

Abstract
Carbohydrates play a pivotal role in intercellular communication processes. In particular, glycan antigens are key for sustaining homeostasis, helping leukocytes to distinguish damaged tissues and invading pathogens from healthy tissues. From a structural perspective, this cross‐talk is fairly complex, and multiple membrane proteins guide these recognition processes, including lectins and Toll‐like receptors. Since the beginning of this century, lectins have become potential targets for therapeutics for controlling and/or avoiding the progression of pathologies derived from an incorrect immune outcome, including infectious processes, cancer, or autoimmune diseases. Therefore, a detailed knowledge of these receptors is mandatory for the development of specific treatments. In this review, we summarize the current knowledge about four key C‐type lectins whose importance has been steadily growing in recent years, focusing in particular on how glycan recognition takes place at the molecular level, but also looking at recent progresses in the quest for therapeutics.
Keywords: drug discovery, glycans, lectins, molecular recognition
Fine specificity: The interaction of glycans with C‐type lectin receptors (CLRs) mediates the dissemination of infections and is strongly related to the immune response. In this review, we emphasize the molecular recognition features of four of the most studied CLRs of group II: DC‐SIGN, Langerin, MGL and LSECtin
1. Introduction
The term “lectin” was firstly coined in 1954 and, since 1972, it has been systematically used to refer to all those known and newly discovered proteins and glycoproteins which have the ability to interact with carbohydrates. [1] Lectins are ubiquitous in nature, and are found in microorganisms, plants and animals at different cellular locations. They function as fundamental information mediators in a wide variety of molecular recognition processes, interacting with specific carbohydrate epitopes found on endogenous or exogenous oligosaccharides, glycoproteins and glycolipids, without modifying them (nonenzymatic). It is worth noting that their heterogeneity in many aspects, including their function, structure, specificity, cellular location and phylogenetic distribution makes it difficult to establish general classification criteria. For instance, in some cases, plant lectins have been typically subdivided into groups according to their distribution among similar species and common structural features: monocot mannose‐binding lectins (MMBL), jacalin‐related lectins, legume lectins, chitin‐binding lectins (hevein domains), etc. [2a] However, a strictly structural classification has been also applied in other cases. In Animalia, there are different categories defined according to both characteristic structural signatures and also their specific physiological roles and subcellular location (Figure 1).[ 2b , 2c ] As examples, galectins orchestrate multiple immunological responses, chiefly participating in glycan crosslinking at the extracellular matrix; [3] L‐type lectins are located in the lumen of the endoplasmic reticulum and Golgi, where they take part in trafficking and sorting of maturing proteins; [4] and siglecs are distributed on the cell surface and mediate cell‐cell adhesion processes by interacting with endogenous sialic acid residues. [5]
Figure 1.
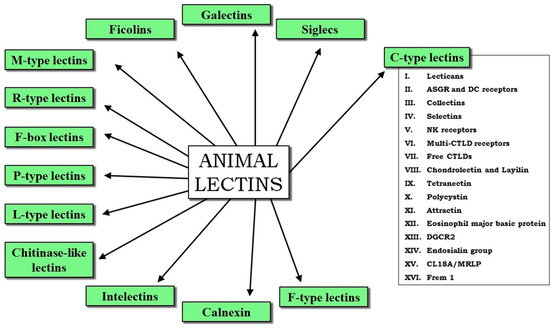
General classification of animal lectins.[ 2b , 2c ] The 16 groups of C‐type lectins are on the right.[ 7 , 10 ]
Among animal lectins, C‐type lectin receptors (CLRs) are particularly important in immunity, as most of them are expressed by different leukocytes and play a great variety of roles in host defense and maintenance of homeostasis, including cell‐cell and host‐pathogen adhesion, antigen uptake and complement activation. [6] From a structural perspective, all these lectins contain one or more carbohydrate recognition domains which share a series of common structural elements highly conserved among species (Figure 2).[ 6 , 7 ] The most important one is the presence of a calcium ion at the binding site, which is mandatory for the sugar interaction (calcium‐dependent binding, “C‐type”). Paradoxically, the C‐type superfamily currently includes other members with the ability to target carbohydrates without the assistance of calcium ions, [8] and even lectins whose substrates are proteins or lipids. [9] Nevertheless, the term C‐type lectin has been maintained due to the high structural similarity of these CRDs with the canonical ones (Figure 2B). Other authors have simply proposed to use the term C‐type lectin‐like domain (CTLD) as generic name for C‐type CRDs.[ 7a , 10 ]
Figure 2.
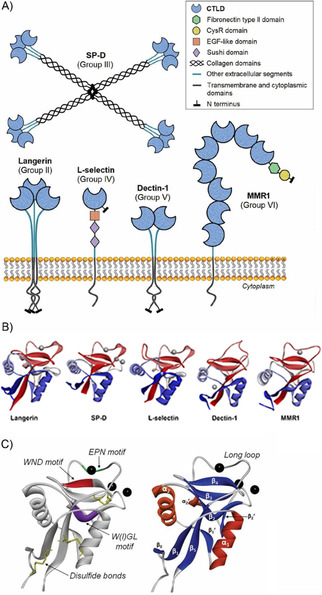
Characteristic structural features of C‐type lectins. A) Cartoon representation of five representative CLRs belonging to five different C‐type lectin groups (II, III, IV, V and VI).[ 6 , 10 ] B) Structural comparison between the CRDs of the same five lectins: human langerin (PDB ID: 5G6U), human surfactant protein D (PDB ID: 4E52), human L‐selectin (PDB ID: 3CFW), murine dectin‐1 (PDB ID: 2CL8), and human macrophage mannose receptor 1 (CRD2, PDB ID: 5XTS). Calcium ions are depicted in each case. C) Common structural motifs present in the CTLD fold (model: DC‐SIGN CRD, PDB ID: 1SL5). Right: the main secondary structure elements; left: typical conserved residues among different CTLDs and species. Calcium ions are shown as black spheres.[ 7a , 11 ]
As defining structural elements, all CTLDs present a central core essentially consisting of six to seven β‐strands organized into two β‐sheets and flanked by two α‐helices. [11] Up to four calcium sites have been described that may be occupied or not depending on the particular amino acid sequence (Figure 2B).[ 7a , 11 ] In canonical CTLDs, calcium site 2 is always occupied as it is the locus for sugar binding. This calcium site is composed of residues from the β4‐strand and the opposite loop. Note, some residues participating in the metal coordination sphere are well conserved, namely, the WND motif on the β4‐strand and the EPN/QPD motif at the loop (Figure 2C). Interestingly, the central proline in the latter case displays a cis conformation in most of cases. Historically, the EPN and QPD protein motifs have been respectively associated to mannose‐binding and galactose‐binding specificities, although the current knowledge has evidenced that these specificities are wider (roughly, EPN for Man/Fuc/Glc and QPD for Gal/GalNAc).[ 10 , 11 , 12 ] Regarding the recognition of sugars, the monosaccharide itself also coordinates the calcium ion through two vicinal hydroxy groups to establish the primary interaction. Typically, this interaction is fairly weak (in the millimolar range), although higher affinities result from additional contacts beyond the conserved residues at the secondary sites and from the multivalent architecture presentation of the lectin, which can lead to the generation of sugar‐lectin clusters.
The C‐type lectin superfamily is subdivided in at least 16 groups, considering other structural aspects and differences related to their CTLD organization and other protein domains, as well as their cell location and roles (Figures 1 and 2A).[ 7b , 10 ] Over the last decades, many CLRs have been discovered to act as central mediators in the dissemination and survival of many pathogens causing infections, as well as in the development and progression of certain cancer types and autoimmune diseases through the recognition of self‐glycans. As a result, these lectins have become potential targets to fight high mortality worldwide diseases such as those caused by Mycobacterium tuberculosis, human immunodeficiency virus (HIV), Ebola virus (EBOV), severe acute respiratory syndrome coronavirus (SARS‐CoV), or cancer and diabetes. It is worth noting that many of these examples involve CLRs from antigen presenting cells such as dendritic cells (DCs) and macrophages (MΦ), which are key in innate immunity and subsequent guiding of the adaptive response. As examples, soluble collectins (group III) like mannose binding lectin (MBL) and surfactant protein A (SP‐A) may be implicated in autoimmune disorders, such as allergy or diabetes.[ 13 , 14 ] Selectins (group IV) are fundamental in leukocyte trafficking and the acquisition of immunological memory, [15] but have been also described to facilitate lymphatic metastasis.[ 16 , 17 ] The dectin‐1 cluster (group V) includes receptors such as myeloid inhibitory C‐type lectin‐like (MICL), lectin‐like oxidized LDL receptor‐1 (LOX‐1), macrophage antigen H (CLEC‐12B or MAH), and the proper dectin‐1, whose implication in different diseases (leukemia, keratitis, rheumatoid arthritis, ulcerative colitis…) has been recently reviewed. [18]
Group II is very varied and includes many transmembrane DC‐ and MΦ‐related CLRs that have been demonstrated to be also relevant in development and progression of different diseases. Some of them, like macrophage‐inducible C‐type lectin (Mincle) or dectin‐2, have been recently reviewed[ 19 , 20 , 21 ] and are progressively gaining relevance in the therapeutic field due to their implication in immune suppression against Leishmania major and Fonsecaea spp., and autoimmune pathologies as atherosclerosis or arthritis. [22] Others, like the dendritic cell immunoreceptor (DCIR) or macrophage C‐type lectin (MCL), are still under study as they could be targets of interest for certain diseases as well. [23] Herein, we provide a detailed picture of the current knowledge on the molecular recognition features of four of the most thoroughly studied CLRs from group II for the last 20 years, namely DC‐SIGN, Langerin, MGL and LSECtin (Table 1).
Table 1.
General information on the four C‐type lectins reviewed in this text.
|
Human C‐type lectin and oligomeric state |
Relevant interacting monosaccharides and glycans |
Murine orthologues and identity [%] for the CRD[b] |
Reported 3D‐structures by X‐ray crystallography or cryoEM |
|---|---|---|---|
|
DC‐SIGN (tetramer) |
Man, Fuc, High Man N‐glycans (inner Man), Le‐type, ABO antigens |
SIGNR1‐SIGNR5 (65–70 %), SIGNR6‐SIGNR8 |
1K9I, 2XR5, 2XR6, 1SL4, 1SL5, 2IT5, 2IT6, 6GHV, 2B6B (cryoEM) |
|
langerin (trimer) |
Man, Fucα1‐2, GlcNAc, 6SGal, High Man N‐glycans (outer Man), GAGs |
langerin (77 %) |
3C22, 5G6U, 3KQG, 3P7F, 3P7G, 3P7H, 4N32, 4N33, 4N34, 4N35, 4N36, 4N37, 4N38, 3P5D, 3P5E, 3P5F, 3P5G, 3P5H, 3P5I |
|
MGL (trimer) |
terminal GalNAc |
MGL1‐MGL2 (64–68 %) |
n.a. |
|
LSECtin (dimer)[a] |
|
LSECtin (71 %) |
n.a. |
2. DC‐SIGN
Dendritic cell‐specific ICAM‐3‐grabbing non‐integrin, abbreviated as DC‐SIGN (CD209), is undoubtedly one of the most studied CLRs over the last two decades. This protein is mainly expressed in immature dendritic cells (DCs) from dermis, lymph nodes and tonsils, [24] and belongs to the heterogeneous group of ASGPR‐related and endocytic DC membrane receptors, [7] playing essential roles in innate immunity. [25] It was first described in the early nineties, [26] but became a target of great interest after its direct implication in HIV infectivity was uncovered. [27] The HIV viral particles interact with DC‐SIGN, which promotes their internalization assisted by LPS1 to guide its further degradation at lysosomal compartments. [28] Alternatively, the virus is able to establish a complex interplay between some DC co‐receptors and its own glycoproteins, particularly gp120, eventually impairing DC activity in a DC‐SIGN‐dependent manner to enhance its proliferation.[ 29 , 30 ] In this regard, HIV is also known to use DCs as vehicles to infect T cells, [31] increasing DC‐SIGN expression and CD4 targeting. [32] After DC‐SIGN recognition, the viral particles may be also shuttled to low‐pH endosomes, where they can survive for prolonged periods of time without replication. [33] Moreover, they can trigger infection of DCs themselves via simultaneous cross‐talk through DC‐SIGN and Toll‐like receptor 8, [34] and eventually induce cell apoptosis. [35]
Besides HIV, this C‐type lectin has been proved to be a crucial anchor in the development of multiple pathogen diseases, what has enormously increased its general interest as a therapeutic target. Some of those pathogens, such as Ebola [36] and dengue,[ 37 , 38 ] are still currently important health risks and hence have been investigated to a greater extent. Ebola virus internalization can be driven by either DC‐SIGN or L‐SIGN (the liver/lymph node‐specific homologue of DC‐SIGN), [39] and subsequent research have unveiled further details about the entry routes triggered by this pathogen.[ 40 , 41 ] The interaction of dengue virus with DC‐SIGN has been thoroughly described at the molecular level by cryoEM. [42] Apart from these, DC‐SIGN has also been described to take part in other viral infections including hCMV, [43] hepatitis C virus,[ 44 , 45 ] KSHV, [46] phlebovirus, [47] measles, [48] and SARS‐CoV. [50] It has been recognized that some coronaviruses bind to the ACE2 receptors located on alveolar cell membranes to initiate infectious processes. [49] In addition, SARS‐CoV was already known to use DC‐SIGN for infecting DCs and enhance viral transmission as well. [50] Besides ACE2, and given the current context of COVID‐19 outbreak, DC‐SIGN and other lectin receptors are under intense research to understand their possible roles in infectivity and viral spread of the novel SARS‐CoV‐2, which is now causing a worldwide pandemic. [51] A recent work points to a relationship between the levels of DC‐SIGN, L‐SIGN and ACE2 expression of the host and the infection risk, concluding that DC‐SIGN, in particular, is expressed at higher levels in Caucasian elder people (above 60) and in lungs from smokers, especially former smokers. [52]
Aside from viruses, DC‐SIGN also participates in bacterial and fungal infections.[ 53 , 54 ] Similar to HIV, Mycobacteria use DC‐SIGN as attachment point [55] and modulate DC functions through several co‐receptors. [56] As a result, the bacteria interfere with the normal production of cytokines and the phagocytic activity, [57] hampering DC maturation and blocking protective immune pathways. [58] In regard to fungi species, Candida albicans is the most studied one.[ 54 , 59 ] At a cellular level, it may use several different CLRs for holding on to the cell membrane, including DC‐SIGN.[ 60 , 61 ] In all these cases, the DC‐SIGN counterparts are exposed mannose‐containing biopolymers including glycoproteins and glycolipids. [62] Besides Man, DC‐SIGN also recognizes fucose (Fuc), [63] a common sugar present in endogenous glycan motifs such as blood group (ABO) and Lewis‐type antigens. [64] DC‐SIGN recognition of these sugars (Man, Fuc) is calcium‐dependent and takes place similarly, exhibiting low millimolar affinities. However, from the biological perspective, each sugar drives the immune response to a different outcome. [65] Thus, as exemplified above, Man‐targeting is usually mediated by DC‐SIGN and assisted by Toll‐like receptors, among others, which jointly stimulate secretion of pro‐inflammatory cytokines. [66] In contrast, Fuc‐mediated binding leads to the recognition of self‐glycoproteins like ICAM‐2 [67] or ICAM‐3, [68] thereby aiding DCs to move, communicate and/or guide the actions of other cells. [69] In some cases, DC‐SIGN also triggers an immunological response through Fuc recognition, for instance, in bacterial infections initiated by Helicobacter pylori [70] or in parasitic infections provoked by Schistosoma mansoni. [71] These organisms display surface glycan motifs very similar or identical to those found on mammalian cells (as LDNF or LeX), [72] exerting modulating effects that allow them to shape the immune response. [73] Similarly, proper DC activation can be impaired by Fuc‐containing self‐motifs, allowing malignant cells to escape from immune detection as occurred through Lewis‐type antigens in colorectal cancer. [206] Altered fucosylation patterns have been also ascribed to an incorrect DC‐SIGN‐mediated regulation of the immune outcome in certain cases, for instance contributing to brain damage in multiple sclerosis (MS). [231]
As other related lectins, DC‐SIGN displays an extracellular domain (ECD) divided into the CRD and the neck region. [7b] The latter is composed of eight amino acid repeats, seven of them comprising 23 residues and an extra truncated repeat (15 residues) at the N terminus. [74] The neck domains trigger the assembly of four protomers into a highly‐structured coiled‐coil tetramer. Notably, tetramerization is exclusively neck‐dependent and requires at least the presence of six repeats to maintain it as the dominant oligomeric species. [75] Although there are no crystallographic models describing the full ECD, partial X‐ray data, SAXS and computational modeling have allowed depicting suitable arrangements for the tetramer. In these models, each repeat forms an α‐helix which places its six aliphatic residues facing the inner core, supporting the hydrophobic packing of four α‐helix coils. Moreover, pH‐driven destabilization of the coiled‐coil stalk suggests that there could be also electrostatic interactions involved in tetramer stability. [76] According to modeling data, the four CRDs can display two types of spatial arrangements, referred to as “open flower” and “closed flower”. Crystallographic data obtained for the close homologue L‐SIGN [74] have pointed towards an “open flower” disposition, in which the last neck repeat forces the CRDs to adopt a configuration of a dimer of dimers.[ 76 , 77 ] In contrast, SAXS data better support a preferential “closed flower” for DC‐SIGN. Experimental evidences additionally agree that CRDs may freely modify their relative conformation to dock multivalent complex glycans, resulting in the transmission of these conformational changes to the neck regions. [78] Overall, a unique 3D‐model might not fully explain the dynamic observations, and moreover, both flexible CRDs and binding‐mediated neck changes fit with the high avidity noticed for this lectin [79] and the role of the neck regions in antigen endocytosis pathways. [80] Finally, the neck domains take part in the association of multiple tetramers at the cell surface, creating dispersed nanoclusters that are thought to magnify cell recognition of the pathogenic entity. [81]
Regarding its sugar recognition profile, DC‐SIGN is the paradigmatic case of ligand binding promiscuity. Its primary calcium site is surrounded by a relatively uniform flat surface, very exposed to the solvent and with few protruding side chains. This particular geometry easily enables the accommodation of a wide variety of glycans. As mentioned above, DC‐SIGN preferentially interacts with Man and Fuc, but the plasticity of the sugar interaction also makes possible the coordination to Glc, GlcNAc and ManNAc. [63] The recognition profile of its close homologue L‐SIGN is substantially similar, except for few differences regarding the interaction with Lewis‐type antigens.[ 63 , 85 , 100 ] Even Gal is recognized by both lectins as well, although the dissociation constant is substantially weaker. According to recent reports using fluorinated analogues, sugar positions 3 and 4 seem to be essential for binding of Fuc, Glc and Gal to DC‐SIGN, whereas only position 3 is important in Man, as it can tolerate fluorination at C4.[ 82 , 83 ] Indeed, Man has been demonstrated to generate up to three different binding poses, two of them through hydroxy groups OH3 and OH4, and an additional one, very stable, through hydroxy groups OH2 and OH3. [82] In natural highly‐mannosylated glycans, the outer Man residues are often α1‐2‐linked to the previous sugars, disabling recognition mediated by the OH2‐OH3 pair. Otherwise, the other two binding poses have been identified in crystallographic structures, and actually co‐exist in solution for the Manα1‐2Man fragment.[ 79 , 84 ] In larger glycans, the presence of multiple surrounding sugars gives rise to other stabilizing interactions that are typically more favored in one of the possible binding modes. Hence, in most cases, X‐ray sugar‐lectin complexes show that the primary Man epitope adopts the same orientation, with its OH2 group close to the long loop and its OH6 next to Val351.[ 79 , 85 , 86 ] Remarkably, in all cases, these structures always display an inner α1‐3‐linked Man moiety at the calcium site, placing the nonreducing end on top of the long loop, while the upstream scaffold is perfectly accommodated at the secondary site, flanking Phe313 (Figure 3A and B). Besides α1‐3‐linked Man moieties, DC‐SIGN targets α1‐2‐linked and α1‐6‐linked Man residues as well, always located at inner positions of the Man9 scaffold. The two first types are the preferred ones, whereas the recognition mediated by α1‐6‐linked mannoses seems to be weaker.[ 87 , 88 ] These observations highlight that interactions provided by outer mannoses might not be so relevant, likely due to the lack of important secondary noncovalent contacts. Therefore, the resulting binding strength is often mainly determined by the number of available inner Man units, justifying in many cases the enhanced affinities noticed for high‐Man ligands, probably as a result of “statistical rebinding” effects.[ 89 , 90 ]
Figure 3.
