

Abstract
A bright spot in the SARS‐CoV‐2 (CoV‐2) coronavirus pandemic has been the immediate mobilization of the biomedical community, working to develop treatments and vaccines for COVID‐19. Rational drug design against emerging threats depends on well‐established methodology, mainly utilizing X‐ray crystallography, to provide accurate structure models of the macromolecular drug targets and of their complexes with candidates for drug development. In the current crisis, the structural biological community has responded by presenting structure models of CoV‐2 proteins and depositing them in the Protein Data Bank (PDB), usually without time embargo and before publication. Since the structures from the first‐line research are produced in an accelerated mode, there is an elevated chance of mistakes and errors, with the ultimate risk of hindering, rather than speeding up, drug development. In the present work, we have used model‐validation metrics and examined the electron density maps for the deposited models of CoV‐2 proteins and a sample of related proteins available in the PDB as of April 1, 2020. We present these results with the aim of helping the biomedical community establish a better‐validated pool of data. The proteins are divided into groups according to their structure and function. In most cases, no major corrections were necessary. However, in several cases significant revisions in the functionally sensitive area of protein–inhibitor complexes or for bound ions justified correction, re‐refinement, and eventually reversioning in the PDB. The re‐refined coordinate files and a tool for facilitating model comparisons are available at https://covid-19.bioreproducibility.org.
Database
Validated models of CoV‐2 proteins are available in a dedicated, publicly accessible web service https://covid-19.bioreproducibility.org
Keywords: coronavirus, COVID‐19, spike glycoprotein, structure‐guided drug discovery, viral proteases
The experimental 3D structure models of SARS‐CoV‐2 proteins and their complexes in the PDB were analyzed, validated, and, where necessary, re‐refined and made publicly available via a dedicated webserver.
Abbreviations
- ACE2
angiotensin‐converting enzyme‐2
- ASU
asymmetric unit
- BMA
β‐D‐mannose
- CoV‐2
coronavirus 2
- COVID‐19
coronavirus disease 2019
- EM
electron microscopy
- HIV
human immunodeficiency virus
- HR
heptad repeat
- MERS
Middle East respiratory syndrome
- NAG
N‐acetyl‐D‐glucosamine
- NDG
2‐(acetylamino)‐2‐deoxy‐α‐D‐glucopyranose
- PanDDA
Pan‐Dataset Density Analysis
- PDB
Protein Data Bank
- RBD
receptor‐binding domain
- rmsd
root‐mean‐square deviation
- SARS
severe acute respiratory syndrome
Introduction
Motto: We should rather light candles than curse the darkness (after Adlai Stevenson’s eulogy for Eleanor Roosevelt).
Structural biology in general, and protein crystallography in particular, has played a crucial role in modern drug discovery. Currently, the development of many new drugs relies on experimentally determined structures of the targeted macromolecules and their complexes with functional ligands or potential inhibitors [1], or by assembling such molecules through fragment screening [2, 3]. Historically, the first spectacular success of structure‐based drug design, achieved in a situation of a looming global pandemic, was the development of retroviral protease inhibitors as drugs for the treatment of HIV infections [4, 5]. Since then, structural biology has repeatedly responded to emerging global threats by furnishing detailed structures of drug targets for pathogens such as SARS [6], MERS [7], Zika [8], and Ebola [9]. It is highly edifying that in reaction to the current pandemic created by the outbreak of the new SARS‐CoV‐2 (hereinafter CoV‐2) coronavirus, structural biologists and other specialists responded instantaneously [10, 11], quickly producing structure models of numerous CoV‐2 components using cryo‐electron microscopy (cryo‐EM) and X‐ray crystallographic techniques [12, 13, 14, 15, 16, 17]. All models of the proteins encoded by CoV‐2 became almost immediately available as drug‐design targets through their deposition into the Protein Data Bank (PDB) [18], without any embargo period and before publication. These models have been subsequently used in large‐scale docking experiments [19], accompanied by extensive media coverage (https://futurism.com/neoscope/fastest‐supercomputer‐finds‐potential‐covid‐treatments; https://www.iflscience.com/technology/worlds‐fastest‐supercomputer‐joins‐the‐battle‐against‐covid19; https://thehill.com/changing‐america/well‐being/prevention‐cures/488627‐the‐worlds‐fastest‐supercomputer‐is‐taking‐on).
There were over 100 CoV‐2‐related deposits released by the PDB on or before April 1, 2020 (including PanDDA groups, see below). A summary of those structures, as well as of selected structures from other coronaviruses that were analyzed in this work, is available at https://covid‐19.bioreproducibility.org/. However, sometimes swiftness in pursuit of scientific discovery—while fully justified in a situation of global emergency—can also impart negative consequences, such as mistakes and errors of different severity, with the ultimate danger of creating false or irreproducible results [20, 21, 22, 23, 24, 25]. Irreproducibility has a detrimental effect on drug discovery and subsequent research efforts by confounding the subject as well as diverting human and other resources in the wrong direction. (https://www.wsj.com/articles/coronavirus‐lessons‐from‐the‐asteroid‐that‐didnt‐hit‐earth‐11585780465).
There are many examples of such errors generated in the past by negligence, lack of supervision, or lack of proper validation of results, which were later corrected, sometimes with the contribution of the present authors [20, 21, 22, 23, 24, 26]. A well‐known case is part of the race to discover the structure of retroviral protease, where a model with incorrectly folded C terminus thwarted proper understanding of the autoexcision process [27]. Scientific urgency, coupled with the competition to be the first, sometimes leads to cutting corners and insufficient attention to detail. Paradoxically, the development of more powerful software had enabled acceptance criteria previously considered good to be achieved for suboptimal models, giving people a false sense of security and even leading to high‐profile retractions. This in turn prompted the development of modern model‐validation tools.
Results
Re‐examination of structure models
In the present work, we have re‐examined the CoV‐2 protein structures deposited in the PDB, with focus on validating the small‐molecule ligands modeled in those structures. If there were reasons to believe that a model could be revised in order to help subsequent biomedical research, we re‐refined the structure, with the intention of redepositing it together with the original authors in the PDB. Temporarily, and in the interest of time, the corrected models have been made publicly available in a dedicated database. Whenever necessary and possible, we intend to start our re‐analysis from reprocessing of the original diffraction images [28]. However, quite often we were unable to obtain the diffraction data despite the IUCr recommendation [29] and an earnest appeal from the community to make diffraction data related to CoV‐2 public (http://phenix‐online.org/pipermail/phenixbb/2020‐March/024556.html). In one case, our request resulted not in deposition of the diffraction data but in redeposition by the original authors of a revised model, 6YB7, superseding the PDB deposit 6Y84. Unfortunately, the superseded entry is still present in the PDB and neither structure is supported by publicly available diffraction data.
Currently, several groups are working to make structural results related to COVID‐19 more easily accessible to biomedical researchers. This work includes, but is not limited to, validation of structural models, highlighting small molecule‐binding sites and protein–protein interactions, reprocessing and re‐refinement of selected SARS‐CoV‐2 models, and computational modeling and simulations. Beyond obvious institutions like the PDB and PDBe, the most notable are Jane and David Richardson’s group at Duke University (http://kinemage.biochem.duke.edu/), Andrea Thorn’s group at University of Würzburg (https://github.com/thorn‐lab/coronavirus_structural_task_force), Gerard Bricogne’s group at Global Phasing (https://www.globalphasing.com/buster/wiki/index.cgi?Covid19), Yang Zhang’s group at University of Michigan (https://zhanglab.ccmb.med.umich.edu/COVID‐19/), Adam Godzik’s group at University of Riverside (https://coronavirus3d.org/), and others including the authors of the present paper (https://covid‐19.bioreproducibility.org/). Roughly 27% depositors of CoV‐2 PDB entries have submitted experimental diffraction images to the Integrated Resource for Reproducibility in Macromolecular Crystallography (https://proteindiffraction.org/search/?q=COVID‐19), thereby making it possible for other crystallographers to verify the accuracy of their models. The involvement of many groups in structure validation/assessment and the urgency of the pandemic have created an unusual situation. Many authors of the CoV‐2‐related deposits have submitted a second or even third version of their coordinates. Some of the groups listed above have suggested these changes in different selections, and the original depositors introduced other changes. Sometimes a request for diffraction data motivated the original authors to re‐investigate and subsequently re‐refine/redeposit the structural model. Every Wednesday, multiple corrections appear to structures that have already been released to the public. The PDB annotates 15 reasons for changes of an existing PDB deposit and divides them into two main categories: major and minor revisions. For example, the deposit 6LU7 has been updated six times, including one major revision. Analysis of the PDB shows that between January 24 and April 27, around 22% of the 45 CoV‐2 structures (excluding PanDDA deposits) required a major revision (10 deposits), usually due to changes in the coordinates of the protein or functional ligands. In the same period, only 12 of 3226 structures that were not related to CoV‐2 (i.e., around 0.4%) had any major revisions. One can draw many, sometimes contradictory, conclusions out of these numbers, and we would like to leave them to the readers of this paper. For reasons explained below, we excluded PanDDA deposits from these statistics.
