

Abstract
The limited delivery of chemotherapy agents to cancer cells and the nonspecific action of these agents are significant challenges in oncology. We have previously developed a customizable drug delivery and activation system in which a nucleic acid functionalized gold nanoparticle (Au-NP) delivers a drug that is selectively activated within a cancer cell by the presence of an mRNA unique to the cancer cell. The amount of drug released from sequestration to the Au-NP is determined by both the presence and the abundance of the cancer cell specific mRNA in a cell. We have now developed this technology for the potent, but difficult to deliver, topoisomerase I inhibitor SN-38. Herein, we demonstrate both the efficient delivery and selective release of SN-38 from gold nanoparticles in Ewing sarcoma cells with resulting efficacy in vitro and in vivo. These results provide further preclinical validation for this novel cancer therapy and may be extendable to other cancers that exhibit sensitivity to topoisomerase I inhibitors.
Graphical Abstract
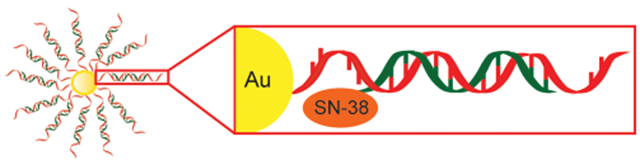
INTRODUCTION
Many cancers remain incurable despite maximally intensive cytotoxic chemotherapy. Furthermore, chemotherapy is often associated with significant short- and long-term morbidities. To address this need for more efficacious and less toxic cancer therapies, we previously developed a novel, customizable drug delivery and activation system. In this approach, a gold nanoparticle (Au-NP) delivers a drug that is selectively activated within the cancer cell by the presence of an mRNA unique to the cancer cell (Figure 1).1,2 The amount of drug released from sequestration to the Au-NP is determined by both the presence and the abundance of the cancer cell specific mRNA in a cell. We showed both the efficient delivery and the selective release of the kinase inhibitor dasatinib from Au-NPs in leukemia cells. Furthermore, these Au-NPs exhibited efficacy in vitro and in vivo against leukemia cells but reduced toxicity against hematopoietic stem cells and T cells relative to free dasatinib.
Figure 1.
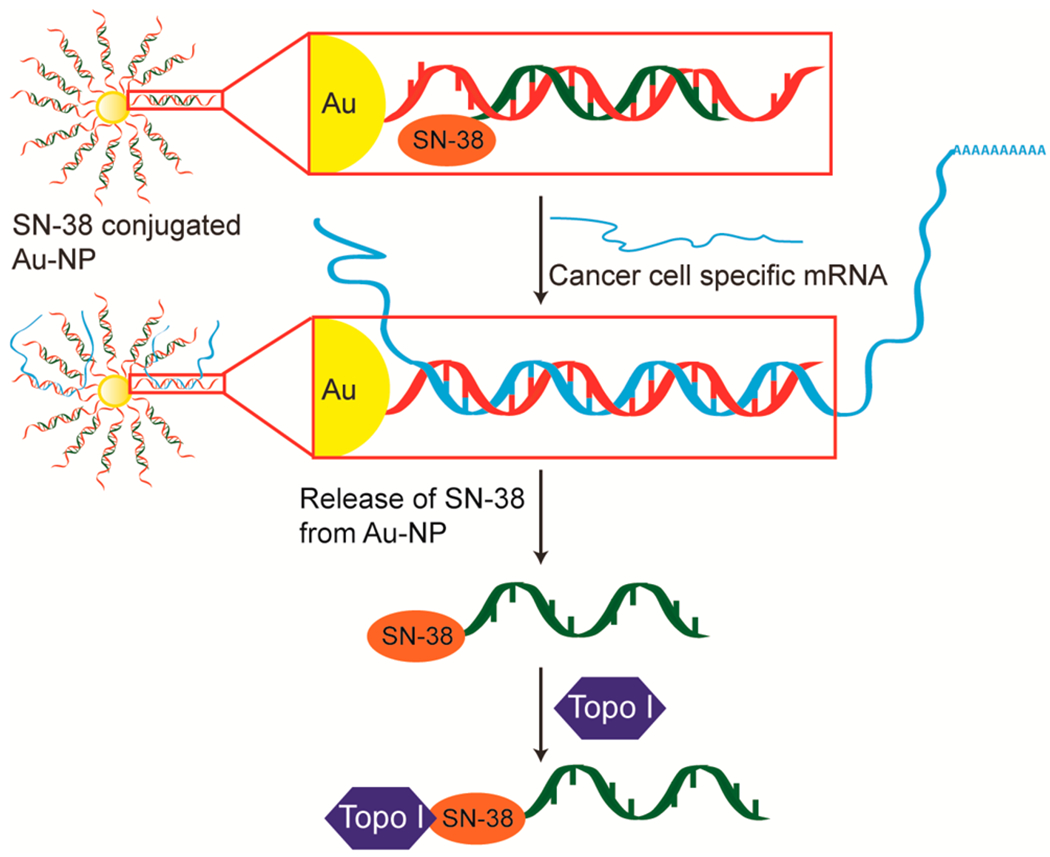
Au-NP system for selective SN-38 activation in cancer cells mediated by cancer cell specific mRNA. In this approach, each gold particle is conjugated to oligonucleotides (red) complementary (antisense) to an mRNA that is either overexpressed in or unique to cancer cells. A shorter, complementary SN38-conjugated oligonucleotide (SN38—orange; oligonucleotide—green) is annealed via base pairing to the antisense oligonucleotide to generate SN38-DNA Au-NPs. In a cancer cell, the SN38-conjugated oligonucleotide is released from sequestration to the Au-NP and can inhibit topoisomerase I by the binding of the targeted mRNA (blue). The amount of SN38-conjugated oligonucleotide released from the Au-NP is proportional to the amount of cancer cell specific mRNA present in the cell.
