

Abstract
Background:
Tubulointerstitial inflammation (TII) in lupus nephritis (LN) is associated with a worse prognosis. Vimentin, a filamental antigen, is commonly targeted by in situ activated B-cells in TII. The prognostic importance of high serum anti-vimentin antibodies (AVAs) in LN and their relationship with common lupus autoantibody specificities is unknown. Herein we investigated associations between AVA isotypes, other autoantibodies, and response to mycophenolate mofetil (MMF) in the presence or absence of rituximab.
Methods:
The Translational Research Inititative in the Department of Medicine (TRIDOM) cross-sectional cohort of 99 lupus patients was assayed for IgG-, IgA- and IgM- AVAs, lupus associated and rheumatoid arthritis (RA) associated antibodies, and hierarchically clustered. Serum from baseline, 26 and 52 weeks from 132 LUNAR trial enrolled LN patients was also analysed and correlated with renal function up to week 78.
Results:
In TRIDOM, AVAs, especially IgM AVAs, clustered with IgG anti-dsDNA and away from anti-Sm and -RNP and RA associated antibodies. In LUNAR at baseline, AVAs correlated weakly with anti-dsDNA and more strongly with anti-cardiolipin titres. Regardless of treatment, IgG-, but not IgM- or IgA-, AVAs were higher at week 52 than at baseline. In contrast, anti-dsDNA titres declined, regardless of therapeutic regime. High IgG AVA titres at entry predicted less response to therapy.
Conclusion:
AVAs, especially IgG AVAs, are unique in distribution and response to therapy compared to other commonly measured autoantibody specificities. Furthermore, high-titre IgG AVAs identify LN patients resistant to conventional therapies. These data suggest that AVAs represent an independent class of prognostic autoantibodies.
Keywords: Vimentin, autoantibodies, Systemic Lupus Erythematosus, lupus nephritis, prognosis
INTRODUCTION
Disease heterogeneity makes managing lupus nephritis (LN) difficult. Standard treatment for LN involves high-dose corticosteroids in combination with either cyclophosphamide (CyP) or mycophenolate mofetil (MMF). Biological therapies, including rituximab (a chimeric anti-CD20 monoclonal antibody), are often efficacious in refractory cases of Systemic Lupus Erythematosus (SLE)[1–3] and as induction therapies[4, 5], and are thus included in the American College of Rheumatology (ACR) and the Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplantation Association (EULAR/ERA-EDTA) LN guidelines [6, 7]. Despite anecdotal successes, a phase III randomized, double blind, placebo controlled multi-center trial in patients with International Society of Nephrology(ISN)/Renal Pathology Society (RPS) class III or IV proliferative LN (the Lupus Nephritis Assessment with Rituximab study, “LUNAR”), did not demonstrate an increase in incidence of complete renal response at one year when rituximab was added to MMF and corticosteroids [8]. This was despite greater improvements in IgG anti-double stranded DNA (dsDNA) titre and serum C3 and C4 with rituximab. Other autoantibodies in LN have different sensitivities to rituximab. For example, rituximab does not reduce anti-histone, -Sjögren’s-syndrome-related antigen A (SSA) and -ribonucleoprotein (RNP) antibodies[9]. Antibody specificities predictive of response to standard LN therapies, or rituximab [10, 11] have not been identified.
In a predominantly African American cohort, tubulointerstitial inflammation (TII) was more prognostic of kidney failure than proliferative classes of glomerulonephritis (GN)[12]. The likely contribution of the active in situ adaptive immune response to loss of renal function was further highlighted in a predominantly white European cohort[13], in whom elevated renal gene expression signatures of B-cells, T-cells and antigen presentation were found to be more associative with impaired renal function than GN class or routinely assayedserological features (including anti-dsDNA and C3)[13]. Unlike GN, TII is often associated with tertiary lymphoid organogenesis (TLO), comprising plentiful T cells, dendritic cells, B-cells and plasma cells[14–17]. Moreover, B-cells in TII are often activated, express antibodies with extensive somatic hypermutations and are clonally restricted[14, 18]. Activated B-cells in TII are distinct from classic B-cells described in lupus, which express antibodies to nuclear antigens. Monoclonal antibodies (mAbs) cloned from TII sorted CD19+CD27+CD38+ plasmablasts, or clonally expanded laser captured Ki-67+ or CD38+ cells, preferentially target cytoskeletal and/or cytoplasmic antigens[18]. Most commonly, these antibodies target vimentin[18], a type III intermediate filamental protein. It is expressed by lymphocytes[19] and macrophages[20] and upregulated during epithelial to mesenchymal transition[21].
Moreover it can be found on the extracellular side of the plasma membrane, and is secreted by cells including macrophages and endothelial cells[20, 22, 23]. Therefore, vimentin is readily available to soluble and B-cell surface IgG (sIg) at various inflammatory sites including the kidney[18], joints[24], lung[25] and nervous system[26]. Notably, high serum IgG AVA titres (measured using purified bovine antigen on superepoxy protein array slides) correlate with severe TII on renal biopsy[18]. Differences in respective antigens driving selection, and niches in which they are produced, suggest that when compared to autoantibodies associated with lupus GN, those selected in TII would differ in regulation and sensitivity to conventional therapies.
MATERIALS AND METHODS
Patient and Public Involvement
Ninety-nine patients satisfying the ACR criteria for SLE [27] , selected from the Translational Research Inititative in the Department of Medicine (TRIDOM) patient database at the University of Chicago, constituted the cross-sectional cohort (table 1). Aliquots of archived serum samples from 132 patients enrolled in the LUNAR trial ([8] and Supplementary Materials and Methods) were analysed at baseline, and (excluding discontinuing patients) weeks 26 and 52. Patient information was further collected up until week 78. Ethical approval for collection and usage of samples at the University of Chicago was granted by IRB15065B.