We have included in our analysis all PDB models of CoV‐2 proteins released by April 1, 2020, and extended this set to include selected proteins from other coronaviruses, to create a basis for comparative and differential analyses. Although our main focus is on X‐ray crystallographic models and their agreement with electron density maps, we have also surveyed the cryo‐EM structures to verify their stereochemical correctness and agreement with cryo‐EM maps. Since for drug discovery the most relevant portion of the models lies at the protein–ligand or protein–receptor interface, we pay special attention to ligand modeling.
Dissemination of results
Many recently deposited CoV‐2‐related structural models are not accompanied by a peer‐reviewed publication. Faced with a global health crisis, scientists proactively take action and allow others to access their models without the usually attached publication citation reward. As an unintended consequence, many structural models can be classified only by the title of the PDB deposition, and the lack of descriptive detail may overwhelm researchers searching for models of validated target structures. To enable quick dissemination of our results, we created a dedicated webservice (https://covid‐19.bioreproducibility.org) presenting the outcome of our assessment in a readily accessible form for researchers who are not necessarily structural biology experts. The website has several filtering functions that provide access to selected structures. Moreover, in cases of re‐refined structures, users can easily compare the re‐refined models with the original structures by using Molstack sessions [30, 31] and downloading the re‐refined structure models. Molstack is an online tool for easy visualization and comparison of multiple sets of atomic models and electron density maps in stacks of dual, synchronized side‐by‐side interactive graphics windows (Fig. 1). The re‐refined structures will ultimately be deposited in the PDB, preferably together with the authors of the original deposits.
Fig. 1.
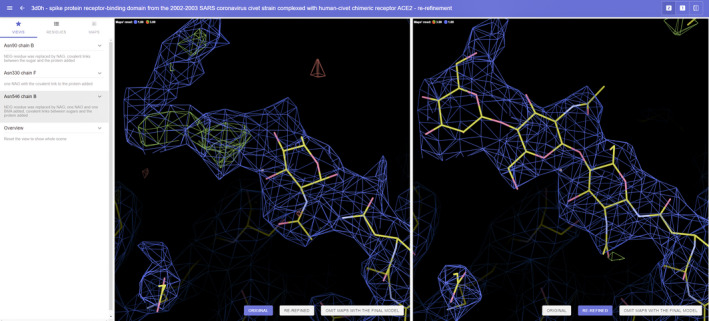
An example of the use of Molstack. This figure depicts the carbohydrate moieties attached to Asn546B of the PDB entry 3D0H. The left stack represents the original structure with only one sugar unit, which was not chemically linked to Asn546B; this stack is labeled ORIGINAL, as shown in the blue box at the bottom of the stack. The right stack represents the corrected model with three chemically linked carbohydrate molecules (NAG‐NAG‐BMA) connected to Asn546B; this stack is labeled RE‐REFINED. The user can change the maps and model shown in each stack by clicking on any of the three options shown in the colored boxes at the bottom of each stack (the third option OMIT MAPS WITH THE FINAL MODEL is not selected in any of the two stacks in this figure). The original and re‐refined models and the respective maps are superimposed; both stacks are always centered at identical location in the unit cell. The user can move around the model as in Coot. Three locations in the models are available on the left side of the window under the VIEWS menu. The electron density map levels are scrollable using the mouse wheel; separate adjustment of each map is available on the MAPS menu that can be found on the upper left. For a detailed description of Molstack, see [31]. This figure was generated by Molstack under the project https://molstack.bioreproducibility.org/project/view/WrI2XslE978LiF95PQYo/.
Validation and re‐refinement of SARS‐CoV‐2 drug target models
Why validate? The goal of model validation is to provide assessment of its plausibility in view of both, the actual experimental evidence, and the agreement with independently established prior knowledge. Both terms should be objectively convincing and free of contradictions. Appropriate validation thus allows the user of such models to make an informed decision whether the model can be useful—or not—for a specific purpose. In the case of small‐molecule ligands complexed with macromolecules of interest, the goal is often identification of lead compounds [2], or in later stages, structure‐guided lead optimization [32]. Experimentally determined ligand structures are also used to train and evaluate docking and virtual ligand screening algorithms [33]. Given the massive resources needed for drug development [34], an informed decision whether or not to use a specific target for further research is crucial. Picking for drug development an invalid or incorrect structure model results in wasted resources, while rejecting a valid ligand model because other parts of the model score low, leads to missed opportunities.
Validation is not evaluation
The distinction between validation (i.e., assessment of technical correctness and physical plausibility of a structure model) versus evaluation (i.e., assessment of usability of the model for a defined purpose) is an important one. The validator can only attest to the former aspect, whereas evaluation of a model for a specific purpose remains the responsibility of the ultimate user.
Evidence
Validation relies primarily on the evaluation of the available experimental evidence. In crystallography, this is not as straightforward as it may appear, because the experimental diffraction intensities or structure‐factor amplitudes do not include critical information necessary for the electron density reconstruction, namely the phase of each X‐ray reflection. During standard PDB validation, these phases are calculated from the model [35] and thus bias the electron density maps and consequently the model itself toward—and never against—any part already included in the model. It is also possible to deposit in the PDB extended data files with phased Fourier coefficients for electron density map generation. In this case, the information how these Fourier coefficients were generated is of fundamental importance for the validation process. Unfortunately, the origin of the Fourier coefficients is generally not sufficiently documented, and their genesis is neither reproducible nor assessable without deeper understanding of the procedures used to calculate those maps. In any case, a ligand model should be supported by the experimental evidence of untainted electron density that closely resembles the shape of the ligand claimed to be present.
Prior knowledge
The second term contributing to the joint posterior model likelihood is the prior probability, judging the model against independently established plausibility criteria. The stereochemistry of the protein part of a macromolecular model is very well understood, and a purported ligand should obey known stereochemical rules, as well as exhibit reasonable molecular contacts within the binding site, with no clashes—physical or chemical—with its environment. An important caveat, however, applies to the sole reliance on stereochemical target validation: At very low resolution (> 3 Å), the tight geometric restraints required to keep the refinement stable impart that the model will reflect to a high degree the restraint targets. However, the backbone (Ramachandran) torsion angles are normally not restrained and effectively act as geometry cross‐validation, absorbing and revealing problems with the overall model tracing. If these backbone torsion angles are also restrained, the model cannot be validated by geometry or clash violations anymore: Stereochemical validation of the practically idealized geometry becomes then self‐fulfilling.
Both these terms—Primary evidence (data likelihood) and compatibility with independently established prior expectations (prior probability)—determine the posterior model likelihood.
Cases
Structures of the complexes of the coronavirus spike protein with ACE2, its cellular receptor
Relevance. The trimeric spike glycoprotein on the viral surface mediates the entry of the virus into host cells. The receptor‐binding domain (RBD) of the spike protein (S) interacts with the protease domain of the angiotensin‐converting enzyme 2 (ACE2). Interference with the interactions between these proteins might provide a path to the development of antivirals that would prevent cell fusion and entry, as has been shown in the case of HIV through the development of peptidic and small‐molecule fusion inhibitors [36]. For that reason, accurate description of the interaction between the RBD and ACE2 is very important.
Analysis. Data sets released by the PDB by April 1, 2020, include 10 medium‐resolution crystal structures of complexes of various forms of the proteolytic domain of ACE2 with the RBD of CoV (2AJF, 3SCI, 3SCK, 3SCL, 3D0H, 3D0I, 3D0G) and CoV‐2 spike protein RBDs (6LZG, 6M0J, 6VW1), as well as a cryo‐EM structure (6M17) of the full‐length human ACE2 in complex with complete CoV‐2 spike (Table 1). Two of the three recent crystal structures of the complexes with the CoV‐2 RBD (6LZG and 6M0J) are fully isomorphous and located identically in the P41212 unit cell, whereas the third one (6VW1) was crystallized in space group P21. That structure is isomorphous with the previously determined crystal structures of complexes of the spike RBDs from CoV viruses infecting humans or civets with a hybrid human‐civet ACE2.
Table 1.