While dasatinib-conjugated Au-NPs showed proof-of-principle for this approach, dasatinib is a well-tolerated and orally dosed drug. We now demonstrate the broad applicability and potential clinical relevance of this approach using SN-38 conjugated Au-NPs activated by mRNAs unique or overexpressed in Ewing sarcoma cells. SN-38 is a potent topoisomerase I inhibitor that exhibits activity in the laboratory against multiple cancer types including Ewing sarcoma, neuroblastoma, rhabdomyosarcoma, lung cancer, and breast cancer.3 However, SN-38 is unable to be given in the clinic due to poor solubility and toxicity. Rather, SN-38 is administered as a more soluble pro-drug, irinotecan, which is converted by endogenous hepatic carboxylesterases to SN-38. However, only a small fraction of irinotecan is metabolized to SN-38 and the extent of activation can vary significantly between patients. As an alternative to irinotecan, SN-38 has been successfully conjugated to other biomolecules or encapsulated to enhance solubility without perturbing its efficacy.3–7 We hypothesized that the significant potency, poor solubility and delivery challenges, and potential for chemical modification without diminishing efficacy make SN-38 an excellent candidate drug for further developing our Au-NP system.
We selected Ewing sarcoma for testing our SN-38 conjugated Au-NPs because Ewing sarcoma is the second leading bone cancer arising in children and adolescents and is often sensitive to irinotecan.8,9 There is also a significant need for novel therapies in the management of Ewing sarcoma, as despite combining intensive cytotoxic chemotherapy with surgery and radiation therapy the overall survival of patients with metastatic and nonmetastatic disease are ~20% and ~70%, respectively.8 Furthermore, gene expression profiling has identified mRNAs unique (EWS-FLIl) or overexpressed (survivin) in Ewing sarcoma tumors relative to normal tissues that can be targeted for drug activation in our Au-NP system.10–12 The EWS-FLIl gene, generated by the t(11;22) chromosome translocation, occurs in >85% of Ewing’s sarcomas and is a driver oncogene in Ewing sarcoma.13 The sequence encompassing the breakpoint of the EWS-FLI1 gene is unique to Ewing sarcoma cancer cells. Although not unique, the pro-survival gene survivin (BIRC5) is overexpressed, and a validated therapeutic target, in many different types of cancers, including Ewing sarcoma.10–12 We have also previously developed and validated drug conjugated Au-NPs activated by the survivin mRNA.1
RESULTS
Conjugation of SN38 to Oligonucleotides.
The C20 hydroxyl on SN-38 has been previously modified with a variety of chemical moieties.3,4,14,15 Accordingly, we tethered an alkyl azide to the C20 hydroxyl of SN-38 through an ester linkage and the pendant azide was then reacted using “click chemistry” with commercially available oligonucleotides containing a 5′-alkyne functional group to generate SN38-oligonucleotide conjugates (Figure 2). We then used lipofectamine to transduce the SN-38 conjugated oligonucleotide into Ewing sarcoma cells sensitive to SN-38 (Figure S1A). The SN-38-oligonucleotide exhibits significant toxicity against Ewing Sarcoma cells at picomolar concentrations (Figure S1B). This result supports that conjugation of an oligonucleotide to the C20 position of SN-38 does not compromise its ability to target topoisomerase I.
Figure 2.