Table 1.
TRIDOM Mixed Lupus Cross-Sectional Cohort (n=99)
| Age, median (range) | 38 (18-87) |
| Frequency, n(%) | |
| Female Gender, | 95 (96.0) |
| Ethnicity | |
| African-American | 91 (91.9) |
| European-American | 4 (4.0) |
| Hispanic | 4 (4.0) |
| Organ Involvement | |
| Lupus Nephritis* | 22 (22.2) |
| Mucocutaneous | 22 (22.2) |
| Musculoskeletal | 9 (9.1) |
| Neuropsychiatric | 0 (0) |
| Vascular | 1 (1.0) |
| Cardiovascular & Respiratory | 2 (2.0) |
| Immunologic | 55 (55.6) |
| Hematologic | 15 (15.2) |
LN details, and serological associations with different organ involvements are in Supplementary File Tables S1–S2
Vimentin cloning and protein purification
Full length (aa 1-466) and a C-terminal (aa 259-466) truncation of human vimentin were cloned into expression vectors for overexpression in E.coli. Proteins were purified as described (Supplementary Materials and Methods and [25, 28]).
ELISAs
Anti-vimentin antibodies were titrated using a custom ELISA (see Supplementary Materials and Methods). Two-fold dilutions of standards (frozen aliquots of pooled AVAhi lupus serum (assigned a value of 1000 arbitrary units [AU]) were added to generate standard curves for titre interpolation. Catalogue references for clinical ELISAs are given in the Supplementary Materials and Methods. Non-AVA antibody titres in the LUNAR cohort were acquired during and subsequent to the original 52 week multi-center study[8].
Statistical analysis
GraphPad Prism v.7 (GraphPad Software, Inc., La Jolla, CA, USA) was used for titre interpolations, graphs, heat maps and analyses unless otherwise stated, and dendrograms constructed using Rstudio ([29, 30]). Longitudinal analyses on the effects of AVAhigh status on clinical paramenters at respective time points was performed by ANOVA and over 78 weeks using a linear mixed effect model (Supplementary Materials and Methods).
RESULTS:
IgG AVAs are distinct from anti-dsDNA, -RNP, -Sm and rheumatoid factor
Our previous lupus study[18] measured reactivity of mAbs cloned from in situ activated B-cells in TII, and serum IgG, using a custom protein array coated with bovine tissue expressed vimentin. Here, we assayed vimentin reactivity with a custom high throughput ELISA using hexa-His-tagged recombinant vimentin purified from E. coli (Supplementary Figure S1). Five representative TII mAbs previously demonstrated to have bovine vimentin reactivity[18], herein reacted with bound recombinant human antigen and had diminished reactivity with in vitro citrullinated antigen (Supplementary Figure S1). Consistent with our initial study, three other HEp-2 negative TII mAbs, without bovine vimentin reactivity on protein array [18], had undetectable reactivity with the vimentin by ELISA. Therefore, AVAs are commonly selected in situ in TII and are different than the anti-citrullinated protein antibodies that are associated with rheumatoid arthritis (RA).
Common Ig isotypes were subsequently assayed for recombinant human vimentin reactivity using lupus serum. Firstly, ninety-nine serum samples from a cross-sectional Lupus cohort (22% LN), The Translational Research Inititative in the Department of Medicine (“TRIDOM”, Table 1 and Supplementary Table S1), were used to determine whether AVA titres in a broad range of lupus cases positively correlated (Spearman’s rho correlation) with routinely assayed lupus associated IgG antibodies, anti-dsDNA, - RNP and -Smith (Sm) (Supplementary Table S2). Although the titres of IgG to RNP and Sm co-varied (rho = 0.70, 95% C.I = 0.58 to 0.79, p < 0.0001), and IgG anti-dsDNA positively correlated with anti-RNP (rho = 0.30, 95% C.I = 0.10 to 0.48, p = 0.003) and -Sm (rho = 0.28, 95% C.I = 0.08 to 0.46, p = 0.0056), no significant positive correlations were observed between any AVA isotype titres and these lupus-associated autoantibody specificities. These data support AVAs being from B-cells differentially regulated (and/or selected) to those secreting common ANA specificities. We next determined whether AVA titre distributions were similar to another antibody type, rheumatoid factor (RF), which is associated with RA and can be selected in inflamed synovium [31]. Both IgM (rho = 0.56, 95% C.I: 0.40 to 0.69, p < 0.0001) and IgA (rho = 0.29, 95% C.I: 0.09 to 0.47, p = 0.0040) RF isotypes, but not IgG, positively correlated with AVA titres. These data suggest that IgG AVAs are independent of these commonly assayed autoantibody specificities.
Antibody titres were next hierarchically clustered. In addition, we titrated anti-cyclic citrullinated peptide (-CCP) antibodies, predicted to cluster with RF. Indeed, anti-RNP and -Sm clustered most closely amongst the lupus associated autoantibodies and the RA associated antibodies RF and anti-CCP clustered as a separate tree (Figure 1). IgM AVA titres clustered most tightly with IgG anti-dsDNA, whereas IgG and IgA isotypes did not cluster tightly with either lupus or RA-associated antibodies. No differences were observed between nephritic and non-nephritic patients when analysed separately (data not shown). These data suggest that IgG AVAs capture different features from commonly assayed autoantibody specificities.
Figure 1.