Selected structures of complexes of coronavirus spike glycoproteins or their fragments with the ACE2 receptor that were analyzed in this work. Abbreviation civ refers to the virus from civet, h to human, and hc to hybrid human‐civet proteins.
| PDB ID | Resol (Å) | R free | Space gr. | a (Å) | b (Å) | c (Å) | β (°) | Spike | Enzyme | Action |
|---|---|---|---|---|---|---|---|---|---|---|
| 6LZG | 2.5 | 0.214 | P41212 | 104.4 | 104.4 | 229.8 | CoV‐2 | hACE2 | – | |
| 6M0J | 2.45 | 0.227 | P41212 | 104.7 | 104.7 | 228.7 | CoV‐2 | hACE2 | – | |
| 6VW1 | 2.68 | 0.229 | P21 | 80.4 | 118.0 | 112.1 | 93.1 | CoV‐2 | hACE2 | Achesym, minor |
| 3SCI | 2.9 | 0.283 | P21 | 81.4 | 118.3 | 111.9 | 93.1 | SARS | hACE2 | Achesym, minor |
| 3SCK | 3.0 | 0.285 | P21 | 81.2 | 119.3 | 113.2 | 92.2 | SARS/civ | hcACE2 | Achesym, minor |
| 3SCL | 3.0 | 0.292 | P21 | 81.7 | 119.5 | 113.5 | 92.5 | SARS | hcACE2 | Achesym, minor |
| 2AJF | 2.9 | 0.275 | P21 | 82.3 | 119.4 | 113.2 | 92.0 | SARS | hACE2 | Achesym |
| 3D0H | 3.1 | 0.302 | P21 | 80.4 | 119.8 | 109.4 | 95.9 | SARS/civ | hcACE2 | Achesym, some |
| 3D0I | 2.9 | 0.278 | P21 | 80.4 | 119.8 | 109.7 | 95.5 | SARS/civ | hcACE2 | Achesym, some |
| 3D0G | 2.8 | 0.279 | P21 | 80.0 | 119.8 | 108.8 | 96.2 | SARS | hcACE2 | Achesym, some |
| 6M17 | 2.9/3.5 | cryo‐EM | CoV‐2 | hACE2 | – |
According to the criteria utilized in molprobity [37], the three crystal structures of CoV‐2 spike protein complexes, determined at similar resolution close to 2.5 Å, are of relatively high quality when compared to other structures at similar resolution. The structure 6LZG [38] scores best both in terms of clashscore and overall molprobity score, although flipping the side chain of Asn487 in chain B is necessary. Whereas 6M0J is in no need of amide group flips, His374 of its ACE2 component had to be flipped, as it interacts with a Zn2+ cation through a carbon atom. This was also noticed by the deposition authors, and the original PDB deposit was appropriately modified while this manuscript was under review. The corresponding Zn2+ cations in the 6VW1 deposit have abnormally high B factors (135 and 196 Å2) and do not make any contacts with their environment that would indicate proper coordination. Otherwise all three structures seem to be in good agreement with the electron density and are much better than 2AJF, the first structure in that series. This is expected due to improved resolution, software, and contribution from prior knowledge.
The three medium‐resolution structures of the RBDs from various human and civet SARS‐CoV strains in complex with a chimeric ACE2 bearing the critical N‐terminal helix from civet and the remaining peptidase domain from human (3D0H, 3D0I, 3D0G) were solved to provide structure‐based assessment of species barrier between humans and civets for SARS‐CoV infections [39]. These structures share a common problem of an unlikely assignment of the carbohydrate moieties bound to asparagine side chains, modeled as 2‐(acetylamino)‐2‐deoxy‐α‐D‐glucopyranose (NDG), rather than the expected β‐linked N‐acetyl‐D‐glucosamine (NAG). Chemical bonds between the carbohydrate moieties and the asparagine side chains were not present in the model, resulting in large distances between the atoms that should be chemically bound. Moreover, the electron density maps clearly indicate that several additional carbohydrate moieties could be modeled. In addition, there are two Zn2+ ions in each of these three structures, modeled in an analogous way, but not quite satisfactorily. Each Zn2+ ion is in good contact with His374 and His378. The two nearby carboxylates from Glu375 and Glu402 are positioned at too long distances to be effectively coordinated by the metal cations. Moreover, on one side of the Zn2+ ions there are no ligands, leading to ambiguity about the actual coordination geometry. Unfortunately, the low resolution of the electron density maps does not provide any features suggesting how to correctly model these ions.
These three structures were further re‐refined by us. NDG residues were replaced by NAG residues, and the β(1–4) chemical bonds were added. Moreover, several carbohydrate moieties were added to those already present, as well as to a few additional asparagine side chains (up to three linked sugar molecules per residue), expanding the list of glycosylated residues to Asn90A/B, Asn322A/B, Asn546A/B, and Asn330E/F in all three structures. Some of the carbohydrate corrections can be inspected interactively using Molstack via the following link https://molstack.bioreproducibility.org/project/view/hnlY2nbqOGN6OoG7s3qz/. An example of a Molstack visualization of one of the carbohydrate corrections is presented in Fig. 1. We have also added several water molecules and fixed multiple side‐chain outliers. For example, in 3D0H, the following side chains were corrected: Gln89A/B, Leu143A, Val212A/B, Thr334A, Leu410B, Leu439B, Leu444A/B, Lys476A, Leu503B, Glu536B, Leu585B, Lys333E, Glu341E, and Glu341E/F.
The other three structures in this series with the same primary citation [39] (3SCI, 3SCK, and 3SCL) do not include carbohydrate moieties, although the same problems with placement of the Zn2+ ions as in the previous three structures are still present. These six structures are almost isomorphous in the space group P21 and originate from the same laboratory, but the molecules are positioned in three different locations in the unit cell, with respect to different definition of the cell origin, floating along the y‐axis in this space group (Fig. 2). During our analysis, all these structures were placed in a consistent location in the unit cell, with the updated models and maps available at https://covid‐19.bioreproducibility.org/.
Fig. 2.
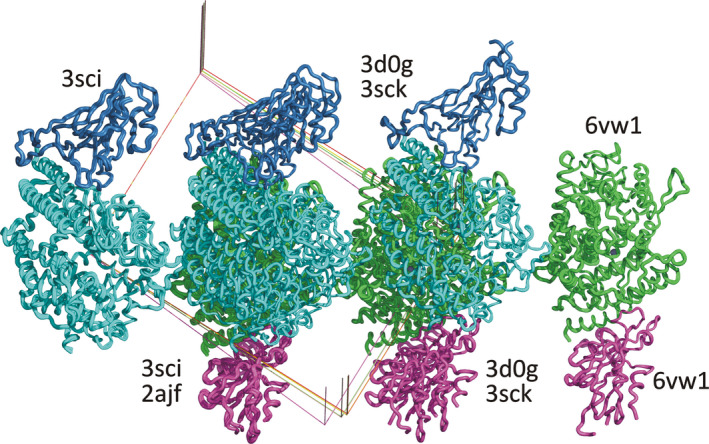
Lack of uniformity in presentation of structures in the PDB. Placement of selected models of the complexes of the receptor‐binding domains of CoV and CoV‐2 spike proteins with the proteolytic domain of ACE2 in isomorphous unit cells in space group P21. The individual structures are identified by their PDB codes. The chains of ACE2 are colored cyan/green, and the RBDs of the spike proteins blue/magenta for the two complexes present in each asymmetric unit. Figure generated with pymol.
The fragments of the CoV and CoV‐2 spike proteins in the general area of interaction with ACE2 differ in their sequences by 28 amino acid residues, with about 12 of them involved in direct interactions with the receptor. The electron density is convincing in all these cases for the whole interaction areas of both proteins, allowing credible interpretation of the differences between the modes of interaction of these two closely related viruses with their common cellular receptor.
A cryo‐EM structure of the CoV‐2 spike protein interacting with full‐length human ACE2 as well as with a neutral amino acid transporter B0AT1 was deposited in the PDB as entry 6M17 [12]. When the deposited map is overlaid with the atomic coordinates, the coordinates need to be shifted by ~ 1.5 Å in order to match the density. When the ACE2 molecule A from the crystal structure 6M0J is superimposed on molecule B of 6M17, the rmsd for the Cα atoms is 0.93 Å, a result that is not surprising in view of the fact that crystallographic models are frequently used to interpret cryo‐EM maps. The reported overall resolution of the cryo‐EM map is 2.9 Å, with the local resolution of the spike RBD‐ACE2 interface of only 3.5 Å, so it is not surprising that the side chains of the residues in that area do not fit the map particularly well and the map is not as clear as the corresponding X‐ray electron density map. The model includes an analogous mistake in the environment of the Zn2+ cation as in 6M0J, except that in this case it is His378 that interacts with it through a carbon atom.