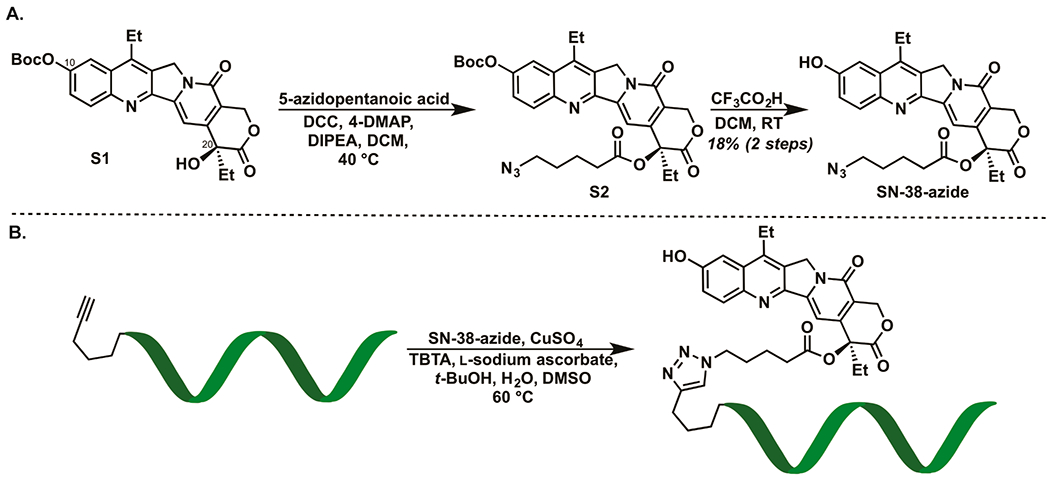
Schematic illustrating the conjugation of SN-38 to an oligonucleotide. A. An alkyl azide was tethered to the C20 hydroxyl of SN-38 through an ester linkage. B. The azide was then reacted with a commercially available oligonucleotide containing a terminal alkyne using copper-catalyzed azide–alkyne cyclo-addition chemistry to yield a SN38-conjugated oligonucleotide.
Highly Efficient Uptake and Specificity of Au-NPs.
Although many different cell types take up oligonucleotide conjugated Au-NPs,16,17 we next examined whether Ewing sarcoma cells take up oligonucleotide conjugated Au-NPs. Oligonucleotide-conjugated Au-NPs were fluorescently labeled with Cy5-PEG-thiol and added to Ewing sarcoma cell lines A673, EW8, TC32, and TC71. Au-NP uptake was assessed by flow cytometry after 3, 5, and 20 h. As shown in Figure 3A,B, highly efficient uptake (>99%) of fluorescently labeled oligonucleotide-conjugated Au-NPs occurred in all cell lines tested in a time-dependent fashion. We also observed highly efficient (>99%) uptake of Au-NPs by Ewing sarcoma cells even when forced to compete with up to a 1000-fold excess of normal, murine bone marrow cells (Figure 3C). Finally, we confirmed efficient uptake of oligonucleotide-conjugated Au-NPs in vivo using murine xenografts containing subcutaneous TC71 tumors. As shown in Figure 3D,E, over 60% of Ewing cells were positive for fluorescently labeled Au-NPs 24 h after a single intratumoral injection.
Figure 3.
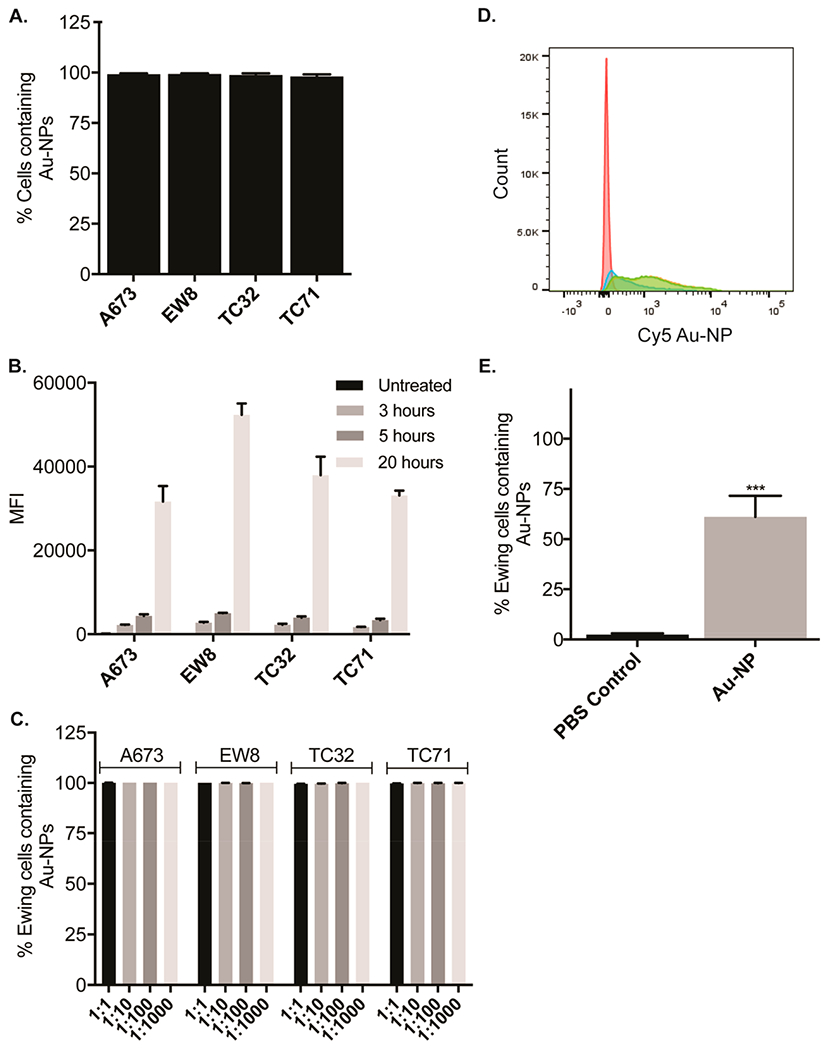
Efficient uptake of Au-NPs by Ewing sarcoma cells. A. Percentage of Ewing sarcoma cells containing DNA Au-NPs labeled with Cy5 (1 nM) was assessed by flow cytometry after an overnight incubation. Percentages are the mean ± s.e.m. from three independent experiments. B. Ewing sarcoma cells were incubated with DNA Au-NPs labeled with Cy5 (1 nM) for 3, 5, or 20 h. The median fluorescence intensities (MFI) were assessed by flow cytometry and are the mean ± s.e.m. from three independent experiments. C. Percentage of Ewing sarcoma cells containing DNA Au-NPs labeled with Cy5 (1 nM) was assessed by flow cytometry after an overnight incubation in the presence of varying ratios of normal murine bone marrow cells. Percentages are the mean ± s.e.m. from three independent experiments. Flow cytometry histograms (D) and quantitation (E) of TC71 Ewing sarcoma cells containing Au-NPs after subcutaneous flanks tumors were either injected with Cy5 labeled Au-NPs (blue, green, orange) or mock (PBS) injected (red). ***, < 0.001.
