict the associations between drugs and side effect terms. In drug space, several kernels are constructed from the chemical substructure fingerprint and known side effect-associated subnet. The side effect kernels are built from the known drug-associated subnet. The kernel functions include GIP, COS, Corr, and MI. Above kernels are combined by KTA-MKL in drug and side effect space, respectively. The integrated kernel matrices (including drug and side effect) are Low Rank Approximation in the TMF model. Our model (TMF) is tested on three benchmark datasets of drug-side effect association. Compared with other excellent methods, TMF achieves the best results (5-CV) on Pauwels's dataset (AUPR: 0.677), Mizutani's dataset (AUPR: 0.685), and Liu's dataset (AUPR: 0.680), respectively. In addition, our model is also compared with CMF, GRMF, and NRLMF under 5 local CV. The best AUPRs are achieved on Pauwels's dataset (AUPR: 0.392), Mizutani's dataset (AUPR: 0.399), and Liu's dataset (AUPR: 0.401). However, our method does not consider the topological relationship of drugs or side effects. In the future, a graph- or hypergraph-embedded MF-based model will be developed to improve the predictive performance of drug-side effect association.
Acknowledgments
This work is supported by a grant from the National Science Foundation of China (NSFC 61772362, 61902271, and 61972280) and the Natural Science Research of Jiangsu Higher Education Institutions of China (19KJB520014). The authors also thank professor Wen Zhang for kindly providing the datasets on his website.
Contributor Information
Yan Yu, Email: rush19830127@163.com.
Yijie Ding, Email: wuxi_dyj@163.com.
Fei Guo, Email: fguo@tju.edu.cn.
Data Availability
The datasets, codes and corresponding results are available at https://figshare.com/s/10ee9c07123304a0ef82.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Pauwels E., Stoven V., Yamanishi Y. Predicting drug side-effect profiles: a chemical fragment-based approach. BMC Bioinformatics. 2011;12(1):169–181. doi: 10.1186/1471-2105-12-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang L., Xiao Y., Ding Y., Tang J., Guo F. FKL-Spa-LapRLS: an accurate method for identifying human microRNA-disease association. BMC Genomics. 2018;19(Supplement 10):p. 911. doi: 10.1186/s12864-018-5273-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeng X., Liu L., Lü L., Zou Q. Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics. 2018;34(14):2425–2432. doi: 10.1093/bioinformatics/bty112. [DOI] [PubMed] [Google Scholar]
- 4.Zhao Q., Yang Y., Ren G., Ge E., Fan C. Integrating bipartite network projection and KATZ measure to identify novel circRNA-disease associations. IEEE Transactions on NanoBioscience. 2019;18(4):578–584. doi: 10.1109/tnb.2019.2922214. [DOI] [PubMed] [Google Scholar]
- 5.Jia C., Zuo Y., Zou Q. O-GlcNAcPRED-II: an integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique. Bioinformatics. 2018;34(12):2029–2036. doi: 10.1093/bioinformatics/bty039. [DOI] [PubMed] [Google Scholar]
- 6.Wei L., Luan S., Nagai L. A. E., Su R., Zou Q. Exploring sequence-based features for the improved prediction of DNA N4-methylcytosine sites in multiple species. Bioinformatics. 2019;35(8):1326–1333. doi: 10.1093/bioinformatics/bty824. [DOI] [PubMed] [Google Scholar]
- 7.Zou Q., Xing P., Wei L., Liu B. Gene2vec: gene subsequence embedding for prediction of mammalian N6-methyladenosine sites from mRNA. RNA. 2019;25(2):205–218. doi: 10.1261/rna.069112.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding Y., Tang J., Guo F. Protein crystallization identification via fuzzy model on linear neighborhood representation. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2019;1 doi: 10.1109/TCBB.2019.2954826. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y., Ding Y., Tang J., Dai Y., Guo F. CrystalM: a multi-view fusion approach for protein crystallization prediction. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2019;1 doi: 10.1109/TCBB.2019.2912173. [DOI] [PubMed] [Google Scholar]
- 10.Wang H., Ding Y., Tang J., Guo F. Identification of membrane protein types via multivariate information fusion with Hilbert–Schmidt independence criterion. Neurocomputing. 2020;383:257–269. doi: 10.1016/j.neucom.2019.11.103. [DOI] [Google Scholar]
- 11.Shen Y., Ding Y., Tang J., Zou Q., Guo F. Critical evaluation of web-based prediction tools for human protein subcellular localization. Briefings in Bioinformatics. 2019 doi: 10.1093/bib/bbz106. [DOI] [PubMed] [Google Scholar]