Other structures of fragments and complexes of the coronavirus spike protein
Relevance. The S2 subunit of the spike protein plays a key role in mediating virus fusion with and entry into the host cell, in which the heptad repeat 1 (HR1) and heptad repeat 2 (HR2) can interact to form a six‐helix bundle (6‐HB), thereby bringing viral and cellular membranes into close proximity for fusion [40]. There are three cryo‐EM structures of the prefusion CoV‐2 spike glycoprotein in different conformational states (open, closed, and with a single receptor‐binding domain up) with the reported resolution ranging from 2.8 to 3.46 Å (6VXX, 6VYB, 6VSB). All three structures have multiple loops that were not modeled due to disorder and several covalently bound carbohydrate residues. In addition, there are two X‐ray crystal structures of different fragments of the spike CoV‐2 glycoprotein and multiple structures of spike CoV glycoprotein, as described below.
Analysis. The PDB entry 6LXT represents the postfusion structure of HR2 of the CoV‐2 spike glycoprotein S2 subunit (fragment 912–1202 according to the alignment with CoV sequence). The asymmetric unit contains two α‐helix bundles consisting of six helices each. However, most of the protein is outside of the unit cell. Overall, the structure is of good quality. It has one Ramachandran outlier, which is justified, and no missing side chains. There are several side‐chain rotamer outliers, which is a common problem at low resolution. A Zn2+ ion is modeled in several places; its modeling is justified by the presence of 200 mm zinc acetate in the crystallization buffer and it does not necessarily have any physiological relevance. A metal cation (Zn2+) is missing between Asp1199 of chain B and Asp1199 of chain F. At the interface between Asp1163 and Asp1165 of chain B and a symmetry mate of Glu1188 and Asp1184 of chain A (crystallographic interface between two six‐helix bundles), a molecule of PEG is modeled instead of, most probably, one or two Zn2+ ions. This PEG molecule is hilariously mismodeled: It has severe clashes with the protein residues, all contacts are polar‐to‐nonpolar, and it is awkwardly curled up.
The PDB entry 6LVN represents the structure of HR2 of the CoV‐2 spike glycoprotein (fragment 1150–1185 according to the alignment with CoV sequence). It has no ligands bound. The asymmetric unit (ASU) of this structure consists of four α‐helices of the same sequence bundled together. Unfortunately, these four helices are not placed in the ASU as a single oligomer, but as two independent dimers. Other than that, this structure is of high quality. The difference electron density map does not show any peaks above 5σ. The model is within the standard unit cell, it has no Ramachandran outliers, no rotamer outliers, and no missing side chains.
The PDB contains dozens of additional structures of CoV and MERS spike glycoproteins and their fragments and complexes. Notably, there are also structures of fragments of spike glycoprotein from other coronaviruses. For example, the PDB deposit 5ZUV represents the structure of the HR1 motif of the human coronavirus HCoV‐229E complexed with a universal peptidic inhibitor, designed by modification of the sequence of a fragment of the spike from HCoV‐OC43. The HR1 motif and the inhibitory peptide were expressed as a fusion protein interconnected by a peptide linker, which is visible in the electron density maps of two out of three molecules in the 5ZUV crystal structure. This fusion molecule is represented in the deposit as a single protein chain with consecutive residue numbering, sowing confusion about the nature of the complex and the factual presence of the inhibitory peptide. The same inhibitor is also present, but this time clearly marked as a separate protein chain, in the two other structures reported in the primary citation [41] for 5ZUV: 5ZVK (crystal structure of the human coronavirus MERS HR1 motif in complex with pan‐CoV inhibitor EK1), and 5ZVM (crystal structure of the human coronavirus SARS HR1 motif in complex with pan‐CoV inhibitor EK1). All three deposits have most of the protein residues outside of the reference unit cell, but only 5ZUV was modified by us. We re‐assigned the inhibitory peptides in 5ZUV as separate protein chains and renumbered the residues in the same way as in the other two deposits, thus making them compatible and directly comparable. We also repositioned the coordinates with ACHESYM [42] and improved the model by adding residue Leu8 in chain C (inhibitor peptide), correcting conformation of 13 residues (Lys23B, Gln788B/A, Glu789A, Asn790B, Gln791A/B, Leu794A, Val806C, Gln839C, Thr848B, Gln856A/B), and adding 43 water molecules.
Antibody structures. The crystal structure of CR3022, a neutralizing antibody previously isolated from a convalescent SARS patient, was determined at 3.1 Å resolution in complex with the receptor‐binding domain of the CoV‐2 spike protein (PDB ID 6W41; [15]). This is a comparatively low‐resolution structure that is highly idealized. The structure was re‐refined by us after shifting the coordinates into the reference unit cell with ACHESYM [42] and after flipping two peptide bonds (Tyr380 of the spike fragment and Ile98 of the antibody heavy chain). There is some evidence of the presence of another NAG group bound to Asn343 of the spike protein, but we did not attempt to model it. While this manuscript was being revised, another structure of a similar complex (PDB ID 6YLA; unpublished) has been released by the PDB. The latter structure was determined at considerably higher resolution (2.42 Å), with two complexes in the asymmetric unit, but it was not further analyzed in this paper.
Structures of the 3CLpro main protease and its inhibitor complexes
Relevance. The main protease of CoV‐2 (3CLpro) is a chymotrypsin‐like enzyme with a polypeptide chain consisting of 306 amino acid residues. However, unlike in chymotrypsin, its catalytic residue is cysteine (Cys145) instead of serine, and the protein is a symmetric homodimer. The amino acid sequence of CoV‐2 3CLpro is 96% identical to that of SARS‐CoV 3CLpro, but only 50% identical to the sequence of MERS‐CoV 3CLpro. We identified in the PDB 28 structures of the SARS‐CoV 3CLpro protein, either in a ligand‐free or inhibited form, as well as four structures of MERS 3CLpro. A few of these structures were analyzed here with the aim of providing a reference for the CoV‐2 enzymes, but structures from other related viruses were not considered. The inhibition of virus proteases is a standard model for antiviral treatment strategies.
Analysis. One of the structures of an inhibitor complex of CoV‐2 3CLpro (6Y7M) is isomorphous with at least two previously determined structures of CoV 3CLpro complexes (2ALV and 2QIQ), although also in this case it was necessary to bring the coordinates to a common location in the unit cell. Although the latter structures were determined at the resolution of 1.9 Å, the maps indicated a need for improvement of the models; thus, they were both re‐refined. During the re‐refinement, minor changes were applied to some side chains of the protein residues of both structures. However, a significant change to the inhibitor molecule within the 2QIQ structure was made: While a large central part of the inhibitor is well defined in the electron density, the ethyl and t‐butyl ester groups of the inhibitor molecule lack any electron density. The inhibitor was thus re‐refined after truncation to terminal carboxyl groups, assuming that the ester groups had been eliminated by hydrolysis, a supposition that is supported by the well‐defined and abrupt limits of the electron density at the ends of both carboxyl groups. Notably, a similar situation was observed for the structure 6LU7 of a peptidic inhibitor complex of CoV‐2 3CLpro, where the terminal benzyl group of the inhibitor had negative difference electron density, again suggesting elimination of this group by hydrolysis (Fig. 3A). Removing the benzyl group from the inhibitor and re‐refinement of the model resulted in a drop of R and R free, by 2.3 and 1.0%, respectively.
Fig. 3.
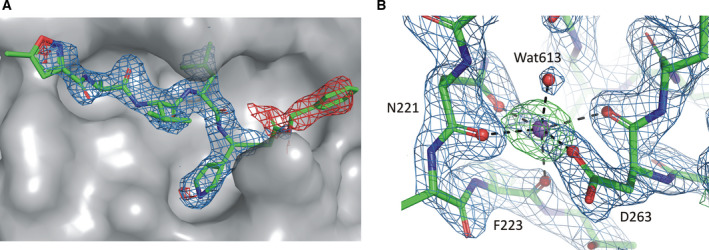
Examples of significant problems with the deposited models that required reinterpretation. (A) Peptidic inhibitor in the substrate‐binding site of the 6LU7 structure. The 2mFo−DFc electron density (blue) is shown at the contour level of 1.5σ. The presence of negative mFo−DFc difference electron density (contoured in red at the − 3σ level) for the terminal benzyl group of the inhibitor may indicate elimination of this group by hydrolysis. The electron density and the model with and without the benzyl group can be inspected interactively in Molstack at https://molstack.bioreproducibility.org/project/view/x9jJQnlGN25TRBSRdxJm/. (B) The highest positive peak (9.7σ) on the difference electron density map of the 6Y2F structure with a sodium cation added during re‐refinement. The environment of the new Na+ site is shown in the original electron density maps: 2mFo−DFc (contoured at 1.5σ, blue) and positive difference mFo−DFc map (contoured at 3.0σ, green) with coordination bonds (black dash lines) between the Na+ cation (violet sphere) and oxygen atoms (red spheres) arranged in an octahedral fashion.