Our group and others have shown the specificity of the Au-NPs targeting survivin.1,18 To demonstrate the specificity of the Au-NPs targeting EWS-FLI1, we synthesized Au-NPs targeting EWS-FLI1 in which the noncovalently linked DNA strand contained a terminal Cy5 fluorophore rather than SN-38. When annealed to its complementary oligonucleotide conjugated to the Au-NP, the fluorophore is quenched by the gold but fluoresces when displaced from the Au-NP by the binding of the target mRNA.18,19 We then added these Au-NPs to HEK293T cells engineered to express the EWS-FLI1 mRNA in the presence of doxycycline. Supporting the specificity of the Au-NPs, there was increased release of the fluorophore-conjugated DNA oligonucleotide in the presence relative to the absence of doxycycline (Figure S2).
In vitro Efficacy of SN38-conjugated Au-NPs.
We next synthesized Au-NPs functionalized with SN38-conjugated oligonucleotides. 37 ± 5 SN38-oligonucleotides were annealed to each Au-NP (Figure S3). We selected oligonucleotide sequences that target the human BIRC5 (survivin) mRNA, the breakpoint of the t(11;22) chromosomal translocation (EWS-FLI1) that is specific to Ewing sarcoma cells, or a scrambled control sequence. Ewing sarcoma cell lines A673, EW8, TC32, and TC71 all express survivin and EWS/FLI1 mRNAs (Figure S4).20–22 As an additional control, we also utilized the osteosarcoma cell line U-2 OS. U-2 OS cells exhibit sensitivity to SN-38 and, similar to Ewing sarcoma cells, efficiently take up Au-NPs (Figure S5A–D). However, while U-2 OS cells express survivin they lack the t(11;22) translocation and EWS-FLI1 mRNA (Figure S4). The Ewing sarcoma and osteosarcoma cells were then either untreated or exposed to SN38-conjugated Au-NPs with DNA sequences targeting survivin, EWS-FLI1, or a sequence scrambled control mRNA for 48 h before assessing apoptosis by annexin-V staining and flow cytometry. The viability of all cells treated with SN38-survivin Au-NPs was significantly diminished when compared to the no treatment control and the SN38-scrambled Au-NPs (Figure 4A,B). Moreover, the Ewing sarcoma cells also showed diminished viability when treated with SN38-EWS/FLI1 Au-NPs, while the viability of U-2 OS, which lacks the EWS/FLI1 mRNA, was not significantly affected (Figure 4A,B). The moderate toxicity of SN38-scrambled Au-NPs is likely caused by the nonspecific release of SN-38 conjugated oligonucleotides from the Au-NPs. Measuring Ewing sarcoma viability using the CellTiter-Glo Luminescent Cell Viability Assay yielded similar results (Figure S6). Finally, SN38-conjugated Au-NPs also significantly inhibited the growth of Ewing sarcoma cells in long-term clonogenic growth assays (Figure 5A,B).
Figure 4.