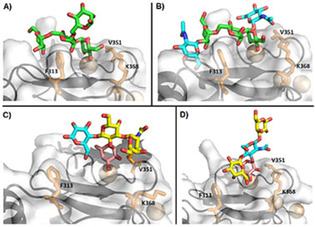
Binding poses experimentally described for the DC‐SIGN CRD and four typical mannosylated and fucosylated oligosaccharides. Structures A, B and D have been solved by X‐ray crystallography. Model C is a representative structure obtained by MD and supported by NMR data. Sugars are colored as follows: Man: green, Fuc: magenta, Gal: yellow, Glc: blue. Residues F313, V351 and K368 are shown as sticks in all cases. A) Man4 (PDB ID: 1SL4). B) GlcNAc2Man3 (PDB ID: 1K9I). C) Blood group A type VI. [83] D) Lacto‐N‐fucopentaose III (PDB ID: 1SL5).
The scenario completely changes when it comes to complex multi‐antennary glycans decorated with other sugars, usually Glc, Gal and sialic acid. Of note, the Man9GlcNAc2 undecasaccharide has two inner Man3 cores, but only the α‐linked one can establish noticeable interactions with DC‐SIGN. [85] Interestingly, the GlcNAc2Man3GlcNAc2 heptasaccharide is recognized, and the smaller GlcNAc2Man3 fragment has been actually crystallized and analyzed (Figure 3B),[ 86 , 91 ] thus suggesting that the two nonreducing GlcNAc moieties might exert a stronger positive effect in the recognition, or at least compensate the unfavorable accommodation of the β‐Man residue. As noted, the interplay existing between both stabilizing interactions and steric effects eventually determines the absence or presence of binding. Recent reports have thoroughly screened these effects making use of large glycan arrays, evidencing striking affinity changes sometimes arising from remote chemical modifications.[ 91 , 92 , 93 ] As examples, the presence of terminal sialylation completely abrogates recognition, [92] as well as the elongation of the GlcNAc2Man3GlcNAc2 scaffold with Galβ1‐4, although the negative effect is asymmetric, given that the presence of one unique Gal moiety at the α1‐6 branch still permits a weak binding (Figure 4, left).[ 91 , 93 ] Similarly, bisecting residues and double core fucosylation at the reducing end of the chitobiose disaccharide also produce an affinity loss in general. Instead, the attachment of more nonreducing Man or GlcNAc residues tends to enhance DC‐SIGN recognition in many cases, but the observed affinity highly depends on the overall glycan geometry. Ligand presentation is thus a crucial aspect underlying the suitable recognition of complex glycans architectures in in vitro assays and this is also reflected in the biological responses. [94] Thus, despite Candida albicans exhibiting several classes of complex mannans (N‐linked, O‐linked, phospho‐Man), DC targeting is essentially supported by N‐glycans, although phosphomannans might be also indirectly involved in conformational modulation of the N‐glycan part.[ 62 , 95 ] Alternatively, Mycobacterium species are differently recognized depending on the presence and distribution of Man caps in their lipoarabinomannans. [96]
Figure 4.
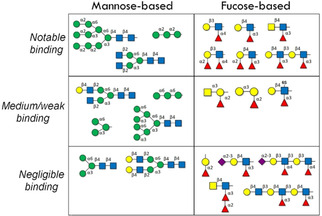
Representative Man‐ and Fuc‐containing epitopes and their recognition by DC‐SIGN according to published array data (refs. [85, 89–91, 93, 98]).
DC‐SIGN interaction with Fuc‐containing oligosaccharides has been intensely studied as well. Similarly, glycan array studies have provided a great amount of information about those geometrical aspects governing binding preferences. However, these results may sometimes result confusing, as there is still not enough structural data to clarify discrepant affinities from a solid molecular basis. In turn, such details are useful since Fuc, as Man, may be analogously exploited in the development of sugar mimetics. [97] In general terms, DC‐SIGN is able to recognize an extensive plethora of Fuc‐containing antigens, including the Lewis‐type motifs (LeA, LeB, LeX, LeY) and blood group determinants (A, B, H).[ 89 , 90 , 98 ] Lewis‐type and ABO antigens differ from each other in the sugar composition and the configuration of the glycosidic linkages. Interestingly, at molecular level, both LeX and blood group antigens have been found to target the calcium site in the same way. In both cases, the Fuc moiety coordinates the metal ion by means of OH3 and OH4 groups, creating a very stable hydrophobic contact with the nearby Val351 residue (H1 and H2; Figure 3C and D). However, the other neighboring sugars play different roles in each case. In the LeX‐DC‐SIGN complex, the nonreducing Gal is accommodated at the secondary site, providing noncovalent interactions close to Phe313.[ 85 , 99 ] Moreover, the central GlcNAc also establishes an aliphatic interaction with Val351. In contrast, for the blood groups, the nonreducing Gal/GalNAc moiety packs against the aliphatic Val351 side chain along with Fuc, enhancing the hydrophobic interaction with this protein residue. [83] It is worth mentioning the role that Val351 has in DC‐SIGN selectivity towards fucosylated antigens. When this side chain is removed (V351G) or its aliphatic nature is changed (V351S), Man‐mediated binding still occurs, whereas Fuc‐mediated binding substantially worsens. [100] In fact, L‐SIGN possesses a polar serine side chain (S363) at the equivalent position and hence, a recognition profile very similar to the V351S mutant of DC‐SIGN: it cannot recognize LeX and LeY structures, while binding of the LeA and LeB analogues is still present, although rather weak.[ 85 , 100 ] In any case, studies with LeX mimetics have also highlight the notable contribution of secondary contacts around Phe313 for generating stable complexes with DC‐SIGN.[ 97 , 101 ]
Taking as a reference the models commented above, some data from arrays may have a partial explanation, whereas other observations remain neither clarified nor supported at all, for which specific structural studies are still necessary. For instance, the recognition of LDNF or LeY can be accounted for on the basis that both ligands can be superimposed on the LeX scaffold, such that their particular structural differences do not preclude the described binding pose (Figure 4, right).[ 89 , 102 ] Analogously, terminal sialylation (sialyl‐LeX, sialyl‐LeA) almost abrogates binding, [98] but the reason is not fully understood yet. [103] Finally, this issue also extends to multivalent presentations: the effect of various Fuc epitopes in the same oligosaccharide is usually rather unpredictable. In general, Lewis‐type repeats exhibit increased affinities (statistical rebinding), but not all the Fuc residues equally sustain the binding. In the same line, the LeB and LeY antigens display higher potencies, suggesting that both fucoses likely participate in the recognition event. [89] In contrast, difucosylated LDN‐DF antigens from S. mansoni have been reported to be poorly recognized by DC‐SIGN (Figure 4, right). [71a] All these cases underline the enormous importance of undertaking detailed structural studies to unveil the fine details that modulate ligand presentation. As a proof‐of‐concept, in the structural study with blood antigens, the fine selectivity of DC‐SIGN towards the B antigen has been found to arise from a slight rebinding effect originated by the existence of a minor binding pose, in which sugar attachment is Gal‐driven. Fittingly, the same binding mode is not possible for the A antigen, whose terminal GalNAc cannot directly coordinate the calcium ion in any way, as demonstrated by STD NMR experiments. [83]
Over the years, the relevance of sugars in pathogen infections, autoimmune diseases, and cancer has been progressively unraveled, and lectin targeting has become an imperative strategy for the development of therapeutics, along with anti‐glycan vaccines. [104] Consequently, the increasing amount of structural information on DC‐SIGN and its biological relevance have remarkably encouraged the design of specific ligands with increased potencies. In this regard, most of research efforts have been focused on exploiting the Man scaffold for either improving or creating new secondary interactions, and also on displaying them in a multivalent fashion. [105] The paradigmatic case is the Manα1‐2Man disaccharide, whose binding modes have been exhaustively described, as mentioned above. Over the last 15 years, sequential modifications have been stepwise introduced in both Man moieties, leading to compounds with affinities two‐three orders of magnitude higher (<100 μM) and total selectivity for DC‐SIGN (disabling langerin recognition; Figure 5). To note, the substitution of the reducing pyranose by a cyclohexane ring already produces a slight improvement, especially with respect to drug‐like properties. [106] Such a structure could be anticipated to bind through the outer Man, mimicking the minor mode described for Manα1‐2Man, [107] but X‐ray and NMR data interestingly supported the opposite orientation, in which the “reducing” aliphatic ring is located on top of Val351. [108] Following these observations, the subsequent chemical modifications were placed to increase the ligand contacts with the long loop [109] and with the secondary site (Phe313), by adding molecular fragments at C2 or C6 of the nonreducing Man.[ 110 , 111 ] In addition, these modifications also take advantage of the shallower and more acidic binding area of DC‐SIGN to hinder langerin recognition, thereby enhancing DC‐SIGN selectivity. [111] The elongation of the mannobioside with a third reducing Man improves the affinity as well, although the binding pose and contacts were found to be identical to those for the mannobiose analogue as revealed by X‐ray crystallography. [112] This is a fairly particular case, as this ligand displays two available Man residues and hence its higher affinity expectedly arises from multiple binding (Figure 5, bottom). Indeed, the observed STD NMR data can only be explained by considering two binding modes, being the major one that observed in the crystal structure. [113] Hydrodynamic measurements showed however that the important gain in affinity arises from the ability of the mannotriose moiety to bridge two ECDs in solution (“receptor clustering”). [112] Overall, the cyclohexane moiety always maintains the key Man residue coordinated in the same orientation, underlining that the alternative one might be unfavorable or not stable enough. Conversely, the alternative orientation has been achieved by changing the aliphatic ring by more flexible functionalized polar glycerol chains, successfully enabling secondary van der Waals contacts at the proximal β‐sheet. [114] Notably, the incorporation of naphtyl and phenyl moieties allows efficient hydrophobic interactions with Phe313, substantially lowering the dissociation constants (low μM).
Figure 5.
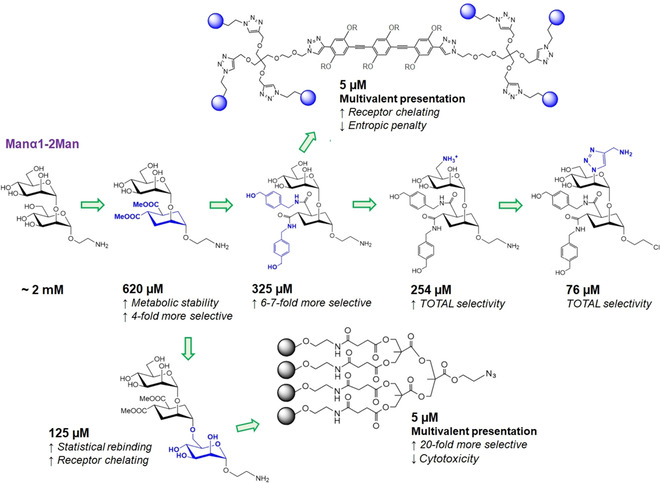
Synthetic modifications progressively introduced in the Manα1‐2Man scaffold in order to increase receptor affinity and DC‐SIGN selectivity over langerin. Also, two examples of low millimolar multivalent structures bearing these mimetics are shown.
As C‐type lectins are intrinsically weak receptors, the most evident way to exponentially raise the inhibitory potencies implies using multivalent scaffolds. [115] For DC‐SIGN in particular, this has been also a field of intense research. Apart, the efficient exploitation of multivalent interactions requires controlling several complex factors operating at once, such as the nature of the ligand, its individual presentation or the spatial distribution of epitopes throughout the multivalent platform (ligand density). Structures with multiple exposed sugar units can compete with natural glycans, even using the simple Man as single epitope (Table 2). Thus, dendrons carrying few tens of mannoses can already inhibit viral infections with IC50 values in the low‐micromolar range. [116] The replacement of Man by better epitopes, for instance Manα1‐2Man, in principle aids to enhance potencies as well.[ 117 , 118 ] In this line, the aforementioned mimetics have also proven to be effective for inhibition of HIV and dengue infections when conjugated to dendrimer‐like structures, exhibiting low‐micromolar IC50 as well as improved drug‐like properties (low cytotoxicity, selectivity against langerin).[ 112 , 119 ] It is worth mentioning that the ligand presentation always plays the dominant role: in some cases, affinity changes have been no longer noticed when increasing the dendrimer generation, indicating that the maximum effective ligand density on the surface has been already reached.[ 117 , 120 ] Man‐coated gold nanoparticles are an illustrative example of such an effect, since the best potencies often correspond to intermediate percentages of epitope occupancy.[ 118 , 121 ] In these cases, linear scaffolds such as synthetic glycopolymers [122] or DNA/RNA templates [123] may show an alternative advantage: the epitope density is fixed regardless the chain length, whereby the best affinity can be always achieved for 100 % occupancy as long as epitope spacing is optimized. [124] Additionally, chelating effects are easier to generate. As last alternative, the attachment mode can be also considered for tuning the epitope presentation. Usually, sugars are O‐linked through the anomeric carbon, but C‐glycosides have been demonstrated to be beneficial in terms of affinity and multivalence, likely due to their higher conformational dynamics. [125] Also, the linkage might be created at position 6, as long as the coordination to the primary site remains unperturbed. [126]
Table 2.
Examples of Man‐coated multivalent scaffolds successfully used to inhibit DC‐SIGN binding to potent Man‐based biological epitopes, as viral glycoproteins or viral strains. Gray zig‐zag lines represent synthetic linkers.
|
Type of multivalent platform |
Single epitope and copy number |
Competition model |
IC50 [nM] |
Ref. |
|---|---|---|---|---|
|
Dendrimers |
|
|
|
|
|
|
120 Man |
Jurkat T cells infected with Zaire EBOV‐pseudotyped recombinant viruses |
0.67 |
[130] |
|
Nanoparticles |
|
|
|
|
|
|
22 Manα(1‐2)Man[a] |
Raji‐DC‐SIGN+ cells infected with HIV‐1 (JR‐Renilla R5) |
2.04 |
[118] |
|
Neoglycoproteins |
|
|
|
|
|
|
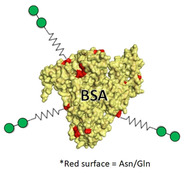
36 Manα(1‐2)Man |
DC‐SIGN targeted by Eu‐DTPA‐labeled Man51‐BSA |
0.8 |
[132] |
|
Linear glycopeptides |
|
|
|
|
|
|
17 Man[a] |
DC‐SIGN targeted by gp120 (HIV) |
48 |
[124] |
|
Glycolipid dendrons and sugar‐coated micelles |
|
|
|
|
|
|
9 Man (per dendron)[b] |
MAGI‐CCR5 infected with HIV‐1 (NL4‐3 R5) |
500 |
[143] |
[a] On average. [b] Autoassembly in micelles at 109 μM (r H=39 nm).
Ligand density can also change the accessibility to alternative binding poses for certain epitopes, sometimes provoking steric hindrance problems which lead these ligands to exhibit smoother affinity enhancements.[ 112 , 127 ] As example, Man‐coated gold nanoparticles display higher affinities for DC‐SIGN when they are functionalized with the Manα1‐2Man disaccharide rather than with Manα1‐2Manα1‐2Man or Man5. [121] To reach very high inhibitory potencies, the exposed surface should be increased such that the available residues grow while maintaining an optimal epitope density and accessibility. In this regard, the combination of rigid elements and flexible spacers must be thoroughly controlled.[ 128 , 129 ] In sugar‐functionalized fullerenes, flexible long spacers have been efficiently used to exponentially increase the amount of exposed residues up to 120 Man, enhancing inhibitory potencies below the nanomolar range. [130] The quest for better inhibitors can be further focused on reducing the flexibility of the sugar‐coated structure, thereby minimizing the entropic penalty. This has been recently noticed for hexavalent scaffolds with either rigid rod‐shape cores or completely flexible chains (Figure 5, top). [128] Alternatively, rigid sugar glycoclusters, as those based on calyx[4]arenes, may help to concentrate the sugar epitopes at specific positions, enhancing sugar rebinding phenomena and likely contributing to receptor clustering. [131]
Neoglycoproteins are another source of large sugar‐coated structures, also utilized as lectin receptors.[ 89 , 132 ] As notable breakthrough, the capsid protein of the Qβ bacteriophage has been applied for building glyconanoparticles coated with more than 1000 Man residues. [133] Each protein possesses a modified amino acid which harbors a nonavalent dendron. At the same time, these capsid components self‐assemble into 180 unit spherical particles. [134] The resulting structures can strongly inhibit EBOV infection with IC50 values within the low nanomolar range. [133]
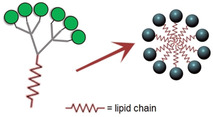
The aforementioned rigid hexavalent structures carrying the dimannoside mimetic can be relatively quickly internalized at physiological temperature, driving DC differentiation and maturation through production of cytokines (IL‐6, TNFα). [135] In contrast, recently reported star‐shaped glycopolymers cannot activate DCs via DC‐SIGN unless they are co‐stimulated with LPS and IFN‐γ. [136] In other cases, ligand presentation directly affects antigen internalization. [137] To exemplify, dimannoside‐containing clusters have been noticed to be preferentially shuttled inside DCs than the trimannoside‐based analogues. [138] Similarly, DC‐SIGN promotes uptake of fucosylated oligolysine scaffolds, but not of the mannosylated ones. [139] In all these cases, it is always important to consider the different responses that might be driven by similar CLRs, like MR or langerin.[ 137 , 139 , 140 ] Frequently, a common strategy to trigger an efficient DC‐mediated immune response consists of combining sugar epitopes for CLRs and other ligands to co‐stimulate Toll‐like receptors, achieving for instance effective antitumor responses.[ 138 , 141 ] DC‐SIGN internalization capabilities have been also exploited for intracellular delivery of therapeutic agents, [142] and for this purpose, sugar‐coated liposomes are ideal platforms. In general, micelles and liposomes are quick pathways to afford enormous multivalent structures from suitable functionalized lipids. For instance, the conjugation of Manα1‐2Man nonavalent dendrons to fully saturated aliphatic chains has been successfully applied to inhibit HIV trans‐infection of DCs. [143] In the same line, liposomes coated with Lewis‐type antigens stimulate DC activity and cross‐talk with T cells. [144] Recently, these platforms have been efficiently used for the delivery of antigenic mRNA in mice, which can induce specific tumor repression. [145] It is worth mentioning that cyclodextrins functionalized with sugar chains can work as carriers as well. However, in such cases, the loading of lipophilic compounds may be hampered by the external polar sugar layer. [146]
Overall, the design of potent DC modulators is still challenging. Although multivalence is in general the most employed strategy to overcome the poor intrinsic affinities of C‐type lectins, computational and screening studies have gradually directed the attention to other novel DC‐SIGN ligands.[ 147 , 148 ] Indeed, previous works have already found non‐sugar fragments with remarkably low affinities compared to monosaccharides (μM; Figure 6A and B), [149] and some of them are able to efficiently trigger cell signaling when conjugated to a protein scaffold. [150] Fragment screening analyses have identified other potentially druggable sites in the CRD architecture, [151] which could be exploited for the design of more potent DC‐SIGN modulators (Figure 6C).
Figure 6.
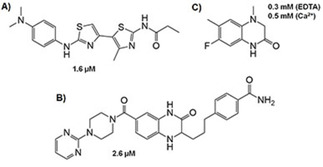
Some non‐sugar inhibitors described for DC‐SIGN and their affinities. A) and B) Active compounds found by fluorescent assays. C) Active fragment detected and validated by NMR. Note, it still binds to DC‐SIGN in the absence of Ca2+, thus suggesting that the interaction is not established at the primary lectin site.
3. Langerin
Langerin (CD207) is another type II C‐type lectin receptor involved in the attachment and uptake of invading pathogens during the first stages of the immune response.[ 152 , 153 ] As defining feature, the expression of this receptor is almost exclusively limited to Langerhans cells (LCs), a subset of immature dendritic cells originated from myeloid precursors that finish their migration at the epidermis and mucosal epithelium. [154] Langerin is known to drive the formation of the so‐called Birbeck granules (BGs), particular organelles found on LCs and constituted by several zippered membranes forming rod‐like structures, which probably take part in the endosomal recycling pathways. [155] As other CLRs, this receptor has been reported to act as anchoring and internalizing factor in many infectious processes, involving bacteria, [156] fungi [157] and viruses. [158] Remarkably, at low viral load, langerin is able to promote the efficient internalization and degradation of HIV particles, protecting LCs from infection and avoiding viral transmission to lymphocytes. [159] However, there is still some controversy regarding the exact roles of langerin in HIV infectivity. [160] Subsequent works have evidenced the efficient infection of LCs orchestrated by langerin and the further transmission of the viral particles to T cells. [161] Certainly, HIV susceptibility may depend on other complex factors besides the viral load, including the cell models used in the assays and the infection phases. [162] In this regard, good LC models are generally difficult to reproduce and the published data maintain this discussion opened yet. Additionally, murine models have shown that LCs preserve their phenotype and normal functions in the complete absence of langerin, just lacking the ability to generate the BGs [163] and feeding even more the mystery about the exact physiological role of this lectin. Nowadays, the scope of study and the importance of this receptor are progressively becoming broader. Recently, langerin has been proposed, along with DC‐SIGN, as a relevant receptor in oral cancer episodes through the recognition of highly‐fucosylated glycans on cell surfaces. [164] Also, similar to DC‐SIGN and L‐SIGN, [165] langerin can act as an entry receptor for influenza A virus (IAV), even in the absence of surface sialylation, [166] thereby confirming it as the major mediator in IAV infections occurring on LCs. This “duality” turns this protein into a challenging target for the development of therapeutics, as it might promote infectivity in some cases while exerting a protective role in some others, triggering the formation of BGs. Hence, a better understanding of this receptor is still pursued to date, especially regarding the existing disparities with DC‐SIGN in HIV transmission. In this aspect, the development of highly specific drugs discriminating DC‐SIGN from langerin is highly desirable, as they should not affect the natural defense created by langerin.
