

Key Points
Δ9-THC increases IL-10 and reduces IL-6 and CCL2 in endotoxemic mice via CB1R.
Δ9-THC decreases lung inflammation and organ injury in endotoxemic mice.
Monocytic-MDSCs mediate Δ9-THC–induced IL-10 upregulation in endotoxemic mice.
Visual Abstract
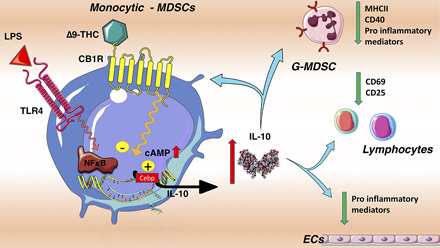
Abstract
Cannabis sativa and its principal components, Δ9-tetrahydrocannabinol (Δ9-THC) and cannabidiol, are increasingly being used to treat a variety of medical problems, including inflammatory conditions. Although studies suggest that the endocannabinoid system has immunomodulatory properties, there remains a paucity of information on the effects of cannabinoids on immunity and on outcomes of infection and injury. We investigated the effects and mechanism(s) of action of cannabinoid receptor agonists, including Δ9-THC, on inflammation and organ injury in endotoxemic mice. Administration of Δ9-THC caused a dramatic early upregulation of plasma IL-10 levels, reduced plasma IL-6 and CCL-2 levels, led to better clinical status, and attenuated organ injury in endotoxemic mice. The anti-inflammatory effects of Δ9-THC in endotoxemic mice were reversed by a cannabinoid receptor type 1 (CB1R) inverse agonist (SR141716), and by clodronate-induced myeloid-cell depletion, but not by genetic invalidation or blockade of other putative Δ9-THC receptors, including cannabinoid receptor type 2, TRPV1, GPR18, GPR55, and GPR119. Although Δ9-THC administration reduced the activation of several spleen immune cell subsets, the anti-inflammatory effects of Δ9-THC were preserved in splenectomized endotoxemic mice. Finally, using IL-10–GFP reporter mice, we showed that blood monocytic myeloid-derived suppressive cells mediate the Δ9-THC–induced early rise in circulating IL-10. These results indicate that Δ9-THC potently induces IL-10, while reducing proinflammatory cytokines, chemokines, and related organ injury in endotoxemic mice via the activation of CB1R. These data have implications for acute and chronic conditions that are driven by dysregulated inflammation, such as sepsis, and raise the possibility that CB1R-signaling may constitute a novel target for inflammatory disorders.
Introduction
Since ancient times, people have made use of plants containing phytocannabinoids (1) for therapeutic, recreational, and religious purposes (2). In the United States, cannabis was widely used and referenced in the Pharmacopoeia from 1850 to the early 20th century. However, in 1937, federal restrictions were put in place that made it difficult to perform studies on cannabinoids in human health and disease (3). The endocannabinoid system includes cannabinoid receptors (CBRs) and vanilloid receptors. CBRs are a group of evolutionarily conserved G protein–coupled receptors (GPCRs) (4, 5) that are expressed by primitive vertebrates as well as more advanced species, including humans, and in numerous tissues and cellular subsets (6). CBRs recognize heterologous cannabinoids, including phytocannabinoids such as Δ(9)-tetrahydrocannabinol (Δ9-THC), and endocannabinoids such as anandamide (AEA) or 2-arachidonoylglycerol (2-AG). Cannabinoids are putative ligands for CBRs type 1 (CB1R) or 2 (CB2R) and additional GPCRs, including GPR18, GPR119, and GPR55 (7, 8), as well as ligand-gated ion-channels such as the transient receptor-potential vanilloid-1 (TRPV1) (9, 10). CBRs and vanilloid receptors are expressed in the nervous system, as well as in a variety of other tissues and cells, including dendritic cells, monocytes/macrophages, T cells and B cells (11, 12). This finding suggests that their function in the body may extend well beyond a role in neuronal function.
A number of immune cell subsets express CBRs, and cannabinoids have been reported to affect cytokine production in immune cells. However, the vast majority of these publications studied cells in vitro or ex vivo (13), and there is little information on the effects of cannabinoids in vivo in acute inflammation or on the regulation of the endocannabinoid system during inflammatory illnesses such as sepsis.
In the current studies, we demonstrate that Δ9-THC has strong and sustained anti-inflammatory properties in mice with acute inflammation and decipher its mechanisms of action. Our results show that in endotoxemic mice, Δ9-THC increases the secretion of the anti-inflammatory cytokine IL-10 by monocytic myeloid-derived suppressive cells (Mo-MDSCs) in a CB1R-dependent manner. Δ9-THC also dramatically reduces levels of proinflammatory mediators, immune cell activation, and potentially alleviates organ injury.
Materials and Methods
Mice
Experiments used 8- to 13-wk-old male and female mice. The following mouse strains were used: wild-type C57BL/6J (WT), B6.129 × 1-Trpv1tm1Jul/J (Trpv1−/−), B6.129P2-Cnr2tm1Dgen/J (Cnr2−/−), FVB.Cg-Tg(HIV-EGFP, Luc)8Tsb/J-NGL (NF-κB–GFP-luciferase reporter mouse with an enhancer-promoter construct containing two NF-κB binding sites driving a GFP-luciferase protein expression (14, 15) and B6(Cg)-Il10tm1.1Karp/J [IL-10 reporter mouse with IL-10 VertX: fluorescent (eGFP) fusion protein placed downstream exon 5 of the Il-10 gene (16)]. Mice were purchased from Jackson Laboratory and bred in the University of California, San Francisco (UCSF) animal facilities. Gpr18−/− were available at UCSF. For splenectomy, under isoflurane anesthesia, we performed left lateral laparotomy, exteriorized the spleen, and ligated its vascular pedicle twice before splenectomy. We waited 10 d post splenectomy before experimentation. The UCSF Institutional Animal Care and Use Committee approved all animal studies. Experiments were performed in accordance with the Public Health Service Policy on the Humane Care and Use of Laboratory Animals. Results are reported in concordance with the ARRIVE guidelines (17). For all studies, mice were given unrestricted access to food and water.
Endotoxemia models, clinical monitoring, and drugs
WT and genetically modified mice were injected i.v. with LPS (1 or 3 mg/kg in 5 μl/g, LPS–Escherichia coli O111:B4; Sigma-Aldrich) or the same volume of 0.9% saline (carrier for LPS). Mice were challenged with LPS or carrier, followed immediately by treatment with GPCR or TRPV1 agonists and antagonists (Δ9-THC, SR-141716, Extendin- 3 [3–39], JWH-133, arachidonoyl-2′-chloroethylamide [ACEA], arachidonoyl cyclopropylamide [ACPA], Hu-210, Win 55212-2, 2-AG, AEA, N-arachidonoyl dopamine [NADA], and CID 16020046 [Cayman Chemical]) i.v. in 4 μl/g. The σ-1R antagonist, BD1047-dihydrobromide (Tocris), was injected i.p. at 20 mg/kg. Drugs, initially in acetonitrile (ACN) (Δ9-THC) or EtOH (other chemicals), were prepared under sterile conditions, and solvents were eliminated by vacuum evaporator and diluted to end up with a solution containing 5% Tween 20 in PBS. The appropriate vehicle was used for each drug. To study the role of IL-10 in vivo, we used an IL-10R blocking Ab (Ultra-LEAF Purified Anti-Mouse CD210 Ab, 500 μg per mouse i.p.; BioLegend). Monocyte depletion was achieved by treating mice with clodronate liposomes (10 μl/g, i.v.) or neutral liposomes (Formumax) 20 h prior to experimentation. In each experiment, the following vital signs of mice were monitored at intervals: heart rate, pulse index, oxygen saturation (SaO2), and respiratory rate using the MouseOx Plus (STARR Life Sciences), and core temperatures (Thermalert TH-5; Physitemp Instruments) were recorded. We assessed illness severity by measurement of the murine sepsis score (MSS) (18) by a third party investigator that was blinded to the treatment conditions.
