

Abstract
Peptides provide an attractive modality for targeting challenging drug targets such as intracellular protein-protein interactions. Unfortunately, peptides are generally impermeable to the cell membrane and inherently susceptible to proteolytic degradation in vivo. Macrocyclization of peptides greatly increases their proteolytic stability and in some cases the cell-penetrating activity. Conjugation of peptidyl cargoes to cyclic cell-penetrating peptides has resulted in potent, cell-permeable, and metabolically stable macrocyclic peptides against intracellular protein targets. Proper conjugation/integration of a peptidyl cargo with a cyclic cell-penetrating peptide is critical to retain the activity of each component and generate a biologically active macrocyclic peptide. This chapter describes the different conjugation strategies that have been developed (including endocyclic, bicyclic, and reversible cyclization methods) and the detailed protocols for their preparation.
Keywords: Bicyclic peptides, cyclic peptides, cyclic cell-penetrating peptides, protein-protein interaction, reversible cyclization
1. Introduction
Traditional drug discovery has largely been dedicated to the discovery and optimization of small-molecule drugs that adhere to the Rule of Five (Ro5) guidelines (1,2). These small molecules have the ability to cross biological membranes by passive diffusion (3). Unfortunately, molecules within the Ro5 boundary are generally ineffective against protein-protein interactions (PPIs), which represent the largest untapped class of therapeutic opportunities but usually do not contain well-defined, hydrophobic pockets required for small molecules to bind (4). Biological drugs (e.g., monoclonal antibodies) have found considerable success in treating a wide range of diseases but cannot cross the cell membrane, limiting their usage to extracellular targets. An estimated ~80% of therapeutically relevant drug targets are currently undruggable by small molecules or biologics and most of them are intracellular (5). Clearly, alternative drug modalities that effectively penetrate the cell membrane are needed.
Peptides have emerged as a promising new drug modality because they have the ability to recapitulate the exceptional affinity and selectivity of proteins, while retaining some of the attributes of small molecules (e.g., synthetic accessibility, lower cost of production, and lower risk of immune response) (6). However, traditional peptide therapeutics suffer from issues inherent to the class, such as rapid proteolytic degradation in vivo and lack of cell-permeability (7). To improve the metabolic stability of peptides, researchers have explored a variety of macrocyclization methods as well as incorporation of unnatural amino acids (7). In general, cyclic peptides of small- and medium-sized rings (≤10 aa) are relatively resistant to proteolytic degradation. Some of these cyclic peptides have already become successful therapeutic agents, clinical candidates, and/or valuable biological probes, including cyclic RGD peptides (8), disulfide cyclized linaclotide (9), and hydrocarbon-stapled peptides against the MDM2-p53 interaction (10). An ongoing and far more challenging effort is to improve the cell-permeability of peptides (including cyclic peptides). Broadly speaking, researchers have been pursuing two different strategies to design cell-permeable cyclic peptides, corresponding to two different cellular entry mechanisms: passive diffusion vs endocytosis. This chapter focuses on the latter approach, which involves conjugation or integration of a peptidyl cargo with a cell-penetrating peptide (CPP).
1.1. Cyclic Cell-Penetrating Peptides
The discovery of the Tat peptide (derived from HIV transactivator of transcription protein) as a CPP in the early 1990s led to the birth of a new field (11, 12). Since then, a large number of CPPs have been discovered, including nonaarginine (R9) and penetratin (Antp), and have been used to deliver a wide range of cargoes in vitro and in vivo (13). However, linear CPPs have encountered several challenges which have so far prevented their translation into the clinic (14), including rapid proteolytic degradation and poor pharmacokinetic properties (15), lack of biodistribution (16), and poor cytosolic delivery efficiencies (17, 18). To overcome these challenges, Pei and colleagues explored cyclic peptides as CPPs and discovered cyclo(Phe-Nal-Arg-Arg-Arg-Arg-Gln) (cyclic CPP1, where Nal is L-2-nathphthylalanine) as a significantly improved CPP (19) (Fig. 1). In addition to its excellent proteolytic stability, CPP1 showed 3- to 12-fold higher cytosolic delivery efficiency than Tat, R9, and Antp. Subsequent SAR and optimization led to the discovery of cyclic CPP9 [cyclo(phe-Nal-Arg-arg-Arg-arg-Gln)] and CPP12 [cyclo(Phe-phe-Nal-Arg-arg-Arg-arg-Gln)] as exceptionally active CPPs, having cytosolic delivery efficiencies of 62% and 120%, respectively (100% is defined as equal cargo concentration in the extracellular medium and cytosol) (20). For comparison, Tat and R9 showed cytosolic delivery efficiencies of 2.0% and 4.4%, respectively, under the same assay condition. Cyclic CPPs bind directly to the plasma membrane phospholipids and enter cells by endocytosis. While inside the early endosome, the cyclic CPPs bind to the endosomal membrane and induce membrane curvature and budding of small CPP-enriched vesicles into the cytosol. Subsequent collapse of the unstable vesicles resulted in the release of the vesicular contents (including the CPPs) into the cytosol.
Figure 1.