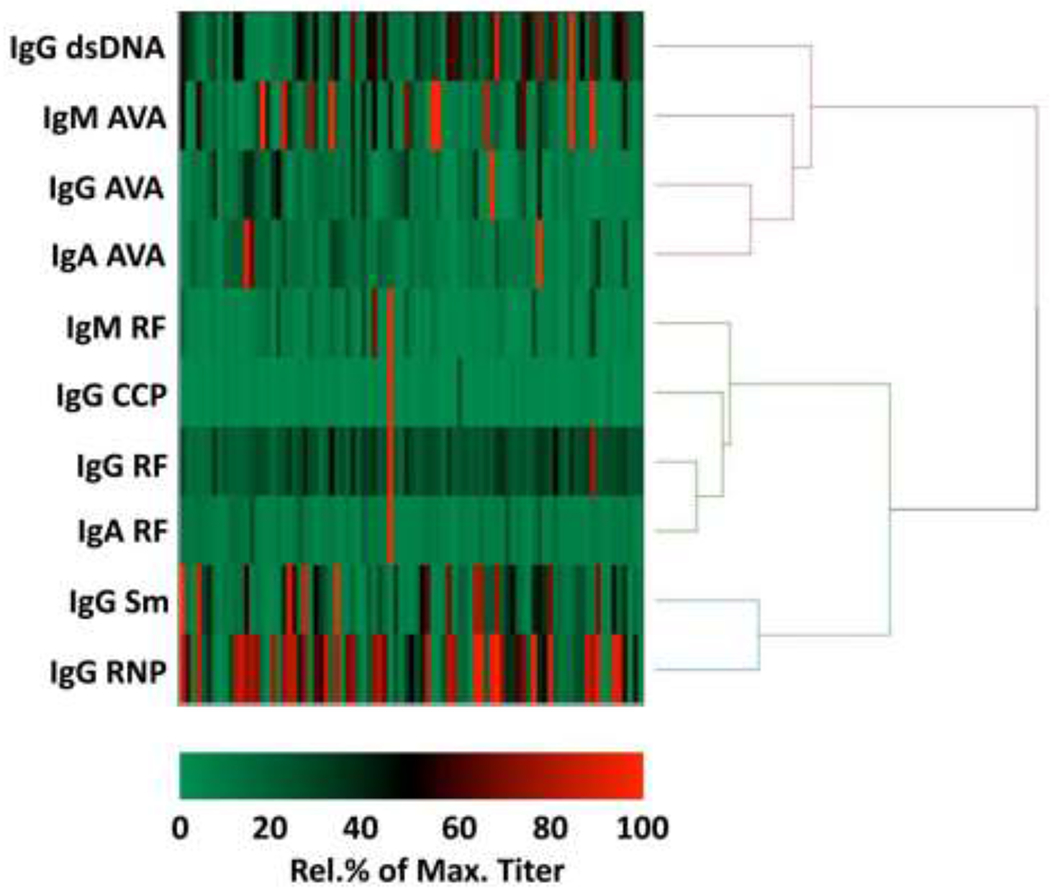
Clustered heat map of autoantibody titres in serum samples from the TRIDOM lupus cross-sectional cohort. Heat map cells represent individual patient serum sample titres, colored (see key below heat map) as percentages, relative to the maximum titre for the respective antibody specificity in the cohort. Titres of antibodies commonly assayed in the diagnosis of SLE (IgG anti-dsDNA, -RNP, -Sm) and RA (IgG anti-CCP, IgG RF, IgM RF and IgA RF) were compared to IgG, IgA and IgM isotypes of AVAs. A hierarchical cluster analysis was performed between the respective antibody specificities, using Ward’s minimum variance method and corresponding dendrogram displayed. Only patients for whom all indicated antibodies were titrated were included in the heat map. AVA=anti-vimentin antibody; CCP= cyclic citrullinated peptide 3; dsDNA=double-stranded DNA; RF=rheumatoid factor; RNP=ribonucleoprotein; Sm=Smith.
In proliferative LN, AVAs cluster weakly with lupus-associated antibodies
We then focused specifically on proliferative LN (using sera from 132 patients from the LUNAR trial) and how AVAs associated with a greater range of routinely measured lupus-associated antibodies (Figure 2 and Supplementary Table S3). Baseline titres of AVAs and other lupus-associated antibodies were again clustered (Figure 2). As in the TRIDOM cohort, IgG-anti-RNP and -Sm strongly co-varied (Spearman’s rho = 0.77, 95% C.I = 0.69 to 0.83, p < 0.0001), as did IgG- and IgA-AVAs (rho = 0.49, 95% C.I = 0.34 to 0.61, p < 0.0001). As predicted, titres of IgG anti-cardiolipin (ACA) and IgG anti-β2 glycoprotein I also strongly co-varied (rho = 0.571 , 95% C.I =0.438 to 0.679, p < 0.0001), forming a separate dendrogram branch.
Figure 2.