SN38-oligonucleotide Au-NPs exhibit toxicity against multiple Ewing sarcoma cell lines. A. Ewing sarcoma cell lines were treated with SN38-DNA Au-NPs (3 nM) and after 48 h apoptosis and cell death assessed by staining with annexin-V antibody and a viability dye followed by flow cytometry. The annexin-V and annexin-V/viability dye positive populations were then used to calculate the percentage of viable cells and normalized to either untreated control cells (A) or cells treated with the SN38-Scrambled Au-NPs (B) and are the mean ± s.e.m. from three independent experiments. *, P < 0.05; **, P < 0.005; ***, P < 0.001 when comparing untreated control cells (A) or cells treated with the SN38-Scrambled Au-NPs (B) and cells treated with SN-38 conjugated Au-NPs.
Figure 5.

SN38-oligonucleotide Au-NPs inhibit Ewing sarcoma clonogenic growth. EW8 and TC71 cells were treated with SN38-conjugated Au-NPs for 4 h prior to plating 500 cells/well in a 6-well plate. Colonies were imaged (A-D) and counted (E) 10−14 days after plating. The data are normalized to untreated control cells and are the mean ± s.e.m. from three independent experiments. *, P < 0.05; **, P < 0.001; ***, P < 0.0001 when comparing untreated control cells and cells treated with SN38-conjugated Au-NPs.
In Vivo Efficacy of SN38-conjugated Au-NPs.
Finally, we examined the effect of SN38-conjugated Au-NPs in vivo using murine xenografts containing subcutaneous TC71 tumors. When subcutaneous tumors became palpable they were treated with a single injection of PBS, survivin Au-NP (lacking SN-38), SN38-survivin Au-NP, or SN38-EWS/FLI1 Au-NP. After 48 h, the mice were euthanized, tumors excised, and apoptosis assessed by annexin-V staining and flow cytometry. The viability of the tumors treated with SN38-survivin and SN38-EWS/FLI1 Au-NPs was significantly diminished in comparison to the tumors injected with PBS or Au-NPs lacking SN-38 (Figure 6).
Figure 6.
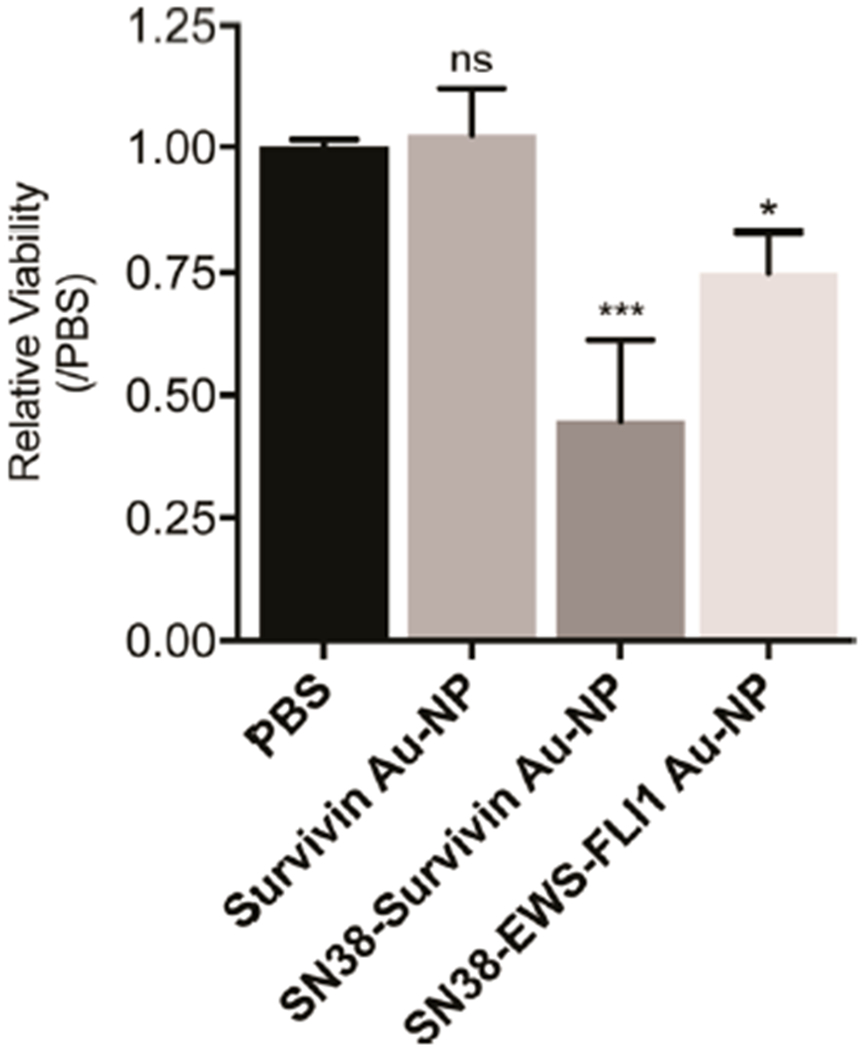
SN38-oligonucleotide Au-NPs exhibit toxicity against Ewing sarcoma cells in vivo. TC71 xenotransplanted tumors were injected with PBS (n = 4), Survivin Au-NPs 75 nM (n = 4), SN38-Survivin Au-NPs 75 nM (n = 5), or SN38-EWS/FLI1 Au-NPs 75 nM (n = 5). After 48 h, the tumors were excised, dissociated, and stained with annexin-V antibody and a viability dye followed by flow cytometry. The annexin-V and annexin-V/viability dye positive populations were then used to calculate the percentage of viable TC71 cells. The data are normalized to tumors injected with PBS. ***, P < 0.0001 and *, <0.05 when comparing PBS with SN38-Survivin Au-NP and SN38-EWS/FLI1 Au-NP injected tumors, respectively.