Early studies with mouse langerin already showed that this lectin can recognize mannose‐capped glycans. [167] Its structure is essentially composed of a cytoplasmic domain, a transmembrane segment, and an extracellular portion including the neck repeats and the Ca2+‐dependent CRD. [168] Accordingly, its CRD presents the typical structural elements of a C‐type lectin‐like domain, including the EPN motif commonly associated with Man/Fuc selectivity.[ 7b , 11 , 169 ] Its sugar preferences are fairly similar to those of DC‐SIGN: [63] Langerin recognizes Man, Fuc, Glc and GlcNAc monosaccharides with low millimolar affinities, as well as high‐Man glycans (Figure 7). Conversely, it is unable to interact with complex multi‐antennary glycans, either capped with sialic acid or not. [170] In addition, its corresponding mouse homologue was surprisingly discovered to interact with dextran sulphate and with epitopes containing terminal 6S‐Gal as well. [171] This exquisite selectivity is likewise observed in the human variant and further complemented with some differences regarding the recognition of Lewis‐type antigens. In contrast to DC‐SIGN, langerin cannot bind to LeX and LeA antigens, while recognition of LeY and LeB, as well as the blood group antigens A and B, is maintained (Figure 7).[ 90 , 172 ] These findings point out a limited ability for the exclusive recognition of terminal α1‐2‐linked Fuc residues, suggesting a possible protective role of the natural LeX‐containing antigens as selective binders for DC‐SIGN, given that these epitopes are abundant in human milk. [173]
Figure 7.
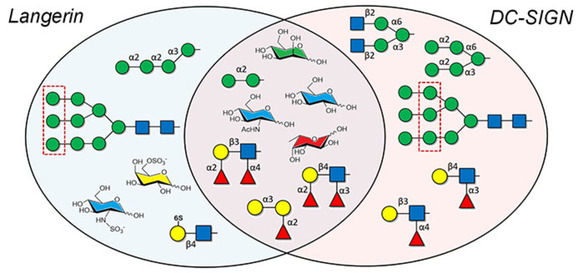
Qualitative comparison between the sugar preferences of DC‐SIGN and langerin. The central area displays those epitopes similarly recognized by both receptors. To clarify, Gal has not been included as free sugar, as the dissociation constants are rather high in both cases. Only langerin recognizes sulfated moieties, including Gal and Glc, although it preferentially targets outer Man residues in complex glycans. Also, DC‐SIGN interacts with a wider range of fucosylated structures and more easily accommodates Man residues from highly branched scaffolds.
The fact that langerin can mediate recognition processes with sulfated sugars immediately raised the question about its interaction with glycosaminoglycans (GAGs) and its potential biological role in such a context. Langerin has been reported to recognize sulfated Gal and GlcNAc residues, both present in keratan sulfate (KS) and heparin‐related GAGs, respectively (Figure 9). Interestingly, the studies with sulfated LacNAc disaccharides have evidenced the existence of certain structural requirements underlying the stability of these interactions.[ 174 , 175 ] Thus, binding takes place when the 6S group is placed at the nonreducing Gal, whereas the presence of sulfate groups at the inner GlcNAc moiety seems to be irrelevant. Moreover, the interaction is abrogated after terminal sialylation (sulfated sialyl‐LacNAc) or when the sulfate group is attached at position 3. [176] It is worth noting that 6S‐GlcNAc can be actually recognized as free monosaccharide. All these findings underline the crucial role of the sulfate group at position 6 in the Gal scaffold, making possible the recognition of this sugar by an EPN lectin. From the protein perspective, two lysine residues (K299 and K313) exclusively present at the secondary site of langerin, and not at other C‐type lectins, are responsible for the stabilization of the negatively charged SO4 − group through a double salt bridge (Figure 8C).[ 174 , 177 ] Despite the restricted binding to terminal sulfated sugars, linear KS chains [178] have been found to exhibit enhanced affinities (ca. 100‐fold lower K D values), which additionally correlate with their SO4 − content, suggesting the existence of avidity effects typical from systems with several available epitopes. Otherwise, recently reported synthetic polymers capped with terminal 6S‐LacNAc units have been shown to display very potent affinities (nM) and are able to exert anti‐inflammatory activities in mouse models with chronic obstructive pulmonary disease (COPD).[ 175 , 179 ] These results certainly represent a new frontier in the treatment of COPD in cigarette smokers, as Langerhans cells have been reported to accumulate in broncoalveolar tissues of COPD patients. [180]
Figure 9.
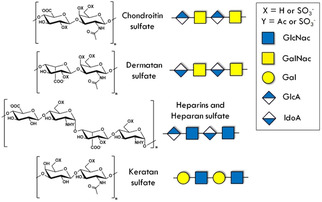
Schematic structure of the most relevant glycosaminoglycans (GAGs). All GAGs consist of a repeated disaccharide unit constituted by an acetylated sugar and an uronic acid. Chondroitin sulfate (CS) and dermatan sulfate (DS) share a GalNAc unit that can be sulfated at O4 and/or O6. Keratan sulfate (KS) displays a sulfated Gal instead of an uronic unit. Heparins and heparan sulfate (HS) are fairly heterogeneous, they can contain variable amounts of IdoA and GlcA. Often, heparins are highly sulfated (up to three sulfate groups per disaccharide) and preferentially contain IdoA.
Figure 8.
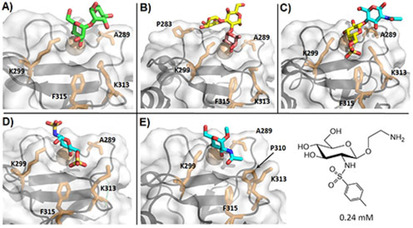
Crystallographic models obtained for langerin interacting with different mono‐ and oligosaccharides. Sugars are colored as follows: Man: green, Fuc: magenta, Gal: yellow, Glc: blue. Residues A289, K299, K313, F315, and V351 are shown as sticks in all structures, other relevant amino acids are labeled in particular cases. A) Man2 (PDB ID: 3P5F). B) Blood group B trisaccharide (PDB ID: 3P5G). C) 6S‐LacNAc (PDB ID: 3P5I). D) GlcNS6S (PDB ID: 5G6U). E) α‐OMe‐GlcNAc (PDB ID: 4N32); on the right is a mimetic scaffold based on the binding pose of GlcNAc, bearing an aromatic moiety to establish aliphatic contacts with nearby side chains (F315).
As for other C‐type lectins, the langerin‐mediated recognition of Man‐containing oligosaccharides has been exhaustively investigated from the structural perspective, especially using X‐ray crystallography. Interestingly, in most structures (Man2, Man4, Man5)[ 177 , 181 ] the Ca2+‐coordinated Man residue has been found to display the opposite ring orientation to that observed for the analogous DC‐SIGN structures (Man2, Man4, Man6).[ 79 , 85 ] In such an orientation, the axial OH2 targets the polar side chain of the nearby K299 and the remaining glycan structure, although not modeled, should point away from the lectin surface (Figure 8A). These models intriguingly suggest that langerin probably would not bind to the inner Man residues of the Man9GlcNAc2 glycan, since this orientation would preclude the accommodation of the outer Man moiety at the secondary site, due to the protruding lysine side chains. Even so, the Manα1‐2Man disaccharide displays the two alternative presentations in the crystal with similar occurrence (ca. 50 %). In this case, although the alternative orientation enables binding through the reducing Man, the accommodation of the extended glycan structure would be expectedly more limited than that reported for DC‐SIGN. In any case, recent NMR data [182] using the same disaccharide also support that langerin preferentially binds to the nonreducing Man residues, additionally suggesting that the neighboring moiety provides stabilization through packing against the Ala289 side chain.
Regarding other sugars, several X‐ray diffraction structures have provided insights into binding of Glc‐containing scaffolds to langerin. The complexes with maltose (Glcα1‐4Glc) [181] and laminaritriose (Glcβ1‐3Glcβ1‐3Glc) [177] highlight that the primary epitope is always the nonreducing Glc, which coordinates the calcium ion through hydroxy groups at C3 and C4. Remarkably, the Glc ring adopts the same orientation than the Man analogue described above, but creating a stabilizing polar contact with K299 through OH6 instead of OH2. The scenario is identical for α‐OMe‐GlcNAc, with the additional contribution of the N‐acetyl group, which establishes water‐mediated polar contacts with K299 and N297 and also van der Waals contacts between the methyl group and the aliphatic side chain of P310 (Figure 8E). [183] Recently, langerin has been crystallized bound to GlcNS6S, and the model displays the sugar in the opposite orientation (Figure 8D). [110] This alternative binding mode suitably justifies the increased affinity observed for sulfated GlcNAc, as the sulfate group at position 6 is crucial to target K313, creating a stable electrostatic contact. In this case, K299 is placed further away and does not seem to be as important as reported for 6S‐Gal. Indeed, mutation studies have confirmed this hypothesis. [110] Finally, only one X‐ray crystallographic structure of langerin has been described in the presence of the Fuc‐containing B antigen (Figure 8B). [177] In the crystal, the Fuc ring coordinates the metal ion through positions C2 and C3, as opposed to the OH3‐OH4‐mediated calcium‐binding described for selectins [103] and for DC‐SIGN.[ 83 , 85 ] Besides the primary epitope, the nonreducing Gal moiety also contributes to the stabilization of the complex, providing polar contacts with P283 and N287.
Given the importance of the amino acids surrounding the calcium binding site for the fine selectivity of langerin, several investigations have thoroughly analyzed the impact of single nucleotide polymorphisms (SNPs) in the overall functionality of this lectin. Some mutations slightly affect to the CRD stability without decreasing the sugar affinities, as A278 V. In contrast, the N288D and A300P variants give rise to a tenfold lower affinity for Man. [184] Also, the W264R mutation is a rare variation that completely abrogates the recognition of any sugar and also precludes the formation of BGs. [185] By thermal shift assays, this mutation has been recently proven to disrupt the entire CRD folding. [186] The already mentioned N288D variant has particularly attracted more attention given that it is more recurrent (ca. 11 %) and often appears associated to another mutation: K313I. This latter structural change has a dual consequence: the ability to stabilize sulfated glycans is completely lost, while the affinity for GlcNAc seems to increase. [183] The second effect is justified by a higher hydrophobic stabilization of the N‐acetyl group jointly contributed by P310 and I313. The recognition of high‐Man glycans and histo blood group antigens is in principle maintained in the single mutant, whereas the affinity drop observed in the double mutant (N288D, F313I) could be driven by the disruption of the normal H‐bond network around the calcium ion.[ 183 , 184 ] The study of the murine homologue of langerin has also helped to clarify how the structural differences on key amino acids lead to noticeable changes in specificities. At the monosaccharide level, the sugar preferences of murine langerin are fairly conserved. Surprisingly, it recognizes 6S‐Gal in spite of lacking both K313 and K299 (N316 and R302, respectively), [182] which has been attributed to the presence of a shallower and wider secondary site able to accommodate bulky groups or other substituents from larger structures, similar to DC‐SIGN. Moreover, X‐ray data suggest that the oligomeric structure is more flexible for the murine homologue, enabling higher avidity effects in the presence of multivalent scaffolds. In fact, glycan arrays have evidenced that murine langerin can bind to a broader range of microbial complex oligosaccharides, although human langerin still exhibits an exclusive ability to recognize some epitopes from Yersinia pestis, Shigella flexneri or Escherichia coli. [182] From a general perspective, all these findings reflect fairly well the complex interplay between the primary interactions, the secondary epitopes, and the multivalent phenomena, which in turn shape the fine specificity of a given receptor. Finally, with regard to multivalent interactions, the F241L mutation has been described to cause an abnormal formation of the BG structures, although the sugar recognition ability is kept. [186] Certainly, the interaction with sugars does not change for the CRD, but there is a noticeable drop in the expected affinity of the oligomeric extracellular domain. A closer inspection of the X‐ray models has revealed the loss of certain contacts between secondary structure elements (α1 and β0) at the N‐terminal regions of the CRD, which would eventually contribute to disrupt the correct 3D arrangement of the whole oligomer.
In solution, the ECD of langerin exists associated into trimers which remain stable even at acidic pH values (ca. 4.0). [170] AUC and SAXS measurements, combined with modeling tools, have revealed that the neck repeats constitute a highly structured coiled‐coil only interrupted at two regions (around residues 100 and 150).[ 186 , 187 ] These observations contrast with the more flexible arrangement found, for instance, for DC‐SIGN or L‐SIGN tetramers,[ 75a , 76 ] where each neck repeat forms an independent α‐helix within a single protomer. Additionally, a truncated version of the trimer could be crystallized, unveiling key neck‐CRD interactions within the same protomer and between neighboring protomers. [176] Taken together, all these data underline that both CRD and neck regions may synergistically contribute to define the 3D shape of the oligomer, eventually resulting in trimers that adopt a fairly rigid conformation. Such an arrangement has been also noticed for other receptors from the collectin family (Group III)[ 188 , 189 ] and could explain the weaker avidity effects generally seen for the langerin ECD in the presence of high‐Man glycans. [182] Another factor to consider is the influence of pH: C‐type lectins often act as endocytic receptors, and the cargo release takes place in the early endosome under acidic conditions. These conditions usually lead to a decreased Ca2+ affinity, which in turn hampers the normal attachment of the sugar, allowing its release.[ 190 , 191 ] In the case of langerin, molecular dynamics simulations supported by experimental NMR data have evidenced that dynamics of the short and long loop regions surrounding the Ca2+ site are connected and regulated by a complex allosteric network involving multiple amino acids. [192] A thorough analysis of the CRD dynamics using different mutants have allowed checking the strong robustness of such a network, which is meant to downregulate the Ca2+ affinity in acidic environments. In particular, H294 has been found as a potential pH sensor helping in this process, although other relevant residues are supposed to participate as well.
As commented on above, the interaction of langerin with GAGs was unexpected at first, although it was structurally well justified taking into account secondary interactions provided at neighboring sites. However, along the last decade, the recognition of long heparin chains (>6 kDa) has been surprisingly found to happen in a Ca2+‐independent fashion. [193] In fact, it seems that this type of interaction is barely supported by the sole CRD, whereas the trimeric ECD can strongly bind to these heparins either with or without Ca2+. Heparins are anionic polysaccharides frequently sulfated, with a high structural variability, and are involved in many relevant biological functions. [194] Their ability to target langerin even in the absence of calcium strongly brought forward the idea that these GAGs might be involved in langerin‐mediated functions at the early endosome, such as BG formation or regulation of the cargo release. However, to date there is not yet a clear consensus about the key structural aspects underlying the recognition of large GAGs. From a general perspective, langerin preferentially binds to heparin and heparan sulfate (HS) over chondroitin (CS) and dermatan sulfate (DS).[ 193 , 195 ] Such preferences seem to be governed by both the amount of sulfate groups and their specific distribution pattern (Figure 9). Thus, GlcNAc moieties play the most important role, as desulfation at either O6 or N2 negatively affects to the interaction, whereas sulfation at the uronate moiety (IdoA) does not seem to be as relevant. [195] Fittingly, significant binding has also been reported for CS‐C, the CS subtype with major sulfate content at positions C6 and N2. DS has been observed to maintain a weaker but still noticeable level of binding as well, in spite of its low sulfation rate at the mentioned positions. [193] This latter finding has suggested a possible role of the IdoA moiety in the stabilization of the complex, although this hypothesis remains partially unclear due to the lack of structural data.
Coming back to heparins, an open debate still exists on how or where long heparins do exactly bind to Langerin and whether Ca2+ is really needed for such a binding. The length of the heparin chain clearly influences both the mode of binding and the affinities. Around six sugar units, the affinities start to subtly increase faster, while the improvement becomes significantly sharper above 16 sugar units. [195] It could be hypothesized that the second increase is driven by avidity effects (ECD), whereas the first one better correlates with the parallel observation that Ca2+‐independent binding already appears for medium chain lengths. NMR data have evidenced that heparin trisaccharides are recognized at the calcium site, and that the interaction is exclusively mediated by the nonreducing GlcNS regardless the sulfation pattern, as long as position N2 remains sulfated. [196] In addition, STD and trNOESY experiments have demonstrated that neither the glycosidic angles nor the ring conformations of the IdoA pyranose substantially change upon binding, compared to those described in the free state. [197] Conversely, the STD‐NMR values collected for heparin hexasaccharides display a more homogeneous saturation profile. [196] From the protein perspective, these hexasaccharides curiously tend to mostly affect the allosteric network previously described in the modulation of the Ca2+ affinity.[ 192 , 198 ] Taken together, these results point out the existence of a completely unique interaction between heparins and langerin. A docking model was first proposed using a ten‐residue heparin chain, in which the entire oligosaccharide establishes electrostatically‐driven interactions with the positively charged interface between two CRDs of the trimer. [193] In principle, this model would reasonably fulfill the subsequently published data, including the STD profiles, [196] the CSPs found by HSQC‐based titrations [198] and the essentially electrostatic nature of the interaction (disrupted with NaCl).[ 195 , 199 ] However, there are still some findings which require further justification. For instance, the proposed docking models suggest a more important role for sulfate groups at N2 than at C6 (salt bridges), [193] but recent reports have highlighted that the Ca2+‐independent binding can be perfectly sustained without N2 sulfation. Also, some degree of Ca2+ dependence co‐exists during the binding event, although it changes with the sulfation pattern as well. [198] So far, the interplay between the classic Ca2+‐mediated and the alternative sugar binding for heparin‐langerin complexes is not fully understood.
Finally, it is worth mentioning the reported advances for developing specific langerin‐binding sugar mimetics. Certainly, langerin features a more reduced and particular specificity than related lectins such as DC‐SIGN or Dectin. [200] It cannot recognize LeA and LeX antigens and presents a limited ability to target inner Man residues in highly branched scaffolds, in part due to the two protruding charged lysine side chains near the calcium site. These two side chains have been actually exploited for the design of bulky and positive substituents which can preclude binding to langerin while still targeting DC‐SIGN, thereby improving the selectivity for the latter.[ 110 , 111 ] Even so, the development of novel specific and potent sugar mimetics remains challenging given that the intrinsic CRD architecture displays very exposed and polar surfaces that are rather undruggable, as already suggested by computational methods. [147] Of course, screening techniques are suitable to reduce the time costs in this quest, but require from the appropriated setup to undertake a reliable and fast enough evaluation of large libraries. Recently, a promising screening method has been developed and simultaneously tested with DC‐SIGN and langerin. [148] The method enables a better evaluation of the binding potencies under physiological conditions, directly on the CLR‐containing cells (Figure 10A). Detection of hits is achieved by direct competition between a reference fluorescent ligand (FITC‐dextran) and the tested fragments. Expectedly, a low percentage of hits were identified for both lectins (5.6 % for langerin and 12.3 % for DC‐SIGN). In both cases, some hits were subsequently validated by orthogonal assays, employing SAR analyses and the fast NMR‐based 19F‐T2 and HSQC setups previously designed and tested for the studied lectins.[ 151 , 201 ]
Figure 10.
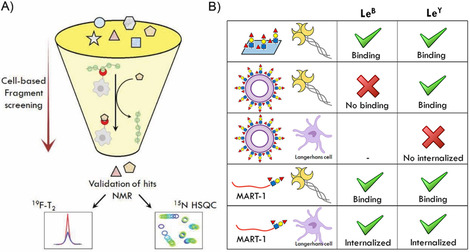
A) Summary of the cell‐based fragment screening assay (Cell‐Fy) developed to directly screen compounds against lectin‐expressing cells. [148] As depicted, the detected hits are further validated by NMR techniques. B) Langerin binding to LeB and LeY using different ligand formulations. [137] Langerin ECD‐Fc can recognize both antigens coated on a plate and linked to the tumor‐associated peptide MART‐1. Moreover, the latter are successfully internalized by LCs. In contrast, only LeY‐coated glycoliposomes are targeted by langerin but no internalization by LCs is observed.