Histology
Tissue was collected at sacrifice, fixed with 4% paraformaldehyde, and sent to HistoWiz for embedding in serial 5-μm sections and H&E staining; then the tissue was anonymized for blind analysis. For lung sections, endothelial injury, epithelial injury, intra-alveolar hemorrhage, alveolar edema, and neutrophil infiltration were blindly scored using a semiquantitative scale ranging from 0 to 4. Neutrophil infiltration was blindly double-checked by an experienced pathologist. Lung condensation was automatically quantified with Image J software and score from 0 (aeration >55%) to 4 (aeration <20%).
Flow cytometry
Blood, bone marrow, and spleen were collected at sacrifice, and cells were labeled with several Ab mixes to explore innate and adaptive immune response in its different compartments, including the following: Pacific Blue–anti-Cd11b (M1/70.15), allophycocyanin (blood) or PECy7 (spleen)–anti-Ly6G (1A8), allophycocyanin–anti-CD19 (1D3), PercpCy5.5–anti-CD25 (PC61.5), PECy7–anti-Foxp3 (FJK-16s), FITC–anti-CD4 (GK1.5), AF700–anti-CD8 (53–6.7), PEcy5–anti-CD40 (1C10), PE–anti-CD69 (N418), and allophycocyanin–anti-GFAP (GA5) from InVitrogen; FITC–anti-CD3 (17A2), PE–anti-Ly6C (HK1.4), and allophycocyanin–anti–MHC II (M5/114.15.2) from BioLegend; BV605–anti-Nk1.1 (PK136), Amcyan–anti-B220 (RA3-6B2), and Percp–anti-CD45 (30F11) from BD Biosciences; and PE–anti-CD11c (HL3) and AF700–anti-F4/80 (BM8) from eBiosciences. For staining of circulating leukocytes, erythrocytes were lysed using BD Fluorescence-Activated Cell Sorter Lysing Solution (BD Biosciences). For intranuclear staining, surface staining was performed and then permeabilized using Foxp3 Staining Buffer Kit (eBiosciences) before intranuclear staining. Single-cell suspensions stained with fluorophore-conjugated Abs and cell count estimation were acquired the day of sacrifice using flow cytometry (LSRII Fortessa; BD Biosciences) and analyzed with FlowJo software (Miltenyi Biotec). Myeloid-derived suppressive cells were identified as follows: CD11b+Ly6G−Ly6Chigh for Mo-MDSCs and CD11b+Ly6G+ for granulocytic myeloid-derived suppressive cells. Nonclassical monocytes were gated as CD11b+Ly6G− Ly6Low. Regulatory T cells (Tregs) were considered as CD3+CD4+CD25highFoxp3+. B cells (CD19+ in or B220+), CD4+, CD8+, NKT (CD3+NK1.1+), and NK cell (NK1.1+ CD3−) lymphocyte subsets were also analyzed.
In vivo imaging of NF-κB activity
NF-κB–GFP-luciferase mice received i.p. injection of d-luciferin–potassium salt (Invitrogen) at 150 mg/kg in 5 μl/g 10 min prior to each luminescence acquisition with a Xenogen In vivo Imagining System Spectrum (PerkinElmer). At each time point, a group of five mice was simultaneously recorded including a “control” mouse that received saline but no LPS to normalize the emitted luminescence within each group.
Δ9-THC pharmacokinetic analysis
At t = 0 h, mice were injected i.v. with Δ9-THC, 5 mg/kg. Blood was collected at 1, 2, 5, 15, 30, and 60 min and then at 2, 6, and 24 h. Δ9-THC was quantified using selective, multiple-reaction monitoring liquid chromatography with tandem mass spectrometry (Cayman Chemicals). Briefly, aliquots of internal standard solution and blank 1:1 ACN/H2O (10 μl each) were added before gently mixing the tube contents. The diluted plasma was extracted with 60 μl of methyl tert-butyl ether (MtBE), thoroughly vortex mixed for 30 s, then phase separated by centrifuging for 30 s at 16,100 relative centrifugal force. The aqueous phases were frozen by placing the samples into −80°C freezer for∼2 min; then the organic phases were drawn off and placed in glass autosampler vials. The MtBE was dried under speed-vac centrifuge for 15 min, after which, 50 μl 1:1 can/H2O was added back to the vial to reconstitute the contents for injection onto the instrument (Waters Acquity I-Class UPLC with Xevo TQ-S micro tandem mass spectrometry).
Statistics
Data were analyzed using a two-tailed Mann–Whitney U test two-sided, and p values <0.05 were considered statistically significant. Statistics and graphical representations were performed using Prism 8.0 (Graph Pad Software). Results are reported as means (±SEM). Group sizes are indicated in the figure legends for each experiment. Experiments were repeated at least twice.
Results
Among cannabinoid agonists, Δ9-THC displays tremendous anti-inflammatory properties
We first tested the effect of a single dose of several endogenous and exogenous cannabinoids on inflammation induced early in the course of endotoxemia. WT mice were concurrently treated with a single dose of LPS (endotoxin, 1 mg/kg i.v.) and the following cannabinoids: ACEA, a synthetic highly selective CB1R agonist; JWH-133, a synthetic CB2R agonist; NADA, an endocannabinoid acting on CB1R and TRPV1; and the phyto-cannabinoid Δ9-THC. We observed that NADA and Δ9-THC potently increased IL-10 and decreased IL-6 in the plasmas of mice at 2 h after LPS challenge, and that Δ9-THC had more potent anti-inflammatory effects as compared with NADA (Fig. 1A). As the IL-6/IL-10 ratio has been reported as an integrative marker of the proinflammatory balance associated with poor outcome in critical illnesses (19, 20), Δ9-THC seemed to be a powerful anti-inflammatory agent. We tested the effects of escalating doses of Δ9-THC (0.1, 1 and 5 mg/kg i.v.) or vehicle alone and quantified plasma cytokines serially over 6 h. Administration of Δ9-THC 5 mg/kg i.v. significantly reduced plasma levels of IL-6 and CCL-2 at 2 and 6 h (p < 0.05) and led to profound upregulation of plasma IL-10 at 2 h in endotoxemic mice (Fig. 1B) (p < 0.05). Notably, plasma IL-6, CCL-2, and IL-10 were at baseline in mice treated with the CBR agonists alone, (data not shown). Our pharmacokinetic study in mice showed that Δ9-THC (5 mg/kg) has a half-life of 2 min in the plasma after i.v. injection (Fig. 1C). We also found that posttreatment with Δ9-THC 2 h after challenge with LPS-augmented plasma IL-10 at 4 and 6 h (Fig. 1D) (p = 0.007, p = 0.03). Similarly, repeated injections of Δ9-THC at t = 0, 2, and 4 h after challenge with LPS led to sustained high plasma levels of IL-10 (Fig. 1E) (p = 0.01). Δ9-THC had an anti-inflammatory effect in male and female endotoxemic mice (Supplemental Fig. 1). Notably, Δ9-THC had a more substantial impact on IL-10 upregulation in female as compared with male mice (Supplemental Fig. 1). We used in vivo imaging (Xenogen In vivo Imagining System) to assess NF-κB activation in NF-κB–GFP-luciferase–transgenic mice. We observed a reduction in total body NF-κB activation in endotoxemic mice treated with Δ9-THC (Fig. 1E) at 2 h (p < 0.01) and 6 h (p = 0.01).
FIGURE 1.