DISCUSSION
Despite diverse cancer therapies that include conventional cytotoxic agents, molecularly targeted drugs, antibody-based drugs, and immunotherapies, many cancers remain incurable. Moreover, the toxicities from current therapies are significant. This is particularly true for Ewing sarcoma where overall survival of patients with metastatic disease is only ~20% despite intensive, and often highly toxic, multimodal therapies.8 To address this shortcoming, we developed a novel approach to cancer therapy in which a nucleic acid functionalized gold nanoparticle (Au-NP) delivers a drug that is selectively activated within the cancer cell by the presence of a mRNA unique to the cancer cell (Figure 1).1,2 Further supporting the potential clinical applications of this approach, nucleic acid functionalized Au-NPs exhibit favorable therapeutic properties including internalization by multiple cell types including Ewing sarcoma (Figure 1), stability in biological environments, resistance to nucleases, minimal cell toxicity, and low immunogenicity.16,17 Additionally, Au-NPs carrying siRNA or DNA antisense oligonucleotides have exhibited in vivo efficacy following intravenous injection when tested in murine models of gastric and brain tumors.17,23,24
In our prior study, we selected dasatinib to demonstrate proof-of-principle because it could be chemically modified without perturbing its activity and it has well-defined kinase targets that facilitated testing and characterization of the dasatinib–oligonucleotide conjugate. We have now extended this approach to the highly potent, but poorly soluble, topoisomerase I inhibitor SN-38.3 Supported by extensive literature describing the chemical modification and/or encapsulation of SN-38, we hypothesized that conjugation of SN-38 to an oligonucleotide and sequestration to Au-NPs would enhance its solubility and delivery.3,4,14 Importantly, conjugation of SN-38 to an oligonucleotide did not perturb its ability to inhibit topoisomerase I and induce cell death (Figure S1B).
In our system, the binding of a complementary mRNA to the Au-NP displaces the SN-38 conjugated oligonucleotide. Accordingly, we synthesized SN-38 conjugated Au-NPs activated by the survivin and EWS-FLI1 mRNAs overexpressed in or unique to Ewing sarcoma cells, respectively. Survivin mRNA is highly expressed in many cancers, including Ewing sarcoma, relative to differentiated tissues and has been previously targeted by nucleic acid functionalized Au-NPs.1,10–12,18 Accordingly, SN38-Survivin Au-NPs could be used in the therapy of other SN-38 sensitive cancers that also overexpress survivin. However, SN38-Survivin Au-NPs may cause more toxicity than SN38-EWS-FLI1 Au-NPs as survivin can be expressed in normal tissues, albeit at lower levels than in cancer cells.10,25 In contrast to survivin, the breakpoint of the EWS-FLI1 gene is specific to Ewing sarcoma cells. This approach of targeting the breakpoint of fusion proteins may be generalizable to other cancers as recurrent chromosomal translocations occur in many malignancies.13 For example, the t(2;13) translocation, resulting in a PAX3-FKHR fusion protein, is detected in ~70% of pediatric alveolar rhabdomyosarcomas (RMS).26 Furthermore, RMS is sensitive to irinotecan and there is a need for novel therapies as the current EFS is <40% for metastatic RMS.27–29 However, one potential limitation to this approach is that multiple breakpoints have been described for EWS and FLI1.30–32 Thus, Au-NPs targeting EWS-FLI1 would need to be tailored to the particular breakpoint sequence for each patient. However, the most common fusion (type 1), which we have targeted with our SN-38 conjugated Au-NPs, occurs in ~60% of patients.30
SN-38 conjugated Au-NPs activated by both survivin and EWS-FLI1 mRNA exhibited toxicity against Ewing sarcoma cells in vitro and in vivo (Figures 4–6). The lack of efficacy of the SN38-EWS/FLI1 Au-NP relative to SN38-Scramble Au-NP in U-2 OS cells support the mRNA sequence dependent release of SN-38 from the Au-NP as U-2 OS is an osteosarcoma cell line that lacks the EWS-FLI1 translocation. The moderate toxicity of SN38-scrambled Au-NPs is likely caused by the nonspecific release of SN-38 conjugated oligonucleotides from the Au-NPs or cleavage of the ester linking SN-38 and the oligonucleotide (Figures 1B and 3A). Although the in vivo delivery of SN-38 conjugated Au-NPs was via intratumoral injection, scaling of the SN-38 oligonucleotide conjugate synthesis would also allow for systemic injection. While systemic injection of SN-38 conjugated Au-NPs would expose more normal tissues to the drug, the risk for increased toxicity may be offset by the enhanced permeability and retention (EPR) effect seen in solid tumors.33 In support of systemic delivery, intravenously administered nucleic acid functionalized gold nanoparticles (siRNA and molecular beacons) showed minimal toxicity to normal tissues while accumulating in tumors.23,24
In summary, we have developed Au-NPs that deliver and activate the topoisomerase I inhibitor SN-38 in Ewing sarcoma cells. With further optimization and preclinical testing, this approach could represent a new therapeutic approach for Ewing sarcoma as well as other cancers currently being treated with irinotecan. More broadly, this approach has the potential to improve the therapeutic efficacy of a drug while being highly tailorable with respect to both the cancer cell specific mRNAs targeted and drugs activated.