As recent breakthrough, the GlcNS monosaccharide has been successfully used for the development of an improved scaffold which can be covalently linked to a multivalent system. Although the affinity barely varies, there is a 63‐fold increase in the selectivity respect to DC‐SIGN. The resulting structure bears an aromatic system on the sulfonamide group which presumably provides stabilizing hydrophobic contacts with Phe315 (Figure 8E). [202] In this regard, NMR data supports that the sulfonamide substituent is located at the region surrounding Lys299, where the C2 substituent additionally establishes polar interactions with Asn307 as well as the cited aliphatic contact. In contrast, Lys313 and Pro310 do not seem to play any substantial role in the recognition. Subsequent linking of these mimetics to lipid chains enabled the development of sugar‐coated liposomes, which are successfully internalized by Langerhans cells in epidermal cell suspensions from skin biopsies. [202] To note, these liposomes also exhibit low cytotoxicity levels and the binding avidity can be controlled through the surface density of the single mimetic. Interestingly, they are internalized in any case, in contrast to other LeY‐coated liposomes tested before. [137] These findings highlight the importance of both the liposome formulation and the chosen epitope, as the nature of the latter one may exert different effects on multivalent lectin targeting depending on its surface density (Figure 10B). Eventually, langerin‐specific liposomes can serve as straightforward methods for transcutaneous vaccination via LC targeting,[ 141b , 203 ] with some advantages with respect to mABs‐based vaccines, especially regarding the final cargo release.[ 80b , 204 ]
4. Macrophage Galactose Lectin (MGL)
The human macrophage galactose lectin (also dubbed CD301 or CLEC10A) is a calcium‐dependent transmembrane receptor included within the group II of C‐type lectins. [7a] In contrast to DC‐SIGN and langerin, its structure displays the characteristic QPD motif at the long loop region, typically associated to galactose (Gal/GalNAc) specificity.[ 11 , 169 ] In the biological context, this receptor has progressively gained much attention over the last two decades due to its involvement in tumor development and progression.[ 6 , 205 , 207 ] In a physiological environment, MGL is known to participate in activateion of dendritic cells for undertaking subsequent T cell downregulation or even inducing apoptosis of effector T cells. [208] The upregulation of this receptor specially takes place on tolerogenic APCs (DCs and macrophages) [209] and, in principle, it is thought to have a protective role in persistent inflammations and autoimmune diseases, preventing excessive tissue damaging and alternatively allowing their remodeling.[ 208 , 210 ] In the same line, tumor cells may escape from immune clearance through MGL targeting via the specific recognition of tumor‐associated glycan motifs, which are not present in healthy tissues. [211] For this reason, MGL has become a potential target for the development of biomarkers, therapeutics and vaccines against cancer.
The sugar preferences of this receptor have been largely studied to better understand its biological roles in the establishment of self‐tolerance, especially in tumor tissues. Over the last decades, several tumor‐associated carbohydrate antigens (TACAs) have been postulated as MGL counterparts and their corresponding recognition processes have been investigated in detail. One of the most important TACAs is the Tn‐antigen (GalNAc‐α‐1‐O‐Ser/Thr), whose structure was already unveiled in the seventies. [212] Its presenceinvolvement in human tumors has been largely described [213] and it characteristically appears as distinctive motif in aberrant glycoforms of MUC1 proteins (Figure 11).[ 214 , 215 ] Similarly, MGL is able to recognize other related antigens present in tumor tissues including the Neu5Ac−Tn antigen and the Neu5Gc−Tn antigen.[ 216 , 217 ]
Figure 11.
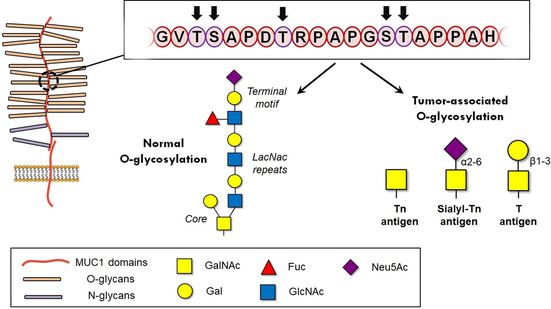
Schematic overview of the MUC‐1 structure.[243] On the top, the 20‐amino acid sequence that constitute one tandem repeat. On the bottom, the most common tumor‐associated antigens as compared to normal glycosylation. Copyright: 2014, Elsevier Ltd.
MGL was first purified from transfectants and the first assays suggested that it could recognize both Gal and GalNAc residues. [218] Later, it was discovered to oligomerize in solution, forming homotrimers. [219] The access to pure recombinant forms of this protein has allowed a better and detailed inspection of its ligand specificity, using the glycan array developed by the Consortium for Functional Glycomics. [220] Importantly, the high specificity of this lectin for α‐GalNAc and the Tn antigen (GalNAc‐α‐1‐O‐Ser/Thr) was confirmed, whereas α/β‐Gal is barely recognized. MGL is also able to interact with β‐GalNAc, suggesting that both configurations can be in principle accommodated at the recognition site, as further seen in larger glycans. In contrast, no binding has been reported for other mono‐ or oligosaccharides devoid of GalNAc moieties, as LacNAc, Lewis X or the glycosphingolipids GM3 and GD3. These studies have also evidenced the MGL fine selectivity towards GalNAc‐containing entities, depending on the geometry and/or configuration of the surrounding sugars. As terminal motif, GalNAc enables MGL targeting mediated by LDN and LDNF epitopes, commonly found on Schistosoma mansoni SEAs, [221] and the glycosphingolipids GM2 and GD2. Conversely, the attachment of other sugars in the GalNAc scaffold seems to be limited to position 6 (O‐glycan core 6, 6‐sialyl‐Tn, 6‐sulfo‐Tn), while binding is totally abrogated after substitution at position 3 (cores 1–4, 3‐sialyl‐Tn).
The exclusive specificity of MGL is clearly different from that of the related ASGPR receptor, found on the sinusoidal surface of hepatocytes. [222] This latter CLR has been extensively studied since its discovery in the early sixties, [223] and is known to bind to either terminal Gal or GalNAc residues in highly branched structures.[ 224 , 225 ] In contrast, MGL has been less investigated even in comparison with other related C‐type lectins as DC‐SIGN[ 26 , 27 ] or MMR, [226] whose biological relevance was guessed at the same moment. The lack of crystallographic models for this lectin may explain in part this slower progression from the molecular perspective, as these models often speed up the interpretation of data from other sources and help to hypothesize about the recognition of other unknown ligands. In such a scenario, NMR has emerged as the primordial tool to unveil the structural details underlying the recognition of GalNAc‐containing glycopeptides by MGL. STD experiments have permitted to closely evaluate the ligand epitope of the primary monosaccharides, α‐Gal and α‐GalNAc, using the recombinant MGL‐ECD domain. [227] As reported for other galactose‐specific lectins, [228] binding takes place through coordination of hydroxy groups at C3 and C4 to the calcium ion. However, two orientations are possible for the sugar ring considering the hydroxy positioning. As no X‐ray models of MGL are available, the evaluation of these poses by docking is still performed by using a homology structure based on an ASGPR crystal. [229] For GalNAc, two possible orientations were postulated to explain the experimental STD‐NMR data, whereas experiments with Gal better adjusted to the existence of an unique binding mode. Interestingly, STD‐NMR data from Tn‐derived mucin‐like peptides support the involvement of the peptide sequence closer to the GalNAc attachment in secondary contacts with the protein surface. In any case, the non‐glycosylated peptides do not provide any STD signal, assessing the role of the sugar as primary epitope for MGL.
In view of these results, new questions were opened concerning other effects, such as the nature and influence of the nearby sugars or the proper peptide chain. MD calculations have validated the importance of the acetyl group of GalNAc in the binding, thereby explaining the 75‐fold better affinity for GalNAc (12 μM) than for Gal (ca. 900 μM). [227] However, GalNAc‐bearing peptides display similar dissociation constants in spite of the additional stabilizing contacts arising from the peptide chain, as deduced by STD‐NMR (Figure 12G). Although merely speculative, these discrepancies may arise from enthalpy‐entropy compensation phenomena. [230] In parallel, the effects of additional sugar residues in GalNAc‐mediated binding to MGL have been also studied for other relevant sugar epitopes. STD‐NMR analyses performed for Tn and sialyl‐Tn antigens have evidenced saturation profiles and affinities rather similar to the α‐OMe‐GalNAc epitope, in the low‐micromolar range. [217] Both antigens display important STD NMR effects at H2, H3, H4 and the acetyl group, although this latter yields higher STD percentages for the sialylated antigen. It is worth noting that the sialic moiety provides additional STDs at H3ax and its acetyl group (Figure 12B). However, SPR measurements have shown a slightly reduced affinity for sialyl‐Tn, presumably arising from a slower k on rate. Recently, the interaction of recombinant MGL‐ECD with other four GalNAc‐containing epitopes has been thoroughly investigated by NMR, assisted by MD simulations using the homology model. [232] Three of these oligosaccharides (BgA, Forsmann and GM2) have been already reported as tumor‐associated antigens, [233] and hence might act as MGL ligands contributing to tumor surveillance. To highlight, 15N HSQC‐based titrations on a 15N‐labeled MGL monomer revealed interesting findings. In all cases, including the four studied ligands and the simplest epitope (α‐OMe‐GalNAc), the binding takes place in a slow‐exchange regime in the NMR chemical shift timescale, suggesting substantially long residence times for all these ligands at the binding site (small k off). Moreover, upon ligand addition, some protein crosspeaks appear or get sharper, especially those from the loop regions close to the binding site. These observations actually prove the conformational stability gain that the flexible regions of MGL experience as a result of glycan binding, similar to that observed for other lectins. [83] For the blood group A trisaccharide, no significant differences were found with respect to α‐OMe‐GalNAc and indeed, only the terminal GalNAc residue provided relevant STD NMR effects (Figure 12A and C). The scenario is similar for the Forsmann antigen, for which the STD‐NMR results underline again the lack of close lectin contacts with the central and reducing saccharide moieties (Figure 12D). In deep contrast, both GM2 and asialo‐GM2 ligands display better affinity constants and strong STD NMR effects, several of them located even at the reducing‐end sugars (Figure 12E and F). Fittingly, some lectin crosspeaks from the binding region were actually affected in a different fashion in the 15N HSQC spectra depending on the ligand added (Lys264, His286). Although the results from MD should be considered as merely approximated, given the homology‐model‐based starting geometry employed, His286 provides a stable H‐bond with the N‐acetyl group of the attached GalNAc and a CH‐π contact with the reducing end as well, pointing out the role that this residue may play in the stabilization of certain extended epitopes. Indeed, a recent report has evidenced the loss of binding provoked by a mutation of such a residue using the MGLshort splice variant (H259 instead of H286). [234] The mutant H259T is unable to recognize diverse MGL ligands including GM2, GD2, LDNF, or sialyl‐Tn, whereas binding to α‐OMe‐GalNAc or the O‐glycan core 6 is preserved.
Figure 12.
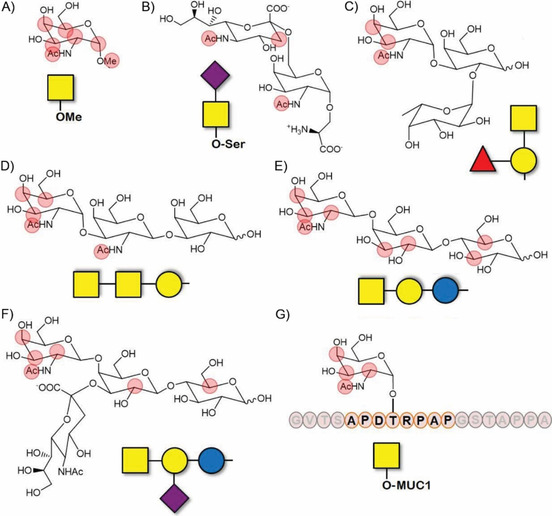
Schematic representation of the STD profiles described for different MGL ligands.[217,227,232] To clarify, for each antigen only those STDs above 50 % in relative scale are depicted as red circles. In all cases, recombinant soluble MGL‐ECD has been used for data recording (in B, it was additionally tagged with myc and associated to anti‐myc AB and streptavidin). A) α‐OMe‐GalNAc. B) sialyl‐Tn antigen. C) Blood group A trisaccharide. D) Forsmann antigen. E) asialo‐GM2. F) GM2. G) GalNAc linked to a MUC1 repeat. In this case, the peptide residues displaying weaker STD effects are also highlighted in orange.
The quest for biological epitopes that could be targeted by MGL has recently led to evaluate the ability of this lectin to recognize the novel GalNAc‐Tyr antigen. Since 2011, several proteins have been found to present this particular motif, consisting of a GalNAc monosaccharide attached to the phenolic OH group of a tyrosine residue. [235] The biological significance of this glycosylation remains still unclear, as it is rather difficult to produce and detect. In any case, analogously to the Tn antigen, MGL has been evidenced to recognize it as well. [236] Using short glycopeptides loaded on a BSA scaffold, the affinities of the Tn antigen and the GalNAc‐Tyr antigen have been checked to be very similar, especially when the ligand densities on the BSA surface are high. In both cases, the best affinity values have been found around 30–40 nM. Apparent K d values show no relevant preference for α‐ or β‐GalNAc‐Tyr, and both configurations are similarly bound especially at higher BSA glycan coating. [236] To note, the configuration of the sugar linkage in natural GalNAc‐Tyr antigens has not been yet clarified, as it has only experimentally assessed in one case so far (“α” in the Aβ1‐15 peptide). [237] Finally, in vitro cultures using the homologue receptor mMGL2, which has a ligand selectivity very similar to hMGL, [238] suggest that uptake of GalNAc‐Tyr‐containing entities by human DCs could be likewise driven by MGL. [236] STD‐NMR experiments using the most potent glycopeptide confirmed the possible participation of the nearby amino acid side chains in the extended epitope, as reported for the Tn antigen. [227] In the same line, other recent works have focused on identifying those specific glycoproteins that might act as MGL counterparts in cell‐cell cross‐talk. In particular, pull‐down experiments with three different colorectal cancer (CRC) cell lines have led to identify up to 85 membrane glycoproteins that are specifically recognized by MGL‐Fc. [239] Most of them include cell surface signaling receptors and integrins. Additionally, subsequent analyses of the glycan content by MS/MS have revealed the presence of the LacDiNac (LDN) epitope in several cases (ITGA3, PTK7) as well as the Tn antigen (DAG1). Strikingly, other glycoproteins were found to bind MGL in spite of carrying glycan structures that were not expected to be MGL ligands according to the reported data so far, for instance high‐Man scaffolds or sialyl‐T antigens. Given the plasticity widely known for these type of lectins,[ 82 , 240 ] it is likely that weak binding of individual ligands might be largely enhanced by multivalent effects (or high epitope densities), thus explaining the results commented above. Conversely, the lack of binding for known ligands often comes from unfavorable geometrical factors imposed by the epitope environment. In this regard, recent works have attempted to shed light into these questions, not only studying the effect of the glycan density, but also how the number and distribution of Tn and related epitopes in mucin peptides modulate MGL binding.[ 241 , 242 ] The MUC1 N‐terminal peptide is constituted by repeats of 20 amino acids, being five of them serine and threonine residues susceptible to O‐glycosylation (T3, S4, T8, S14 and T15; Figure 11). [243] MGL has been described to bind slightly better to the threonine‐containing Tn antigen, although the affinities for mono‐GalNAc MUC1 repeats are rather similar regardless the type of Tn epitope and its location on the peptide sequence.[ 234 , 241 ] In contrast, interesting differences have been observed for di‐ and tri‐Tn MUC1 repeats. [241] In these cases, MGL can clearly recognize di‐Tn peptides at the same level, although the simultaneous presence of Tn at the central Thr (T8) and the initial GVTS region (T3/S4) seems to negatively affect the recognition event. To note, the binding is maintained with two consecutive GalNAc motifs on either the GVTS or the GSTA regions. This fact suggests that the adjacent Tn antigen is suitably accommodated or at least does not disturb the binding of the other Tn moieties. Overall, the presence of more Tn antigens does not significantly contribute to improve the affinities. Indeed, binding to MGL can be sustained by MUC1 chains with multiple epitopes, but in these cases, affinities tend to progressively worsen as the glycan crowding increases. Parallel studies using synthetic linear GalNAc‐containing glycopolymers have proven that the dissociation constant subtly decreases within the 1.0–0.1 μM range when exposing the MGL trimer to different GalNAc contents, from 13 to 100 sugar units. [242] Interestingly, the same experiments give rise to a noticeable enhancement in the affinities for the related ASGPR lectin. From the receptor perspective, these findings agree with the existence of “cluster effects” [115] that could be readily engaged for the MGL ensemble, but not for the ASGPR receptor. Broadly speaking, these results probably reflect that the MGL trimer is flexible enough to independently accommodate a GalNAc epitope in any of the CRDs taking advantage of the high local epitope concentration. In contrast, for the more rigid ASGPR ensemble, a suitable valence‐dependent targeting is needed for improving its weaker affinities.
Aside from tumor‐related epitopes, other recent investigations have underlined the possible role that MGL could play in bacterial infections as well. One example is found in Staphylococcus aureus, one of the most prominent causes of health care‐associated pneumonia. [244] Usually, the exposed wall teichoic acid (WTA) chains in the cell wall of the pathogen consist in a poly‐ribitolphosphate backbone decorated with single α‐GlcNAc residues. [245] However, certain lineages can alternatively decorate the negatively charged backbone with α‐GalNAc units (Figure 13). In particular, the GalNAc‐WTA‐expressing S. aureus lineage ST395 has been reported to bind to MGL through these exposed GalNAc residues. [246] The specificity of the recognition process has been assessed by both observing the lack of binding to GlcNAc‐WTA‐expressing strains and also, the loss of binding after impairing the correct formation of the GalNAc‐decorated WTA structure (mutant devoid of GalNAc‐transferases). In addition, in vitro studies with moDCs and this S. aureus lineage have shown that MGL can induce the production of certain pro‐inflammatory cytokines (IL6, IL12p70), although the host‐bacteria interaction is not totally inhibited by blocking MGL, suggesting the simultaneous participation of other membrane receptors. [247] Similarly, a more detailed study has permitted to depict the possible glycan epitope that MGL could target in the truncated bacterial lipopolysaccharide exhibited by Gram‐negative bacteria. This shorter version of the LPS is often named LOS (lipooligosaccharide), and constituted by two cores, one consisting of heptoses (Kdo, hepMan) and the outer one containing hexoses (Glc, Man, Gal; Figure 14). [248] In particular, STD‐NMR experiments have evidenced the ability of MGL to target those LOS from E. coli R1 strains, which contain the terminal disaccharide Galα1‐2Gal. [249] As suggested by the observed STD signals, the entire outer core (five residues) is involved in close contacts with the lectin surface, especially the cited disaccharide portion. The primary epitope (nonreducing Gal) is fairly similar to that of the GalNAc‐containing glycosphingolipid scaffolds GM2 and asialo‐GM2, suggesting that calcium coordination probably occurs in the same way. [232] The docking structures with the homology‐model geometry support the participation of two Glc residues in additional H‐bonds with distal amino acids of the protein (Glu242), whereas the inner core is completely devoid of protein contacts.
Figure 13.
Schematic cartoon of the Gram‐positive bacterial cell wall displaying WTA and LTA chains. The GalNAc‐containing WTA structure is depicted at the top.
Figure 14.
Structure of the LOS decasaccharide of E. coli R1.
5. L‐Sectin
The liver and lymph node sinusoidal endothelial cell C‐type lectin (LSECtin) was described for the first time in 2004 by Liu et al. [250] Its expression was initially described to be restricted to liver and lymph node sinusoidal endothelial cells, although it has also been found in Kupffer cells, [251] in ex vivo peripheral blood, thymic DCs, and in monocyte‐derived macrophages in vitro. [252] Gramberg et al. found LSECtin tissue expression limited to lymph node, liver, and also bone marrow sinusoids. [253] The gene encoding LSECtin (CLEC4G) is located at chromosome 19p13.3, a cluster that also contains sequences codifying the closely related C‐type lectins CD23, DC‐SIGN and DS‐SIGNR. LSECtin is a type II trans‐membrane protein, formed by a short intracellular NH2‐terminal tail of 31 amino acids, a trans‐membrane domain of 22 amino acids, and a long extra‐cellular domain (ECD). This ECD is composed by a neck region of 110 residues and a C‐type carbohydrate‐recognition domain (CRD) of 129 amino acids at the carboxyl terminus. From the structural perspective, LSECtin was first detected in solution in different oligomeric states from monomers to tetramers, [250] with a molecular weight of 40 kDa for the monomeric unit. Afterwards, it was observed the lectin mainly forms dimers (and probably tetramers in small amount), through formation of disulfide bonds using cysteine residues in the neck region. [254] Analogously to other lectins of its gene cluster, LSECtin possesses two N‐glycosylation sites in the neck region at positions 77 and 159. [250] Indeed, glycosylation was reported to be a requirement for efficient expression on the cell surface. [255] The intracellular Y6SKW and E14E motifs from the cytoplasmic tail of the lectin have been related to ligand internalization,[ 253 , 255 ] thus suggesting that the lectin might promote the uptake of antigens in an immune response.
The biological role of LSECtin is rather diverse. A study in mice with T‐cell‐mediated acute liver injury showed that LSECtin is able to modulate hepatic T‐cell activation. In particular, the disease is accelerated in the absence of LSECtin, leading to an increased immune response by T‐cells. However, the exogenous administration of recombinant LSECtin protein or plasmid resulted in a protective effect and amelioration of the damage by decreasing accumulation of T‐cells, revealing its possible therapeutic use for treatment of acute liver injury. [256] LSECtin is expressed in human melanoma tissue, where it facilitates tumor cells escape from the immune system by inhibiting tumor‐specific T‐cell responses. The coregulatory molecule LAG‐3 on T‐cells was identified as its binding partner. [257] Moreover, LSECtin has also been found to be involved in tumor progression in breast cancer. [258] Specifically, it is highly expressed by tumor‐associated macrophages in human breast cancer tissue, where it is able to interact with the breast cancer cell‐intrinsic BTN3A3 receptor to promote tumor stemness.