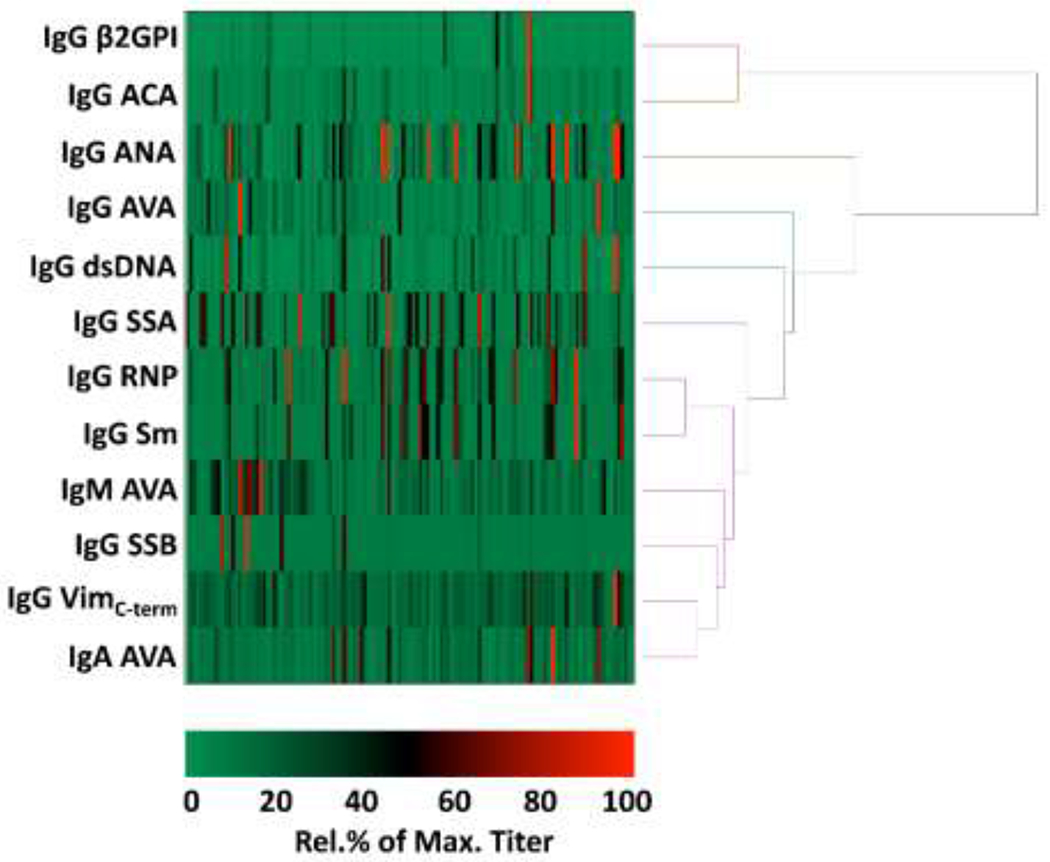
Clustering of lupus associated autoantibody titres LUNAR baseline serum samples. Heat maps and dendrograms were plotted as for Figure 1, but for a larger cohort of lupus patients (and more extensive range of lupus associated autoantibodies) with class III and/or IV proliferative nephritis enrolled in the LUNAR trial. ACA=anti-cardiolipin; ANA=anti-nuclear antibody; 2GPI= 2 glycoprotein I; SSA=Sjögren’s syndrome antigen A, SSB=Sjögren’s syndrome antigen B; VimC-term=recombinant vimentin C-terminus (residues 260-466).
Of the lupus-associated IgG autoantibodies, IgG AVAs correlated most closely with anti-cardiolipin antibody titres (rho = 0.30, 95% C.I = 0.13 to 0.45, p = 0.0005) and less with anti-dsDNA (rho = 0.25, 95% C.I = 0.07 to 0.40, p = 0.0047). Antibodies reactive with residues 259-466 of the Vimentin C-terminus (anti-VimC-term) have previously been shown to be at higher concentrations in the lungs of sarcoidosis patients who are HLA-DRB1*03+[25]. In the LUNAR cohort these IgG antibodies co-varied most with IgG AVA (rho = 0.53, 95% C.I = 0.40 to 0.65, p < 0.0001), and IgA AVA (rho = 0.41, 95% C.I = 0.25 to 0.55, p < 0.0001), but poorly with the other lupus associated antibodies (Supplementary Table S3).
IgG, but not IgA or IgM, AVA titres rise during treatment
We next determined whether AVA titres were suppressed by MMF and corticosteroids alone (placebo group) or in combination with rituximab. As opposed to anti-dsDNA titres, which fall following either treatment[8], IgG AVAs at week 52 substantially increased among patients in the placebo group (median change 195%) whereas only a modest increase (28%) from baseline was observed in the rituximab group (p < 0.0001, Figure 3a–b, Supplementary Figure S3ai). In comparison, IgA AVAs did not markedly change in the placebo group (median change −6%) but decreased mildly in the rituximab group (−22%, p = 0.005 for comparison between treatments), where the change from baseline achieved statistical significance at week 52 (p = 0.0012 vs. baseline). For IgM AVAs, absolute titres did not significantly differ between treatment groups (Supplementary Figure S3c) or between baseline and 52 weeks (Figure 3b). In summary, rituximab blunted the increase in IgG AVAs that was observed in the placebo group, and led to greater reduction in IgA AVAs at week 52. When IgG anti-VimC-term titres were considered separately, a moderate reduction from baseline was observed in the rituximab group only (median relative percentage decrease of 21% from baseline to week 52, change in IgG AVA vs change in IgG anti-VimC-term at week 52: p<0.0001 (Wilcoxon matched-paired test) Supplementary Figure S4a–c). This data demonstrates that although IgG anti-Vimc-term are moderately more sensitive to Rituximab than IgG AVA, generally IgG antibodies reactive with various vimentin epitopes are not profoundly altered even with the addition of Rituximab.
Figure 3.