EXPERIMENTAL PROCEDURES
Cell Culture.
TC71 cells were cultured in IMDM (GIBCO Life Technologies) supplemented with 10% FCS (Seradigm) and penicillin/streptomycin (GIBCO Life Technologies). TC32 cells were grown in RPMI (GIBCO Life Technologies) supplemented with 10% FCS and penicillin/streptomycin. A673, EW8, and U2OS cells were grown in DMEM (GIBCO Life Technologies) supplemented with 10% FCS and penicillin/streptomycin. TC71 We generated Ewing sarcoma cells expressing GFP by transducing TC71 cells with MSCV PIG (Puro-IRES-GFP; Addgene plasmid #18751) retrovirus and selection with puromycin. All cell lines were cultured in a humidified incubator with 5% CO2 at 37 °C.
Synthesis of SN38-Oligonucleotides.
General Synthesis Methods and Materials.
Unless noted, reactions were performed under a nitrogen or argon atmosphere and stirred with a Teflon-coated magnetic stir bar. Liquid reagents and solvents were transferred via syringe and cannula using standard techniques. Dichloromethane (DCM) was dried by passage over a column of activated alumina using a solvent purification system (MBraun). All other chemicals were used as received unless noted. Reaction temperatures above 23 °C refer to oil bath temperature, which was controlled by a temperature modulator. Reaction progress was monitored by thin layer chromatography using EMD Chemicals Silica Gel 60 F254 glass plates (250 μm thickness) and visualized by UV irradiation (at 254 nm) and/or KMnO4 stain. Silica gel (SiO2) chromatography was performed on a Teledyne-Isco Combiflash Rf-200 instrument utilizing Redisep Rf High Performance silica gel columns (Teledyne-Isco).
Synthesis of SN-38-Azide.
10-OBoc-protected SN-38 (Figure 2; compound S1) was synthesized as previously reported.34 Next, 10-OBoc-SN-38 (S1; 23.5 mg, 0.048 mmol) was dissolved in DCM (5 mL), followed by the addition of 4-dimethylaminopyridine (3.0 mg, 0.024 mmol), 5-azidopentanoic acid (13.7 mg, 0.095 mmol), N,N-diisopropylethylamine (8 μL, 0.05 mmol), and N,N′-dicyclohexylcarbodiimide (19.7 mg, 0.095 mmol). The reaction was heated to 40 °C for 4 h. The reaction solution was cooled to room temperature and then concentrated in vacuo. The crude product was purified on SiO2 (0–20% gradient MeOH in DCM) to obtain 10-OBoc-SN-38-azide (Figure 2; compound S2) with an impurity (45.9 mg), which was immediately used in the next step. 10-OBoc-SN-38-azide (45.9 mg, 0.074 mmol) was dissolved in DCM (7 mL) and trifluoroacetic acid (3 mL) was added and the reaction was stirred at room temperature for 6 h. The reaction was quenched with NaHCO3 (saturated aqueous) and extracted with DCM (3X, 20 mL). The combined organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The resulting crude mixture was purified on SiO2 (0–80% gradient ethyl acetate in hexanes) to obtain the desired SN-38-azide (6.9 mg, 18% yield for two steps). 1H NMR (500 MHz, DMSO-d6) δ 10.32 (s, 1H), 8.02 (d, J = 8.9 Hz, 1H), 7.42–7.40 (m, 2H), 6.93 (s, 1H), 5.48 (app t, J = 17.1 Hz, 2H), 5.29 (dd, J = 18.6, 3.4 Hz, 2H), 3.33 (t, J = 6.4 Hz, 2H, after adding D2O), 3.08 (q, J = 7.6 Hz, 2H), 2.60–2.53 (m, 2H), 2.14 (ddd, J = 12.3, 7.5, 3.7 Hz, 2H), 1.62–1.50 (m, 4H), 1.29 (t, J = 6.0, 3H), 0.92 (t, J = 6.0, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.3, 167.8, 157.3, 157.1, 149.1, 147.4, 145.9, 144.1, 143.3, 132.0, 128.8, 128.5, 122.9, 118.3, 105.3, 94.2, 76.2, 66.8, 50.7, 50.0, 33.0, 30.6, 28.0, 22.8, 22.1, 13.8, 8.0. MS-ESI+ m/z [M + H]+ calculated for C27H28N5O6, 518.2; found 518.3.