Two crystal structures of complexes of the CoV‐2 protease with the same inhibitor are presented in two different crystal forms: monoclinic (6Y2F, space group C2) and orthorhombic (6Y2G, space group P212121). In the former structure, the asymmetric unit contains only one subunit, and the protease dimer is formed by two identical protomers related by crystallographic symmetry. The latter structure has a dimer composed by two independent subunits, which, in principle, could adopt slightly different conformations. However, the subunits of these two crystal forms are very similar with an rmsd range of 0.36–0.45 Å for all Cα pairs. The structures were refined at resolution 1.95 (6Y2F) and 2.2 Å (6Y2G), and the protein chains are modeled well, with some minor issues with side‐chain rotamers and a couple of solvent molecules that are too far from the nearest protein chain or ligand molecule.
During the re‐refinement of the monoclinic structure 6Y2F, the highest positive peak of 9.7σ on the difference electron density map was detected close to the protein surface between residues Asn221, Phe223, and Asp263 (Fig. 3B). It was coordinated by their three backbone carbonyl groups, two side‐chain oxygen atoms of the Asn and Asp residues, and one water molecule. The distinctive octahedral coordination and oxygen distances preclude modeling of a water molecule at this position. A sodium cation has been, therefore, modeled at this site and validated by the CheckMyMetal web server [25]. Subsequent re‐refinement of this model with two corrected side‐chain rotamers (Arg298 and Phe294), three water molecules added, and application of TLS parameters led to a significant drop of R free by 1.3%.
The unit cell of the crystal structure 6W63, representing a complex of CoV‐2 3CLpro with a noncovalently bound inhibitor, is different from the unit cells of other complexes of this enzyme. In this case, the placement of the molecule in the asymmetric unit is reasonable and the electron density maps do not indicate any need for further improvement.
Four structures of unliganded CoV‐2 3CLpro were deposited in the PDB as of April 1, 2020, by three different research groups (Table 2) [16]. Whereas the crystals used for structure determination and refinement were practically all isomorphous in space group C2, the atomic coordinates of the models were placed differently in the deposited data sets. Our analysis commenced with moving all four sets of coordinates to a common origin with the program achesym [42], followed by re‐refinement that utilized the deposited structure factors. The two highest resolution structures (6YB7 at 1.25 Å and 6Y84 at 1.39 Å) were deposited by the authors of a vast number of PanDDA structures of potential weak ligand complexes, thus they may have been useful as sets of reference in that process. Although the two structures were refined with the same program buster [43], the refinement protocols were different: only isotropic B factors for 6YB7 and a single TLS group for 6Y84. Re‐refinement of 6YB7 with individual anisotropic B factors, well justified at the near‐atomic resolution, has led to substantial improvement of the refinement statistics and better electron density map, but with rather minimal changes to the model itself. Re‐refinement of 6Y84 and 6Y2E basically reproduced the results from the original deposits and the main reasons for performing this step were to standardize model placement in the unit cell. However, our re‐refinement of the PDB structure 6M03 resulted in an increase of R free by almost 3%. We noticed that the number of reflections reported in the header of the PDB file, as well as in the validation report, is almost twice the number expected at this resolution, whereas the number of structure factors used to calculate maps is consistent with the expectation. Our conclusion is that the anomalous Bijvoet pairs of reflections may have been used in the refinement separately, without good reason, and that this fact is not accurately reflected in the deposited data and their description.
Table 2.
Selected structures of 3CLpro main protease analyzed in this work. The Inh? column indicates presence/absence of an inhibitor.
| PDB ID | Resol (Å) | R free original | R free re‐refined | Space group | a (Å) | b (Å) | c (Å) | β (°) | Virus | Inh? | Action |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 6LU7 | 2.16 | 0.235 | 0.225 | C2 | 97.9 | 79.5 | 51.8 | 114.6 | CoV‐2 | Y | Achesym, some |
| 6Y2F | 1.95 | 0.219 | 0.206 | C2 | 98.1 | 80.9 | 51.7 | 114.8 | CoV‐2 | Y | Achesym, some |
| 6M03 | 2.0 | 0.246 | 0.275 | C2 | 114.0 | 53.4 | 45.0 | 101.8 | CoV‐2 | N | Achesym, minor |
| 6Y2E | 1.75 | 0.222 | 0.219 | C2 | 115.0 | 53.8 | 44.8 | 101.2 | CoV‐2 | N | Achesym, minor |
| 6YB7 | 1.25 | 0.192 | 0.163 | C2 | 112.4 | 52.8 | 44.6 | 103.0 | CoV‐2 | N | Achesym, minor |
| 6Y84 | 1.39 | 0.200 | 0.202 | C2 | 112.8 | 52.9 | 44.6 | 103.2 | CoV‐2 | N | Achesym, minor |
| 6Y2G | 2.2 | 0.247 | 0.240 | P212121 | 68.6 | 101.6 | 103.7 | CoV‐2 | Y | minor | |
| 6Y7M | 2.2 | 0.258 | 0.256 | C2 | 107.9 | 82.2 | 53.1 | 104.5 | CoV‐2 | Y | Achesym, minor |
| 2ALV | 1.9 | 0.315 | 0.295 | C2 | 107.1 | 83.2 | 53.7 | 104.4 | SARS | Y | Achesym, some |
| 2QIQ | 1.9 | 0.275 | 0.253 | C2 | 108.3 | 82.2 | 53.7 | 104.7 | SARS | Y | Achesym, significant |
| 2DUC | 1.7 | 0.221 | P21 | 52.3 | 96.3 | 67.8 | 102.9 | SARS | N | – | |
| 3D62 | 2.7 | 0.363 | P21212 | 106.7 | 45.2 | 54.0 | SARS | Y | – | ||
| 4MDS | 1.6 | 0.212 | C2 | 118.8 | 56.2 | 53.1 | 111.1 | SARS | Y | – | |
| 4RSP | 1.62 | 0.203 | 0.213 | C2 | 106.5 | 57.3 | 48.9 | 112.8 | MERS | Y | Achesym, minor |
| 6W63 | 2.1 | 0.221 | P21212 | 45.05 | 63.8 | 106.6 | CoV‐2 | Y | – |
The highest resolution (1.7 Å) structure of unliganded CoV 3CLpro is 2DUC. Unlike the CoV‐2 structures discussed above, in which the obligatory dimer was created by crystal symmetry, the asymmetric unit of 2DUC contains a complete dimer. Nevertheless, the structures are very similar, with superposition of all 306 Cα atoms of the 6YB7 model on molecule A of 2DUC yielding an rmsd of 1.05 Å. An analogous superposition of the high resolution (1.62 Å) structure of an inhibitor complex of MERS 3CLpro (4RSP) yielded an rmsd of 1.55 Å for 304 Cα pairs.
Papain‐like protease (PLpro)
Relevance. The MERS and SARS coronaviruses contain a second, papain‐like protease (PLpro), which is responsible for processing the cleavage sites in the N‐terminal part of the polyproteins, a process that is essential for viral replication. PLpro can also act as a deubiquitinating enzyme involved in regulation of the innate immune response to the viral infection [44]. The amino acid sequence CoV‐2 PLpro is 83% identical to SARS‐CoV PLpro and only 31% to MERS PLpro. There are 30 deposited CoV PLpro structures in the PDB (16 from MERS and 13 from SARS) but only one of CoV‐2 PLpro (6W9C).
Analysis. The PLpro protein from CoV‐2 is highly homologous to SARS‐CoV PLpro, with rmsd of ~ 1.0 Å for all Cα pairs, when compared to any of the CoV PLpro structures. The 6W9C structure was determined in the monoclinic C2 space group at the nominal resolution of 2.7 Å, but the completeness of the experimental data was very low (57.3%). The protease structure contains three independent molecules in the ASU, each with a classical catalytic triad composed of Cys111‐His272‐Asp286. Because of lack of interpretable electron density in the regions of C‐terminal zinc‐finger‐like domains (consisting of two β‐hairpins), the four cysteine residues which coordinate a Zn2+ ion with tetrahedral geometry (Cys189, Cys192, Cys224, Cys226) were not modeled properly. The three independent molecules of PLpro in the crystal structure form a trimer stabilized by a zinc ion coordinated by Cys270 from each subunit.
Re‐refinement of 6W9C resulted in a significant drop of R/R free by 0.7/1.1%, which allowed rebuilding of some parts of the model. However, because of poor quality of the electron density maps, further improvement of the model was not possible.