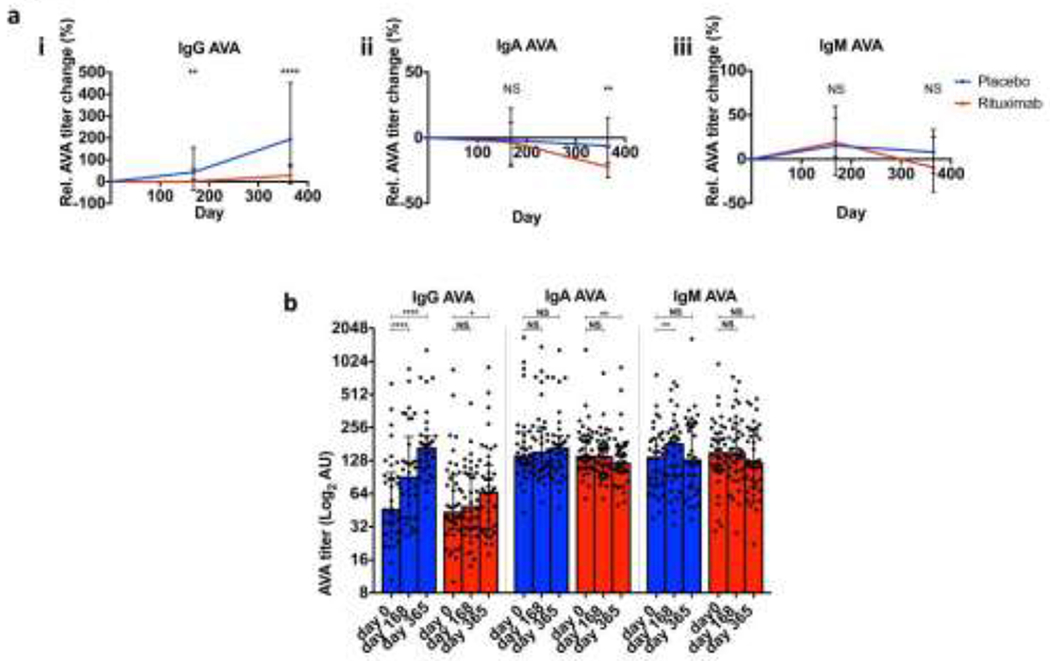
Effect of different treatment regimens on titres of AVAs. (a) Relative (percentage) change in labelled isotypes of serum AVA from baseline in LUNAR patients. Medians and interquartile ranges (IQRs) from each group of patients’ titres are plotted for respective treatments. Mann Whitney tests were performed between serum titres in treatment groups for each time point. (b) Raw AVA titres at different time points for respective treatments (medians and IQRs are plotted, and Wilcoxon matched pairs signed rank tests performed between baseline and the indicated time points). Each dot represents an individual patient’s AVA titre. 0.01< *p <0.05, 0.01< **p <0.001, 0.001< ***p < 0.0001, ****p <0.0001, NS=non-significant.
Baseline AVA titres predict worse response to therapy
We next examined how baseline AVA titres correlated with subsequent clinical response using mixed effects linear regression. Patients were divided into AVAhigh and AVAlow groups based on baseline AVA titres (above or below the median for each isotype, Supplementary Table S4). When comparing AVAhigh versus AVAlow subsets, trends for change in urine protein/creatinine ratio (UPCR) over time did not appear to qualitatively differ by treatment group (Supplementary Figure S5). Patients from both treatment groups were therefore pooled for subsequent comparisons (Figure 4). Less reduction in proteinuria from baseline over 78 weeks was observed for patients with baseline high titres of IgG AVA (p < 0.0001 for overall effect; mean difference in change of UPCR between high and low groups at week 78 = 1.42g/g), IgA AVA (p = 0.0069; mean difference: 0.88g/g), and IgG anti-VimC-term AVA (p = 0.0011; mean difference = 0.82g/g). In comparison, baseline IgM AVA status was not associated with subsequent difference in change of UPCR (p = 0.75) (Supplementary Table S5).
Figure 4.
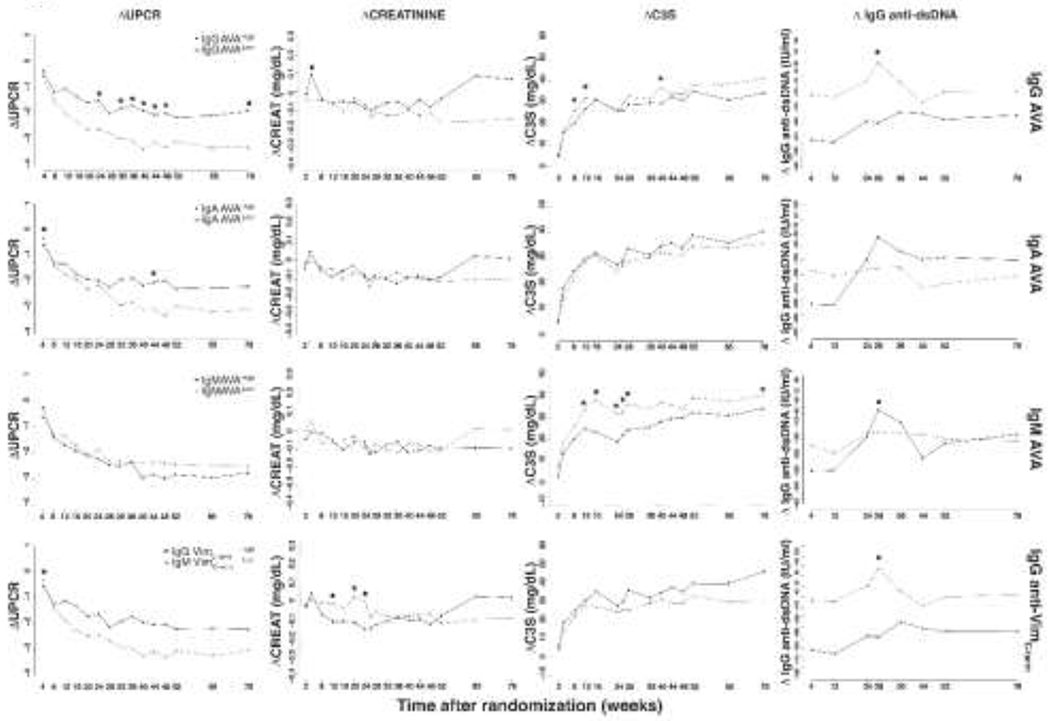
Disease activity changes (Δ) from baseline in LUNAR patients with high or low AVA titres. For each AVA subtype (sparated by row, labelled on the right hand side), all patients (both treatment regimens combined) were divided into AVA high (above median baseline AVA subtype titre) or AVA low (below median baseline AVA subtype titre) groups. ANOVA tests were performed between groups at each time point, and where significant indicated. 0.01<* p <0.05. UPCR=Urine protein/serum creatinine ratio; CREAT=serum creatinine; C3S=soluble complement factor C3. For each row, legends in the left most graph indicate the serological stratification of LUNAR patients.