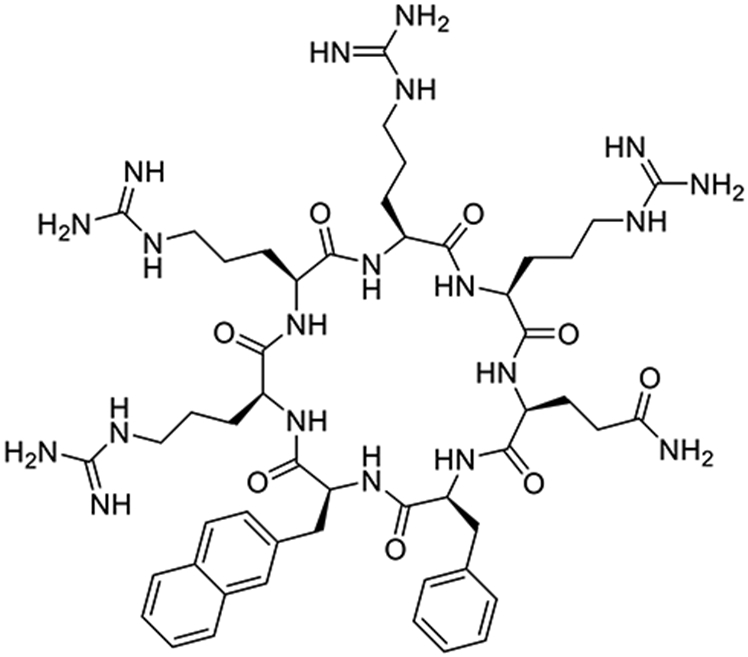
Structure of cyclic CPP1.
1.2. Integration of Peptidyl Cargo with Cyclic CPP
Cyclic CPPs have no known biological activity on their own; their utility lies in their ability to transport biologically active but membrane-impermeable cargoes into the cytosol of mammalian cells. Their mechanism of action predicts that cyclic CPPs should be cargo agnostic, although the nature of the cargo can affect the cellular entry efficiency via several different mechanisms. For example, the cargo may bind to the plasma membrane and enhance the endocytic uptake of the CPP-cargo conjugate, or to the endosomal membrane and enhance endosomal escape of the conjugate. The cargo may increase or decrease the binding of cyclic CPP to serum proteins during circulation, affecting the cellular uptake kinetics and/or efficiency. Finally, the cargo may physically interact with the CPP, either intramolecularly or intermolecularly, and decrease the activity of the cyclic CPP or the cargo. Therefore, proper conjugation of a given cargo with a cyclic CPP is critical to ensure that the activity of the CPP and/or the cargo is maintained or, in some cases, enhanced. So far, cyclic CPPs have been used to successfully deliver small molecules (e.g., fluorescent dyes), linear peptides, cyclic peptides, proteins, and nucleic acids (21). Below, we describe the different strategies that have been developed to conjugate cyclic CPPs with peptidyl cargoes, including endocyclic, exocyclic, bicyclic, and reversible cyclization delivery methods (Fig. 2)
Figure 2.

Schemes showing the different methods by which peptidyl cargoes may be conjugated with a cyclic CPP.
1.2.1. Endocyclic Conjugation
Endocyclic conjugation involves direct insertion of a cargo sequence into a cyclic CPP ring and the resulting conjugate is a monocyclic peptide (19). This conjugation method is limited to relatively small cargoes (i.e., peptides of ≤5 aa). Longer peptide cargoes result in larger rings, which have progressively poorer cellular entry efficiencies, likely because increasing conformational flexibility decreases the entropic advantages offered by macrocyclization. Since the target-binding sequence and the CPP sequence are in the same ring, negatively charged cargo sequences may interact electrostatically with the cationic CPP and negatively impact the cellular uptake efficiency. On the other hand, the proximity of the cargo and CPP sequences makes it possible to design cyclic peptides in which the same residue(s) serves the dual function of cellular entry and target engagement. This can significantly reduce the size of macrocycles and make them more “drug-like”. By using this strategy, Bedewy et. al (22) designed a cycloheptapeptidyl inhibitor against peptidyl–prolyl cis-trans isomerase Pin1 (Fig. 3a). Despite the presence of a negatively charged phospho-D-threonine as a key Pin1-binding motif, the macrocycle is cell-permeable and inhibited the Pin1 activity in HeLa cells. To our knowledge, this is the smallest macrocyclic peptide reported, which enters mammalian cells by an endocytic mechanism and elicits biological activity. Upadhyaya et al. designed a combinatorial library of monocyclic peptides by integrating a CPP-like motif and randomized peptide sequences (23). Screening of this library for binding to G12V mutant K-Ras followed by optimization led to a potent, cell-permeable cycloundecapeptidyl K-Ras inhibitor, which selectively blocked Ras-GTP from binding to its downstream effector proteins such as Raf and PI3 kinases and induced apoptosis of mutant K-Ras driven cancer cells.
Figure 3.

Structures of cell-permeable Pin1 inhibitors generated by endocyclic (a) and bicyclic conjugation methods (b).
1.2.2. Bicyclic Conjugation
When the biologically active cargo is also a cyclic peptide, one can fuse the cyclic peptide with a cyclic CPP to form a bicyclic peptide, in which one ring ensures efficient cellular entry while the other ring binds to the target of interest (or bicyclic conjugation). A simple and yet general, highly effective method of bicyclization is to fuse the CPP and cargo sequences to form a linear peptide containing two side-chain amine-containing amino acids [e.g., lysine or 2,3-diaminopropionic acid (Dap)], one at the C-terminus and the other at the CPP-cargo junction. The resulting peptide is then converted into a bicyclic peptide by reacting the N-terminal amine and the two sidechain amines with trimesic acid (Fig. 2c) (24). This rigid scaffold helps preorganize the peptide into productive binding conformations, increasing their binding affinity and specificity for protein targets. The planar scaffold also orients the CPP and cargo rings away from each other, minimizing their mutual interference. Most importantly, bicyclic conjugation can in principle accommodate cargoes of any size or sequence, since changes in the cargo ring does not affect the CPP ring or its cellular entry efficiency. By utilizing this strategy, Jiang and Pei designed a bicyclic peptide library aimed at finding a potent, cell-permeable non-phosphorylated bicyclic peptidyl inhibitor against Pin1 (25). In this library, one ring of the bicyclic peptides featured a fixed CPP motif, while the other ring consisted of a degenerate peptide sequence (Fig. 2c). Screening of the library against Pin1 led to the identification of a potent and highly selective Pin1 inhibitor (Fig. 3b) which bound to Pin1 with KD = 0.12 μM and inhibited Pin1 activity in human cancer cells. The library approach was also applied to discover cell-permeable and biologically active inhibitors against protein-tyrosine phosphatases 1B (24) and TCPTP (26), as well as K-Ras (27).