Among cannabinoid agonists, the endogenous-cannabinoid NADA and the phytocannabinoid Δ9-THC display anti-inflammatory features in endotoxemic mice. (A) Δ9-THC and NADA limit the proinflammatory response in LPS-treated mice. WT mice (8- to 10-wk-old male) were challenged with LPS (1 mg/kg, i.v.) and immediately thereafter were treated with a single dose of cannabinoid (CB) agonists (5 mg/kg), including ACEA, JW-133, NADA, and Δ9-THC (n = 3–5 per group). Plasma levels of IL-6 and IL-10 were measured at 2 h. The individual CB agonists alone, in the absence of LPS, did not affect baseline levels of the cytokines (data not shown). *p < 0.05, **p < 0.01, ***p < 0.001, LPS alone versus LPS + CB agonist. (B) Δ9-THC reduces circulating IL-6 and CCL2 and increases IL-10 levels in endotoxemic mice. WT mice (10-wk-old female, n = 5 per group) were challenged with LPS (1 mg/kg, i.v.) and immediately thereafter were treated with Δ9-THC (1 or 5 mg/kg, i.v.) or vehicle (i.v.). IL-10, IL-6, and CCL2 were quantified in plasma at 2, 4, and 6 h. Baseline plasma levels of the cytokines were measured at t = 0 in three untreated mice matched for sex and age without any stimulation. *p < 0.05, LPS-treated mice in the 5 mg/kg Δ9-THC group versus vehicle at each time point. (C) Pharmacokinetic study of i.v. Δ9-THC. Plasma levels of Δ9-THC were quantified in WT mice (9-wk-old male) at 1, 2, 5, 10, 15, 30, 60, and 120 min, 6 and 24 h after i.v. administration of 5 mg/kg of Δ9-THC (n = 4 mice per time point). Plasmas underwent an MtBE-based liquid-liquid extraction method followed by liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis. The concentrations shown are the ratios of integrated areas of the chromatographic peaks (ion count versus time) corresponding to Δ9-THC to the integrated areas of designated internal standards. (D) Delayed administration of Δ9-THC after LPS injection still provokes the rise of IL-10. WT mice (7- to 8-wk-old female) were challenged with LPS (1 mg/kg, i.v.) at t = 0 (red arrow) and then were treated Δ9-THC (5 mg/kg, i.v., n = 5) or vehicle (n = 5) at t = 2 h (blue arrow). IL-10 was quantified in plasma at t = 2, 4, and 6 h. *p < 0.05, **p < 0.01, Δ9-THC versus carrier treatment. (E) Repeated doses of Δ9-THC maintain higher IL-10 plasma levels. WT mice were challenged with LPS (1 mg/kg, i.v.) at t = 0 (red arrow) and then treated with Δ9-THC (5 mg/kg, i.v.) or vehicle (i.v.) single dose at t = 2 h, or multiple doses at t = 0 (blue arrow), and again at 2 and 4 h (purple arrows). Plasma IL-10 was quantified at t = 2, 4, and 6 h (10-wk-old male, n = 5 + 5 + 4 per group). *p < 0.05 comparing LPS-treated mice + 5 mg/kg Δ9-THC single-dose versus vehicle, #p < 0.05 comparing LPS-treated mice + 5 mg/kg Δ9-THC single-dose versus multiple-dose. (F) Δ9-THC reduces LPS-induced NF-κB activity in vivo in early endotoxemia. Luminescence measurements in 8- to 12-wk-old NF-κB reporter mice at t = 2, 4, 6, and 24 h after challenge with i.v. saline or LPS (1 mg/kg) followed immediately by i.v. administration of vehicle or Δ9-THC (5 mg/kg). The plots in the left panel represent bioluminescence signal quantification of mice cohort under each treatment condition (n = 9 + 20 + 22 per group, respectively saline, LPS + vehicle, and LPS + Δ9-THC). Mean values are represented as relative values of photon flux per second normalized to the saline group value for each group of five mice simultaneously acquired. Error bars indicate SEM. *p < 0.05, ***p < 0.001 comparing LPS-treated mice vehicle versus Δ9-THC. The right panel shows representative images of the bioluminescence signal observed at t = 2 h after challenge with saline, LPS, or LPS + Δ9-THC. Mice are FVB.Cg-Tg(HIV-EGFP, Luc)8Tsb/J-NGL (NF-κB binding sites driving a GFP-luciferase protein expression).
Δ9-THC administration reduces the severity of endotoxemic shock
Next, we investigated the effect of Δ9-THC in sublethal endotoxemic shock (LPS 3 mg/kg i.v.). At this dosage, mice develop signs of critical illness that peaks in severity at∼12 h, after which they gradually recover. We observed that Δ9-THC improved the clinical status of endotoxemic mice at 12 (p = 0.01) and 26 h (p < 0.01), as assessed by measuring the “MSS” (18) (Fig. 2A1–3). We observed that Δ9-THC treatment upregulated IL-10 at 2 h, but not at 12 or 26 h (p < 0.001), and reduced IL-6 and CCL-2 levels at 2 h (p < 0.01, p = 0.05) and 26 h (Fig. 2B) (p = 0.02, p = 0.05). These anti-inflammatory effects in endotoxemic mice that received Δ9-THC were accompanied by fewer indices of organ injury, less severe lung injury histologically (Fig. 2C) (p < 0.01), and reduced lung mRNA for inflammatory markers such as LCN2 and IL-6 (Supplemental Fig. 2A1). Liver histology did not show any obvious pathology, but liver expression of mRNAs for iNOS and IL-6 by quantitative PCR was reduced in Δ9-THC–treated mice (Supplemental Fig. 2B1–3) (p = 0.01). Levels of aspartate aminotransferase, urea, hemoglobin, and platelet counts were similar in both groups (Supplemental Fig. 2B2, 2C).
FIGURE 2.

Early administration of Δ9-THC improves MSS, decreases proinflammatory cytokines, and reduces organ injury late in the course of endotoxemia. WT mice were challenged with LPS (3 mg/kg, i.v.) and immediately thereafter were treated with Δ9-THC (5 mg/kg, i.v.) or vehicle (12-wk-old male, n = 10, 10 + 10, 10 + 10, 10 + 8 per group and per time point). (A) Δ9-THC reduces the severity of endotoxemic shock at t = 12 and 26 h after challenge with LPS. (A1) Shows MSSs longitudinally over 26 h; (A2) and (A3) show diagram representations of average score for each items in the mouse sepsis score (from 0 [healthy] to 4 [very sick]). (B) Plasma IL-10 levels were significantly higher only at t = 2 h, whereas IL-6 and CCL2 were significantly lower at t = 2 and 26 h. *p < 0.05, **p < 0.01, ****p < 0.0001, endotoxemic mice treated with vehicle versus Δ9-THC, two-tailed Mann–Whitney U test at each time point. (C) Representative H&E-stained sections of lungs (original magnification ×20) collected from mice 24 h after challenge with saline, LPS + vehicle, or LPS + Δ9-THC. Lung injury was assessed by analysis of epithelial and endothelial injury, alveolar hemorrhage, alveolar edema, PMN infiltration, and condensation. **p < 0.01, ***p < 0.001.
Δ9-THC modulates expression of activation markers by spleen leukocyte subsets
We used flow cytometry to assess for activation of immune cells in different compartments (blood, bone marrow, spleen) at 2, 12, and 26 h in endotoxemic mice treated with Δ9-THC. We did not observe any significant divergence in the blood or bone marrow leukocytes subset in endotoxemic mice receiving Δ9-THC versus vehicle (Supplemental Fig. 3A, 3B). Nonetheless, the spleens of mice that received Δ9-THC maintained a sustained reduction of activation markers expressed on several innate and adaptive leukocyte subsets. In particular, splenic neutrophils expressed less MHC class II and CD40 at 12 and 26 h post-LPS in Δ9-THC–treated mice (Supplemental Fig. 4A). Moreover, Δ9-THC treatment reduced activation of splenic B cells and CD4+ T cells (CD25 and CD69 expression, Supplemental Fig. 4B, 4C). Notably, no changes were observed in Tregs (Foxp3+CD25+CD4) or dendritic cells in the spleen.