Change in serum creatinine did not substantially differ by 52 weeks by baseline AVA status (Figure 4). Small increases in serum creatinine were observed among IgG AVAhigh, IgA AVAhigh, IgM AVAlow, and IgG anti-VimC-termhigh patients at weeks 65 and 78. However, these differences did not achieve statistical significance among the pooled treatment groups (Figure 4). Upon subgroup analysis by treatment (Supplementary Table S6), the greatest difference was observed for IgG AVA status, and was only significant (p < 0.05) in the rituximab group (IgG AVAhigh versus IgG AVAlow mean change from baseline at week 78 of +0.22mg/dL versus −0.18mg/dL with rituximab, compared with −.10mg/dL versus −.10mg/dL in its absence). A similar pattern, but of insignificant difference was also seen within the rituximab treated patients with IgA AVAhigh or IgG anti-VimC-term AVAhigh titres. In contrast, high titres of IgM AVA, but not IgG-, IgA-, or , IgG anti-VimC-term, were associated with less improvement in serum C3 over 78 weeks (p = 0.0101 for overall effect; Figure 4). Finally, as reported previously, anti-dsDNA antibody titres decreased over time irrespective of baseline AVA status, and baseline IgG AVAhigh or IgG anti-VimC-termhigh status was associated with greater anti-dsDNA titre reduction over 78 weeks (p < 0.0001 for both).
DISCUSSION
We have previously observed that high AVA titres are associated with severe TII [18]. Herein, we demonstrate that the IgG isotype of these TII-associated antibodies identifies a unique subset of LN patients poorly responsive to MMF and steroids, in the presence, or absence, of rituximab. Whether high IgG AVA titres also associate with poor response to cyclophosphamide awaits study. An absence of strong correlation was seen between AVA titres and routinely titrated lupus anti-nuclear autoantibodies. Furthermore, the class of antibodies that clustered most closely with AVAs, anti-dsDNA antibodies, behaved very differently in response to therapy. This suggests that each broad class of autoantibody, nuclear and filamental, arises from B cells that are selected and/or maintained by different mechanisms.
Indeed, in the LUNAR trial, AVA titres behaved differently in response to therapy than other routinely assayed lupus associated autoantibody specificities. In both treatment groups, IgG AVA titres increased by week 52, though much less so among patients treated with rituximab. In contrast, IgG anti-dsDNA titres decreased in both groups, more so with rituximab [8]. Importantly, the association with less improvement in proteinuria in the IgG AVAhigh patient group did not appear to be accounted for by anti-dsDNA titre changes, as the average decrease in IgG anti-dsDNA titres was greater in the IgG AVAhigh patient group. These data indicate that IgG AVA titres at baseline, and not anti-dsDNA titres, or changes in anti-dsDNA titres with therapy, may be most prognostically meaningful. IgG anti-RNP have been reported as unchanged during therapy [9], and are more helpful in defining disease subsets [32] than for assessing disease activity. In contrast, AVAs were both resistant to therapy and prognostically meaningful. We propose that this is because AVAs reflect TII activity [18], an LN manifestation resistant to current therapies. Unfortunately, in the LUNAR trial there was no centralized repository for entry biopsies and repeat biopsies were not obtained. Therefore, correlations of the degree of TII with AVA titres could not be made. However, our previous cross-sectional study[18] demonstrated a strong correlation of high serum IgG AVA with TII severity. We do not know if persistence of IgG AVAs reflects a persistence or progression of TII. That severe TII patients are more resistant to current therapies[12] suggests that this is the case. It will be critical to relate AVA titres, serial biopsies and renal function in future clinical trials.
At week 52 of the LUNAR trial, rituximab was associated with lower titres of both IgG- and IgA-AVAs compared with MMF and corticosteroids alone, with the difference being most pronounced for IgG AVAs. The apparent responsiveness of IgM-, and to a lesser degree, IgA-AVAs, to MMF and corticosteroids alone suggests they arise from distinct B cell populations compared with IgG AVAs. Increased IgG AVA titres following rituximab withdrawal, versus maintained non-IgG AVA, anti-RNP, -SSA and –Histone [9], and reduced anti-dsDNA titres [8] suggests that the B cell populations secreting these specificities are fundamentally different.
How IgG AVA secreting B cells are different is not known. It is possible that these cells rapidly repopulate following rituximab clearance and/or they are long lived plasma cells that reside in an anatomical niche resistant to therapy. In addition, they could express lower levels of CD20 and/or internalize rituximab more effectively. Indeed, Reddy et.al demonstrated that rituximab is internalized more readily by lupus B-cells than those from RA patients or healthy controls. Furthermore, internalization was inversely proportional to B-cell depletion efficacy[33]. It is not known if differential internalization correlates with AVA titres or the presence of specific B cell subsets.
Vimentin, a multimeric filament forming molecule[34] could, due to its antigenic polyvalency, cross-link surface Ig (sIg) thereby providing a strong B cell activation stimulus. It has also been suggested that vimentin inherently stimulates via the innate pathway, as a danger associated molecular pattern (DAMP), analogous to f-actin via DNGR-1[35, 36]. One candidate DAMP receptor (DAMPR) for vimentin is Dectin-1[37]. Furthermore, molecules that associate with vimentin, either directly or as cellular debris [25], might stimulate B cells through other DAMPRs. Therefore, through multiple mechanisms, vimentin might potently activate AVA secreting B cells.
Vimentin expression is induced by TNF-α via a PKC dependent[20] mechanism. Therefore, vimentin is commonly upregulated in the context of inflammation [18, 24–26, 38]. Indeed, AVAs are also relatively common and have been reported in multiple states including rheumatoid arthritis[39], transplantation rejection[40–42], hepatitis, [43] pulmonary sarcoidosis[25], idiopathic pulmonary fibrosis [44], and infection (including acute viral hepatitis)[45]. We, propose that vimentin is a molecular pattern and potent antigen of inflammation. Whether AVAs are inflammatory, or serve to limit inflammation, is likely dependent upon multiple factors including immunoglobulin Fc region sialylation[46].