- 12.Wei L., Ding Y., Su R., Tang J., Zou Q. Prediction of human protein subcellular localization using deep learning. Journal of Parallel and Distributed Computing. 2018;117:212–217. doi: 10.1016/j.jpdc.2017.08.009. [DOI] [Google Scholar]
- 13.Liu B., Jiang S., Zou Q. HITS-PR-HHblits: protein remote homology detection by combining PageRank and hyperlink-induced topic search. Briefings in Bioinformatics. 2020;21(1):298–308. doi: 10.1093/bib/bby104. [DOI] [PubMed] [Google Scholar]
- 14.Qu K., Guo F., Liu X., Lin Y., Zou Q. Application of machine learning in microbiology. Frontiers in Microbiology. 2019;10:p. 827. doi: 10.3389/fmicb.2019.00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ru X., Li L., Zou Q. Incorporating distance-based top-n-gram and random forest to identify electron transport proteins. Journal of Proteome Research. 2019;18(7):2931–2939. doi: 10.1021/acs.jproteome.9b00250. [DOI] [PubMed] [Google Scholar]
- 16.Ding Y., Tang J., Guo F. Identification of drug-target interactions via fuzzy bipartite local model. Neural Computing and Applications. 2019 doi: 10.1007/s00521-019-04569-z. [DOI] [Google Scholar]
- 17.Ding Y., Tang J., Guo F. Identification of drug-side effect association via multiple information integration with centered kernel alignment. Neurocomputing. 2019;325:211–224. doi: 10.1016/j.neucom.2018.10.028. [DOI] [Google Scholar]
- 18.Ding Y., Tang J., Guo F. Identification of drug-side effect association via semi-supervised model and multiple kernel learning. IEEE Journal of Biomedical and Health Informatics. 2019;23(6):2619–2632. doi: 10.1109/JBHI.2018.2883834. [DOI] [PubMed] [Google Scholar]
- 19.Ding Y., Tang J., Guo F. Predicting protein-protein interactions via multivariate mutual information of protein sequences. BMC Bioinformatics. 2016;17(1):p. 398. doi: 10.1186/s12859-016-1253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding Y., Tang J., Guo F. Identification of protein-protein interactions via a novel matrix-based sequence representation model with amino acid contact information. International Journal of Molecular Sciences. 2016;17(10):p. 1623. doi: 10.3390/ijms17101623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu H., Ren G., Chen H., Liu Q., Yang Y., Zhao Q. Predicting lncRNA–miRNA interactions based on logistic matrix factorization with neighborhood regularized. Knowledge-Based Systems. 2020;191:p. 105261. doi: 10.1016/j.knosys.2019.105261. [DOI] [Google Scholar]
- 22.Yamanishi Y., Pauwels E., Kotera M. Drug side-effect prediction based on the integration of chemical and biological spaces. Journal of Chemical Information and Modeling. 2012;52(12):3284–3292. doi: 10.1021/ci2005548. [DOI] [PubMed] [Google Scholar]
- 23.Cheng F., Li W., Wang X., et al. Adverse drug events: database construction and in silico prediction. Journal of Chemical Information and Modeling. 2013;53(4):744–752. doi: 10.1021/ci4000079. [DOI] [PubMed] [Google Scholar]
- 24.Mizutani S., Pauwels E., Stoven V., Goto S., Yamanishi Y. Relating drug-protein interaction network with drug side effects. Bioinformatics. 2012;28(18):i522–i528. doi: 10.1093/bioinformatics/bts383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu M., Wu Y., Chen Y., et al. Large-scale prediction of adverse drug reactions using chemical, biological, and phenotypic properties of drugs. Journal of the American Medical Informatics Association. 2012;19(1):28–35. doi: 10.1136/amiajnl-2011-000699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W., Zou H., Luo L., Liu Q., Wu W., Xiao W. Predicting potential side effects of drugs by recommender methods and ensemble learning. Neurocomputing. 2016;173(P3):979–987. doi: 10.1016/j.neucom.2015.08.054. [DOI] [Google Scholar]
- 27.Zhang W., Liu F., Luo L., Zhang J. Predicting drug side effects by multi-label learning and ensemble learning. BMC Bioinformatics. 2015;16(1):365–375. doi: 10.1186/s12859-015-0774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W., Yue X., Liu F., Chen Y., Tu S., Zhang X. A unified frame of predicting side effects of drugs by using linear neighborhood similarity. BMC Systems Biology. 2017;11(6):23–34. doi: 10.1186/s12918-017-0477-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y., Wu M., Miao C., Zhao P., Li X.-L. Neighborhood regularized logistic matrix factorization for drug-target interaction prediction. PLoS Computational Biology. 2016;12(2, article e1004760) doi: 10.1371/journal.pcbi.1004760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng X., Ding H., Mamitsuka H., Zhu S. Collaborative matrix factorization with multiple similarities for predicting drug-target interactions. ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 2013. pp. 1025–1033.