CB1R mediates Δ9-THC–induced IL-10 upregulation during TLR4 acute inflammation independently of σ-1R signaling
In addition to CB1R and CB2R, Δ9-THC is a putative ligand of other GPCRs (i.e., GPR18, GPR55, and GPR119) and the vanilloid receptor, TRPV1. We used pharmacologic antagonist and mice with genetic deficiencies of cannabinoid receptors to identify the receptors involved in Δ9-THC/TLR4–induced early upregulation of IL-10. Gene deficiency of functional Cnr2−/−, Trpv1−/−, or Gpr18−/− did not reverse the Δ9-THC–induced upregulation during endotoxemia (Fig. 3A). We also treated endotoxemic mice with Δ9-THC in the presence and absence of antagonists for CB1R, GPR55, and GPR119. Treatment with a GPR55 antagonist (CID-16020046) or with a GPR119 antagonist (Extendin-3 [3–39]) did not reverse the Δ9-THC–induced upregulation of IL-10 (Fig. 3B). In contrast, the CB1R inverse agonist/antagonist SR141716 (Rimonabant) almost entirely counteracted the Δ9-THC–induced IL-10 increase in LPS-treated mice (Fig. 3B, 3C). Simultaneous administration of SR141716 with Δ9-THC also abrogated the reduction in NF-κB activation in early hours following LPS injection when mice received Δ9-THC (data not shown). These data suggest that Δ9-THC augments TLR4-dependent upregulation of IL-10 via CB1R.
FIGURE 3.

CB1R mediates the TLR4/Δ9-THC–induced upregulation of IL-10 (A) TRPV1, CB2R, and GRPR18 are not responsible for Δ9-THC–induced IL-10 upregulation in LPS-induced inflammation. n = 7 + 8 + 11 WT male mice, 7+7 Trpv1−/− male mice, 4 + 8 Cnr2−/− male mice, and 2 + 3 male and 2 + 3 female mice Gpr18−/− (all 8–14 wk old). (B) Pharmacological blockade of CB1R by SR141617 abrogates Δ9-THC–induced IL-10 upregulation in LPS-induced inflammation, whereas GPR119 and GPR55 blockade have no effect (8- to 11-wk-old WT male mice, n = 2 + 3 + 8+ 7 + 3 + 4 per group). **p < 0.01, ***p < 0.001, LPS + Δ9-THC–treated mice versus LPS + vehicle or LPS + Δ9-THC + SR141716. (C) CB1 antagonist SR141716 abrogates Δ9-THC–induced anti-inflammatory response in LPS-treated mice. Levels of IL-10 were quantified in plasma at t = 2, 4, and 6 h after LPS challenge (10-wk-old male, n = 5 per group). *p < 0.05, **p < 0.01, LPS + vehicle versus LPS + Δ9-THC and LPS + vehicle versus LPS + Δ9-THC + SR141617, two-tailed Mann–Whitney U test at each time point. (D) Among CB1R agonists only Win5522-2, Hu-210, and NADA also upregulate IL-10 at t = 2 h after LPS challenge (9-wk-old male, n = 40). **p < 0.01, ***p < 0.001, LPS + vehicle versus LPS + CB1R agonist. (E) σ1R blockade with BD1047 does not affect Δ9-THC–induced reduction in proinflammatory cytokines and IL-10 upregulation. Mice were pretreated with BD1047 IP 20 mg/,g 30 min before LPS (1 mg/kg i.v.) ± Δ9-THC (5 mg/kg i.v.). Levels of cytokines were quantified in plasma after at t = 2 h (8-wk-old female, n = 1 + 5 + 5 + 5 per group). *p < 0.05, **p < 0.01, ***p < 0.001, LPS + vehicle versus LPS + Δ9-THC or LPS + Δ9-THC + BD1047.
To corroborate a possible role for CB1R in Δ9-THC–induced upregulation of IL-10, we tested the effects of other CB1R agonists in endotoxemic mice. Similar to Δ9-THC, Hu-210, Win 55,212-2, and NADA (all at 5 mg/kg) upregulated IL-10 plasma levels in endotoxemic mice (Fig. 3D). Conversely, AEA, 2-arachidonoyl glycerol (2A-G), ACPA, and ACEA did not augment LPS-induced IL-10 production at the dose of 5 mg/kg. Interactions have been visualized between CB1R and σ-1R in the CNS (21, 22), and σ-1R–specific agonists have been reported to enhance LPS-induced systemic release of IL-10 while simultaneously inhibiting secretion of proinflammatory cytokine (23). We, therefore, tested the hypothesis that Δ9-THC upregulates IL-10 in endotoxemia via σ-1R. We observed that treatment with a σ-1R pathway inhibitor (BD1047) had no effect on Δ9-THC–induced upregulation of IL-10 in endotoxemic WT mice, indicating that the anti-inflammatory effects of Δ9-THC during endotoxemia are independent of σ-1R signaling (Fig. 3E).
Mo-MDSCs from the bone marrow are responsible for Δ9-THC- induced IL-10 upregulation during endotoxemia
To investigate the cells responsible for the initial boost of IL-10 production induced by Δ9-THC, we treated IL-10–GFP reporter [B6(Cg)-Il10tm1.1Karp/J] mice with LPS (1 mg/kg, i.v.) and Δ9-THC or the vehicle alone. We performed flow cytometry to assess IL-10 expression by different lineages of cells from multiple compartments (blood, bone marrow, spleen, liver, brain). We identified the blood Mo-MDSCs (CD11b+Ly6G-Ly6Chi) (24) as the cell subset responsible for the Δ9-THC–induced IL-10 upregulation in endotoxemic mice. LPS challenge by itself provokes the emergence of an IL-10+ subset among CD11b+Ly6G-Ly6Chi cells. But in mice receiving Δ9-THC treatment, we observed an increase in IL-10 expression by Mo-MDSCs at 1 and 2 h (Fig. 4A, 4B) (p = 0.004, p = 0.01). The upregulation of IL-10 expression was not present in other cell subsets from blood, bone marrow, or in spleen leukocytes, including splenic Tregs, B regulatory cells (strictly B-10 spleen B lymphocytes) or conventional dendritic cells (Supplemental Fig. 5A–C). Similarly, IL-10 was not upregulated in liver Kupffer cells (CD45+, CD11b+, F4/80+) or brain astrocytes (CD45−, GFAP+), both of which express CB1R and potentially produce IL-10 (Supplemental Fig. 5D, 5E).
FIGURE 4.

Mo-MDSCs are responsible for Δ9-THC–induced IL-10 upregulation in acute inflammation. Flow cytometry analysis of blood leukocytes shows that Mo-MDSCs (CD11b + LY6Chi monocytes) display increased IL-10 expression after LPS challenge (1 mg/kg i.v.) in IL-10–GFP reporter [B6(Cg)-Il10tm1.1Karp/J] mice. Simultaneous Δ9-THC (5 mg/kg i.v.) treatment significantly increases mean fluorescence intensity and percentage of IL-10 + Mo-MDSCs (12-wk-old male, n = 4 + 5 + 5). *p < 0.05, **p < 0.01, LPS + vehicle versus LPS + Δ9-THC, two-tailed Mann–Whitney U test (A1, A2, and B).
We corroborated these data suggesting that Mo-MDSCs are the source of IL-10 in endotoxemic mice treated with Δ9-THC by depleting monocytes using clodronate 20 h prior to inducing endotoxemia (Fig. 5A). Δ9-THC did not augment serum IL-10 levels in endotoxemic mice that had undergone clodronate depletion of their monocytes (Fig. 5B). Because clodronate depletes bone marrow–derived monocytes as well as resident macrophages, at this point, we could not rule out a role for spleen monocytes/macrophages in this result (Supplemental Fig. 6). We, therefore, investigated the anti-inflammatory effect of Δ9-THC in splenectomized mice and observed unequivocally that the absence of spleen did not abrogate the anti-inflammatory effects of Δ9-THC (Fig. 5C).
FIGURE 5.