1.2.3. Reversible Cyclization
For some target proteins (e.g., PDZ domains), the peptide ligand must be in its linear, extended form to be biologically active. This type of linear peptidyl cargoes are not suitable for either the endocyclic or bicyclic conjugation method. Simply conjugating a linear peptide to the side chain of a cyclic CPP is often inadequate, because linear peptides, especially those consisting of primarily proteinogenic acids, are susceptible to rapid proteolytic degradation in vivo. To this end, Qian et. al. introduced a reversible cyclization strategy which enhances both the proteolytic stability and the cell-permeability of linear peptide cargoes (28). In this strategy, the CPP sequence is fused with a peptidyl cargo sequence and cyclized through a disulfide bond. Cyclization improves both resistance to proteolysis and cellular uptake efficiency. Upon successful entry into the cytosol, the disulfide bond is reduced by intracellular thiols to liberate the biologically active, linear peptide for engagement of the desired intracellular target (Fig. 2d). Qian et al. have applied this strategy to generate cell-permeable peptide substrates for real-time detection of intracellular caspase activities (28). The same investigators also developed a disulfide-cyclized peptidyl inhibitor against the CFTR-associated ligand CAL-PDZ domain as a potential treatment for cystic fibrosis (28) (Fig. 4a).
Figure 4.
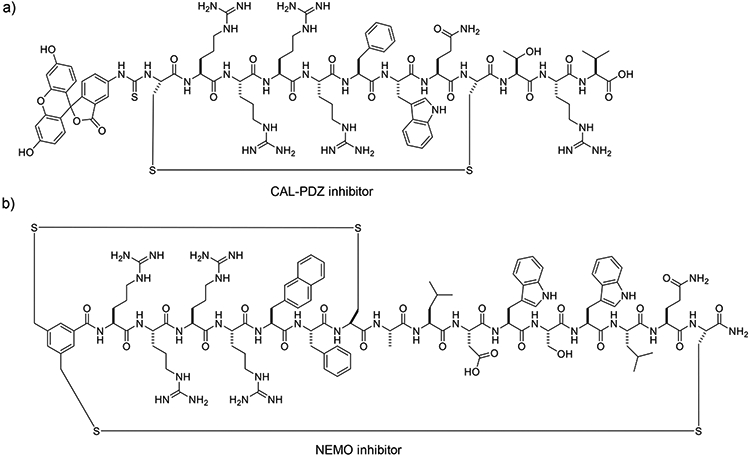
Structures of reversibly cyclized cell-permeable peptidyl inhibitors. (a) A monocyclic peptidyl inhibitor against the CFTR-CAL interaction, and b) a bicyclic peptidyl inhibitor against the NEMO-IKK interaction.
1.2.4. Reversible Bicyclization
Like endocyclic conjugation, disulfide-mediated cyclization of CPP-cargo fusion peptides is limited to relatively small ring sizes. Reversible bicyclization provides an alternative approach to introducing additional conformational rigidity into the macrocyclic peptide. Qian et al. first demonstrated this strategy by designing a cell-permeable inhibitor against NF-κB essential modulator (NEMO) (29). They fused the NEMO-binding domain (NBD) of IκB kinase β (ALDWSWLQ) with a short CPP motif (RRRRΦF, where Φ is 2-naphthylalanine) and adding two cysteine residues, one at the C-terminus and the other in between the NBD and CPP sequences. The resulting peptide was cyclized into a bicycle by forming two pairs of disulfide bonds between the cysteines and an N-terminal 3,5-bis(mercaptomethyl)benzoyl moiety (Fig. 4b). Relative to a monocyclic peptide, bicyclization reduces the size of each ring to approximately half of the original size, greatly increasing the conformational rigidity of the macrocycle. This in turn greatly improves the cellular uptake efficiency as well as resistance to proteolysis. The bicyclic NEMO inhibitor blocked the interaction between NEMO and IκB kinases and inhibited TNFα-induced NF-κB signaling in cell culture.
2. Materials
2.1. Endocyclic Conjugation
Rink amide resin LS (see Note 1)
Coupling reagents: 4 eq. 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU), 4 eq. N-hydroxybenzotriazole (HOBt), 8 eq. N,N-diisopropylethylamine (DIPEA) as solids or as prepared as stock solutions in DMF (see Note 2)
Deprotection reagents: 20% piperidine in DMF (v/v) (see Note 3)
Synthesis vessels: Pierce unpacked chromatography columns (Cat# 89898) (see Note 4).
Amino acids: 4 eq. of the desired Fmoc-protected amino acids dissolved in DMF, can be stored at 4 °C (see Note 5).
Synthesis solvents: DCM and DMF.
Cyclization reagents: (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP), HOBt and DIPEA, dissolved in 1:1 DCM:DMF (v/v) (see Note 6).