Regarding infections, LSECtin has reported to be an attachment factor for the spike protein of the SARS coronavirus. [255] The presence of GlcNAc terminating glycans in this glycoprotein has been demonstrated, which is consistent with their recognition by LSECtin. [254] For both SARS and Zaire Ebolavirus (ZEBOV), LSECtin is able to enhance viral infection, in contrast to the observations for hepatitis C virus and HIV infection, even though it is able to interact with the surface glycoprotein of the latter. [255] Moreover, LSECtin delays the clearance of both adenovirus and hepatitis B virus from blood and infected hepatocytes. [259] In the case of a hepatotropic adenovirus, the lack of LSECtin is translated in an increase of intrahepatic effector CTLs, which generate cytokines and cytotoxic factors with antiviral activity. In the case of hepatitis B virus, the absence of LSECtin reduces the amount of hepatitis B virus‐specific IFN‐γ‐producing cells.
Additionally, it has been shown that LSECtin plays a key role in the maintenance and regeneration of the intestinal epithelial barrier during dextran sulfate sodium‐induced colitis. [260] It was found that LSECtin stimulated dead cell clearance phagocytosis by macrophages, which at the same time makes the macrophages produce more tissue repairing factors, ultimately promoting intestinal healing after damage. Furthermore, it has been observed that LSECtin interacts, in cultured cells and brain tissue in mice, with the β‐site amyloid precursor protein cleaving enzyme‐1 (BACE1), a transmembrane protein crucial in Alzheimer disease. [261] The lectin is able to negatively regulate BACE1 function, which leads to a decrease in amyloid‐β peptide production. The results suggest that the interaction does not take place through the bisecting GlcNAc residues on the glycosylated enzyme, as initially hypothesized. Therefore, further studies are necessary to unveil the mechanism of this interaction.
In the molecular recognition context, LSECtin is able to bind Man, Fuc, Glc, and GlcNAc in a calcium‐dependent manner, as it was shown by employing column‐binding assays. [250] Binding to Gal moieties was not observed. These experiments were performed using a chimeric LSECtin fused to the Fc region of human IgG1, LSECtin‐Fc. Additionally, a sugar competition assay showed that Man and GlcNAc were slightly tighter binders than Fuc. Thus, these binding preferences strongly suggest that the C3 and C4 hydroxy groups, oriented in an equatorial disposition, bind the Ca2+ ion at the lectin binding site in a similar fashion of other CLRs. More recently, it has been observed that neither Gal, GlcNAc, Fuc, nor Man, when incubated together with LSECtin expressing cells infected with ZEBOV, were able to block the interaction of the lectin with the virus glycoproteins. [255] Nevertheless, the differences in the sugar recognition profiles between these studies can be attributed to the different experimental approaches employed. In fact, it has been observed that there is not necessarily any correlation between the capability of LSECtin to bind virus glycoproteins in solution and its ability to effectively capture viral particles, as described for HIV‐1 virus. [253]
Mass spectrometry analysis on glycans released from the viral glycoprotein GP1 of the Ebola virus surface suggest that N‐linked glycans carrying the GlcNAc residue at the terminal part are responsible for LSECtin recognition. [254] In particular, by employing glycan arrays, it was revealed that LSECtin preferentially binds to GlcNAcβ1‐2Man of terminal N‐linked glycoproteins with high affinity and specificity. [254] A K d of about 3.5 μM was estimated for the disaccharide‐lectin binding. More recently, another glycan‐array‐based study revealed further features on the recognition preferences of the lectin towards branched N‐glycans. In particular, the lectin prefers the GlcNAcβ1‐2Man epitope when presented at the 1–3 over its presentation at the 1–6 branch, thus displaying an exquisite branch specificity. [91]
6. Summary and Outlook
C‐type lectin receptors (CLRs) are fundamental mediators in the development of efficient immune responses. As highlighted herein, they all are characterized by a very broad recognition profile which can be finely tuned as the glycan complexity increases. This is a particular feature of receptors from innate leukocytes such as DCs and LCs, and improves their ability to quickly detect a wide variety of strange substrates and initiate a more specific adaptive response. However, the same feature turns the quest for specific therapeutics into a challenging task. Langerin and DC‐SIGN are the paradigmatic cases of overlapped specificities which lead to different outcomes: whereas Langerin targets gp120 of HIV and promotes viral clearance, DC‐SIGN recognition results in efficient trans infection of T cells via DCs. Interestingly, DC‐SIGN exhibits a marked ligand promiscuity and conversely, langerin stands out by its uncommon ability to recognize sulfated glycans (GAGs). Thus, the inefficient recognition of LeX by langerin could be used in favor of DC‐SIGN targeting, and similarly, GAGs can be exploited for modulation of LC functions via langerin without affecting DCs. A rational understanding about the structural features governing the fine specificity in each case has been also proven to be effective for achieving lectin specificity. Following this strategy, potent Man‐based compounds have resulted highly specific for DC‐SIGN as well.
MGL is involved in the recognition of tumor‐related MUC1 variants, being a promising target for cancer therapy. In the last years, important advances have been achieved concerning those structural aspects underlying the recognition of GalNAc‐containing glycans, especially by NMR. The influence of extended epitopes has set a solid basis to clarify the involvement of certain TACAs in tumor surveillance via MGL. In this regard, ligand presentation has been shown to influence the ability of MGL to distinguish MUC1 variants according to their glycosylation pattern and even the presence of neighboring MUC1 amino acids. LSECtin might be another focus in cancer therapy and disease progression. Its exquisite ability to target the GlcNAcβ1‐2Man disaccharide may be a starting point for the design of specific substrates avoiding cross‐reactivity with DC‐SIGN, for instance. However, it should be noted that, for both MGL and LSECtin, more structural data is still needed to delineate their respective sugar profiles, especially regarding extended epitopes and multivalent scaffolds.
So far, the intrinsic geometry of CLRs notably has hampered the rapid development of sugar mimetics with better affinities, and multivalence has become a common solution to overcome this fact. Importantly, lectin targeting at the cell membrane must be thoroughly analyzed, as multivalent scaffolds provide higher affinities but introduce other factors that should be taken into account, including ligand density and geometry. Moreover, lectin specificity might be mandatory in certain cases, whereas in other cases, as demonstrated, simultaneous CLR targeting can result useful for achieving cell internalization and/or stimulation of the desired immune response.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Pablo Valverde studied chemistry at the University of Salamanca and obtained his MsC degree in drug discovery at the University Complutense of Madrid. In 2015, he obtained an FPU grant to join the chemical glycobiology group at CIC bioGUNE for his PhD on the study of glycan‐lectin interactions by NMR. In 2018, he obtained a mobility grant to work with Prof. B. Linclau on the synthesis of fluorosugars at the University of Southampton (UK). His research aims to understand sugar‐mediated cell‐cell and host‐pathogen communication from a structural perspective by using different experimental techniques and computational tools, especially NMR.

Biographical Information
J. Daniel Martínez obtained his MsC in drug discovery from the University Complutense of Madrid in 2015, then worked on NMR structural elucidation at the Janssen Drug Discovery Center in Toledo. In 2017, he started his PhD at CIC bioGUNE as an FPI fellow, focusing on sugar‐lectin recognition by NMR and molecular modeling. During this period, he also worked at David Baker's lab at the Institute for Protein Design (Seattle). His research interests include sugar and protein chemistry, NMR methodology, and the development of computational and data analyses tools to efficiently assess the experimental results.

Biographical Information
Francisco Javier Cañada received his Ph.D in organic chemistry from the University of the Basque Country (Bilbao) in 1985. After postdoctoral periods at the Center for Molecular Biology (Madrid) and Harvard Medical School (Boston), he moved to the Institute for Organic Chemistry‐CSIC (Madrid) to work with Prof. Martín‐Lomas focusing on sugar molecular recognition. He moved to CIB Margarita Salas (CSIC, Madrid) in 2002, where he is Vice‐director. His group develops NMR strategies for studying ligand‐protein interactions and collaborates with a pharmaceutical company on carbohydrate‐related vaccines. He is currently President of the Carbohydrate Division of the Royal Society of Chemistry of Spain.

Biographical Information
Ana Ardá studied chemistry at the University of A Coruña, receiving her doctorate in 2006. Between 2006 and 2008 she did postdoctoral work with Prof. Hans Kamerling at Utrecht University, where she started to work with carbohydrates. In 2008 she moved to CIB‐CSIC in Madrid to join Prof. Jiménez‐Barbero's group. In 2014 she moved to CIC bioGUNE in the Basque Country, and since December 2015 she has been a Ramón y Cajal Fellow. Her research focuses on the study of glycan‐protein interactions through a multidisciplinary approach, with a special focus on the development and application of NMR techniques.

Biographical Information
Jesús Jiménez‐Barbero has been Ikerbasque Research Professor and Scientific Director of CIC bioGUNE since 2014. He received his PhD in 1987 at CSIC in Madrid. After postdoctoral stays at Zürich, Mill Hill, and Pittsburgh, he returned to CSIC to work on protein‐carbohydrate interactions. In 2002 he became a CSIC Research Professor at CIB‐CSIC, before moving to Bilbao. His scientific interest is focused on the field of molecular recognition and chemical glycobiology, employing a multidisciplinary approach that combines synthesis, biochemistry, molecular biology, molecular modeling, and especially NMR, thanks to a wide network of scientific collaborations worldwide.

Acknowledgements
We thank the European Research Council (RECGLYCA NMR, advanced grant no. 788143), and the Agencia Estatal de Investigación (Spain) for grants RTI2018‐094751‐B‐C21 and B‐C22, Ramón y Cajal contract to A.A. and the Severo Ochoa Excellence Accreditation (SEV‐2016‐0644).
P. Valverde, J. D. Martínez, F. J. Cañada, A. Ardá, J. Jiménez-Barbero, ChemBioChem 2020, 21, 2999.
Dedicated to Prof. Antonio Echavarren on the occasion of his 65th birthday.
References
- 1.
- 1a. Boyd W. C., Shapleigh E., J. Lab. Clin. Med. 1954, 44, 235–237; [PubMed] [Google Scholar]
- 1b. Sharon N., Lis H., Science 1972, 177, 949–959. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Peumans W. J., Van Damme E. J., Barre A., Rougé P., Adv. Exp. Med. Biol. 2001, 491, 27–54; [DOI] [PubMed] [Google Scholar]
- 2b. Dodd R. B., Drickamer K., Glycobiology 2001, 11, 71R–79R; [DOI] [PubMed] [Google Scholar]
- 2c.http://www.imperial.ac.uk/research/animallectins/ctld/lectins.html.
- 3. Kaltner H., Toegel S., Caballero G. G., Manning J. C., Ledeen R. W., Gabius H. J., Histochem. Cell Biol. 2017, 147, 239–256. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Etzler M. E., Surolia A., Cummings R. D., Taylor M. E. in Essentials in Glycobiology, 2nd ed. (Eds.: Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., Seeberger P. H.), Cold Spring Harbor, 2009, Chapter 29; [Google Scholar]
- 4b. Kamiya Y., Kamiya D., Yamamoto K., Nyfeler B., Hauri H. P., Kato K., J. Biol. Chem. 2008, 283, 1857–1861; [DOI] [PubMed] [Google Scholar]
- 4c. Hauri H. P., Kappeler F., Andersson H., Appenzeller C., J. Cell Sci. 2000, 113, 587–596. [DOI] [PubMed] [Google Scholar]
- 5. Duan S., Paulson J. C., Annu. Rev. Immunol. 2020, 38, 365–395. [DOI] [PubMed] [Google Scholar]
- 6. Brown G. D., Willment J. A., Whitehead L., Nat. Rev. Immunol. 2018, 18, 374–389. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Zelensky A. N., Gready J. E., FEBS J. 2005, 272, 6179–6217; [DOI] [PubMed] [Google Scholar]
- 7b. Taylor M. E., Drickamer K. in Glycoscience: Biology and Medicine, 1st ed. (Eds. Taniguchi N., Endo T., Hart G. W., Seeberger P. H., Wong C.-H.), Springer, Japan, 2015, Chapter 125. [Google Scholar]
- 8. Brown J., O′Callaghan C. A., Marshall A. S., Gilbert R. J., Siebold C., Gordon S., Brown G. D., Jones E. Y., Protein Sci. 2007, 16, 1042–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Natarajan K., Dimasi N., Wang J., Mariuzza R. A., Margulies D. H., Annu. Rev. Immunol. 2002, 20, 853–885; [DOI] [PubMed] [Google Scholar]
- 9b. Sano H., Kuroki Y., Honma T., Ogasawara Y., Sohma H., Voelker D. R., Akino T., J. Biol. Chem. 1998, 273, 4783–4789. [DOI] [PubMed] [Google Scholar]
- 10. Cummings R. D., McEver R. P. in Essentials of Glycobiology, 3rd ed. (Eds.: Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., Seeberger P. H.), Cold Spring Harbor, New York, 2015, Chapter 34. [PubMed] [Google Scholar]
- 11. Nagae M., Yamaguchi Y., Curr. Top. Microbiol. Immunol. 2019, 1–30. doi: 10.1007/82_2019_181. [DOI] [PubMed] [Google Scholar]
- 12. Moriuchi H., Unno H., Goda S., Tateno H., Hirabayashi J., Hatakeyama T., Biochim. Biophys. Acta. 2015, 1850, 1457–1465. [DOI] [PubMed] [Google Scholar]
- 13. Axelgaard E., Ostergaard J. A., Thiel S., Hansen T. K., J. Diabetes Res. 2017, 6368780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salazar F., Sewell H. F., Shakib F., Ghaemmaghami A. M., J. Allergy Clin. Immunol. 2013, 132, 27–36. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. McEver R. P., Cardiovasc. Res. 2015, 107, 331–339; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Raffler N. A., Rivera-Nieves J., Ley K., Drug Discovery Dev. Today Ther. Strateg. 2005, 2, 213–220. [Google Scholar]
- 16. Ku W., Muhitch J. B., Powers C. A., Diehl M., Kim M., Fisher D. T., Sharda A. P., Clements V. K., O′Loughlin K., Minderman H., Messmer M. N., Ma J., Skitzki J. J., Steeber D. A., Walcheck B., Ostrand-Rosenberg S., Abrams S. I., Evans S. S., eLife 2016, 5, e17375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kedmi R., Peer D., Sci. Transl. Med. 2017, 8, 345 fs11. [DOI] [PubMed] [Google Scholar]
- 18. Tone K., Stappers M. H. T., Willment J. A., Brown G. D., Eur. J. Immunol. 2019, 49, 2127–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haider M., Dambuza I. M., Asamaphan P., Stappers M., Reid D., Yamasaki S., Brown G. D., Gow N. A. R., Erwig L. P., PLoS One 2019, 14, e0220867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu X., Nagata M., Yamasaki S., Int. Immunol. 2018, 30, 233–239. [DOI] [PubMed] [Google Scholar]
- 21. Feinberg H., Jégouzo S. A. F., Rex M. J., Drickamer K., Weis W. I., Taylor M. E., J. Biol. Chem. 2017, 292, 13402–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Iborra S., Martínez-López M., Cueto F. J., Conde-Garrosa R., Del Fresno C., Izquierdo H. M., Abram C. L., Mori D., Campos-Martín Y., Reguera R. M., Kemp B., Yamasaki S., Robinson M. J., Soto M., Lowell C. A., Sancho D., Immunity, 2016, 45, 788–801; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Wuthrich M., Wang H., Li M., Lerksuthirat T., Hardison S. E., Brown G. D., Klein B., Eur. J. Immunol. 2015, 45, 2542–2552; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22c. Cáliz R., Canet L. M., Lupiañez C. B., Canhão H., Escudero A., Filipescu I., Segura-Catena J., Soto-Pino M. J., Expósito-Ruiz M., Ferrer M. A., García A., Romani L., González-Utrilla A., Vallejo T., Pérez-Pampin E., Hemminki K., Försti A., Collantes E., Fonseca J. E., Sainz J., PLoS One 2013, 8, e72732; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22d. Clement M., Basatemur G., Masters L., Baker L., Bruneval P., Iwawaki T., Kneilling M., Yamasaki S., Goodall J., Mallat Z., Circulation 2016, 134, 1039–1051. [DOI] [PubMed] [Google Scholar]
- 23. Guo J., Wu X., Too C. L., Yin F., Lu X., He J., Li R., Liu X., Murad S., Padyukov L., Li Z., PLoS One 2012, 7, e41228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. van Vliet S. J., García-Vallejo J. J., van Kooyk Y., Cell Biol. Int. 2008, 86, 580–587; [DOI] [PubMed] [Google Scholar]
- 24b. Figdor C. G., van Kooyk Y., Adema G. J., Nat. Rev. Immunol. 2002, 2, 77–84. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Garcia-Vallejo J. J., van Kooyk Y., Immunity 2015, 42, 983–985; [DOI] [PubMed] [Google Scholar]
- 25b. Monteiro J. T., Lepenies B., Viruses 2017, 9, E59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Curtis B. M., Scharnowske S., Watson A. J., Proc. Natl. Acad. Sci. USA 1992, 89, 8356–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Geijtenbeek T. B., Kwon D. S., Torensma R., van Vliet S. J., van Duijnhoven G. C., Middel J., Cornelissen I. L., Nottet H. S., KewalRamani V. N., Littman D. R., Figdor C. G., van Kooyk Y., Cell 2000, 100, 587–597; [DOI] [PubMed] [Google Scholar]
- 27b. Pöhlmann S., Baribaud F., Lee B., Leslie G. J., Sanchez M. D., Hiebenthal-Millow K., Munch J., Kirchhoff F., Doms R. W., J. Virol. 2001, 75, 4664–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Smith A. L., Ganesh L., Leung K., Jongstra-Bilen J., Jongstra J., Nabel G. J., J. Exp. Med. 2007, 204, 421–430; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Moris A., Nobile C., Buseyne F., Porrot F., Abastado J. P., Schwartz O., Blood 2004, 103, 2648–2654. [DOI] [PubMed] [Google Scholar]
- 29. Mason C. P., Tarr A. W., Molecules 2015, 20, 2229–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.
- 30a. Ribeiro C. M., Sarrami-Forooshani R., Setiawan L. C., Zijlstra-Willems E. M., van Hamme J. L., Tigchelaar W., van der Wel N. N., Kootstra N. A., Gringhuis S. I., Geijtenbeek T. B., Nature 2016, 540, 448–452; [DOI] [PubMed] [Google Scholar]
- 30b. Sarkar R., Mitra D., Chakrabarti S., PLoS One 2013, 8, e59073; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30c. Hodges A., Sharrocks K., Edelmann M., Baban D., Moris A., Schwartz O., Drakesmith H., Davies K., Kessler B., McMichael A., Simmons A., Nat. Immunol. 2007, 8, 569–577. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Kwon D. S., Gregorio G., Bitton N., Hendrickson W. A., Littman D. R., Immunity 2002, 16, 135–144; [DOI] [PubMed] [Google Scholar]
- 31b. McDonald D., Wu L., Bohks S. M., KewalRamani V. N., Unutmaz D., Hope T. J., Science 2003, 300, 1295–1297. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Hijazi K., Wang Y., Scala C., Jeffs S., Longstaff C., Stieh D., Haggarty B., Vanham G., Schols D., Balzarini J., Jones I. M., Hoxie J., Shattock R., Kelly C. G., PLoS One 2011, 6, e28307; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32b. Sol-Foulon N., Moris A., Nobile C., Boccaccio C., Engering A., Abastado J. P., Heard J. M., van Kooyk Y., Schwartz O., Immunity 2002, 16, 145–155. [DOI] [PubMed] [Google Scholar]
- 33. Miyauchi K., Kim Y., Latinovic O., Morozov V., Melikyan G. B., Cell 2009, 137, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gringhuis S. I., van der Vlist M., van den Berg L. M., den Dunnen J., Litjens M., Geijtenbeek T. B., Nat. Immunol. 2010, 11, 419–426. [DOI] [PubMed] [Google Scholar]
- 35. Chen Y., Hwang S. L., Chan V. S., Chung N. P., Wang S. R., Li Z., Ma J., Lin C. W., Hsieh Y. J., Chang K. P., Kung S. S., Wu Y. C., Chu C. W., Tai H. T., Gao G. F., Zheng B., Yokoyama K. K., Austyn J. M., Lin C. L., PLoS Pathog. 2013, 9, e1003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alvarez C. P., Lasala F., Carrillo J., Muñiz O., Corbí A. L., Delgado R., J. Virol. 2002, 76, 6841–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu P., Ridilla M., Patel P., Betts L., Gallichotte E., Shahidi L., Thompson N. L., Jacobson K., Traffic 2017, 18, 218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alen M. M., Dallmeier K., Balzarini J., Neyts J., Schols D., Antiviral Res. 2012, 96, 280–287. [DOI] [PubMed] [Google Scholar]
- 39. Simmons G., Reeves J. D., Grogan C. C., Vandenberghe L. H., Baribaud F., Whitbeck J. C., Burke E., Buchmeier M. J., Soilleux E. J., Riley J. L., Doms R. W., Bates P., Pöhlmann S., Virology 2003, 305, 115–123. [DOI] [PubMed] [Google Scholar]
- 40. Carette J. E., Raaben M., Wong A. C., Herbert A. S., Obernosterer G., Mulherkar N., Kuehne A. I., Kranzusch P. J., Griffin A. M., Ruthel G., Dal Cin P., Dye J. M., Whelan S. P., Chandran K., Brummelkamp T. R., Nature 2011, 477, 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aleksandrowicz P., Marzi A., Biedenkopf N., Beimforde N., Becker S., Hoenen T., Feldmann H., Schnittler H. J., J. Infect. Dis. 2011, 204, S957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pokidysheva E., Zhang Y., Battisti A. J., Bator-Kelly C. M., Chipman P. R., Xiao C., Gregorio G. G., Hendrickson W. A., Kuhn R. J., Rossmann M. G., Cell 2006, 124, 485–493. [DOI] [PubMed] [Google Scholar]
- 43. Plazolles N., Humbert J. M., Vachot L., Verrier B., Hocke C., Halary F., J. Leukocyte Biol. 2011, 89, 329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pöhlmann S., Zhang J., Baribaud F., Chen Z., Leslie G. J., Lin G., Granelli-Piperno A., Doms R. W., Rice C. M., McKeating J. A., J. Virol. 2003, 77, 4070–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lozach P. Y., Lortat-Jacob H., de Lacroix de Lavalette A., Staropoli I., Foung S., Amara A., Houles C., Fieschi F., Schwartz O., Virelizier J. L., Arenzana-Seisdedos F., Altmeyer R., J. Biol. Chem. 2003, 278, 20358–20366. [DOI] [PubMed] [Google Scholar]
- 46. Kerur N., Veettil M. V., Sharma-Walia N., Sadagopan S., Bottero V., Paul A. G., Chandran B., Virology 2010, 406, 103–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Léger P., Tetard M., Youness B., Cordes N., Rouxel R. N., Flamand M., Lozach P. Y., Traffic 2016, 17, 639–656. [DOI] [PubMed] [Google Scholar]
- 48.