Clodronate-mediated depletion of phagocytic myeloid cells, but not splenectomy abolishes TLR-4/Δ9-THC–induced IL-10 upregulation. (A) Pretreatment of mice with clodronate liposomes 20 h prior to LPS challenge achieves a 90% depletion in blood Mo-MDSCs. (B) Clodronate pretreatment fully abolish the Δ9-THC–induced IL-10 upregulation and partially reverse the Δ9-THC–induced reduction of IL-6 and CCL-2 at t = 2 h after LPS challenge (12-wk-old male, n = 4 + 4 + 5 + 5 + 5 + 5). (C) Splenectomy does not abrogate the anti-inflammatory effect of Δ9-THC. These results suggest that Mo-MDSCs producing IL-10 in response to LPS/Δ9-THC cosimulation are derived from the bone marrow rather than the spleen. Plasma cytokines were quantified in plasma at t = 2 h after LPS challenge (1 mg/kg i.v.) ± Δ9-THC (5 mg/kg i.v.) (9-wk-old female, n = 3 + 5 + 6 sham, 1 + 5 + 6 splenectomy). *p < 0.05, **p < 0.01, ***p < 0.001.
The anti-inflammatory effects of Δ9-THC in endotoxemia are not exclusively dependent on IL-10
We tested the hypothesis that IL-10 mediates the attenuated proinflammatory response induced by Δ9-THC in endotoxemic mice, using IL-10R Ab to block IL-10 signaling. We found that in endotoxemic WT mice that did not receive Δ9-THC, IL-10R Ab led to worse clinical status, vital parameters, and lung injury, and higher IL-10 (p < 0.001), IL-6 (p = 0.001), and CCL-2 (p = 0.001) levels at least during the first 12 h (Supplemental Fig. 7). Nevertheless, in IL-10R Ab–treated endotoxemic mice, Δ9-THC still augments IL-10 (p = 0.01) and reduces IL-6 (p = 0.02) and CCL-2 levels (p < 0.001) (Fig. 6). This result suggests that Δ9-THC reduces IL-6 and CCL-2 expression via IL-10 independent mechanisms.
FIGURE 6.

IL-10 upregulation is not the exclusive mechanism of Δ9-THC anti-inflammatory effect in acute inflammation IL-10 signaling blocking does not fully abrogate anti-inflammatory effects of Δ9-THC (12-wk-old male, n = 2 + 8 + 10 + 10 + 5). *p < 0.05, ***p < 0.001, LPS + Δ9-THC + isotype versus LPS + vehicle + isotype. ##p < 0.01, ###p < 0.001, LPS + vehicle + isotype versus LPS + isotype + IL-10R Ab. $p < 0.05, $$p < 0.01, $$$p < 0.001, LPS + Δ9-THC + isotype versus LPS + Δ9-THC + IL-10R Ab. Two-tailed Mann–Whitney U test at each time point.
Non-immunoinflammatory side effects of Δ9-THC
We observed several noteworthy effects of Δ9-THC treatment on the behavior and physiology of mice at baseline and during endotoxemic shock. Mice receiving Δ9-THC displayed substantial but transient neurologic alteration. Immediately after injection, they were slow to awaken from anesthesia, appeared “lethargic” or sleepy, and moved and breathed slowly. In response to a stimulus, and occasionally without apparent provocation, they became hyperactive, with chaotic jumps and agitation. This acute phase lasted for∼45 min and was followed by a period of slow motion lasting for∼2 h. Hu-210 and Win5522-2 provoked axial rigidity, hypertonia, and coma. These neurologic effects were not elicited by treatment with other CB1R agonists, including ACPA, ACEA, AEA, or 2A-G.
Δ9-THC also had substantial effects on the mouse physiology in endotoxemic mice. It aggravated LPS-induced hypothermia during the first 6 h (Supplemental Fig. 8A) (p < 0.001) (25) and reduced respiratory rate and heart rate during the first 2 h (Supplemental Fig. 8B, 8C). Notably, the pulse index was higher in mice that received LPS and Δ9-THC compared with mice that received LPS and the vehicle at 6 and 24 h (Supplemental Fig. 8C) (p < 0.001), suggesting better peripheral perfusion. Pharmacological blockade of CB1R by SR141716 partially reversed those hemodynamics effects (Supplemental Fig. 8D).
Discussion
The current study revealed several novel findings that to our knowledge have not previously been reported. First, we observed in vivo direct anti-inflammatory and immunosuppressive effects of Δ9-THC in acute systemic inflammation associated with reduced organ injury and improved clinical parameters. Second, we found that Δ9-THC strongly induces early systemic production of IL-10 by bone marrow–derived Mo-MDSCs. Third, we demonstrated that CB1R is responsible for the anti-inflammatory effects of Δ9-THC in endotoxemic mice. Despite millennia of humans using cannabis-based products, the understanding of the role of the endocannabinoid system in health and disease is still in its nascence, and there is a dearth of scientific information on the immune effects of cannabinoids in vivo. Available data suggest that, in vitro exposure to LPS modulates the endocannabinoid system via the production of 2-AG and AEA and affects CBRs expression levels (26–28). Studies suggest that CB2R, which is predominantly expressed by leukocytes, has a protective role in endotoxemia and sepsis (29, 30). The strong anti-inflammatory effects of Δ9-THC in the current studies, in conjunction with our prior work on the endocannabinoid, NADA (31), point to a role for the endocannabinoid and vanilloid systems in regulating immune homeostasis. This raises the possibility that the cannabinoids could potentially be beneficial in a variety of acute conditions that are driven by dysregulated inflammation, such as sepsis and ischemia-reperfusion injury. Moreover, they could be beneficial in nonacute conditions in which IL-10 deficiency is believed to play a central role, such as inflammatory bowel disease (32, 33), atherosclerosis (34, 35), multiple sclerosis (36), and neuropathic pain (37).
IL-10 is an essential homeostatic mechanism that controls the intensity and duration of inflammation by inhibiting production of proinflammatory cytokines in monocytes, macrophages, neutrophils, and NK cells and reducing T cell activation in septic and nonseptic critical illness in mice (38) as well as human (39). In this regard, IL-10 gene enhancer/promoter activation in monocytic cells is mostly mediated by cAMP-dependent signaling pathways, including the CREB/activating transcription factor interacting with two cAMP response element motifs (40). CB1R activation by several agonists can also lead to cAMP accumulation via G protein αs activation or reduced activation of G protein αi/o (41). Other signaling pathways could be involved in the anti-inflammatory effects observed. For instance, CB1R activation has been reported to increase extracellular adenosine levels (42). Adenosine promotes resolution of inflammation in LPS-induced lung injury (43), which raises the possibility that adenosine signaling may also contribute to the anti-inflammatory effects of CB1R agonists. Although IL-10 is believed to be a principal mediator of the anti-inflammatory response, experimental studies have demonstrated that only 20% of the genes activated by LPS in macrophages are eventually repressed by IL-10 (44). We, in this study, observed that the anti-inflammatory effect of Δ9-THC is not exclusively mediated via IL-10 but also directly via a reduction in NF-κB activation.
Although the potent anti-inflammatory properties of Δ9-THC may be beneficial in certain inflammatory conditions, there could be detrimental effects, including secondary immune dysfunction and impaired ability to clear infection. Indeed, IL-10 has been reported to impair antimicrobial defenses and activation of adaptive immune cells and to increase susceptibility to secondary infections (45–48). Consistent with this hypothesis, in the present studies, we observed that treatment of endotoxemic mice with Δ9-THC reduced the expression of CD25 and CD69 on CD4+ T and B lymphocytes, and MHC class II and CD40 on neutrophils. Further studies are needed to evaluate the effects of CB1R agonists on bacterial clearance and development of secondary infections. CB1R agonists have also been reported to decrease temperature (25, 49), which could potentially be harmful and exacerbate immune dysfunction in sepsis (50, 51). Conversely, Δ9-THC has been reported to decrease oxygen consumption (52, 53), suggesting that it might be protective in conditions in which there is low oxygen delivery such as septic shock and ischemia-reperfusion injury. We also observed that Δ9-THC reduces LPS-induced tachycardia in the first hours following injection. This is consistent with a report that the CB1R agonist WIN55212-2 induces sympathetic inhibition leading to bradycardia and inhibition of norepinephrine release (54). Recent studies suggest that reducing sympathetic activity may be beneficial in septic shock and point to immune modulatory roles of the sympathetic nervous system (55, 56). Thus, Δ9-THC may also indirectly influence immune function via the sympathetic nervous system.