Deallylation: 0.3 eq. tetrakis(triphenylphosphine)palladium(0) [Pd(PPh3)4] dissolved in dry DCM. Immediately before addition of this solution to the resin, add 10 eq. phenylsilane (see Note 7).
Side-chain deprotection and cleavage: freshly prepare a solution of 95:2.5:2.5 trifluoroacetic acid (TFA)/triisopropylsilane (TIPS)/water (see Note 8).
Inert gas (e.g. nitrogen or argon) and a gas manifold (see Note 9).
Diethyl ether, chilled to −20 °C or below (see Note 10).
2.2. Bicyclic Peptide Library
TentaGel S NH2 resin (90 μm) (see Note 1).
Coupling reagents: 4 eq. HATU, 4 eq. HOBt, 8 eq. DIPEA as solids or as prepared as stock solutions in DMF for standard amino acid couplings. 4 eq. diisopropylcarbodiimide (DIC) (see Note 2).
Deprotection reagents: 20% piperidine in DMF (v/v) (see Note 3)
Synthesis vessels: Pierce unpacked chromatography columns (Cat# 89898) (see Note 4)
Amino acids: 4 eq. of the desired Fmoc-protected amino acids dissolved in DMF, can be stored at 4°C (see Note 5).
Synthesis solvents: DCM and DMF.
1,3,5-benzenetricarboxylic acid diallyl ester (see Note 11).
Cyclization reagents: PyBOP, HOBt and DIPEA, dissolved in 1:1 DCM:DMF (v/v) (see Note 6).
Deallylation: 0.3 eq. Pd(PPh3)4 and 10 eq. triphenylphosphine (PPh3) dissolved in dry DCM. Immediately before addition of this solution to the resin, add 15 eq. of N-methylaniline. (see Note 7). Re-suspend the resin in a solution of 10% sodium dimethyldithiocarbamate (wt/v) in DMF for 10 min to scavenge residual palladium after deprotection.
Side-chain deprotection solution: freshly prepare a solution of 87.5:2.5:2.5:2.5:2.5:2.5 TFA/thioanisole/water/phenol/1,2-ethanedithiol (see Note 8).
Acylation: 10 eq. acetic anhydride and 20 eq. DIPEA combined in DCM immediately before addition to the resin.
2.3. Reversible Bicyclization
Rink amide resin HS (see Note 1).
Coupling reagents: 4 eq. HATU, 4 eq. HOBt, 8 eq. DIPEA as solids or as prepared as stock solutions in DMF (see Note 2).
Deprotection reagents: 20% piperidine in DMF (v/v), 2 M Hg(OAc)2 in DMF (see Note 3).
Synthesis vessels: Pierce unpacked chromatography columns (Cat# 89898) (see Note 4).
Amino acids: 4 eq. of the desired Fmoc-protected amino acids dissolved in DMF, can be stored at 4 °C (see Note 5).
Synthesis solvents: DCM and DMF.
Cyclization reagents: PyBOP, HOBt and DIPEA, dissolved in 1:1 DCM:DMF (v/v) (see Note 6).
Deallylation: 0.3 eq. Pd(PPh3)4 and 10 eq. PPh3 dissolved in dry DCM. Immediately before addition of this solution to the resin, add 15 eq. of N-methylaniline (see Note 7).
Side-chain deprotection and cleavage: freshly prepare a solution of 95:2.5:2.5 TFA/TIPS/water (see Note 8).
Inert gas (e.g. nitrogen or argon) and a gas manifold (see Note 9).
Diethyl ether, chilled to −20 °C or below (see Note 10).
2.4. Reversible Cyclization
Rink amide resin HS (see Note 1).
Coupling reagents: 4 eq. HATU, 4 eq. HOBt, 8 eq. DIPEA as solids or as prepared as stock solutions in DMF (see Note 2).
Deprotection reagents: 20% piperidine in DMF (v/v) (see Note 3).
Synthesis vessels: Pierce unpacked chromatography columns (Cat# 89898) (see Note 4).
Amino acids: 4 eq. of the desired Fmoc-protected amino acids dissolved in DMF, can be stored at 4 °C (see Note 5).
Synthesis solvents: DCM and DMF.
Cyclization reagents: PyBOP, HOBt, and DIPEA, dissolved in 1:1 DCM:DMF (v/v) (see Note 6).
3. Methods
3.1. Endocyclic Conjugation
Swell 100 mg of Rink amide resin in DMF for 20 min (see Note 1).
Drain and add 20% (v/v) piperidine in DMF for 10 min (twice) to remove Fmoc group (see Note 3).
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
Add 4 eq. Fmoc-Glu-OAll, 4 eq. HATU, 4 eq. HOBt, and 8 eq. DIPEA and mixed for 1 h in DMF (see Note 2).
Drain and wash exhaustively with DMF, DCM and DMF.
Repeat steps 2 through 5 to couple the remaining amino acids in the linear sequence.
Add a solution of 0.3 eq. of Pd(PPh3)4, 10 eq. of PhSiH3 in dry DCM to the resin and allow to mix for 15 min. Repeat the procedure 3 times (see Note 7).
Drain and wash exhaustively with DCM and DMF.
Incubate the resin in a solution of 10% sodium dimethyldithiocarbamate in DMF for 10 min (twice).
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
Drain and add 20% (v/v) piperidine in DMF for 10 min (twice) to remove Fmoc group (see Note 3).
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
Incubate resin with 1 M solution HOBt for 15 min. (see Note 12).
Add a solution of 5 eq. of PyBOP, 5 eq. of HOBt and 10 eq. of DIPEA in DMF and mix for 1.5 h (see Note 6). Repeat the procedure once.