Structures of CoV‐2 proteins of relevance for viral RNA binding or processing
Relevance. Several nonstructural proteins of coronavirus interact with RNA or are involved in its processing. As of April 1, 2020, there were several released PDB crystal structures of CoV‐2 proteins belonging to this group. These are three structures of ADP‐ribose phosphatase NSP3 (6W02 in complex with ADP, 6W6Y in complex with AMP, 6VXS apo structure), two structures of endonuclease NSP15 (6VWW, 6W01), two of the RNA‐binding domain of the nucleocapsid phosphoprotein (6VYO, 6M3M), three structures of the NSP16‐NSP10 methyltransferase‐stimulatory factor complex (6W61, 6W4H, 6W75), and the structure of NSP9, annotated as RNA‐binding replicase (6W4B). They were contributed by the Center for Structural Genomics of Infectious Diseases of the NIH (USA), except 6M3M, which was submitted by Chinese researchers. These structures were refined at resolution ranging from 1.5 to 2.2 Å, except two lower resolution structures 6W4B (2.95 Å) and 6M3M (2.7 Å). Except for the NSP3 protein, which was also crystallized with the substrate/product of the enzymatic reaction, they correspond to uncomplexed enzymes but contain various small molecules originating from the crystallization buffers.
Analysis. The structure of the nucleocapsid RNA‐binding domain 6VYO contains a well‐refined model of the protein, but the zinc ions in this structure are not presented satisfactorily. Each of the four independent protein molecules contains a Zn2+ cation with tetrahedral coordination provided by one Asp and two His side chains (one from another protomer), as well as another zinc ion without any ligands. Two of the central Zn2+ ions have assigned occupancies of 0.98 and 0.92, while the apical Zn2+ ions have occupancies of 0.58, 0.55, 0.60, and 0.63, which is unrealistic. The presence of apical Zn2+ ions as ligands of the central Zn2+ ions is unacceptable from the chemical point of view, and the incompatible occupancies make their modeling clearly implausible. Most probably those sites are occupied by Cl− ions, and such constellations can be successfully refined with full occupancy of all atoms. In the 6M3M structure of the same protein, the packing of molecules is different, and the Zn‐binding site is not formed since one of the His residues is not available for metal coordination.
In general, all protein chains of the structures in this group are modeled correctly. There are some residues with side chains lacking clear definition in the electron density maps, especially in the two lower resolution structures, but this is expected and not abnormal. However, there are some issues with how these structures are presented and in the interpretation of certain structural details.
The cell dimensions (a, b, c, α, β, γ, in Å and °) in space group P1 of two of the NSP3 structures are:
These two structures are therefore presented in different unit cells despite being isomorphous and authored by the same group. The structure 6VXS refines equally well against ‐h, ‐k, l reindexed data in a unit cell with supplementary angles α = 95.63° and β = 97.89°.
The two NSP15 structures 6VWW and 6W01 are clearly isomorphous in space group P63 with very close cell dimensions but are presented differently. The diffraction data for these two structures were indexed with the orientation of the polar c‐axis inverted. After k, h, ‐l reindexing of one of the data sets, both models can be refined satisfactorily and presented in the same location in the unit cell. In the structure 6VWW, the site modeled as occupied by a magnesium cation with tetrahedral coordination is most probably containing a water molecule, as Mg2+ ions require strictly octahedral coordination.
Among the NSP16/NSP10 complexes, two (6W61 and 6W4B) are isomorphous in space group P3121. After ‐h, ‐k, l reindexing of one of them, the models can be refined satisfactorily at the same location in the unit cell. All three structures contain well‐defined S‐adenosylmethionine (SAM methyl donor) molecules, with Zn2+ ions and other small molecules and ions adequately supported by electron density.
The NSP9 replicase structure 6W4B is based on diffraction data of 2.95 Å resolution. The protein main chain in this structure follows the electron density, but most of the side chains lack convincing electron density and their orientation is therefore tentative and not well supported by experiment.
A cryo‐EM structure of the RNA‐dependent RNA polymerase from CoV‐2 with bound cofactors NSP7 and NSP8 was released as PDB entry 6M71 (unpublished). The structure was determined at 3.1 Å after homogenous refinement, increased to 2.9 Å after local refinement. The model was derived directly from the analogous 3.1 Å cryo‐EM structure of an equivalent complex from SARS‐CoV (PDB ID 6NUR; [45]); thus, it is not surprising that the rmsd for 1013 aligned Cα atoms is only 0.63 Å. The model of the CoV‐2 enzyme complex includes a single polymerase (NSP12) protein, a single NSP7 protein, and two copies of the NSP8 protein. The model is highly idealized and fits the electrostatic potential map very well in most regions. However, a significant part of one of the NSP8 molecules (residues 1–83 and 123–198 of chain D) were not modeled. The two copies of the NSP8 protein assume distinctly different conformations and the C‐terminal part of chain B is much more complete. Although considerable map density is present in the region presumably corresponding to the C‐terminal part of molecule D, it remained unassigned. The C‐terminal part of the equivalent molecule of the CoV complex seems to be generally covered by the unassigned density, but there are many breaks allowing significantly different tracing. Reinterpretation of that part of the structure is beyond the scope of the present work.
Problems with validation of PanDDA group depositions
A fundamental problem arises with validation or re‐refinement of a model where the primary experimental evidence cannot be reliably reproduced. In this situation, only the second term determining the model likelihood—the prior expectations in the form of binding site environment, contacts, and chemically plausible ligand conformation—remains assessable.
The problem of lack of a path to reproducible primary evidence arises with structure models and data generated by the Pan‐Dataset Density Analysis (PanDDA) procedure [46, 47]. The technical complexity of the PanDDA process and the current inability to adequately deposit in the PDB all necessary data, models, and procedures required to generate the maps make it practically impossible—despite ample supplementary information on the web (https://pandda.bitbucket.io/) routinely inspect the proposed PanDDA ligand models.
Inspection via the PDBe ligand density viewer and our analysis of the PanDDA structures of ligand complexes of the CoV‐2 main protease (3CLpro) suggest limited experimental evidence for the ligands based on the electron density. Direct re‐refinement of one of those structures (5R7Z) against deposited data resulted in 3.4% drop of R free, suggesting significant overall improvements of the model (for comparison of models and maps, see https://molstack.bioreproducibility.org/collection/view/GqjIgzdHOk3yuQaO0ukE/). However, a deeper analysis reveals that a deposited PanDDA model should not be assessed individually because it represents only the ‘event’ state, whereas the original refinement was performed with an ensemble comprised of the ‘event’ state (with a weakly bound ligand) and the ‘ground’ state (without the ligand) averaged over many ‘ground’ state crystals. The deposited structure‐factor files do include Fourier coefficients for the ‘event’ state map, which is used as the primary evidence for the presence and placement of the expected ligand. Unfortunately, displaying these ‘event’ maps requires specialized crystallographic software and is unlikely to be performed by users without appropriate training, who are only interested in confirming the level of confidence in the modeling of the ligand. In addition, the assembly of datasets giving rise to the reference structure (‘ground’ state) with which the individual ligand structures (‘event’ states) are refined in an ensemble is not directly accessible.
In consequence, effective validation of the PanDDA ligands or fragments—which are present with low or very low occupancy—is currently not possible based on the deposited data. This is unfortunate, because it is crucial to clearly distinguish between high‐occupancy ligands with strong experimental support forming the foundations of structure‐based drug lead optimization, as opposed to ligands with low occupancy, which have weak experimental support and should guide only drug lead discovery or target enrichment, and even that with prudence. In view of their limited use due to low ligand occupancy and current impossibility to pass independent tests, it might be beneficial to further develop a separate procedure for handling PanDDA depositions by the PDB. Another problem with PanDDA group depositions is that they can literally flood the PDB with large numbers of low occupancy ligand complexes, thus impeding the search of good ligands.
Discussion
We have carefully analyzed 59 PDB entries corresponding to very recent deposits directly related to CoV‐2 proteins or representing previously deposited, closely related proteins from other coronaviruses. The analyzed proteins were divided into broad classes related to CoV‐2 structure and function, including the main protease 3CLpro and its inhibitor complexes, the papain‐like protease PLpro, the spike protein and its ACE2 receptor complexes, and various proteins either associated with or processing the viral RNA. The main goal has been to provide careful validation of the protein models, and especially of their small‐molecule ligands, that are produced at accelerated speed by first‐line structural research, so that a reliable information database could be created for subsequent drug discovery efforts. It is a gratifying observation that many of the analyzed structures passed our stringent validation criteria or required only minimal corrections, without affecting the biochemical conclusions. In several cases, however, the corrections brought about by our re‐refinement and modeling were significant and required presentation of a revised model.