We identified CB1R as the receptor responsible for the Δ9-THC–induced anti-inflammatory effects. Notably, Δ9-THC and some synthetic CB1R agonists are psychodysleptic molecules, raising concerns for their suitability for clinical application. However, CB1R signaling is complex and multifaceted, and CB1R engagement can activate multiple downstream signaling pathways depending on the ligand (57–60). Should that the anti-inflammatory and neurologic effects be mediated through different pathways, the concept of “biased-agonism” could be used to develop ligands that specifically act on inflammation but not consciousness (61, 62).
Our study also brings new data to the field about the cellular source and the regulation of the “cytokine storm” that occurs in acute critical illness, which to our knowledge have not previously been reported. First, we demonstrated that a very early increase in IL-10 with a peak at 2 h, precedes the peak of the proinflammatory mediators IL-6 or CCL-2. We speculate that this early peak in IL-10 may be involved in limiting the magnitude of the proinflammatory responses, and in initiating inflammatory resolution. Moreover, we demonstrated that IL-10–signaling blockade in LPS-treated mice provokes a continuous increase in proinflammatory mediators at least up to 24 h and worsens functional outcomes of endotoxemia. It suggests that IL-10 is an absolutely essential mediator of the resolution of inflammation, and that other anti-inflammatory mediators such as IL-4, IL-13, or TGF-β may be inoperative in the absence of IL-10. Our data in mice that underwent clodronate depletion of myeloid cells and in IL-10–GFP-reporter mice suggest that the Mo-MDSCs represent the predominant cell in Δ9-THC–induced upregulation of IL-10 during endotoxemia. However, previous reports have demonstrated a role for specific CB1R ligands (Cannabidiol) and CB2R agonist (O-1966) on expansion of Tregs (63, 64) and 2A-G on NK cells activation (65), suggesting that these other cell subsets could also be involved in the anti-inflammatory effects of Δ9-THC. Additionally, we observed, in our myeloid-cell depletion experiments, that in the absence of monocytes/macrophages, IL-6 and CCL-2 levels [two critical mediators and prognostic markers in sepsis (66, 67)] were not significantly reduced during the first hours of endotoxemia. This suggests that cells other than monocytes/macrophages contribute to the production of these cytokines such as endothelial cells, which have been reported to produce substantial amounts of IL-6 and CCL-2 in response to LPS (68).
To date, there is insufficient evidence to solidly support or refute the therapeutic value of cannabis-derived products in acute inflammation (69–71). Our studies demonstrate that Δ9-THC has substantial immunosuppressive and anti-inflammatory properties in TLR4-dependent acute inflammation that are mediated through CB1R. The profound early upregulation in IL-10 by Δ9-THC in endotoxemia has not previously been described, and its relevance in sepsis remains to be determined. These results highlight important interplay between the endocannabinoid system and the immune systems. They suggest that the endocannabinoid system may represent a novel therapeutic target for a number of inflammatory disorders and raise the possibility of using cannabinoids to promote inflammatory resolution and reduce organ injury in sepsis and injury.
Supplementary Material
Acknowledgments
We thank Dr. Jay Levy (UCSF Cancer Research Institute) for critical review of the manuscript and valuable comments, Dr. Jason Cyster (UCSF Department of Microbiology and Immunology) for providing the Gpr18−/− mice, and Dr. Manohar Sharma (UCSF Department of Anesthesia and Perioperative Care) for checking the purity of Δ9-THC by HPLC.
This work was supported by National Institutes of Health (NIH) National Institute of General Medical Sciences Grant R01GM132379 (to J.H.), the University of California, San Francisco (UCSF) Department of Anesthesia and Perioperative Care (to J.H.), a fellowship from the Société de Réanimation de Langue Française (to J.J.), and the Amicale des Anciens Internes en Medicine des Hopitaux de Paris and Assistance Publique, Hopitaux de Paris (to J.J.). The UCSF Flow Cytometry Facility acknowledges Diabetes Research Center Grant NIH P30 DK063720.
The online version of this article contains supplemental material.
- ACEA
- arachidonoyl-2′-chloroethylamide
- ACN
- acetonitrile
- ACPA
- arachidonoyl cyclopropylamide
- AEA
- anandamide
- 2A-G
- 2-arachidonoyl glycerol
- CBR
- cannabinoid receptor
- CB1R
- CBR type 1
- CB2R
- CBR type 2
- GPCR
- G protein–coupled receptor
- Mo-MDSC
- monocytic myeloid-derived suppressive cell
- MSS
- murine sepsis score
- MtBE
- methyl tert-butyl ether
- NADA
- N-arachidonoyl dopamine
- Δ9-THC
- Δ(9)-tetrahydrocannabinol
- Treg
- regulatory T cell
- TRVP1
- transient receptor-potential vanilloid-1
- UCSF
- University of California, San Francisco
- WT
- wild-type C57BL/6J.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Bonini S. A., Premoli M., Tambaro S., Kumar A., Maccarinelli G., Memo M., Mastinu A. 2018. Cannabis sativa: a comprehensive ethnopharmacological review of a medicinal plant with a long history. J. Ethnopharmacol. 227: 300–315. [DOI] [PubMed] [Google Scholar]
- 2.Zias J., Stark H., Sellgman J., Levy R., Werker E., Breuer A., Mechoulam R. 1993. Early medical use of cannabis. Nature 363: 215. [DOI] [PubMed] [Google Scholar]
- 3.Bridgeman M. B., Abazia D. T. 2017. Medicinal cannabis: history, pharmacology, and implications for the acute care setting. P&T 42: 180–188. [PMC free article] [PubMed] [Google Scholar]
- 4.Pertwee R. G. 2006. Cannabinoid pharmacology: the first 66 years. Br. J. Pharmacol. 147(Suppl. 1): S163–S171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pertwee R. G. 2006. The pharmacology of cannabinoid receptors and their ligands: an overview. Int. J. Obes. 30(Suppl. 1): S13–S18. [DOI] [PubMed] [Google Scholar]
- 6.Galiègue S., Mary S., Marchand J., Dussossoy D., Carrière D., Carayon P., Bouaboula M., Shire D., Le Fur G., Casellas P. 1995. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur. J. Biochem. 232: 54–61. [DOI] [PubMed] [Google Scholar]
- 7.Morales P., Reggio P. H. 2017. An update on non-CB1, non-CB2 cannabinoid related G-protein-coupled receptors. Cannabis Cannabinoid Res. 2: 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pertwee R. G., Howlett A. C., Abood M. E., Alexander S. P., Di Marzo V., Elphick M. R., Greasley P. J., Hansen H. S., Kunos G., Mackie K., et al. 2010. International union of basic and clinical pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol. Rev. 62: 588–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janero D. R., Makriyannis A. 2014. Terpenes and lipids of the endocannabinoid and transient-receptor-potential-channel biosignaling systems. ACS Chem. Neurosci. 5: 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown A. J. 2007. Novel cannabinoid receptors. Br. J. Pharmacol. 152: 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanasescu R., Constantinescu C. S. 2010. Cannabinoids and the immune system: an overview. Immunobiology 215: 588–597. [DOI] [PubMed] [Google Scholar]
- 12.Berdyshev E. V. 2000. Cannabinoid receptors and the regulation of immune response. Chem. Phys. Lipids 108: 169–190. [DOI] [PubMed] [Google Scholar]