- 31.Ezzat A., Zhao P., Wu M., Li X.-L., Kwoh C.-K. Drug-target interaction prediction with graph regularized matrix factorization. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2016;14(3):646–656. doi: 10.1109/TCBB.2016.2530062. [DOI] [PubMed] [Google Scholar]
- 32.van Laarhoven T., Nabuurs S. B., Marchiori E. Gaussian interaction profile kernels for predicting drug–target interaction. Bioinformatics. 2011;27(21):3036–3043. doi: 10.1093/bioinformatics/btr500. [DOI] [PubMed] [Google Scholar]
- 33.Nascimento A. C. A., Prudêncio R. B. C., Costa I. G. A multiple kernel learning algorithm for drug-target interaction prediction. BMC Bioinformatics. 2016;17(1):46–61. doi: 10.1186/s12859-016-0890-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Laarhoven T., Marchiori E. Predicting drug-target interactions for new drug compounds using a weighted nearest neighbor profile. Plos One. 2013;8(6, article e66952) doi: 10.1371/journal.pone.0066952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang W., Chen Y., Li D. Drug-target interaction prediction through label propagation with linear neighborhood information. Molecules. 2017;22(12):2056–2069. doi: 10.3390/molecules22122056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerf N. J., Adami C. Information theory of quantum entanglement and measurement. Physica D Nonlinear Phenomena. 1998;120(1-2):62–81. doi: 10.1016/S0167-2789(98)00045-1. [DOI] [Google Scholar]
- 37.Peng H., Long F., Ding C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Transactions on Pattern Analysis and Machine Intelligence. 2005;27(8):1226–1238. doi: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- 38.Cristianini N., Kandola J., Elisseeff A., Shawe-Taylor J. On kernel-target alignment. Advances in Neural Information Processing Systems. 2001;179(5):367–373. [Google Scholar]
- 39.Lanckriet G. R., Cristianini N., Bartlett P., Ghaoui L. E., Jordan M. I. Learning the kernel matrix with semidefinite programming. Journal of Machine Learning Research. 2002;5(1):27–72. [Google Scholar]
- 40.Shi J.-Y., Zhang A.-Q., Zhang S.-W., Mao K.-T., Yiu S.-M. A unified solution for different scenarios of predicting drug-target interactions via triple matrix factorization. BMC Systems Biology. 2018;12(S9) Supplement 9:p. 136. doi: 10.1186/s12918-018-0663-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wishart D. S. Drugbank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Research. 2006;34(Database issue):668–672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuhn M., Campillos M., Letunic I., Jensen L. J., Bork P. A side effect resource to capture phenotypic effects of drugs. Molecular Systems Biology. 2010;6(1):p. 343. doi: 10.1038/msb.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanehisa M., Goto S., Furumichi M., Tanabe M., Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Research. 2010;38(Supplement 1):D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y., Xiao J., Suzek T. O., Zhang J., Wang J., Bryant S. H. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Research. 2009;37(Web Server):W623–W633. doi: 10.1093/nar/gkp456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Q., Cheng T., Wang Y., Bryant S. H. PubChem as a public resource for drug discovery. Drug Discovery Today. 2010;15(23-24):1052–1057. doi: 10.1016/j.drudis.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets, codes and corresponding results are available at https://figshare.com/s/10ee9c07123304a0ef82.