We have paid particular attention to the ligands, especially the inhibitors of the 3CLpro protease, whose complexes are considered the first targets of drug‐design efforts. Also in this area, the results were often satisfactory, although in several cases we were able to propose a chemical modification of the ligand or correct the description of metal binding sites. The latter ones, while often serendipitous, may provide in fact guidance as to the exploitation of metal cations in designing enzyme inhibition strategies.
A separate comment is needed for the ligand complexes presented in the PDB by the PanDDA approach [47]. The incompatibility of the deposition protocol and the very complex PanDDA algorithm make it virtually impossible to independently recreate the results and thus to validate them. In our view, this is particularly worrisome as the algorithm itself is not without methodological questions, and the purported ligands sometimes have less than marginal support from independent experimental evidence.
Our analysis of the deposited structure models of CoV‐2‐related proteins has led to some general observations that might provide guidance for current and future uses of PDB models. Crystal structures can be presented with the macromolecules located in various places of space that are crystallographically equivalent and formally correct from the point of view of crystal symmetry. Indeed, this freedom is seen among multiple submissions of isomorphous structures that are presented in many different ways [48]. This is not a serious issue for trained crystallographers, but for many biomedical researchers that are not necessarily fluent in the principles of crystallography, such structures may appear to be completely different and this may lead to unnecessary misinterpretations and confusion. The publicly available ACHESYM server ([42]; http://achesym.ibch.poznan.pl/) allows the presentation of isomorphous structures in a uniquely standardized way, taking into account the equivalence of the space group symmetry positions. This makes comparisons of analogous structures and their electron density maps very easy, as this task boils down to simple superposition on any graphics system.
Another source of confusion may result from the freedom in selecting the direction of polar axes in some crystallographic symmetry classes. In these cases, unification of the results may require appropriate reindexing of the diffraction data, which also changes the system of coordinates for the model. In such cases, a newly solved crystal structure should be compared with previous isomorphous analogs in the PDB (if any) and, if necessary, reindexed and standardized via ACHESYM. A number of structures analyzed here required such an action for consistency.
The definition of the unit cell and crystal symmetry should abide by the well‐defined crystallographic standards [49]. For example, the unit cell angles of primitive and centered lattices should not exceed 120° and 135°, respectively. It is unfortunate that three of the structures analyzed here (5R7Z, 5R80, 5R83) are expressed in unit cells with one of the angles in excess of 159°, confusing not only people, but also the very robust graphics program coot [50]. For interactive inspection of the original 5R7Z model and the unusually skewed map, see an interactive visualization at https://molstack.bioreproducibility.org/project/view/mWteFUuuplHDabCzP470/.
In the context of efficient and fast communication between scientists focused on an urgent structural goal, such as discovery of drug‐design targets, we must reiterate the appeal that has been voiced numerous times [48, 51], that macromolecular models should be placed in standardized locations in convention‐abiding crystallographic unit cells.
Conclusions and Outlook
The revised models of the CoV‐2‐related proteins that were generated during this project are currently stored in a dedicated, publicly accessible web service (https://covid‐19.bioreproducibility.org). The ultimate goal is, however, to redeposit them in the PDB together with the original authors, using the mechanism of reversioning with clear cross‐references.
The work that we have initiated with this paper will continue as new CoV‐2 structures keep appearing in the PDB. We are also in the process of acquisition of raw diffraction images for the corrected structures, hoping that after expert reprocessing of the diffraction data, the final model might be even better, or described at higher resolution. The reprocessing work is also reflected in the https://covid‐19.bioreproducibility.org webserver as well as in the proteindiffraction.org repository.
Materials and Methods
The basis for the present analysis was the holdings of the Protein Data Bank, release of April 1, 2020. We extracted from the PDB all the deposited protein structures from the CoV‐2 coronavirus, plus a small fraction of homologous structures from closely related coronaviruses, such as SARS‐CoV or MERS‐CoV. For each deposit selected for validation, both the atomic coordinates and structure‐factor files were retrieved from the PDB. Unfortunately, in some cases, such as PanDDA group depositions, retrieval of all information necessary for reproducing the original results was not possible (e.g., the Deposition Group G_1002135 does not specify the model and the structure factors representing the ground state). For each X‐ray crystal structure, the validation procedure started with careful examination of the electron density maps in coot [50]. The maps were either downloaded from the PDB or calculated from the structure‐factor data deposited together with the atomic coordinates in the PDB, using programs from the ccp4 package [52]. If serious deficiencies, omissions, or errors were noticed, the structures were corrected according to the electron density maps during modeling rounds interspersed with structure‐factor refinement in refmac5 v. 5.8.0258 [53], as included in the CCP4 suite [52] or integrated in the HKL‐3000 suite [54] of programs. Reflections for R free testing [55] were the same as in the originally deposited data sets, if the respective flags were present. TLS parameters were generated with the TLSMD server [56] and were used in refinement (even if they were not utilized for the original models) if indicated by improved R free. Wherever possible, we tried to get access to the original diffraction images and start the re‐analysis with reprocessing of the diffraction data. Structure‐quality improvements were additionally monitored using the validation tools available in coot [50] and molprobity [37]. Extraction and ranking of validation metrics for the ligand molecules were carried out using twilight [57]. The achesym server was used for standardized placement of the structural models within the unit cell [42]. Molecular and structural illustrations, including presentation of electron density maps, were prepared in pymol [58]. molstack [30], a web‐based interactive publishing platform, was used for interactive representation of the electron density maps and models online. The re‐refined structures with all accompanying data were deposited in a dedicated public web resource https://covid‐19.bioreproducibility.org created specifically for this project. Ultimately, following all necessary procedures, the rectified structures will be deposited in the PDB, either reversioning the previous entries, or at least creating clear superseding pointers.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
AW, ZD, IGS, BR, WM, and MG analyzed data, discussed results, worked on manuscript. DB and MK created the webserver. MJ conceived and coordinated the project and coordinated manuscript preparation.
Acknowledgments
We thank C.X. Weichenberger, Eurac Bozen, Italy, for PDB data mining with Twilight. We also thank David R. Cooper and Marek Grabowski for their help during our research and manuscript preparation. We are indebted to the anonymous reviewer for the extremely thoughtful and helpful suggestions. This work was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, by NIGMS grant R01‐GM132595. DB acknowledges the support of the Polish National Agency for Academic Exchange under Grant No. PPN/BEK/2018/1/00058/U/00001. BR acknowledges partial funding through FWF (Austrian Science Foundation) grant P 32821. We acknowledge support of the National Science Center (NCN, Poland), grant No. 2020/01/0/NZ1/00134.
References
- 1. Hassan MI (2016) Recent advances in the structure‐based drug design and discovery. Curr Top Med Chem 16, 899–900. [DOI] [PubMed] [Google Scholar]
- 2. Thomas SE, Collins P, James RH, Mendes V, Charoensutthivarakul S, Radoux C, Abell C, Coyne AG, Floto RA, von Delft F et al. (2019) Structure‐guided fragment‐based drug discovery at the synchrotron: screening binding sites and correlations with hotspot mapping. Philos Trans A Math Phys Eng Sci 377, 20180422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Congreve M, Carr R, Murray C & Jhoti H (2003) A 'rule of three' for fragment‐based lead discovery? Drug Discov Today 8, 876–877. [DOI] [PubMed] [Google Scholar]
- 4. Wlodawer A & Jaskolski M (2015) In Inhibitors of HIV protease in Encyclopedia of Cell Biology (Bradshaw RA & Stahl P, eds), pp. 738–745. Waltham, MA: Elsevier. [Google Scholar]
- 5. Ghosh AK, Osswald HL & Prato G (2016) Recent progress in the development of HIV‐1 protease inhibitors for the treatment of HIV/AIDS. J Med Chem 59, 5172–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anand K, Ziebuhr J, Wadhwani P, Mesters JR & Hilgenfeld R (2003) Coronavirus main proteinase (3CLpro) structure: basis for design of anti‐SARS drugs. Science 300, 1763–1767. [DOI] [PubMed] [Google Scholar]
- 7. Hilgenfeld R (2014) From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. Science 281, 4085–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG & Kuhn RJ (2016) The 3.8 Å resolution cryo‐EM structure of Zika virus. Science 352, 467–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR & Saphire EO (2008) Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baker EN (2020) Visualizing an unseen enemy; mobilizing structural biology to counter COVID‐19. Acta Crystallogr F Struct Biol Commun 76, 158–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burley SK (2020) How to help the free market fight coronavirus. Nature 580, 167. [DOI] [PubMed] [Google Scholar]
- 12. Yan R, Zhang Y, Li Y, Xia L, Guo Y & Zhou Q (2020) Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science 367, 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C‐L, Abiona O, Graham BS & McLellan JS (2020) Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science 367, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT & Veesler D. (2020) Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell 181, 281–292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yuan M, Wu NC, Zhu X, Lee C‐CD, So RTY, Lv H, Mok CKP & Wilson IA (2020) A highly conserved cryptic epitope in the receptor‐binding domains of SARS‐CoV‐2 and SARS‐CoV. Science, eabb7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K & Hilgenfeld R. (2020) Crystal structure of SARS‐CoV‐2 main protease provides a basis for design of improved alpha‐ketoamide inhibitors. Science 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A & Li F. (2020) Structural basis of receptor recognition by SARS‐CoV‐2. Nature 581, 221–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burley SK, Berman HM, Christie C, Duarte JM, Feng Z, Westbrook J, Young J & Zardecki C (2018) RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci 27, 316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith M & Smith JC. (2020) Repurposing therapeutics for COVID‐19: supercomputer‐based docking to the SARS‐CoV‐2 viral spike protein and viral spike protein‐Human ACE2 interface. ChemRxiv11871402/.