- 48a. de Witte L., de Vries R. D., van der Vlist M., Yüksel S., Litjens M., de Swart R. L., Geijtenbeek T. B., PLoS Pathog. 2008, 4, e1000049; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48b. Mesman A. W., Zijlstra-Willems E. M., Kaptein T. M., de Swart R. L., Davis M. E., Ludlow M., Duprex W. P., Gack M. U., Gringhuis S. I., Geijtenbeek T. B., Cell Host Microbe 2014, 16, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.
- 49a. Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N. H., Nitsche A., Müller M. A., Drosten C., Pöhlmann S., Cell 2020, doi: 10.1016/j.cell.2020.02.052; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49b. Song Z., Xu Y., Bao L., Zhang L., Yu P., Qu Y., Zhu H., Zhao W., Han Y., Qin C., Viruses 2019, 11, E59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang Z. Y., Huang Y., Ganesh L., Leung K., Kong W. P., Schwartz O., Subbarao K., Nabel G. J., J. Virol. 2004, 78, 5642–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.
- 51a. Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., Tan W., N. Engl. J. Med. 2020, 382, 727–733; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51b. Wang C., Horby P. W., Hayden F. G., Gao G. F., Lancet, 2020, 395, 470–473; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51c. Zhou P., Yang X. L., Wang X. G., Hu B., Zhang L., Zhang W., Si H. R., Zhu Y., Li B., Huang C. L., Chen H. D., Chen J., Luo Y., Guo H., Jiang R. D., Liu M. Q., Chen Y., Shen X. R., Wang X., Zheng X. S., Zhao K., Chen Q. J., Deng F., Liu L. L., Yan B., Zhan F. X., Wang Y. Y., Xiao G. F., Shi Z. L., Nature, 2020, 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cai G., Cui X., Zhu X., Zhou J., Preprints 2020, DOI: 10.20944/preprints202002.0408.v1. [Google Scholar]
- 53. Pawlowski A., Jansson M., Sköld M., Rottenberg M. E., Källenius G., PLoS One 2012, 8, e1002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cambi A., Gijzen K., de Vries I. J., Torensma R., Joosten B., Adema G. J., Netea M. G., Kullberg B. J., Romani L., Figdor C. G., Eur. J. Immunol. 2003, 33, 532–538. [DOI] [PubMed] [Google Scholar]
- 55. Tailleux L., Schwartz O., Herrmann J. L., Pivert E., Jackson M., Amara A., Legres L., Dreher D., Nicod L. P., Gluckman J. C., Lagrange P. H., Gicquel B., Neyrolles O., J. Exp. Med. 2003, 197, 121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Balboa L., Romero M. M., Yokobori N., Schierloh P., Geffner l., Basile J. I., Musella R. M., Abbate E., de la Barrera S., Sasiain M. C., Alemán M., Immunol. Cell Biol. 2010, 88, 716–726. [DOI] [PubMed] [Google Scholar]
- 57.
- 57a. van Kooyk Y., Appelmelk B., Geijtenbeek T. B., Trends Mol. Med. 2003, 9, 153–159; [DOI] [PubMed] [Google Scholar]
- 57b. Vergne I., Gilleron M., Nigou J., Front. Cell. Infect. Microbiol. 2015, 4, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.
- 58a. Ehlers S., Eur. J. Cell Biol. 2010, 89, 95–101; [DOI] [PubMed] [Google Scholar]
- 58b. Geijtenbeek T. B., Van Vliet S. J., Koppel E. A., Sanchez-Hernandez M., Vandenbroucke-Grauls C. M., Appelmelk B., Van Kooyk Y., J. Exp. Med. 2003, 197, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kullberg B. J., Arendrup M. C., N. Engl. J. Med. 2015, 373, 1445–1456. [DOI] [PubMed] [Google Scholar]
- 60. Gow N. A., van de Veerdonk F. L., Brown A. J., Netea M. G., Nat. Rev. Microbiol. 2011, 10, 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gauglitz G. G., Callenberg H., Weindl G., Korting H. C., Acta Derm.-Venereol. 2012, 92, 291–298. [DOI] [PubMed] [Google Scholar]
- 62.
- 62a. Cambi A., Netea M. G., Mora-Montes H. M., Gow N. A. R., Hato S. V., Lowman D. W., Kullberg B.-J., Torensma R., Williams D. L., Figdor C. G., J. Biol. Chem. 2008, 283, 20590–20599; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62b. van Kooyk Y., Geijtenbeek T. B., Nat. Rev. Immunol. 2003, 3, 697–709. [DOI] [PubMed] [Google Scholar]
- 63. Mitchell D. A., Fadden A. J., Drickamer K., J. Biol. Chem. 2001, 276, 28939–28945. [DOI] [PubMed] [Google Scholar]
- 64.
- 64a. Varki A., Glycobiology 2017, 27, 3–49; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64b. Kaltner H., Abad-Rodríguez J., Corfield A. P., Kopitz J., Gabius H. J., Biochem. J. 2019, 476, 2623–2655; [DOI] [PubMed] [Google Scholar]
- 64c. Marionneau S., Cailleau-Thomas A., Rocher J., Le Moullac-Vaidye B., Ruvoën N., Clément M., Le Pendu J., Biochimie 2001, 83, 565–573. [DOI] [PubMed] [Google Scholar]
- 65. Geijtenbeek T. B., Gringhuis S. I., Nat. Rev. Immunol. 2016, 16, 433–448. [DOI] [PubMed] [Google Scholar]
- 66. Gringhuis S. I., den Dunnen J., Litjens M., van der Vlist M., Geijtenbeek T. B., Nat. Immunol. 2009, 10, 1081–1088. [DOI] [PubMed] [Google Scholar]
- 67. Garcia-Vallejo J. J., van Liempt E., da Costa M. P., Beckers C., van het Hof B., Gringhuis S. I., Zwaginga J.-J., van Dijk W., Geijtenbeek T. B., van Kooyk Y., van Die I., Mol. Immunol. 2008, 45, 2359–2369. [DOI] [PubMed] [Google Scholar]
- 68. Bogoevska V., Nollau P., Lucka L., Grunow D., Klampe B., Uotila L. M., Samsen A., Gahmberg C. G., Wagener C., Glycobiology 2007, 17, 324–333. [DOI] [PubMed] [Google Scholar]
- 69.
- 69a. Geijtenbeek T. B., Krooshoop D. J. E. B., Bleijs D. A., van Vliet S. J., van Duijnhoven G. C. F., Grabovsky V., Alon R., Figdor C. G., van Kooyk Y., Nat. Immunol. 2000, 1, 353–357; [DOI] [PubMed] [Google Scholar]
- 69b. van Gisbergen K. P. J. M., Sanchez-Hernandez M., Geijtenbeek T. B., van Kooyk Y., J. Exp. Med. 2005, 201, 1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Geijtenbeek T. B., Gringhuis S. I., Nat. Rev. Immunol. 2009, 9, 465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.
- 71a. van Die I., van Vliet S. J., Nyame A. K., Cummings R. D., Bank C. M. C., Appelmelk B., Geijtenbeek T. B., van Kooyk Y., Glycobiology 2003, 13, 471–478; [DOI] [PubMed] [Google Scholar]
- 71b. Gringhuis S. I., Kaptein T. M., Wevers B. A., van der Vlist M., Klaver E. J., van Die I., Vriend L. E. M., de Jonj M. A. W. P., Geijtenbeek T. B., Nat. Commun. 2014, 5, 5074. [DOI] [PubMed] [Google Scholar]
- 72. Mickum M. L., Prasanphanich N. S., Heimburg-Molinaro J., Leon K. E., Cummings R. D., Front. Genet. 2014, 5, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.
- 73a. Kalantari P., Bunnell S. C., Stadecker M. J., Front. Immunol. 2019, 10, 26; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73b. Meevissen M. J. H., Driessen N. N., Smits H. H., Versteegh R., van Vliet S. J., van Kooyk Y., Schramm G., Deelder A. M., Haas H., Yazdanbakhsh M., Hokke C. H., Int. J. Parasitol. 2012, 42, 269–277. [DOI] [PubMed] [Google Scholar]
- 74. Soilleux E. J., Clin. Sci. 2003, 104, 437–446. [DOI] [PubMed] [Google Scholar]
- 75.
- 75a. Feinberg H., Guo Y., Mitchell D. A., Drickamer K., Weis W. I., J. Biol. Chem. 2005, 280, 1327–1335; [DOI] [PubMed] [Google Scholar]
- 75b. Yu Q. D., Oldring A. P., Powlesland A. S., Tso C. K., Yang C., Drickamer K., Taylor M. E., J. Mol. Biol. 2009, 387, 1075–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tabarani G., Thépaut M., Stroebel D., Ebel C., Vivès C., Vachette P., Durand D., Fieschi F., J. Biol. Chem. 2009, 284, 21229–21240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Feinberg H., Tso C. K., Taylor M. E., Drickamer K., Weis W. I., J. Mol. Biol. 2009, 394, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Menon S., Rosenberg K., Graham S. A., Ward E. M., Taylor M. E., Drickamer K., Leckband D. E., Proc. Natl. Acad. Sci. USA 2009, 106, 11524–11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Feinberg H., Castelli R., Drickamer K., Seeberger P. H., Weis W. I., J. Biol. Chem. 2007, 282, 4202–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.
- 80a. Keller B. G., Rademacher C., Curr. Opin. Struct. Biol. 2019, 62, 31–38; [DOI] [PubMed] [Google Scholar]
- 80b. Tacken P. J., Ginter W., Berod L., Cruz L. J., Joosten B., Sparwasser T., Figdor C. G., Cambi A., Blood 2011, 118, 4111–4119. [DOI] [PubMed] [Google Scholar]
- 81.
- 81a. de Bakker B. I., de Lange F., Cambi A., Korterik J. P., van Dijk E. M., van Hulst N. F., Figdor C. G., Garcia-Parajo M. F., ChemPhysChem 2007, 8, 1473–1480; [DOI] [PubMed] [Google Scholar]
- 81b. Manzo C., Torreno-Pina J. A., Joosten B., Reinieren-Beeren I., Gualda E. J., Loza-Alvarez P., Figdor C. G., Garcia-Parajo M. F., Cambi A., J. Biol. Chem. 2012, 287, 38946–38955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Martínez J. D., Valverde P., Delgado S., Romanò C., Linclau B., Reichardt N.-C., Oscarson S., Ardá A., Jiménez-Barbero J., Cañada F. J., Molecules, 2019, 24, E2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Valverde P., Delgado S., Martínez J. D., Vendeville J.-B., Malassis J., Linclau B., Reichardt N.-C., Cañada F. J., Jiménez-Barbero J., Ardá A., ACS Chem. Biol. 2019, 14, 1660–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Angulo J., Diaz I., Reina J. J., Tabarani G., Fieschi F., Rojo J., Nieto P. M., ChemBioChem 2008, 9, 2225–2227. [DOI] [PubMed] [Google Scholar]
- 85. Guo Y., Feinberg H., Conroy E., Mitchell D. A., Alvarez R., Blixt O., Taylor M. E., Weis W. I., Drickamer K., Nat. Struct. Mol. Biol. 2004, 11, 591–598. [DOI] [PubMed] [Google Scholar]
- 86. Feinberg H., Mitchell D. A., Drickamer K., Weis W. I., Science 2001, 294, 2163–2166. [DOI] [PubMed] [Google Scholar]
- 87. Adams E. W., Ratner D. M., Bokesch H. R., McMahon J. B., O'Keefe B. R., Seeberger P. H., Chem. Biol. 2004, 11, 875–881. [DOI] [PubMed] [Google Scholar]
- 88. Reina J. J., Diaz I., Nieto P. M., Campillo N. E., Paez J. A., Tabarani G., Fieschi F., Rojo J., Org. Biomol. Chem. 2008, 6, 2743–2754. [DOI] [PubMed] [Google Scholar]
- 89. van Liempt E., Bank C. M. C., Mehta P., García-Vallejo J. J., Kawar Z. S., Geyer R., Alvarez R. A., Cummings R. D., van Kooyk Y., van Die I., FEBS Lett. 2006, 580, 6123–6131. [DOI] [PubMed] [Google Scholar]
- 90. Holla A., Skerra A., Protein Eng. Des. Sel. 2011, 24, 659–669. [DOI] [PubMed] [Google Scholar]
- 91. Echeverria B., Serna S., Achilli S., Vivès C., Pham J., Thépaut M., Hokke C. H., Fieschi F., Reichardt N.-C., ACS Chem. Biol. 2018, 13, 2269–2279. [DOI] [PubMed] [Google Scholar]
- 92. Temming A. R., Dekkers G., van de Bovenkamp F. S., Plomp H. R., Bentlage A. E. H., Szittner Z., Derksen N. I. L., Wuhrer M., Rispens T., Vidarsson G., Sci. Rep. 2019, 9, 9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Brzezicka K., Echeverria B., Serna S., van Diepen A., Hokke C. H., Reichardt N.-C., ACS Chem. Biol. 2015, 10, 1290–1302. [DOI] [PubMed] [Google Scholar]
- 94. van Montfort T., Eggink D., Boot M., Tuen M., Hioe C. E., Berkhout B., Sanders R. W., J. Immunol. 2011, 187, 4676–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. te Riet J., Reinieren-Beeren I., Figdor C. G., Cambi A., J. Mol. Recognit. 2015, 28, 687–698. [DOI] [PubMed] [Google Scholar]
- 96. Pitarque S., Herrmann J.-L., Duteyrat J.-L., Jackson M., Stewart G. R., Lecointe F., Payre B., Schwartz O., Young D. B., Marchal G., Lagrange P. H., Puzo G., Gicquel B., Nigou J., Neyrolles O., J. Biochem. 2005, 392, 615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Guzzi C., Angulo J., Doro F., Reina J. J., Thépaut M., Fieschi F., Bernardi A., Rojo J., Nieto P. M., Org. Biomol. Chem. 2011, 9, 7705–7712. [DOI] [PubMed] [Google Scholar]
- 98. Appelmelk B. J., van Die I., van Vliet S. J., Vandenbroucke-Grauls C. M. J. E., Geijtenbeek T. B., van Kooyk Y., J. Immunol. 2003, 170, 1635–1639. [DOI] [PubMed] [Google Scholar]
- 99. Pederson K., Mitchell D. A., Prestegard J. H., Biochemistry 2014, 53, 5700–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. van Liempt E., Imberty A., Bank C. M. C., van Vliet S. J., van Kooyk Y., Geijtenbeek T. B., van Die I., J. Biol. Chem. 2004, 279, 33161–33167. [DOI] [PubMed] [Google Scholar]
- 101.
- 101a. Andreini M., Doknic D., Sutkevičiu̅tė I., Reina J. J., Duan J., Chabrol E., Thépaut M., Moroni E., Doro F., Belvisi L., Weiser J., Rojo J., Fieschi F., Bernardi A., Org. Biomol. Chem. 2011, 9, 5778–5786; [DOI] [PubMed] [Google Scholar]
- 101b. Doknic D., Abramo M., Sutkevičiu̅tė I., Reinhardt A., Guzzi C., Schlegel M. K., Potenza D., Nieto P. M., Fieschi F., Seeberger P. H., Bernardi A., Eur. J. Org. Chem. 2013, 2013, 5303–5314. [Google Scholar]
- 102. Meyer S., van Liempt E., Imberty A., van Kooyk Y., Geyer H., Geyer R., van Die I., J. Biol. Chem. 2005, 280, 37349–37359. [DOI] [PubMed] [Google Scholar]
- 103. Somers W. S., Tang J., Shaw G. D., Camphausen R. T., Cell 2000, 103, 467–479. [DOI] [PubMed] [Google Scholar]
- 104.
- 104a. Rodríguez E., Schetters S. T. T., van Kooyk Y., Nat. Rev. Immunol. 2018, 18, 204–211; [DOI] [PubMed] [Google Scholar]
- 104b. Valverde P., Ardá A., Reichardt N.-C., Jiménez-Barbero J., Gimeno A., MedChemComm 2019, 10, 1678–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.
- 105a. Illescas B. M., Rojo J., Delgado R., Martín N., J. Am. Chem. Soc. 2017, 139, 6018–6025; [DOI] [PubMed] [Google Scholar]
- 105b. Bernardi A., Sattin S., Eur. J. Org. Chem. 2020, DOI: 10.1002/ejoc.202000155. [Google Scholar]
- 106.
- 106a. Mari S., Posteri H., Marcou G., Potenza D., Micheli F., Javier Cañada F., Jimenez-Barbero J., Bernardi A., Eur. J. Org. Chem. 2004, 2004, 5119–5125; [Google Scholar]
- 106b. Bordoni V., Porkolab V., Sattin S., Thépaut M., Frau I., Favero L., Crotti P., Bernardi A., Fieschi F., Di Bussol V., RSC Adv. 2016, 6, 89578–89584. [Google Scholar]
- 107. Reina J. J., Sattin S., Invernizzi D., Mari S., Martinez-Prats L., Tabarani G., Fieschi F., Delgado R., Nieto P. M., Rojo J., Bernardi A., ChemMedChem 2007, 2, 1030–1036. [DOI] [PubMed] [Google Scholar]
- 108. Thépaut M., Guzzi C., Sutkevičiu̅tė I., Sattin S., Ribeiro-Viana R., Varga N., Chabrol E., Rojo J., Bernardi A., Angulo J., Nieto P. M., Fieschi F., J. Am. Chem. Soc. 2013, 135, 2518–2529. [DOI] [PubMed] [Google Scholar]
- 109.
- 109a. Varga N., Sutkevičiu̅tė I., Guzzi C., McGeagh J., Petit-Haertlein I., Gugliotta S., Weiser J., Angulo J., Fieschi F., Bernardi A., Chem. Eur. J. 2013, 19, 4786–4797; [DOI] [PubMed] [Google Scholar]
- 109b. Obermajer N., Sattin S., Colombo C., Bruno M., Svajger U., Anderluh M., Bernardi A., Mol. Diversity 2011, 15, 347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Porkolab V., Chabrol E., Varga N., Ordanini S., Sutkevičiu̅tė I., Thépaut M., García-Jiménez M. J., Girard E., Nieto P. M., Bernardi A., Fieschi F., ACS Chem. Biol. 2018, 13, 600–608. [DOI] [PubMed] [Google Scholar]
- 111. Medve L., Achilli S., Guzman-Caldentey J., Thépaut M., Senaldi L., Le Roy A., Sattin S., Ebel C., Vivès C., Martin-Santamaria S., Bernardi A., Fieschi F., Chem. Eur. J. 2019, 25, 14659–14668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sutkevičiu̅tė I., Thépaut M., Sattin S., Berzi A., McGeagh J., Grudinin S., Weiser J., Le Roy A., Reina J. J., Rojo J., Clerici M., Bernardi A., Ebel C., Fieschi F., ACS Chem. Biol. 2014, 9, 1377–1385. [DOI] [PubMed] [Google Scholar]
- 113.