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
Add a solution of 95:2.5:2.5 (v/v) TFA/H2O/TIPS for 3 h to release the cyclic peptide from the resin and effect side-chain deprotection (see Note 8).
Drain and concentrate the cleavage solution to a semi-solid by gently blowing an inert gas over the solution inside a fume hood (see Note 9).
Add chilled diethyl ether to the concentrated cleavage solution to precipitate the peptide. Centrifuge at 7.5 × 103 rpm for 5 min and remove supernatant with a pipette. Repeat the step three times.
Dissolve the triturated crude peptide in DMF and purify it by reversed-phase HPLC (see Note 13)
3.2. Bicyclic Peptide Library
Swell 1 g of TentaGel S NH2 resin in DMF for 20 min (see Note 1).
Couple a linker sequence (β-Ala-β-Ala-Met) using standard Fmoc/HATU/DIPEA chemistry (see Note 2).
Soak beads in DMF for 30 min.
Drain and soak in 1:1 (v/v) degassed DMF/water mixture for 1 h (twice).
Drain and soak beads in 1:4 (v/v) degassed DMF/water for 1 h (twice).
Drain, wash with degassed water and soak beads in degassed water overnight.
Carefully drain and quickly resuspend beads in 55:45 DCM:diethyl ether (v/v) containing 0.4 eq. of Fmoc-OSu and 1 eq. DIPEA and allow to mix for 30 min.
Drain, wash with DMF, and incubate with a solution of 5 eq. of di-tert-butyl dicarbonate and 0.1 eq. of DMAP in DMF for 30 min.
Drain and wash with DMF. Add 20% piperidine in DMF solution to the resin and incubate for 10 min to remove the Fmoc group from the outer layer.
Drain and wash resin with DMF, DCM, and DMF (2-3 times each) (see Note 14).
Couple 4-(hydroxymethyl)benzoic acid (HMB) to the outer layer using HATU/HOBt/DIPEA (see Note 2).
Couple β-Ala to the HMB linker with Fmoc-β-Ala-OH/DIC/DMAP (5, 5.5, 0.1 eq., respectively) for 2 h.
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
Couple the sequence Dap(alloc)-β-Ala-β-Ala-Pra using standard Fmoc/HATU/HOBt/DIPEA chemistry.
Treat resin with 95/2.5/2.5 (v/v) mixture of TFA/H2O/TIPS to remove Boc group from the inner layer.
Couple the sequence Dap(Mtt)-Phe-Nal-Arg-Arg-Arg-Arg using standard Fmoc/HATU/HOBt/DIPEA chemistry.
Divide the resin into desired number of equal portions corresponding to the number of amino acid building blocks to be incorporated into the sequence (see Note 15)
Couple a different amino acid building block to each aliquot of the resin by standard Fmoc chemistry (see Note 2)
Combine the resin together in the original synthesis vessel.
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
Drain and add 20% (v/v) piperidine in DMF for 10 min (twice) to remove Fmoc group (see Note 3).
Divide the resin again as in Step 17 and repeat Steps 18-20 as needed until the desired number of random residues are coupled.
Treat resin with a solution of 2% (v/v) TFA in DCM for 5 min and repeat until the solution is clear to remove the Mtt group on the internal Dap residue (see Note 16).
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
Add a solution of 3 eq. of Fmoc-OSu and 10 eq. DIPEA to the resin and incubate with mixing for 1 h.
Remove the allyl protecting group on the C-terminal Dap residue by treating the resin with a solution of tetrakis(triphenylphosphine)palladium/triphenylphosphine/N-methylaniline (0.3, 5, 15 eq., respectively) in dry DCM for 12 h (see Note 7).
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
Incubate the resin in a solution of 10% sodium dimethyldithiocarbamate in DMF for 10 min (twice).
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
Add a solution of 3 eq. diallyl trimesic acid, 3 eq. HATU and 6 eq. DIPEA in DMF and incubate for 1 h to acylate the exposed amine (see Note 11).
Remove the allyl protecting groups on trimesic acid by treating the resin with a solution of tetrakis(triphenylphosphine)palladium/triphenylphosphine/N-methylaniline (0.3, 5, 15 eq., respectively) in dry DCM for 12 h (see Note 7).
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
Incubate the resin in a solution of 10% (wt/v) sodium dimethyldithiocarbamate in DMF for 10 min (twice).
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
Remove the Fmoc groups at the N-terminus as well as on the internal Dap residue as described in steps 6 and 7.
Drain and add 20% (v/v) piperidine in DMF for 10 min (twice) to remove Fmoc group (see Note 3).
Incubate with 1 M HOBt for 10 min (see Note 12).
To cyclize the library, add a solution of 5 eq. of PyBOP, 5 eq. of HOBt and 10 eq. of DIPEA in DMF and incubate with mixing for 1.5 h (twice) (see Note 6).
Drain and wash resin thoroughly with DMF followed by DCM.
Add a modified reagent K (TFA/thioanisole/water/phenol/1,2-ethanedithiol, 82.5:5:5:5:2.5 v/v) to the cyclized library and allow to mix for 3 h (see Note 8).
Wash the resulting library thoroughly with DCM, DMF, 5% (v/v) DIPEA in DMF, 1:1 (v/v) DCM/diethyl ether, DMF, and DCM.
3.3. Reversible Bicyclization
Swell 100 mg of Rink amide resin LS in DMF for 20 min (see Note 1)
Drain and add 20% (v/v) piperidine in DMF for 10 min (twice) to remove Fmoc group (see Note 3).