- 13.Croxford J. L., Yamamura T. 2005. Cannabinoids and the immune system: potential for the treatment of inflammatory diseases? J. Neuroimmunol. 166: 3–18. [DOI] [PubMed] [Google Scholar]
- 14.Carlsen H., Alexander G., Austenaa L. M., Ebihara K., Blomhoff R. 2004. Molecular imaging of the transcription factor NF-kappaB, a primary regulator of stress response. Mutat. Res. 551: 199–211. [DOI] [PubMed] [Google Scholar]
- 15.Everhart M. B., Han W., Sherrill T. P., Arutiunov M., Polosukhin V. V., Burke J. R., Sadikot R. T., Christman J. W., Yull F. E., Blackwell T. S. 2006. Duration and intensity of NF-kappaB activity determine the severity of endotoxin-induced acute lung injury. J. Immunol. 176: 4995–5005. [DOI] [PubMed] [Google Scholar]
- 16.Madan R., Demircik F., Surianarayanan S., Allen J. L., Divanovic S., Trompette A., Yogev N., Gu Y., Khodoun M., Hildeman D., et al. 2009. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J. Immunol. 183: 2312–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kilkenny C., Browne W., Cuthill I. C., Emerson M., Altman D. G., NC3Rs Reporting Guidelines Working Group 2010. Animal research: reporting in vivo experiments: the ARRIVE guidelines. J. Gene Med. 12: 561–563. [DOI] [PubMed] [Google Scholar]
- 18.Shrum B., Anantha R. V., Xu S. X., Donnelly M., Haeryfar S. M., McCormick J. K., Mele T. 2014. A robust scoring system to evaluate sepsis severity in an animal model. BMC Res. Notes 7: 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taniguchi T., Koido Y., Aiboshi J., Yamashita T., Suzaki S., Kurokawa A. 1999. Change in the ratio of interleukin-6 to interleukin-10 predicts a poor outcome in patients with systemic inflammatory response syndrome. Crit. Care Med. 27: 1262–1264. [DOI] [PubMed] [Google Scholar]
- 20.de Brito R. C., Lucena-Silva N., Torres L. C., Luna C. F., Correia J. B., da Silva G. A. 2016. The balance between the serum levels of IL-6 and IL-10 cytokines discriminates mild and severe acute pneumonia. BMC Pulm. Med. 16: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sánchez-Blázquez P., Rodríguez-Muñoz M., Herrero-Labrador R., Burgueño J., Zamanillo D., Garzón J. 2014. The calcium-sensitive Sigma-1 receptor prevents cannabinoids from provoking glutamate NMDA receptor hypofunction: implications in antinociception and psychotic diseases. Int. J. Neuropsychopharmacol. 17: 1943–1955. [DOI] [PubMed] [Google Scholar]
- 22.Su T. P., Su T. C., Nakamura Y., Tsai S. Y. 2016. The sigma-1 receptor as a pluripotent modulator in living systems. Trends Pharmacol. Sci. 37: 262–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourrié B., Bribes E., De Nys N., Esclangon M., Garcia L., Galiègue S., Lair P., Paul R., Thomas C., Vernières J. C., Casellas P. 2002. SSR125329A, a high affinity sigma receptor ligand with potent anti-inflammatory properties. Eur. J. Pharmacol. 456: 123–131. [DOI] [PubMed] [Google Scholar]
- 24.Lai D., Qin C., Shu Q. 2014. Myeloid-derived suppressor cells in sepsis. BioMed Res. Int. 2014: 598654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smirnov M. S., Kiyatkin E. A. 2008. Behavioral and temperature effects of delta 9-tetrahydrocannabinol in human-relevant doses in rats. Brain Res. 1228: 145–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varga K., Wagner J. A., Bridgen D. T., Kunos G. 1998. Platelet- and macrophage-derived endogenous cannabinoids are involved in endotoxin-induced hypotension. FASEB J. 12: 1035–1044. [DOI] [PubMed] [Google Scholar]
- 27.Liu J., Batkai S., Pacher P., Harvey-White J., Wagner J. A., Cravatt B. F., Gao B., Kunos G. 2003. Lipopolysaccharide induces anandamide synthesis in macrophages via CD14/MAPK/phosphoinositide 3-kinase/NF-kappaB independently of platelet-activating factor. J. Biol. Chem. 278: 45034–45039. [DOI] [PubMed] [Google Scholar]
- 28.Pestonjamasp V. K., Burstein S. H. 1998. Anandamide synthesis is induced by arachidonate mobilizing agonists in cells of the immune system. Biochim. Biophys. Acta 1394: 249–260. [DOI] [PubMed] [Google Scholar]
- 29.Gui H., Sun Y., Luo Z. M., Su D. F., Dai S. M., Liu X. 2013. Cannabinoid receptor 2 protects against acute experimental sepsis in mice. Mediators Inflamm. 2013: 741303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tschöp J., Kasten K. R., Nogueiras R., Goetzman H. S., Cave C. M., England L. G., Dattilo J., Lentsch A. B., Tschöp M. H., Caldwell C. C. 2009. The cannabinoid receptor 2 is critical for the host response to sepsis. J. Immunol. 183: 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawton S. K., Xu F., Tran A., Wong E., Prakash A., Schumacher M., Hellman J., Wilhelmsen K. 2017. N-arachidonoyl dopamine modulates acute systemic inflammation via nonhematopoietic TRPV1. J. Immunol. 199: 1465–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kühn R., Löhler J., Rennick D., Rajewsky K., Müller W. 1993. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75: 263–274. [DOI] [PubMed] [Google Scholar]
- 33.Schreiber S., Fedorak R. N., Nielsen O. H., Wild G., Williams C. N., Nikolaus S., Jacyna M., Lashner B. A., Gangl A., Rutgeerts P., et al. 2000. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn’s disease. Crohn’s Disease IL-10 Cooperative Study Group. Gastroenterology 119: 1461–1472. [DOI] [PubMed] [Google Scholar]
- 34.Mallat Z., Besnard S., Duriez M., Deleuze V., Emmanuel F., Bureau M. F., Soubrier F., Esposito B., Duez H., Fievet C., et al. 1999. Protective role of interleukin-10 in atherosclerosis. Circ. Res. 85: e17–e24. [DOI] [PubMed] [Google Scholar]
- 35.Pinderski Oslund L. J., Hedrick C. C., Olvera T., Hagenbaugh A., Territo M., Berliner J. A., Fyfe A. I. 1999. Interleukin-10 blocks atherosclerotic events in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 19: 2847–2853. [DOI] [PubMed] [Google Scholar]
- 36.Sedeeq M. S., El-Nahrery E. M. A., Shalaby N., Hussein M., Shehata H., El Aal R. A., Abdel Ghaffar N. F., Mohamed M. M. 2019. Micro-RNA-96 and interleukin-10 are independent biomarkers for multiple sclerosis activity. J. Neurol. Sci. 403: 92–96. [DOI] [PubMed] [Google Scholar]
- 37.Krukowski K., Eijkelkamp N., Laumet G., Hack C. E., Li Y., Dougherty P. M., Heijnen C. J., Kavelaars A. 2016. CD8+ T cells and endogenous IL-10 are required for resolution of chemotherapy-induced neuropathic pain. J. Neurosci. 36: 11074–11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Opal S. M., DePalo V. A. 2000. Anti-inflammatory cytokines. Chest 117: 1162–1172. [DOI] [PubMed] [Google Scholar]
- 39.Chernoff A. E., Granowitz E. V., Shapiro L., Vannier E., Lonnemann G., Angel J. B., Kennedy J. S., Rabson A. R., Wolff S. M., Dinarello C. A. 1995. A randomized, controlled trial of IL-10 in humans. Inhibition of inflammatory cytokine production and immune responses. J. Immunol. 154: 5492–5499. [PubMed] [Google Scholar]
- 40.Brenner S., Prösch S., Schenke-Layland K., Riese U., Gausmann U., Platzer C. 2003. cAMP-induced interleukin-10 promoter activation depends on CCAAT/enhancer-binding protein expression and monocytic differentiation. J. Biol. Chem. 278: 5597–5604. [DOI] [PubMed] [Google Scholar]
- 41.Eldeeb K., Leone-Kabler S., Howlett A. C. 2016. CB1 cannabinoid receptor-mediated increases in cyclic AMP accumulation are correlated with reduced Gi/o function. J. Basic Clin. Physiol. Pharmacol. 27: 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murillo-Rodriguez E., Blanco-Centurion C., Sanchez C., Piomelli D., Shiromani P. J. 2003. Anandamide enhances extracellular levels of adenosine and induces sleep: an in vivo microdialysis study. Sleep 26: 943–947. [DOI] [PubMed] [Google Scholar]