The distribution of variable region mutations in AVAs cloned from lupus kidney TII suggests T-dependent selection[18]. T-dependent immunity might be a common feature of anti-vimentin immune responses. In pulmonary sarcoidosis, the sarcoid lung has elevated anti-VimC-term antibodies and larger TLOs when patients are HLA-DRB1*03+[25]. As HLA-DRB1*03 can present vimentin C-terminal peptide[47] these data suggest that local anti-VimC-term titres are due to a cognate B-:T-cell activation network. Whether HLA-DRB1*03 (associated with susceptibility to SLE and high anti-SSA, -SSB and -Sm titres [48]), or other MHC class II polymorphisms, are associated with high AVA titres in lupus is an open question.
Extensive LN research has focused on GN and the auto-antibodies most closely associated with it, including those against dsDNA. In contrast, TII is associated with anti-filamental antibodies, most notably AVAs. Herein, we demonstrate that titres of these antibodies identify those resistant to current therapies. Understanding the mechanisms that give rise to AVAs holds the promise of new therapeutic targets and more efficacious therapies for those at greatest risk for progressive renal disease.
Supplementary Material
Acknowledgments
FUNDING:
Work conducted at the University of Chicago was funded by NIH Autoimmunity Center of Excellence grants (AI082724 and AR55646). MKO is funded by medical scientist training program grant T32HD007009.
Footnotes
COMPETING INTERESTS:
MC, JD & MJT are employee of Roche/Genentech. LLD is an employee of Janssen Biopharma which is part of the Johnson and Johnson family of companies. All other authors have nothing to disclose
REFERENCES
- 1.Edelbauer M, Jungraithmayr T, and Zimmerhackl LB, Rituximab in childhood systemic lupus erythematosus refractory to conventional immunosuppression: case report. Pediatr Nephrol, 2005. 20(6): p. 811–3. [DOI] [PubMed] [Google Scholar]
- 2.Fra GP, Avanzi GC, and Bartoli E, Remission of refractory lupus nephritis with a protocol including rituximab. Lupus, 2003. 12(10): p. 783–7. [DOI] [PubMed] [Google Scholar]
- 3.van Vollenhoven RF, et al. , Biopsy-verified response of severe lupus nephritis to treatment with rituximab (anti-CD20 monoclonal antibody) plus cyclophosphamide after biopsy-documented failure to respond to cyclophosphamide alone. Scand J Rheumatol, 2004. 33(6): p. 423–7. [DOI] [PubMed] [Google Scholar]
- 4.Condon MB, et al. , Prospective observational single-centre cohort study to evaluate the effectiveness of treating lupus nephritis with rituximab and mycophenolate mofetil but no oral steroids. Ann Rheum Dis, 2013. 72(8): p. 1280–6. [DOI] [PubMed] [Google Scholar]
- 5.Gracia-Tello B, Ezeonyeji A, and Isenberg D, The use of rituximab in newly diagnosed patients with systemic lupus erythematosus: long-term steroid saving capacity and clinical effectiveness. Lupus Sci Med, 2017. 4(1): p. e000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertsias GK, et al. , Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of adult and paediatric lupus nephritis. Ann Rheum Dis, 2012. 71(11): p. 1771–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hahn BH, et al. , American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res (Hoboken), 2012. 64(6): p. 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rovin BH, et al. , Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum, 2012. 64(4): p. 1215–26. [DOI] [PubMed] [Google Scholar]
- 9.Cambridge G, et al. , B cell depletion therapy in systemic lupus erythematosus: effect on autoantibody and antimicrobial antibody profiles. Arthritis Rheum, 2006. 54(11): p. 3612–22. [DOI] [PubMed] [Google Scholar]
- 10.Lal P, et al. , Inflammation and autoantibody markers identify rheumatoid arthritis patients with enhanced clinical benefit following rituximab treatment. Arthritis Rheum, 2011. 63(12): p. 3681–91. [DOI] [PubMed] [Google Scholar]
- 11.Sellam J, et al. , B cell activation biomarkers as predictive factors for the response to rituximab in rheumatoid arthritis: a six-month, national, multicenter, open-label study. Arthritis Rheum, 2011. 63(4): p. 933–8. [DOI] [PubMed] [Google Scholar]
- 12.Hsieh C, et al. , Predicting outcomes of lupus nephritis with tubulointerstitial inflammation and scarring. Arthritis Care Res (Hoboken), 2011. 63(6): p. 865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pamfil C, et al. , Intrarenal activation of adaptive immune effectors is associated with tubular damage and impaired renal function in lupus nephritis. Ann Rheum Dis, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang A, et al. , In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J Immunol, 2011. 186(3): p. 1849–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ko K, et al. , Bcl-2 as a Therapeutic Target in Human Tubulointerstitial Inflammation. Arthritis Rheumatol, 2016. 68(11): p. 2740–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liarski VM, et al. , Cell distance mapping identifies functional T follicular helper cells in inflamed human renal tissue. Sci Transl Med, 2014. 6(230): p. 230ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liarski VM, et al. , Quantifying in situ adaptive immune cell cognate interactions in humans. Nat Immunol, 2019. 20(4): p. 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinloch AJ, et al. , Vimentin is a dominant target of in situ humoral immunity in human lupus tubulointerstitial nephritis. Arthritis Rheumatol, 2014. 66(12): p. 3359–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huet D, et al. , SC5 mAb represents a unique tool for the detection of extracellular vimentin as a specific marker of Sezary cells. J Immunol, 2006. 176(1): p. 652–9. [DOI] [PubMed] [Google Scholar]