- 113a. Guzzi C., Muñoz-García J. C., Enriquez-Navas P. M., Rojo J., Angulo J., Nieto P. M., Pure Appl. Chem. 2013, 85, 1771–1787; [Google Scholar]
- 113b. Guzzi C., Alfarano P., Sutkevičiu̅tė I., Sattin S., Ribeiro-Viana R., Fieschi F., Bernardi A., Weiser J., Rojo J., Angulo J., Nieto P. M., Org. Biomol. Chem. 2016, 14, 335–344. [DOI] [PubMed] [Google Scholar]
- 114.
- 114a. Tomašič T., Hajsek D., Svajger U., Luzar J., Obermajer N., Petit-Haertlein I., Fieschi F., Anderluh M., Eur. J. Med. Chem. 2014, 75, 308–326; [DOI] [PubMed] [Google Scholar]
- 114b. Kotar A., Tomašič T., Živkovid M. L., Jug G., Plavec J., Anderluh M., Org. Biomol. Chem. 2016, 14, 862–875. [DOI] [PubMed] [Google Scholar]
- 115. Cecioni S., Imberty A., Vidal S., Chem. Rev. 2015, 115, 525–561. [DOI] [PubMed] [Google Scholar]
- 116.
- 116a. Lasala F., Arce E., Otero J. R., Rojo J., Delgado R., Antimicrob. Agents Chemother. 2003, 47, 3970–3972; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116b. Tabarani G., Reina J. J., Ebel C., Vivès C., Lortat-Jacob H., Rojo J., Fieschi F., FEBS Lett. 2006, 580, 2402–2408. [DOI] [PubMed] [Google Scholar]
- 117. Blattes E., Vercellone A., Eutamène H., Turrin C.-O., Théodorou V., Majorai J.-P., Caminade A.-M., Prandi J., Nigou J., Puzo G., Proc. Natl. Acad. Sci. USA 2013, 110, 8795–8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Martínez-Ávila O., Bedoya L. M., Marradi M., Clavel C., Alcamí J., Penadés S., ChemBioChem 2009, 10, 1806–1809. [DOI] [PubMed] [Google Scholar]
- 119.
- 119a. Sattin S., Daghetti A., Thépaut M., Berzi A., Sanchez-Navarro M., Tabarani G., Rojo J., Fieschi F., Clerici M., Bernardi A., ACS Chem. Biol. 2010, 5, 301–312; [DOI] [PubMed] [Google Scholar]
- 119b. Berzi A., Reina J. J., Ottria R., Sutkevičiu̅tė I., Antonazzo P., Sanchez-Navarro M., Chabrol E., Biasin M., Trabattoni D., Cetin I., Rojo J., Fieschi F., Bernardi A., Clerici M., AIDS 2012, 26, 127–137; [DOI] [PubMed] [Google Scholar]
- 119c. Varga N., Sutkevičiu̅tė I., Ribeiro-Viana R., Berzi A., Ramdasi R., Daghetti A., Vettoretti G., Amara A., Clerici M., Rojo J., Fieschi F., Bernardi A., Biomaterials 2014, 35, 4175–4184. [DOI] [PubMed] [Google Scholar]
- 120. Wang S.-K., Liang P.-H., Astronomo R. D., Hsu T.-L., Hsieh S.-L., Burton D. R., Wong C.-H., Proc. Natl. Acad. Sci. USA 2008, 105, 3690–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Martinez-Ávila O., Hijazi K., Marradi M., Clavel C., Campion C., Kelly C., Penades S., Chem. Eur. J. 2009, 15, 9874–9888. [DOI] [PubMed] [Google Scholar]
- 122. Zhang Q., Collins J., Anastasaki A., Wallis R., Mitchell D. A., Becer C. R., Haddleton D. M., Angew. Chem. Int. Ed. 2013, 52, 4435–4439; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4531–4535. [Google Scholar]
- 123. Ciobanu M., Huang K.-T., Daguer J.-P., Barluenga S., Chaloin O., Schaeffer E., Mueller C. G., Mitchell D. A., Winssinger N., Chem. Commun. 2011, 47, 9321–9323. [DOI] [PubMed] [Google Scholar]
- 124.
- 124a. Huang J., Zhang Q., Li G.-Z., Haddleton D. M., Wallis R., Mitchell D., Heise A., Becer C. R., Macromol. Rapid Commun. 2013, 34, 1542–1546; [DOI] [PubMed] [Google Scholar]
- 124b. Becer C. R., Gibson M. I., Geng J., Ilyas R., Wallis R., Mitchell D. A., Haddleton D. M., J. Am. Chem. Soc. 2010, 132, 15130–15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.
- 125a. Bertolotti B., Sutkevičiu̅tė I., Ambrosini M., Ribeiro-Viana R., Rojo J., Fieschi F., Dvořáková H., Kašáková M., Parkan K., Hlaváčková M., Nováková K., Moravcová J., Org. Biomol. Chem. 2017, 15, 3995–4004; [DOI] [PubMed] [Google Scholar]
- 125b. Bertolotti B., Oroszová B., Sutkevičiu̅tė I., Kniezo L., Fieschi F., Parkan K., Lovyová Z., Kašáková M., Moravcová J., Carbohydr. Res. 2016, 435, 7–18. [DOI] [PubMed] [Google Scholar]
- 126. Reina J. J., Maldonado O. S., Tabarani G., Fieschi F., Rojo J., Bioconjugate Chem. 2007, 18, 963–969. [DOI] [PubMed] [Google Scholar]
- 127. Luczkowiak J., Sattin S., Sutkevičiu̅tė I., Juan Reina J., Sanchez-Navarro M., Thépaut M., Martinez-Prats L., Daghetti A., Fieschi F., Delgado R., Bernardi A., Rojo J., Bioconjugate Chem. 2011, 22, 1354–1365. [DOI] [PubMed] [Google Scholar]
- 128. Ordanini S., Varga N., Porkolab V., Thépaut M., Belvisi L., Bertaglia A., Palmioli A., Berzi A., Trabattoni D., Clerici M., Fieschi F., Bernardi A., Chem. Commun. 2015, 51, 3816–3819. [DOI] [PubMed] [Google Scholar]
- 129. Brument S., Cheneau C., Brissonnet Y., Deniaud D., Halary F., Gouin S. G., Org. Biomol. Chem. 2017, 15, 7660–7671. [DOI] [PubMed] [Google Scholar]
- 130.
- 130a. Luczkowiak J., Muñoz A., Sánchez-Navarro M., Ribeiro-Viana R., Ginieis A., Illescas B. M., Martín N., Delgado R., Rojo J., Biomacromolecules 2013, 14, 431–437; [DOI] [PubMed] [Google Scholar]
- 130b. Muñoz A., Sigwalt D., Illescas B. M., Luczkowiak J., Rodríguez-Pérez L., Nierengarten I., Holler M., Remy J.-S., Buffet K., Vincent S. P., Rojo J., Delgado R., Nierengarten J.-F., Martín N., Nat. Chem. 2016, 8, 50–57. [DOI] [PubMed] [Google Scholar]
- 131. Morbioli I., Porkolab V., Magini A., Casnati A., Fieschi F., Sansone F., Carbohydr. Res. 2017, 453–454, 36–43. [DOI] [PubMed] [Google Scholar]
- 132. Lee R. T., Hsu T.-L., Huang S. K., Hsieh S.-L., Wong C.-H., Lee Y. C., Glycobiology 2011, 21, 512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ribeiro-Viana R., Sánchez-Navarro M., Luczkowiak J., Koeppe J. R., Delgado R., Rojo J., Davis B. G., Nat. Commun. 2012, 3, 1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kozlovska T. M., Cielens I., Dreilinņa D., Dislers A., Baumanis V., Ose V., Pumpens P., Gene 1993, 137, 133–137. [DOI] [PubMed] [Google Scholar]
- 135. Berzi A., Ordanini S., Joosten B., Trabattoni D., Cambi A., Bernardi A., Clerici M., Sci. Rep. 2016, 6, 35373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Mitchell D. A., Zhang Q., Voorhaar L., Haddleton D. M., Herath S., Gleinich A. S., Randeva H. S., Crispin M., Lehnert H., Wallis R., Patterson S., Becer C. R., Chem. Sci. 2017, 8, 6974–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Fehres C. M., Kalay H., Bruijns S. C. M., Musaafir S. A. M., Ambrosini M., van Bloois L., van Vliet S. J., Storm G., Garcia-Vallejo J. J., van Kooyk Y., J. Controlled Release 2015, 203, 67–76. [DOI] [PubMed] [Google Scholar]
- 138. Li E. R., Hogervorst T. P., Achilli S., Bruijns S. C., Arnoldus T., Vivès C., Wong C. C., Thépaut M., Meeuwenoord N. J., Elst van den H., Overkleeft H. S., Marel van der G. A., Filippov D. V., van Vliet S. J., Fieschi F., Codée J. D., Van Kooyk Y., Front. Chem. 2019, 7, 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Frison N., Taylor M. E., Soilleux E., Bousser M. T., Mayer R., Monsigny M., Drickamer K., Roche A. C., J. Biol. Chem. 2003, 278, 23922–23929. [DOI] [PubMed] [Google Scholar]
- 140. McIntosh J. D., Brimble M. A., Brooks A. E. S., Dunbar P. R., Kowalczyk R., Tomabechi Y., Fairbanks A. J., Chem. Sci. 2015, 6, 4636–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.
- 141a. Silva J. M., Zupancic E., Vandermeulen G., Oliveira V. G., Salgado A., Videira M., Gaspar M., Graca L., Préat V., Florindo H. F., J. Controlled Release 2015, 198, 91–103; [DOI] [PubMed] [Google Scholar]
- 141b. Boks M. A., Ambrosini M., Bruijns S. C., Kalay H., van Bloois L., Storm G., Garcia-Vallejo J. J., van Kooyk Y., J. Controlled Release 2015, 216, 37–46. [DOI] [PubMed] [Google Scholar]
- 142. Glaffig M., Stergiou N., Hartmann S., Schmitt E., Kunz H., ChemMedChem 2018, 13, 25–29. [DOI] [PubMed] [Google Scholar]
- 143. Dehuyser L., Schaeffer E., Chaloin O., Mueller C. G., Baati R., Wagner A., Bioconjugate Chem. 2012, 23, 1731–1739. [DOI] [PubMed] [Google Scholar]
- 144. Unger W. W. J., van Beelen A. J., Bruijns S. C., Joshi M., Fehres C. M., van Bloois L., Verstege M. I., Ambrosini M., Kalay H., Nazmi K., Bolscher J. G., Hooijberg E., de Gruijl T. D., Storm G., van Kooyk Y., J. Controlled Release 2012, 160, 88–95. [DOI] [PubMed] [Google Scholar]
- 145. Le Moignic A., Malard V., Benvegnu T., Lemiègre L., Berchel M., Jaffrès P.-A., Baillou C., Delost M., Macedo R., Rochefort J., Lescaille G., Pichon C., Lemoine F. M., Midoux P., Mateo V., J. Controlled Release 2018, 278, 110–121. [DOI] [PubMed] [Google Scholar]
- 146. Zhang Q., Su L., Collins J., Chen G., Wallis R., Mitchell D. A., Haddleton D. M., Becer C. R., J. Am. Chem. Soc. 2014, 136, 4325–4332. [DOI] [PubMed] [Google Scholar]
- 147. Aretz J., Wamhoff E. C., Hanske J., Heymann D., Rademacher C., Front. Immunol. 2014, 5, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Schulze J., Baukmann H., Wawrzinek R., Fuchsberger F. F., Specker E., Aretz J., Nazaré M., Rademacher C., ACS Chem. Biol. 2018, 13, 3229–3235. [DOI] [PubMed] [Google Scholar]
- 149.
- 149a. Borrok M. J., Kiessling L. L., J. Am. Chem. Soc. 2007, 129, 12780–12785; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149b. Mangolg S. L., Prost L. R., Kiessling L. L., Chem. Sci. 2012, 3, 772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.
- 150a. Garber K. C., Wangkanont K., Carlson E. E., Kiessling L. L., Chem. Commun. (Camb.) 2010, 46, 6747–6749; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150b. Prost L. R., Grim J. C., Tonelli M., Kiessling L. L., ACS Chem. Biol. 2012, 7, 1603–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Aretz J., Baukmann H., Shanina E., Hanske J., Wawrzinek R., Zapol‘skii V. A., Seeberger P. H., Kaufmann D. E., Rademacher C., Angew. Chem. Int. Ed. 2017, 56, 7292–7296; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7398–7402. [Google Scholar]
- 152.
- 152a. McDermott R., Bausinger H., Fricker D., Spehner D., Proamer F., Lipsker D., Cazenave J. P., Goud B., De La Salle H., Salamero J., Hanau D., J. Invest. Dermatol. 2004, 123, 72–77; [DOI] [PubMed] [Google Scholar]
- 152b. Valladeau J., Duvert-Frances V., Pin J. J., Dezutter-Dambuyant C., Vincent C., Massacrier C., Vincent J., Yoneda K., Banchereau J., Caux C., Davoust J., Saeland S., Eur. J. Immunol. 1999, 29, 2695–2704. [DOI] [PubMed] [Google Scholar]
- 153. Banchereau J., Steinman R. M., Nature 1998, 392, 245–252. [DOI] [PubMed] [Google Scholar]
- 154. Ito T., Inaba M., Inaba K., Toki J., Sogo S., Iguchi T., Adachi Y., Yamaguchi K., Amakawa R., Valladeau J., Saeland S., Furuhara S., Ikehara S., J. Immunol. 1999, 163, 1409–1419. [PubMed] [Google Scholar]
- 155.
- 155a. Valladeau J., Ravel O., Dezutter-Dambuyant C., Moore K., Kleijmeer M., Liu Y., Duvert-Frances V., Vincent C., Schmitt D., Davoust J., Caux C., Lebecque S., Saeland S., Immunity. 2000, 12, 71–81; [DOI] [PubMed] [Google Scholar]
- 155b. McDermott R., Ziylan U., Spehner D., Bausinger H., Lipsker D., Mommaas M., Cazenave J. P., Raposo G., Goud B., de la Salle H., Salamero J., Hanau D., Mol. Biol. Cell 2002, 13, 317–335; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155c. Valladeau J., Dezutter-Dambuyant C., Saeland S., Immunol. Res. 2003, 28, 93–107. [DOI] [PubMed] [Google Scholar]
- 156.
- 156a. Hunger R. E., Sieling P. A., Ochoa M. T., Sugaya M., Burdick A. E., Rea T. H., Brennan P. J., Belisle J. T., Blauvelt A., Porcelli S. A., Modlin R. L., J. Clin. Invest. 2004, 113, 701–708; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156b. Yang K., Park C. G., Cheong C., Bulgheresi S., Zhang S., Zhang P., He Y., Jiang L., Huang H., Ding H., Wu Y., Wang S., Zhang L., Li A., Xia L., Bartra S. S., Plano G. V., Skurnik M., Klena J. D., Chen T., Immunol. Cell Biol. 2015, 93, 815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. de Jong M. A., Vriend L. E., Theelen B., Taylor M. E., Fluitsma D., Boekhout T., Geijtenbeek T. B., Mol. Immunol. 2010, 47, 1216–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.
- 158a. Stoitzner P., Romani N., Eur. J. Immunol. 2011, 41, 2526–2529; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158b. de Jong M. A., de Witte L., Taylor M. E., Geijtenbeek T. B., J. Immunol. 2010, 185, 1633–1641. [DOI] [PubMed] [Google Scholar]
- 159. de Witte L., Nabatov A., Pion M., Fluitsma D., de Jong M. A., de Gruijl T., Piguet V., van Kooyk Y., Geijtenbeek T. B., Nat. Med. 2007, 13, 367–371. [DOI] [PubMed] [Google Scholar]
- 160. Mayr L., Su B., Moog C., Trends Microbiol. 2017, 25, 170–172. [DOI] [PubMed] [Google Scholar]
- 161.
- 161a. Peressin M., Proust A., Schmidt S., Su B., Lambotin M., Biedma M. E., Laumond G., Decoville T., Holl V., Moog C., AIDS 2014, 28, 667–677; [DOI] [PubMed] [Google Scholar]
- 161b. Kawamura T., Koyanagi Y., Nakamura Y., Ogawa Y., Yamashita A., Iwamoto T., Ito M., Blauvelt A., Shimada S., J. Immunol. 2008, 180, 3297–3304; [DOI] [PubMed] [Google Scholar]
- 161c. Nasr N., Lai J., Botting R. A., Mercier S. K., Harman A. N., Kim M., Turville S., Center R. J., Domagala T., Gorry P. R., Olbourne N., Cunningham A. L., J. Immunol. 2014, 193, 2554–2564. [DOI] [PubMed] [Google Scholar]
- 162.
- 162a. Smed-Sörensen A., Loré K., Vasudevan J., Louder M. K., Andersson J., Mascola J. R., Spetz A. L., Koup R. A., J. Virol. 2005, 79, 8861–8869; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162b. Turville S. G., Santos J. J., Frank I., Cameron P. U., Wilkinson J., Miranda-Saksena M., Dable J., Stössel H., Romani N., Jr Piatak M., Lifson J. D., Pope M., Cunningham A. L., Blood 2004, 103, 2170–2179. [DOI] [PubMed] [Google Scholar]
- 163. Kissenpfennig A., Ait-Yahia S., Clair-Moninot V., Stossel H., Badell E., Bordat Y., Pooley J. L., Lang T., Prina E., Coste I., Gresser O., Renno T., Winter N., Milon G., Shortman K., Romani N., Lebecque S., Malissen B., Saeland S., Douillard P., Mol. Cell. Biol. 2005, 25, 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Chen J. T., Chen C. H., Ku K. L., Hsiao M., Chiang C. P., Hsu T. L., Chen M. H., Wong C. H., Proc. Natl. Acad. Sci. USA 2015, 112, 13057–13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Gillespie L., Roosendahl P., Ng W. C., Brooks A. G., Reading P. C., Londrigan S. L., Sci. Rep. 2016, 6, 19428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Ng W. C., Londrigan S. L., Nasr N., Cunningham A. L., Turville S., Brooks A. G., Reading P. C., J. Virol. 2015, 90, 206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Takahara K., Omatsu Y., Yashima Y., Maeda Y., Tanaka S., Iyoda T., Clusen B., Matsubara M., Letterio J., Steinman R. M., Matsuda Y., Inaba K., Int. Immunol. 2002, 14, 433–444. [DOI] [PubMed] [Google Scholar]
- 168. Drickamer K., Curr. Opin. Struct. Biol. 1999, 9, 585–590. [DOI] [PubMed] [Google Scholar]
- 169.M. E. Taylor, K. Drickamer, Introduction to Glycobiology, 3rd ed., Oxford University Press, Oxford, 2003.
- 170. Stambach N. S., Taylor M. E., Glycobiology 2003, 13, 401–410. [DOI] [PubMed] [Google Scholar]
- 171. Galustian C., Park C. G., Chai W., Kiso M., Bruening S. A., Kang Y.-S., Steinman R. M., Feizi T., Int. Immunol. 2004, 16, 853–866. [DOI] [PubMed] [Google Scholar]
- 172. de Jong M. A., de Witte L., Santegoets S. J., Fluitsma D., Taylor M. E., de Gruijl T. D., Geijtenbeek T. B., J. Leukocyte Biol. 2010, 87, 637–643. [DOI] [PubMed] [Google Scholar]
- 173.
- 173a. Naarding M. A., Ludwig I. S., Groot F., Berkhout B., Geijtenbeek T. B., Pollakis G., Paxton W. A., J. Clin. Invest. 2005, 115, 3256–3264; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173b. Saeland E., de Jong M. A., Nabatov A. A., Kalay H., Geijtenbeek T. B., van Kooyk Y., Mol. Immunol. 2009, 46, 2309–2316. [DOI] [PubMed] [Google Scholar]
- 174. Tateno H., Ohnishi K., Yabe R., Hayatsu N., Sato T., Takeya M., Narimatsu H., Hirabayashi J., J. Biol. Chem. 2010, 285, 6390–6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Ota F., Hirayama T., Kizuka Y., Yamaguchi Y., Fujinawa R., Nagata M., Ismanto H. S., Lepenies B., Aretz J., Rademacher C., Seeberger P. H., Angata T., Kitazume S., Yoshida K., Betsuyaku T., Kida K., Yamasaki S., Taniguchi N., Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1592–1601. [DOI] [PubMed] [Google Scholar]
- 176. Feinberg H., Powlesland A. S., Taylor M. E., Weis W. I., J. Biol. Chem. 2010, 285, 13285–13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Feinberg H., Taylor M. E., Razi N., McBride R., Knirel Y. A., Graham S. A., Drickamer K., Weis W. I., J. Mol. Biol. 2011, 405, 1027–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Funderburgh J. L., Glycobiology 2000, 10, 951–958. [DOI] [PubMed] [Google Scholar]
- 179. Gao C., Fujinawa R., Yoshida T., Ueno M., Ota F., Kizuka Y., Hirayama T., Korekane H., Kitazume S., Maeno T., Ohtsubo K., Yoshida K., Yamaguchi Y., Lepenies B., Aretz J., Rademacher C., Kabata H., Hegab A. E., Seeberger P. H., Betsuyaku T., Kida K., Taniguchi N., Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L268–L276. [DOI] [PubMed] [Google Scholar]
- 180.