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
To synthesize a disulfide-mediated bicyclic peptides, first synthesize the corresponding linear peptide containing two Acm-protected cysteine residues (one at the C-terminus and one at the CPP-cargo junction) using standard Fmoc/HATU chemistry (see Note 2)
To remove the Acm group, add 2 mL of 2 M mercury (II) acetate in DMF to the resin and incubate with mixing overnight. (see Fig. 5)
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
Incubate the resin with 2 mL of 20% β-mercaptoethanol in DMF for 2 h (twice) to release the free thiol.
Wash exhaustively with DMF to remove all reducing agents.
Incubate the resin overnight with a solution of 1 eq. 3,5-bis((pyridin-2-yldisulfanyl)methyl)benzoic acid in methanol containing 1% acetic acid (v/v) (see Note 17).
Treat resin with 20% piperidine in DMF for 10 min (twice) to remove N-terminal Fmoc protecting group.
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
Add 1 eq. of HATU and 5 eq. of DIPEA in DMF and incubate for 2 h.
Drain and wash resin with DMF, DCM and DMF (2-3 times each).
Deprotect and release peptide by treating the resin with a solution of 85:10:2.5:2.5 (v/v) TFA/DCM/water/TIPS for 2 h (see Note 8).
Drain and concentrate the cleavage solution as described above (see Note 9).
Triturate the crude peptide with chilled diethyl ether and purify by reversed-phase HPLC as described above.
Figure 5.
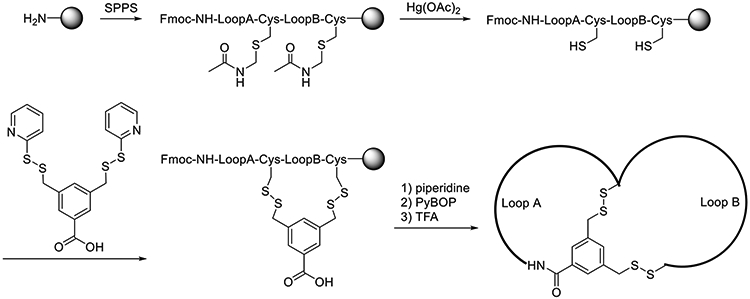
Scheme showing the solid-phase synthesis of disulfide-mediated bicyclic peptides.
3.4. Reversible Cyclization
Swell 100 mg of Rink amide resin LS in DMF for 20 min (see Note 1)
Drain and add 20% (v/v) piperidine in DMF for 10 min. twice to remove Fmoc group (see Note 3).
Drain and wash resin with DMF, DCM, and DMF (2-3 times each).
To synthesize the disulfide-mediated cyclic peptide, first synthesize the corresponding linear peptide containing Cys(Trt) residue at the C-terminus using standard Fmoc/HATU chemistry.
Remove N-terminal Fmoc group using 20% (v/v) piperidine in DMF for 10 min (twice) (see Note 3).
Install N-terminal thiol by treating with 5 eq. of 3,3’-dithiodipropionic acid, 5 eq. N,N’- DIC and 0.1 eq. 4-(dimethylamino)pyridine (DMAP) in anhydrous DCM for 2 h (see Note 18).
Incubate resin with 2 mL of 20% β-mercaptoethanol in DMF for 2 h (twice) to expose the free thiol.
Wash exhaustively with DMF followed by DCM.
Add a solution of 90:2.5:2.5:2.5:2.5 (v/v) TFA/water/phenol/TIPS/1,2-ethanedithiol (EDT) and incubate for 2 h.
Drain and concentrate the cleavage solution as described above (see Note 9).
Triturate the crude peptide with chilled diethyl ether as described above.
Incubate the crude peptide with 5% (v/v) DMSO in PBS (pH 7.4) and mix gently overnight to effect intramolecular cyclization (see Note 19).
Purify the crude mixture on reversed-phase HPLC to isolate the intramolecularly cyclized peptide product.
5. Conclusion and Future Directions
Although not the focus of this article, several powerful combinatorial libraries technologies have been developed over the past decade or so and can be applied to generate macrocyclic peptidyl ligands with antibody-like affinity and specificity against essentially any protein target (30). The recent discovery of cyclic CPPs as powerful intracellular delivery vehicles has now made it possible to deliver these macrocyclic peptidyl ligands into the cytosol and nucleus of mammalian cells. Furthermore, as demonstrated by the examples covered in this article, the two platform technologies (library screening and cyclic CPPs) can be integrated to rapidly discover potent, specific, cell-permeable, and metabolically stable macrocyclic peptides to modulate intracellular targets that are undruggable by current drug modalities (i.e., small molecules and biologics). These advancements have ushered in a new era for peptide therapeutics, which may well become the third major drug modality, occupying a vast and presently barren land bounded by molecular weights of 500-5000. These macrocyclic peptides will also provide powerful tool compounds for biological and biomedical research.
To realize the potential of macrocyclic peptides as the third major drug modality, we believe that research in the following areas will be essential. First, we need to gain a better mechanistic understanding of how cyclic CPPs (and CPPs in general) achieve cytosolic entry. This knowledge will help predict how this new class of molecules behave in vivo as well as further improve the CPPs. Second, the pharmacokinetics, cytotoxicity, and immunogenic activity of this class of compounds need to be systemically evaluated, especially since poor PK properties were considered as the major limitations of peptidyl drugs in the past. Finally, the chemistry for synthesizing macrocyclic peptides will need significant improvements, at both medicinal and process chemistry stages. In particular, commercial availability of a large collection of affordable non-proteinogenic amino acids will greatly facilitate the lead optimization efforts.