- 20.Mor-Vaknin N, et al. , Vimentin is secreted by activated macrophages. Nat Cell Biol, 2003. 5(1): p. 59–63. [DOI] [PubMed] [Google Scholar]
- 21.Zeisberg M and Neilson EG, Biomarkers for epithelial-mesenchymal transitions. J Clin Invest, 2009. 119(6): p. 1429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teshigawara K, et al. , A novel compound, denosomin, ameliorates spinal cord injury via axonal growth associated with astrocyte-secreted vimentin. Br J Pharmacol, 2013. 168(4): p. 903–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu B, et al. , The endothelial cell-specific antibody PAL-E identifies a secreted form of vimentin in the blood vasculature. Mol Cell Biol, 2004. 24(20): p. 9198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tilleman K, et al. , Synovial detection and autoantibody reactivity of processed citrullinated isoforms of vimentin in inflammatory arthritides. Rheumatology (Oxford), 2008. 47(5): p. 597–604. [DOI] [PubMed] [Google Scholar]
- 25.Kinloch AJ, et al. , In Situ Humoral Immunity to Vimentin in HLA-DRB1*03(+) Patients With Pulmonary Sarcoidosis. Front Immunol, 2018. 9: p. 1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Didangelos A, et al. , High-throughput proteomics reveal alarmins as amplifiers of tissue pathology and inflammation after spinal cord injury. Sci Rep, 2016. 6: p. 21607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan EM, et al. , The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum, 1982. 25(11): p. 1271–7. [DOI] [PubMed] [Google Scholar]
- 28.Cha SC, et al. , Nonstereotyped lymphoma B cell receptors recognize vimentin as a shared autoantigen. J Immunol, 2013. 190(9): p. 4887–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galili T, dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics, 2015. 31(22): p. 3718–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu Z, Eils R, and Schlesner M, Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics, 2016. 32(18): p. 2847–9. [DOI] [PubMed] [Google Scholar]
- 31.Egeland T, et al. , Quantitation of cells secreting rheumatoid factor of IgG, IgA, and IgM class after elution from rheumatoid synovial tissue. Arthritis Rheum, 1982. 25(12): p. 1445–50. [DOI] [PubMed] [Google Scholar]
- 32.Hoffman IE, et al. , Specific antinuclear antibodies are associated with clinical features in systemic lupus erythematosus. Ann Rheum Dis, 2004. 63(9): p. 1155–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reddy V, et al. , Internalization of rituximab and the efficiency of B Cell depletion in rheumatoid arthritis and systemic lupus erythematosus. Arthritis Rheumatol, 2015. 67(8): p. 2046–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Traub P and Vorgias CE, Involvement of the N-terminal polypeptide of vimentin in the formation of intermediate filaments. J Cell Sci, 1983. 63: p. 43–67. [DOI] [PubMed] [Google Scholar]
- 35.Ahrens S, et al. , F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity, 2012. 36(4): p. 635–45. [DOI] [PubMed] [Google Scholar]
- 36.Zhang JG, et al. , The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity, 2012. 36(4): p. 646–57. [DOI] [PubMed] [Google Scholar]
- 37.Thiagarajan PS, et al. , Vimentin is an endogenous ligand for the pattern recognition receptor Dectin-1. Cardiovasc Res, 2013. 99(3): p. 494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghosh S, et al. , Association of filamin A and vimentin with hepatitis C virus proteins in infected human hepatocytes. J Viral Hepat, 2011. 18(10): p. e568–77. [DOI] [PubMed] [Google Scholar]
- 39.Bang H, et al. , Mutation and citrullination modifies vimentin to a novel autoantigen for rheumatoid arthritis. Arthritis Rheum, 2007. 56(8): p. 2503–11. [DOI] [PubMed] [Google Scholar]
- 40.Carter V, et al. , Vimentin antibodies: a non-HLA antibody as a potential risk factor in renal transplantation. Transplant Proc, 2005. 37(2): p. 654–7. [DOI] [PubMed] [Google Scholar]
- 41.Jurcevic S, et al. , Antivimentin antibodies are an independent predictor of transplant-associated coronary artery disease after cardiac transplantation. Transplantation, 2001. 71(7): p. 886–92. [DOI] [PubMed] [Google Scholar]
- 42.Wheeler CH, et al. , Characterization of endothelial antigens associated with transplant-associated coronary artery disease. J Heart Lung Transplant, 1995. 14(6 Pt 2): p. S188–97. [PubMed] [Google Scholar]
- 43.Kurki P, et al. , Cytoskeleton antibodies in chronic active hepatitis, primary biliary cirrhosis, and alcoholic liver disease. Hepatology, 1983. 3(3): p. 297–302. [DOI] [PubMed] [Google Scholar]
- 44.Li FJ, et al. , Autoimmunity to Vimentin Is Associated with Outcomes of Patients with Idiopathic Pulmonary Fibrosis. J Immunol, 2017. 199(5): p. 1596–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown C, et al. , Autoantibodies to intermediate filaments in acute viral hepatitis A, B and non-A, non-B are directed against vimentin. J Clin Lab Immunol, 1986. 19(1): p. 1–4. [PubMed] [Google Scholar]
- 46.Kaneko Y, Nimmerjahn F, and Ravetch JV, Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science, 2006. 313(5787): p.670–3. [DOI] [PubMed] [Google Scholar]
- 47.Wahlstrom J, et al. , Autoimmune T cell responses to antigenic peptides presented by bronchoalveolar lavage cell HLA-DR molecules in sarcoidosis. Clin Immunol, 2009. 133(3): p. 353–63. [DOI] [PubMed] [Google Scholar]
- 48.Graham RR, et al. , Specific combinations of HLA-DR2 and DR3 class II haplotypes contribute graded risk for disease susceptibility and autoantibodies in human SLE. Eur J Hum Genet, 2007. 15(8): p. 823–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.