- 180a. Bratke K., Klug M., Bier A., Julius P., Kuepper M., Virchow J. C., Lommatzsch M., Am. J. Respir. Cell Mol. Biol. 2008, 38, 655–660; [DOI] [PubMed] [Google Scholar]
- 180b. Van Pottelberge G. R., Bracke K. R., Demedts I. K., De Rijck K., Reinartz S. M., van Drunen C. M., Verleden G. M., Vermassen F. E., Joos G. F., Brusselle G. G., Respir. Res. 2010, 11, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Chatwell L., Holla A., Kaufer B. B., Skerra A., Mol. Immunol. 2008, 45, 1981–1994. [DOI] [PubMed] [Google Scholar]
- 182. Hanske J., Schulze J., Aretz J., McBride R., Loll B., Schmidt H., Knirel Y., Rabsch W., Wahl M. C., Paulson J. C., Rademacher C., J. Biol. Chem. 2017, 292, 862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Feinberg H., Rowntree T. J., Tan S. L., Drickamer K., Weis W. I., Taylor M. E., J. Biol. Chem. 2013, 288, 36762–36771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Ward E. M., Stambach N. S., Drickamer K., Taylor M. E., J. Biol. Chem. 2006, 281, 15450–15456. [DOI] [PubMed] [Google Scholar]
- 185. Verdijk P., Dijkman R., Plasmeijer E. I., Mulder A. A., Zoutman W. H., Mommaas A. M., Tensen C. P., J. Invest. Dermatol. 2005, 124, 714–717. [DOI] [PubMed] [Google Scholar]
- 186. Chabrol E., Thépaut M., Dezutter-Dambuyant C., Vivès C., Marcoux J., Kahn R., Valladeau-Guilemond J., Vachette P., Durand D., Fieschi F., Biophys. J. 2015, 108, 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Thépaut M., Valladeau J., Nurisso A., Kahn R., Arnou B., Vivès C., Saeland S., Ebel C., Monnier C., Dezutter-Dambuyant C., Imberty A., Fieschi F., Biochemistry 2009, 48, 2684–2698. [DOI] [PubMed] [Google Scholar]
- 188. Head J. F., Mealy T. R., McCormack F. X., Seaton B. A., J. Biol. Chem. 2003, 278, 43254–43260. [DOI] [PubMed] [Google Scholar]
- 189. Wallis R., Shaw J. M., Uitdehaag J., Chen C.-B., Torgersen D., Drickamer K., J. Biol. Chem. 2004, 278, 14065–14073. [DOI] [PubMed] [Google Scholar]
- 190. Onizuka T., Shimizu H., Moriwaki Y., Nakano T., Kanai S., Shimada I., Takahashi H., FEBS J. 2012, 279, 2645–2656. [DOI] [PubMed] [Google Scholar]
- 191. Wragg S., Drickamer K., J. Biol. Chem. 1999, 274, 35400–35406. [DOI] [PubMed] [Google Scholar]
- 192. Hanske J., Aleksić S., Ballaschk M., Jurk M., Shanina E., Beerbaum M., Schmieder P., Keller B. G., Rademacher C., J. Am. Chem. Soc. 2016, 138, 12176–12186. [DOI] [PubMed] [Google Scholar]
- 193. Chabrol E., Nurisso A., Daina A., Vassal-Stermann E., Thépaut M., Girard E., Vivès R. R., Fieschi F., PLoS One 2012, 7, e50722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.
- 194a. Capila I., Linhardt R. J., Angew. Chem. Int. Ed. 2002, 41, 390–412; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 426–450; [Google Scholar]
- 194b. Hacker U., Nybakken K., Perrimon N., Nat. Rev. Mol. Cell Biol. 2005, 6, 530–541. [DOI] [PubMed] [Google Scholar]
- 195. Zhao J., Liu X., Kao C., Zhang E., Li Q., Zhang F., Linhardt L. J., Biochemistry 2016, 55, 4552–4559. [DOI] [PubMed] [Google Scholar]
- 196. Muñoz-García J. C., Chabrol E., Vivès R. R., Thomas A., de Paz J. L., Rojo J., Imberty A., Fieschi F., Nieto P. M., Angulo J., J. Am. Chem. Soc. 2015, 137, 4100–4110. [DOI] [PubMed] [Google Scholar]
- 197. Muñoz-García J. C., López-Prados J., Angulo J., Díaz-Contreras I., Reichardt N., de Paz J. L., Martín-Lomas M., Nieto P. M., Chem. Eur. J. 2012, 18, 16319–16331. [DOI] [PubMed] [Google Scholar]
- 198. Hanske J., Wawrzinek R., Geissner A., Wamhoff E. C., Sellrie K., Schmidt H., Seeberger P. H., Rademacher C., ChemBioChem 2017, 18, 1183–1187. [DOI] [PubMed] [Google Scholar]
- 199.
- 199a. Fraser P. E., Nguyen J. T., Surewicz W. K., Kirschner D. A., Biophys. J. 1991, 60, 1190–1201; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199b. Veldkamp C. T., Peterson F. C., Pelzek A. J., Volkman B. F., Protein Sci. 2005, 14, 1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Medve L., Achilli S., Serna S., Zuccotto F., Varga N., Thépaut M., Civera M., Vivès C., Fieschi F., Reichardt N., Bernardi A., Chem. Eur. J. 2018, 24, 14448–14460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Wamhoff E. C., Hanske J., Schnirch L., Aretz J., Grube M., Varón Silva D., Rademacher C., ACS Chem. Biol. 2016, 11, 2407–2413. [DOI] [PubMed] [Google Scholar]
- 202. Wamhoff E. C., Schulze J., Bellmann L., Rentzsch M., Bachem G., Fuchsberger F. F., Rademacher J., Hermann M., Del Frari B., van Dalen R., Hartmann D., van Sorge N. M., Seitz O., Stoitzner P., Rademacher C., ACS Cent. Sci. 2019, 5, 808–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Zaric M., Lyubomska O., Poux C., Hanna M. L., McCrudden M. T., Malissen B., Ingram R. J., Power U. F., Scott C. J., Donnelly R. F., Kissenpfennig A., J. Invest. Dermatol. 2015, 135, 425–434. [DOI] [PubMed] [Google Scholar]
- 204. O'Reilly M. K., Tian H., Paulson J. C., J. Immunol. 2011, 186, 1554–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Lenos K., Goos J. A., Vuist I. M., den Uil S. H., Delis-van Diemen P. M., Belt E., Stockmann H. B., Bril H., de Wit M., Carvalho B., Giblett S., Pritchard C. A., Meijer G. A., van Kooyk Y., Fijneman R. J., van Vliet S. J., Oncotarget 2015, 6, 26278–26290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Nonaka M., Ma B. Y., Murai R., Nakamura N., Baba M., Kawasaki N., Hodohara K., Asano S., Kawasaki T., J. Immunol. 2008, 180, 3347–3356. [DOI] [PubMed] [Google Scholar]
- 207. van Vliet S. J., Saeland E., van Kooyk Y., Trends Immunol. 2008, 29, 83–90. [DOI] [PubMed] [Google Scholar]
- 208. van Vliet S. J., Gringhuis S. I., Geijtenbeek T. B., van Kooyk Y., Nat. Immunol. 2006, 7, 1200–1208. [DOI] [PubMed] [Google Scholar]
- 209. van Vliet S. J., van Liempt E., Geijtenbeek T. B., van Kooyk Y., Immunobiology 2006, 211, 577–585. [DOI] [PubMed] [Google Scholar]
- 210.
- 210a. Sato K., Imai Y., Higashi N., Kumamoto Y., Onami T. M., Hedrick S. M., Irimura T., Blood 2005, 106, 207–215; [DOI] [PubMed] [Google Scholar]
- 210b. Sato K., Imai Y., Higashi N., Kumamoto Y., Mukaida N., Irimura T., Int. Immunol. 2005, 17, 559–568. [DOI] [PubMed] [Google Scholar]
- 211. Kudelka M. R., Ju T., Heimburg-Molinaro J., Cummings R. D., Adv. Cancer Res. 2015, 126, 53–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.
- 212a. Dahr W., Uhlenbruck G., Gunson H., van der Hart M., Vox Sang. 1975, 29, 36–50; [DOI] [PubMed] [Google Scholar]
- 212b. Gunson H. H., Stratton F., Mullard G. W., Br. J. Haematol. 1970, 18, 309–316. [DOI] [PubMed] [Google Scholar]
- 213.
- 213a. Ju T., Lanneau G., Gautam T., Wang Y., Xia B., Stowell S., Willard M., Wang W., Xia J., Zuna R., Laszik Z., Benbrook D. M., Hanigan M. H., Cummings R. D., Cancer Res. 2008, 68, 1636–1646; [DOI] [PubMed] [Google Scholar]
- 213b. Ju T., Otto V., Cummings R. D., Angew. Chem. Int. Ed. 2011, 50, 1770–1791; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1808–1830. [Google Scholar]
- 214. Patsos G., Corfield A., in The Sugar Code. Fundamentals of Glycosciences (Ed.: Gabius H.-J.), Wiley-VCH, Weinheim, 2009, pp. 111–137. [Google Scholar]
- 215. Fu C., Zhao H., Wang Y., Cai H., Xiao Y., Zeng Y., Chen H., HLA 2016, 88, 275–286. [DOI] [PubMed] [Google Scholar]
- 216. Beatson R., Maurstad G., Picco G., Arulappu A., Coleman J., Wandell H. H., Clausen H., Mandel U., Taylor-Papadimitriou J., Sletmoen M., Burchel J. M., PLoS One, 2015, 10, e0125994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Mortezai N., Behnken H. N., Kurze A. K., Ludewig P., Buck F., Meyer B., Wagener C., Glycobiology 2013, 23, 844–852. [DOI] [PubMed] [Google Scholar]
- 218. Suzuki N., Yamamoto K., Toyoshima S., Osawa T., Irimura T., J. Immunol. 1996, 156, 128–135. [PubMed] [Google Scholar]
- 219. Iida S., Yamamoto K., Irimura T., J. Biol. Chem. 1999, 274, 10697–10705. [DOI] [PubMed] [Google Scholar]
- 220. van Vliet S. J., van Liempt E., Saeland E., Aarnoudse C. A., Appelmelk B., Irimura T., Geijtenbeek T. B., Blixt O., Alvarez R., van Die I., van Kooyk Y., Int. Immunol. 2005, 17, 661–669. [DOI] [PubMed] [Google Scholar]
- 221.
- 221a. Mickum M. L., Prasanphanich N. S., Heimburg-Molinaro J., Leon K. E., Cummings R. D., Front. Genet. 2014, 5, 262; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221b. Hokke C. H., Deelder A. M., Hoffmann K. F., Wuhrer M., Exp. Parasitol. 2007, 117, 275–283. [DOI] [PubMed] [Google Scholar]
- 222. Ashwell G., Harford J., Annu. Rev. Biochem. 1982, 51, 531–554. [DOI] [PubMed] [Google Scholar]
- 223. Roggenbuck D., Mytilinaiou M. G., Lapin S. V., Reinhold D., Conrad K., Auto Immun. Highlights 2012, 3, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Chiu M. H., Tamura T., Wadhwa M. S., Rice K. G., J. Biol. Chem. 1994, 269, 16195–16202. [PubMed] [Google Scholar]
- 225. Ozaki K., Lee R. T., Lee Y. C., Kawasaki T., Glycoconjugate J. 1995, 12, 268–274. [DOI] [PubMed] [Google Scholar]
- 226. O′Brien S. J., Jones I. G., Christie P., Lancet 1998, 351, 906–907. [DOI] [PubMed] [Google Scholar]
- 227. Marcelo F., Garcia-Martin F., Matsushita T., Sardinha J., Coelho H., Oude-Vrielink A., Koller C., André S., Cabrita E. J., Gabius H.-J., Nishimura S.-I., Jiménez-Barbero J., Cañada F. J., Chem. Eur. J. 2014, 20, 16147–16155. [DOI] [PubMed] [Google Scholar]
- 228.
- 228a. Hatakeyama T., Kamiya T., Kusunoki M., Nakamura-Tsuruta S., Hirabayashi J., Goda S., Unno H., J. Biol. Chem. 2011, 286, 10305–10315; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228b. Poget S. F., Legge G. B., Proctor M. R., Butler P. J., Bycroft M., Williams R. L., J. Mol. Biol. 1999, 290, 867–879. [DOI] [PubMed] [Google Scholar]
- 229. Meier M., Bider M. D., Malashkevich V. N., Spiess M., Burkhard P., J. Mol. Biol. 2000, 300, 857–865. [DOI] [PubMed] [Google Scholar]
- 230. Ardá A., Jiménez-Barbero J., Chem. Commun. (Camb.) 2018, 54, 4761–4769. [DOI] [PubMed] [Google Scholar]
- 231. García-Vallejo J. J., Ilarregui J. M., Kalay H., Chamorro S., Koning N., Unger W. W., Ambrosini M., Montserrat V., Fernandes R. J., Bruijns S. C. M., van Weering J. R. T., Paauw N. J., O'Toole T., van Horssen J., van der Valk P., Nazmi K., Bolscher J. G. M., Bajramovic J., Dijkstra C. D., 't Hart B. A., van Kooyk Y., J. Exp. Med., 2014, 211, 1465–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Diniz A., Coelho H., Dias J. S., van Vliet S. J., Jiménez-Barbero J., Corzana F., Cabrita E. J., Marcelo F., Chem. Eur. J. 2019, 25, 13945–13955. [DOI] [PubMed] [Google Scholar]
- 233.
- 233a. Hakomori S., Annu. Rev. Immunol. 1984, 2, 103–126; [DOI] [PubMed] [Google Scholar]
- 233b. Hakomori S., Wang S.-M., Young W. W., Proc. Natl. Acad. Sci. USA 1977, 74, 3023–3027; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233c. Marionneau S., Le Moullac-Vaidye B., Le Pendu J., Glycobiology 2002, 12, 851–856. [DOI] [PubMed] [Google Scholar]
- 234. Marcelo F., Supekar N., Corzana F., van der Horst J. C., Vuist I. M., Live D., Boons G.-J., Smith D. F., van Vliet S. J., J. Biol. Chem. 2019, 294, 1300–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.
- 235a. Halim A., Brinkmalm G., Ruetschi U., Westman-Brinkmalm A., Portelius E., Zetterberg H., Blennow K., Larson G., Nilsson J., Proc. Natl. Acad. Sci. USA 2011, 108, 11848–11853; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235b. King S. L., Joshi H. J., Schjoldager K. T., Halim A., Madsen T. D., Dziegiel M. H., Woetmann A., Vakhrushev S. Y., Wandall H. H., Blood 2017, 1, 429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Gibadullin R., Farnsworth D. W., Barchi J. J., Gildersleeve J. C., ACS Chem. Biol. 2017, 12, 2172–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Wang P., Nilsson J., Brinkmalm G., Larson G., Huang X., Chem. Commun. 2014, 50, 15067–15070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Singh S. K., Streng-Ouwehand I., Litjens M., Weelij D. R., Garcia-Vallejo J. J., van Vliet S. J., Saeland E., van Kooyk Y., Mol. Immunol. 2009, 46, 1240–1249. [DOI] [PubMed] [Google Scholar]
- 239. Pirro M., Rombouts Y., Stella A., Neyrolles O., Burlet-Schiltz O., van Vliet S. J., de Ru A. H., Mohammed Y., Wuhrer M., van Veelen P. A., Hensbergen P. J., Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129513. [DOI] [PubMed] [Google Scholar]
- 240. Ng K. K., Drickamer K., Weis W. I., J. Biol. Chem. 1996, 271, 663–674. [DOI] [PubMed] [Google Scholar]
- 241. Artigas G., Monteiro J. T., Hinou H., Nishimura S. I., Lepenies B., Garcia-Martin F., J. Med. Chem. 2017, 60, 9012–9021. [DOI] [PubMed] [Google Scholar]
- 242. Tanaka J., Gleinich A. S., Zhang Q., Whitfield R., Kempe K., Haddleton D. M., Davis T. P., Perrier S., Mitchell D. A., Wilson P., Biomacromolecules 2017, 18, 1624–1633. [DOI] [PubMed] [Google Scholar]
- 243. Nath S., Mukherjee P., Trends Mol. Med. 2014, 20, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Pozzi C., Olaniyi R., Liljeroos L., Galgani I., Rappuoli R., Bagnoli F., Curr. Top. Microbiol. Immunol. 2017, 409, 491–528. [DOI] [PubMed] [Google Scholar]
- 245. Weidenmaier C., Peschel A., Nat. Rev. Microbiol. 2008, 6, 276–287. [DOI] [PubMed] [Google Scholar]
- 246. Mnich M. E., van Dalen R., Gerlach D., Hendriks A., Xia G., Peschel A., van Strijp J. A. G., van Sorge N. M., Cell. Microbiol. 2019, 21, e13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.
- 247a. van Vliet S. J., Bay S., Vuist I. M., Kalay H., Garcia-Vallejo J. J., Leclerc C., van Kooyk Y., J. Leukocyte Biol. 2013, 94, 315–323; [DOI] [PubMed] [Google Scholar]
- 247b. Heger L., Balk S., Luhr J. J., Heidkamp G. F., Lehmann C. H. K., Hatscher L., Cesnjevar R., Front. Immunol. 2018, 9, 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Heinrichs D. E., Yethon J. A., Whitfield C., Mol. Microbiol. 1998, 30, 221–223. [DOI] [PubMed] [Google Scholar]
- 249. Maalej M., Forgione R., Marchetti R., Bulteau F., Thépaut M., Lanzetta R., Laguri C., Simorre J., Fieschi F., Molinaro A., Silipo A., ChemBioChem 2019, 20, 1778–1782. [DOI] [PubMed] [Google Scholar]
- 250. Tang L., Cui Y., Liu W., Kang G., Zhu Y., Jiang D., Wei H., He F., Zhang G., Gou Z., Guo L., Chen X., J. Biol. Chem. 2004, 279, 18748–18758. [DOI] [PubMed] [Google Scholar]
- 251. Domínguez-Soto Á., Aragoneses-Fenoll L., Gómez-Aguado F., Corcuera M. T., Clária J., García-Monzón C., Bustos M., Corbí A. L., Hepatology 2009, 49, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Colmenares M., Munoz P., Naranjo-Gomez M., Toribio M. L., Delgado R., Martinez-Prats L., Martin-Gayo E., Dominguez-Soto A., Borras F. E., Aragoneses-Fenoll L., Corbi A. L., Zubiaur M., Blood 2007, 109, 5337–5345. [DOI] [PubMed] [Google Scholar]
- 253. Gramberg T., Soilleux E., Fisch T., Lalor P. F., Hofmann H., Wheeldon S., Cotterill A., Wegele A., Winkler T., Adams D. H., Pöhlmann S., Virology 2008, 373, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Powlesland A. S., Fisch T., Taylor M. E., Smith D. F., Tissot B., Dell A., Pöhlmann S., Drickamer K., J. Biol. Chem. 2008, 283, 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Gramberg T., Hofmann H., Möller P., Lalor P. F., Marzi A., Geier M., Krumbiegel M., Winkler T., Kirchhoff F., Adams D. H., Becker S., Münch J., Pöhlmann S., Virology 2005, 340, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Tang L., Yang J., Liu W., Tang X., Chen J., Zhao D., Wang M., Xu F., Lu Y., Liu B., Sun Q., Zhang L., He F., Gastroenterology 2009, 137, 1498–1508.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Xu F., Liu J., Liu D., Liu B., Wang M., Hu Z., Du X., Tang L., He F., Cancer Res. 2014, 74, 3418–3428. [DOI] [PubMed] [Google Scholar]
- 258. Liu D., Lu Q., Wang X., Wang J., Lu N., Jiang Z., Hao X., Li J., Liu J., Cao P., Peng G., Tao Y., Zhao D., He F., Tang L., Cell Res. 2019, 29, 365–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Liu B., Wang M., Wang X., Zhao D., Liu D., Liu J., Chen P.-J., Yang D., He F., Tang L., J. Immunol. 2013, 190, 4185–4195. [DOI] [PubMed] [Google Scholar]
- 260. Yang Z., Li Q., Wang X., Jiang X., Zhao D., Lin X., He F., Tang L., Proc. Natl. Acad. Sci. USA 2018, 115, 11054–11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Kizuka Y., Kitazume S., Sato K., Taniguchi N., FEBS Lett. 2015, 589, 1418–1422. [DOI] [PubMed] [Google Scholar]