Acknowledgement
This work was supported by NIH grant GM122459 to D.P.
Footnotes
There are a large number of different resins that have been developed to facilitate solid-phase peptide synthesis. In general, when an amidated C-terminus is desired, Rink amide resin serves as a good all-purpose choice. In the case of library synthesis, a more hydrophilic support such as TentaGel is desirable and is recommended. For peptides with a free C-terminus, Wang resin and DIC coupling (see Note 2) are recommended.
The uronium-based (e.g., HATU, HBTU, and HCTU) coupling reagents enable cost- and time-effective syntheses with minimal racemization. Phosphonium- (e.g. PyBOP) or carbenium-based (e.g. COMU) reagents are also effective. Carbodiimide-based (e.g., DIC) reagents are not recommended for non-microwave assisted syntheses, as they often cause unacceptable levels of racemization, especially when used in combination with DMAP. However, DIC/DMAP is required for effective coupling of amino acids to alcohols, such as those on Wang resin.
If Fmoc deprotection with 20% piperidine in DMF is incomplete (as revealed by MS or ninhydrin tests), a mixture of 2% 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 2% piperidine in DMF (v/v/v) is recommended. 20% piperazine in DMF is also an acceptable alternative.
The choice of vessel for synthesis is highly specific to the equipment present in each laboratory. In our experience, unpacked chromatography columns provide an economical solution, but these vessels are generally non-reusable and some researchers may prefer to use glass vessels designed specifically for SPPS.
Alternatively, N-methylpyrrolidinone (NMP) may be used in place of DMF to prepare Fmoc-protected AA solutions when solubility issues are encountered.
Uronium-based coupling reagents (e.g., HATU) should not be used for peptide cyclization, as they can react with the peptide N-terminus to form N-guandinylation products. Phosphonium-based reagents do not have this problem and are recommended.
Anhydrous solvents should be used for these steps, as the palladium catalyst is water-sensitive. Facile deprotection can be accomplished by using phenylsilane (PhSiH3) with a typical reaction condition of 0.3 eq. Pd(PPh3)4 and10 eq. PhSiH3 in dry DCM for 3 × 15 min.
The listed condition is for general side-chain deprotection and cleavage from the resin. If peptides contain redox-sensitive amino acids (e.g. Trp, Cys, or Met), addition of 2.5% 1,2-ethanedithol or 2,2’-(ethylenedioxy)diethanethiol is advised to prevent side-chain oxidation during cleavage.
Concentration of peptide solution under inert atmosphere is only required for peptides containing redox-sensitive residues; for all other sequences regular purified air is acceptable.
Diethyl ether used for trituration should be chilled to the lowest, conveniently allowed temperatures (e.g., −20 °C) to minimize unwanted product loss. Multiple trituration is recommended to remove as much of the scavengers as possible. More hydrophobic ethers, such as methyl tert-butyl ether (MTBE), are also acceptable solvents.
Diallyl trimesic acid is synthesized using a two-step protocol, first by refluxing trimesic acid with thionyl chloride in allyl alcohol, followed by hydrolysis with 1 eq. KOH in allyl alcohol.
Incubation and washing with 1 M HOBt ensures complete removal of piperidine from the preceding step. Piperidine can compete with peptide N-terminal amine during peptide cyclization and decrease the reaction yield.
Dissolving the conjugates for RP-HPLC can be difficult for highly hydrophobic sequences. The crude peptide may first be dissolved in DMSO or DMF (if containing redox-sensitive residues). Dilution of the solution is best accomplished by gradually adding the hydrophobic component of the mobile phase (e.g. acetonitrile) to the crude peptide solution and then adding water dropwise until the desired volume is reached. Additional organic solvent (DMSO or DMF) can be added if necessary.
Successful bead segregation is crucial to the quality and usefulness of the library and can be assessed by performing a chloranil test. Remove a small amount of the resin, deprotect the Fmoc group, add the chloranil reagents, and then examine the beads under a light microscope. A reddish ring on the exterior of the bead indicates successful segregation. If segregation is unsuccessful, remove the Boc group in the inner layer by incubation with 50% TFA/DCM solution for 1 h and repeat steps 3 through 10.
Resin splitting is best accomplished volumetrically, by suspending the resin in a known volume of 1:1 DMF/DCM, withdrawing equal aliquots of the suspension, and transferring them into individual synthesis vessels. Washing and deprotection steps can be conveniently performed in the main synthesis vessel as opposed to in each of the smaller positional synthesis vessels by combining the resin after each coupling.
Removal of the Mtt protecting group produces a yellow color due to the resulting methyltrityl cation, providing a visual indication for the reaction progression. 2% TIPS can be included in the deprotection solution to scavenge this cation and prevent reattachment.
The synthetic route to this compound can be found in Ref. 29.
Other thiol-containing acids (e.g. cysteine) can also be used for this purpose.
Intramolecular cyclization can be challenging depending on the sequence. Cyclization can be performed directly with the crude linear peptide following trituration, or with purified peptide under the same conditions. Reaction progress can be monitored by HPLC/MS. If necessary, additional DMSO can be added (up to 10% v/v) or higher pH is used (e.g. NaHCO3, pH 8.5) to facilitate the cyclization. Higher-order disulfide-mediated conjugates (e.g. dimers/trimers) may be separated by HPLC, reduced, and subjected to another round of intramolecular disulfide formation.