- 43.Ehrentraut H., Clambey E. T., McNamee E. N., Brodsky K. S., Ehrentraut S. F., Poth J. M., Riegel A. K., Westrich J. A., Colgan S. P., Eltzschig H. K. 2013. CD73+ regulatory T cells contribute to adenosine-mediated resolution of acute lung injury. FASEB J. 27: 2207–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murray P. J. 2005. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc. Natl. Acad. Sci. USA 102: 8686–8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walton A. H., Muenzer J. T., Rasche D., Boomer J. S., Sato B., Brownstein B. H., Pachot A., Brooks T. L., Deych E., Shannon W. D., et al. 2014. Reactivation of multiple viruses in patients with sepsis. PLoS One 9: e98819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asehnoune K., Roquilly A., Abraham E. 2012. Innate immune dysfunction in trauma patients: from pathophysiology to treatment. Anesthesiology 117: 411–416. [DOI] [PubMed] [Google Scholar]
- 47.Steinhauser M. L., Hogaboam C. M., Kunkel S. L., Lukacs N. W., Strieter R. M., Standiford T. J. 1999. IL-10 is a major mediator of sepsis-induced impairment in lung antibacterial host defense. J. Immunol. 162: 392–399. [PubMed] [Google Scholar]
- 48.Couper K. N., Blount D. G., Riley E. M. 2008. IL-10: the master regulator of immunity to infection. J. Immunol. 180: 5771–5777. [DOI] [PubMed] [Google Scholar]
- 49.Wiley J., Balster R., Martin B. 1995. Discriminative stimulus effects of anandamide in rats. Eur. J. Pharmacol. 276: 49–54. [DOI] [PubMed] [Google Scholar]
- 50.Young P. J., Bellomo R. 2014. Fever in sepsis: is it cool to be hot? Crit. Care 18: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang Q., Cross A. S., Singh I. S., Chen T. T., Viscardi R. M., Hasday J. D. 2000. Febrile core temperature is essential for optimal host defense in bacterial peritonitis. Infect. Immun. 68: 1265–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fitton A. G., Pertwee R. G. 1982. Changes in body temperature and oxygen consumption rate of conscious mice produced by intrahypothalamic and intracerebroventricular injections of delta 9-tetrahydrocannabinol. Br. J. Pharmacol. 75: 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Athanasiou A., Clarke A. B., Turner A. E., Kumaran N. M., Vakilpour S., Smith P. A., Bagiokou D., Bradshaw T. D., Westwell A. D., Fang L., et al. 2007. Cannabinoid receptor agonists are mitochondrial inhibitors: a unified hypothesis of how cannabinoids modulate mitochondrial function and induce cell death. Biochem. Biophys. Res. Commun. 364: 131–137. [DOI] [PubMed] [Google Scholar]
- 54.Niederhoffer N., Szabo B. 1999. Effect of the cannabinoid receptor agonist WIN55212-2 on sympathetic cardiovascular regulation. Br. J. Pharmacol. 126: 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sawa M., Tsurimaki Y., Tsuru T., Shimizu H. 1988. New quantitative method to determine protein concentration and cell number in aqueous in vivo. Jpn. J. Ophthalmol. 32: 132–142. [PubMed] [Google Scholar]
- 56.van Loon L. M., van der Hoeven J. G., Lemson J. 2019. Hemodynamic response to β-blockers in severe sepsis and septic shock: a review of current literature. J. Crit. Care 50: 138–143. [DOI] [PubMed] [Google Scholar]
- 57.Mukhopadhyay S., Howlett A. C. 2001. CB1 receptor-G protein association. Subtype selectivity is determined by distinct intracellular domains. Eur. J. Biochem. 268: 499–505. [DOI] [PubMed] [Google Scholar]
- 58.Felder C. C., Joyce K. E., Briley E. M., Mansouri J., Mackie K., Blond O., Lai Y., Ma A. L., Mitchell R. L. 1995. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol. Pharmacol. 48: 443–450. [PubMed] [Google Scholar]
- 59.Mukhopadhyay S., Howlett A. C. 2005. Chemically distinct ligands promote differential CB1 cannabinoid receptor-Gi protein interactions. Mol. Pharmacol. 67: 2016–2024. [DOI] [PubMed] [Google Scholar]
- 60.Hudson B. D., Hébert T. E., Kelly M. E. 2010. Ligand- and heterodimer-directed signaling of the CB(1) cannabinoid receptor. Mol. Pharmacol. 77: 1–9. [DOI] [PubMed] [Google Scholar]
- 61.Laprairie R. B., Kulkarni P. M., Deschamps J. R., Kelly M. E. M., Janero D. R., Cascio M. G., Stevenson L. A., Pertwee R. G., Kenakin T. P., Denovan-Wright E. M., Thakur G. A. 2017. Enantiospecific allosteric modulation of cannabinoid 1 receptor. ACS Chem. Neurosci. 8: 1188–1203. [DOI] [PubMed] [Google Scholar]
- 62.Laprairie R. B., Bagher A. M., Denovan-Wright E. M. 2017. Cannabinoid receptor ligand bias: implications in the central nervous system. Curr. Opin. Pharmacol. 32: 32–43. [DOI] [PubMed] [Google Scholar]
- 63.Dhital S., Stokes J. V., Park N., Seo K. S., Kaplan B. L. 2017. Cannabidiol (CBD) induces functional Tregs in response to low-level T cell activation. Cell. Immunol. 312: 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robinson R. H., Meissler J. J., Fan X., Yu D., Adler M. W., Eisenstein T. K. 2015. A CB2-selective cannabinoid suppresses T-Cell activities and increases Tregs and IL-10. J. Neuroimmune Pharmacol. 10: 318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kishimoto S., Muramatsu M., Gokoh M., Oka S., Waku K., Sugiura T. 2005. Endogenous cannabinoid receptor ligand induces the migration of human natural killer cells. J. Biochem. 137: 217–223. [DOI] [PubMed] [Google Scholar]
- 66.Kellum J. A., Pike F., Yealy D. M., Huang D. T., Shapiro N. I., Angus D. C., and the Protocol-based Care for Early Septic Shock Investigators (ProCESS) Investigators 2017. Relationship between alternative resuscitation strategies, host response and injury biomarkers, and outcome in septic shock: analysis of the protocol-based care for early septic shock study. Crit. Care Med. 45: 438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bozza F. A., Salluh J. I., Japiassu A. M., Soares M., Assis E. F., Gomes R. N., Bozza M. T., Castro-Faria-Neto H. C., Bozza P. T. 2007. Cytokine profiles as markers of disease severity in sepsis: a multiplex analysis. Crit. Care 11: R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pober J. S., Sessa W. C. 2007. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 7: 803–815. [DOI] [PubMed] [Google Scholar]
- 69.Turgeman I., Bar-Sela G. 2019. Cannabis for cancer - illusion or the tip of an iceberg: a review of the evidence for the use of Cannabis and synthetic cannabinoids in oncology. Expert Opin. Investig. Drugs 28: 285–296. [DOI] [PubMed] [Google Scholar]
- 70.Amato L., Minozzi S., Mitrova Z., Parmelli E., Saulle R., Cruciani F., Vecchi S., Davoli M. 2017. [Systematic review of safeness and therapeutic efficacy of cannabis in patients with multiple sclerosis, neuropathic pain, and in oncological patients treated with chemotherapy]. Epidemiol. Prev. 41: 279–293. [DOI] [PubMed] [Google Scholar]
- 71.Whiting P. F., Wolff R. F., Deshpande S., Di Nisio M., Duffy S., Hernandez A. V., Keurentjes J. C., Lang S., Misso K., Ryder S., et al. 2015. Cannabinoids for medical use: a systematic review and meta-analysis. [Published errata appear in 2015 JAMA 314: 520, 2015 JAMA 314: 837, 2015 JAMA 314: 2308, and 2016 JAMA 315: 1522.] JAMA 313: 2456–2473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.