- 20. Dauter Z, Wlodawer A, Minor W, Jaskolski M & Rupp B (2014) Avoidable errors in deposited macromolecular structures: an impediment to efficient data mining. IUCrJ 1, 179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shabalin I, Dauter Z, Jaskolski M, Minor W & Wlodawer A (2015) Crystallography and chemistry should always go together: a cautionary tale of protein complexes with cisplatin and carboplatin. Acta Crystallogr D Biol Crystallogr D71, 1965–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Minor W, Dauter Z, Helliwell JR, Jaskolski M & Wlodawer A (2016) Safeguarding structural data repositories against bad apples. Structure 24, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rupp B, Wlodawer A, Minor W, Helliwell JR & Jaskolski M (2016) Correcting the record of structural publications requires joint effort of the community and journal editors. FEBS J 283, 4452–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wlodawer A, Dauter Z, Porebski PJ, Minor W, Stanfield R, Jaskolski M, Pozharski E, Weichenberger CX & Rupp B (2018) Detect, correct, retract: How to manage incorrect structural models. FEBS J 285, 444–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng H, Chordia MD, Cooper DR, Chruszcz M, Muller P, Sheldrick GM & Minor W (2014) Validation of metal‐binding sites in macromolecular structures with the CheckMyMetal web server. Nat Protoc 9, 156–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Raczynska JE, Shabalin IG, Minor W, Wlodawer A & Jaskolski M (2018) A close look onto structural models and primary ligands of metallo‐beta‐lactamases. Drug Resist Updat 40, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jaskolski M, Miller M, Mohana Rao JK, Gustchina A & Wlodawer A (2015) Elucidation of the structure of retroviral proteases: a reminiscence. FEBS J 282, 4059–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grabowski M, Langner KM, Cymborowski M, Porebski PJ, Sroka P, Zheng H, Cooper DR, Zimmerman MD, Elsliger MA, Burley SK & et al. (2016) A public database of macromolecular diffraction experiments. Acta Crystallogr D Struct Biol 72, 1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Helliwell JR, Minor W, Weiss MS, Garman EF, Read RJ, Newman J, van Raaij MJ, Hajdu J & Baker EN (2019) Findable Accessible Interoperable Re‐usable (FAIR) diffraction data are coming to protein crystallography. IUCrJ 6, 341–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Porebski PJ, Sroka P, Zheng H, Cooper DR & Minor W (2017) Molstack‐Interactive visualization tool for presentation, interpretation, and validation of macromolecules and electron density maps. Protein Sci 27, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Porebski PJ, Bokota G, Venkataramany BS & Minor W (2020) Molstack: A platform for interactive presentations of electron density and cryo‐EM maps and their interpretations. Protein Sci 29, 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoffer L, Muller C, Roche P & Morelli X (2018) Chemistry‐driven hit‐to‐lead optimization guided by structure‐based approaches. Mol Inform 37, e1800059. [DOI] [PubMed] [Google Scholar]
- 33. Parks CD, Gaieb Z, Chiu M, Yang H, Shao C, Walters WP, Jansen JM, McGaughey G, Lewis RA, Bembenek SD et al. (2020) D3R grand challenge 4: blind prediction of protein‐ligand poses, affinity rankings, and relative binding free energies. J Comput Aided Mol Des 34, 99–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. DiMasi JA, Grabowski HG & Hansen RW (2016) Innovation in the pharmaceutical industry: New estimates of R&D costs. J Health Econ 47, 20–33. [DOI] [PubMed] [Google Scholar]
- 35. Yang H, Peisach E, Westbrook JD, Young J, Berman HM & Burley SK (2016) DCC: a Swiss army knife for structure factor analysis and validation. J Appl Crystallogr 49, 1081–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Suttisintong K, Kaewchangwat N, Thanayupong E, Nerungsi C, Srikun O & Pungpo P (2019) Recent progress in the development of HIV‐1 entry inhibitors: from small molecules to potent anti‐HIV agents. Curr Top Med Chem 19, 1599–1620. [DOI] [PubMed] [Google Scholar]
- 37. Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB et al. (2018) MolProbity: More and better reference data for improved all‐atom structure validation. Protein Sci 27, 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, Lu G, Qiao C, Hu Y, Yuen KY, Wang Q, Zhou H, Yan J & Qi J. (2020) Structural and functional basis of SARS‐CoV‐2 entry by using human ACE2. Cell 181, 894–904.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li F (2008) Structural analysis of major species barriers between humans and palm civets for severe acute respiratory syndrome coronavirus infections. J Virol 82, 6984–6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xia S, Zhu Y, Liu M, Lan Q, Xu W, Wu Y, Ying T, Liu S, Shi Z, Jiang S & et al. (2020) Fusion mechanism of 2019‐nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol Immunol, in press. 10.1038/s41423-020-0374-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xia S, Yan L, Xu W, Agrawal AS, Algaissi A, Tseng C‐TK, Wang Q, Du L, Tan W, Wilson IA et al. (2019) A pan‐coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Science Adv 5, eaav4580‐eaav4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kowiel M, Jaskolski M & Dauter Z (2014) ACHESYM: an algorithm and server for standardized placement of macromolecular models in the unit cell. Acta Crystallogr D Biol Crystallogr 70, 3290–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Blanc E, Roversi P, Vonrhein C, Flensburg C, Lea SM & Bricogne G (2004) Refinement of severely incomplete structures with maximum likelihood in BUSTER‐TNT. Acta Crystallogr D Biol Crystallogr 60, 2210–2221. [DOI] [PubMed] [Google Scholar]
- 44. Baez‐Santos YM, St John SE & Mesecar AD (2015) The SARS‐coronavirus papain‐like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res 115, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kirchdoerfer RN & Ward AB (2019) Structure of the SARS‐CoV nsp12 polymerase bound to nsp7 and nsp8 co‐factors. Nature Comm 10, 2342–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pearce NM, Krojer T & von Delft F (2017) Proper modelling of ligand binding requires an ensemble of bound and unbound states. Acta Crystallogr D Struct Biol 73, 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pearce NM, Krojer T, Bradley AR, Collins P, Nowak RP, Talon R, Marsden BD, Kelm S, Shi J, Deane CM et al. (2017) A multi‐crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat Commun 8, 15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dauter Z & Wlodawer A (2018) Crystallographically correct but confusing presentation of structural models deposited in the Protein Data Bank. Acta Crystallogr D Struct Biol 74, 939–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Henry NFM & Lonsdale K (1969) International Tables for X‐Ray Crystallography. The Kynoch Press, Birmingham, UK. [Google Scholar]
- 50. Emsley P & Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 51. Wlodawer A, Minor W, Dauter Z & Jaskolski M (2013) Protein crystallography for aspiring crystallographers or how to avoid pitfalls and traps in macromolecular structure determination. FEBS J 280, 5705–5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F & Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Minor W, Cymborowski M, Otwinowski Z & Chruszcz M (2006) HKL‐3000: The integration of data reduction and structure solution – from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62, 859–866. [DOI] [PubMed] [Google Scholar]
- 55. Brünger AT (1992) The free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–474. [DOI] [PubMed] [Google Scholar]
- 56. Painter J & Merritt EA (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr 62, 439–450. [DOI] [PubMed] [Google Scholar]
- 57. Weichenberger CX, Pozharski E & Rupp B (2013) Visualizing ligand molecules in twilight electron density. Acta Crystallogr F Struct Biol Commun 69, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. DeLano WL (2002) The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA. [Google Scholar]