References
- 1.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews 23:3–25. [DOI] [PubMed] [Google Scholar]
- 2.Yang NJ, Hinner MJ. (2015) Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. Methods in molecular biology (Clifton, N.J.) 1266:29–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh SS. (2006) Preclinical pharmacokinetics: an approach towards safer and efficacious drugs. Curr Drug Metab 7:165–82. [DOI] [PubMed] [Google Scholar]
- 4.Arkin MR, Tang Y, Wells JA. (2014) Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol 21:1102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar V, Sanseau P, Simola DF, Hurle MR, Agarwal P. (2016) Systematic Analysis of Drug Targets Confirms Expression in Disease-Relevant Tissues. Scientific Reports 6:36205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zorzi A, Deyle K, Heinis C. (2017) Cyclic peptide therapeutics: past, present and future. Curr Opin Chem Biol 38:24–29. [DOI] [PubMed] [Google Scholar]
- 7.Gentilucci L, De Marco R, Cerisoli L. (2010) Chemical modifications designed to improve peptide stability: incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr Pharm Des 16:3185–203. [DOI] [PubMed] [Google Scholar]
- 8.Dechantsreiter MA, Planker E, Matha B, Lohof E, Holzemann G, Jonczyk A, Goodman SL, Kessler H. (1999) N-Methylated cyclic RGD peptides as highly active and selective alpha(V)beta(3) integrin antagonists. J Med Chem 42:3033–40. [DOI] [PubMed] [Google Scholar]
- 9.Corsetti M, Tack J. (2013) Linaclotide: A new drug for the treatment of chronic constipation and irritable bowel syndrome with constipation. United European Gastroenterology Journal 1:7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A and others. (2013) Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A 110:E3445–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frankel AD, Pabo CO. (1988) Cellular uptake of the tat protein from human immunodeficiency virus. Cell 55:1189–93. [DOI] [PubMed] [Google Scholar]
- 12.Green M, Loewenstein PM. (1988) Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 55:1179–88. [DOI] [PubMed] [Google Scholar]
- 13.Bechara C, Sagan S. (2013) Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett 587:1693–702. [DOI] [PubMed] [Google Scholar]
- 14.Reissmann S (2014) Cell penetration: scope and limitations by the application of cell-penetrating peptides. Journal of Peptide Science 20:760–784. [DOI] [PubMed] [Google Scholar]
- 15.Henninot A, Collins JC, Nuss JM. (2018) The Current State of Peptide Drug Discovery: Back to the Future? Journal of Medicinal Chemistry 61:1382–1414. [DOI] [PubMed] [Google Scholar]
- 16.Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. (2008) Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proc Natl Acad Sci U S A 105:12128–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Sayed A, Futaki S, Harashima H. (2009) Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. Aaps j 11:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erazo-Oliveras A, Muthukrishnan N, Baker R, Wang TY, Pellois JP. (2012) Improving the endosomal escape of cell-penetrating peptides and their cargos: strategies and challenges. Pharmaceuticals (Basel) 5:1177–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian Z, Liu T, Liu Y-Y, Briesewitz R, Barrios AM, Jhiang SM, Pei D. (2013) Efficient Delivery of Cyclic Peptides into Mammalian Cells with Short Sequence Motifs. ACS Chemical Biology 8:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qian Z, Martyna A, Hard RL, Wang J, Appiah-Kubi G, Coss C, Phelps MA, Rossman JS, Pei D. (2016) Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 55:2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang F, Wang Y, Zhang X, Zhang W, Guo S, Jin F. (2014) Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J Control Release 174:126–36. [DOI] [PubMed] [Google Scholar]
- 22.Bedewy W, Liao H, Abou-Taleb NA, Hammad SF, Nasr T, Pei D. (2017) Generation of a cell-permeable cycloheptapeptidyl inhibitor against the peptidyl-prolyl isomerase Pin1. Organic & Biomolecular Chemistry 15:4540–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Upadhyaya P, Qian Z, Selner NG, Clippinger SR, Wu Z, Briesewitz R, Pei D. (2015) Inhibition of Ras Signaling by blocking Ras-Effector interactions with cyclic peptides. Angew. Chem. Int. Ed 54:7602–7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lian W, Jiang B, Qian Z, Pei D. (2014) Cell-Permeable Bicyclic Peptide Inhibitors Against Intracellular proteins. J. Am. Chem. Soc 136:9830–9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang B, Pei D. (2015) A Selective, Cell-Permeable Nonphosphorylated Bicyclic Peptidyl Inhibitor against Peptidyl–Prolyl Isomerase Pin1. Journal of Medicinal Chemistry 58:6306–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao H, Pei D. (2017) Cell-permeable bicyclic peptidyl inhibitors against T-cell protein tyrosine phosphatase from a combinatorial library. Organic & Biomolecular Chemistry 15:9595–9598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trinh TB, Upadhyaya P, Qian Z, Pei D. (2016) Discovery of a Direct Ras Inhibitor by Screening a Combinatorial Library of Cell-Permeable Bicyclic Peptides. ACS Comb. Sci 18:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian Z, Xu X, Amacher JF, Madden DR, Cormet-Boyaka E, Pei D. (2015) Intracellular Delivery of Peptidyl Ligands by Reversible Cyclization: Discovery of a PDZ Domain Inhibitor that Rescues CFTR Activity. Angew. Chem. Int. Ed 54:5874–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian Z, Rhodes CA, McCroskey LC, Wen J, Appiah-Kubi G, Wang DJ, Guttridge DC, Pei D. (2017) Enhancing the Cell Permeability and Metabolic Stability of Peptidyl Drugs by Reversible Bicyclization. Angew. Chem. Int. Ed 56:1525–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dougherty PG, Qian Z, Pei D. (2017) Macrocycles as protein-protein interaction inhibitors. Biochem J 474:1109–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]