General Protocol for SN-38-Oligonucleotide Conjugation.
Oligonucleotides were purchased with 5′-hexynyl modifications from Integrated DNA Technologies (IDT) on 1 gmol scale with standard desalting. Each oligonucleotide was dissolved in DNase/RNase free water to a 10 mM stock solution. SN-38-azide was dissolved in DMSO and kept as a 100 mM stock solution. Both materials were stored at −20 °C when not in use. The desired oligonucleotide (500 nmol, 50 μL) and SN-38-azide (12.5 gmol, 125 μL) was added to a 1.5 mL microcentrifuge tube. Then, tBuOH (100 μL) and DMSO (50 μL) were added to the reaction tube. In a separate tube, tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA; 10 mM DMSO stock, 500 nmol, 50 μL), CuSO4 (100 mM stock in ddH2O, 500 nmol, 5 μL), and L-sodium ascorbate (100 mM stock solution in ddH2O, 500 nmol, 5 μL) were combined, vortexed briefly, and added to the solution containing the oligonucleotide and SN-38-azide. The cap was wrapped tightly with parafilm and the tube was heated to 60 °C for 6 h. The reaction was cooled to RT and then filtered through a syringe filter (Agilent, 0.2 μm). The reaction was purified via reverse phase HPLC using an Agilent 1200 series instrument equipped with a diode array detector (260 nm wavelength monitored) and a semipreparative column (Agilent PLRP-S, 8 μm, 100 Å). The purification method was as follows: flow rate, 2.75 mL/min; eluent A, triethylammonium acetate (TEAA, aqueous, 100 mM, pH = 7); eluent B: triethylammonium acetate:MeCN (1:1, 100 mM TEAA, pH = 7). The eluent gradient was as follows: 98:2 A:B (0–5 min), gradient 98:2 to 20:80 A:B (540 min). All UV-active peaks were collected and lyophilized. Samples were desalted using a C18 desalting cartridge (Sephedex G25, GE Healthcare) and lyophilized to dryness until further use. SN-38-oligonucleotide conjugates were characterized using LC-MS (see Supporting Information for characterization data).
Preparation of DNA Conjugated Au-NPs.
3′-Propylthiol and 5′-hexynyl oligonucleotides were obtained from IDT (Iowa City, IA) and purified as recommended. Prior to use, oligonucleotides with a 3′-thiol modification were incubated in 100 mM DTT for 1.5 h at room temperature and then isolated using a NAP-5 column (GE Healthcare). 15 nm Au colloids stabilized with citrate were purchased (Ted Pella). Cy5-PEG-Thiol derivatives were obtained from Nanocs.
Oligonucleotides were conjugated to gold nanoparticles using published protocols.1,19,35 Deprotected alkylthiol-terminated oligonucleotides (IDT; 5 μM) were mixed with citrate-capped 15 nm gold particles (Ted Pella; 15 nM diameter), 10 nM Na-Phosphate pH 8.0, and 0.01% SDS and incubated for 20–60 min at room temperature with gentle rocking. Sodium chloride was then added every ~20 min in 50 mM increments until the final NaCl concentration was 500 mM. The sodium chloride solution contained both SDS and Na-Phosphate buffer to ensure that their concentrations were maintained at 0.01% and 10 mM, respectively. Next, the mixture was sonicated for 10 s and shaken at room temperature overnight. The DNA conjugated Au-NPs were isolated by centrifugation at 14,000 rpm for 25 min × 3 and resuspended in phosphate buffered saline. Next, the complementary oligonucleotide conjugated to SN-38, or control oligonucleotide lacking SN-38, was added at ~100-fold excess to the purified DNA-conjugated Au-NPs. To facilitate annealing, the mixture was heated to 80 °C and then cooled to 4 °C over ~3 h in a PCR machine. After annealing, the Au-NPs were purified by centrifugation at 14,000 rpm for 25 min X 3 at 4 °C, resuspended in PBS, and stored at 4 °C. The concentration of Au-NPs was determined by measuring absorbance at 520 nm. For some experiments, Au-NPs were backfilled with Cy5-PEG-thiol (Nanocs) by incubating Au-NPs with Cy5-PEG-thiol 30 μM for 4 h at room temperature prior to purification by centrifugation as described.
We used published protocols to determine the number of the annealed SN38-oligonucleotide per Au-NP.36 Briefly, we released the SN38-oligonucleotide from its complementary sequence conjugated to the Au-NP by incubating the Au-NPs in 8 M urea heated to 45 °C, followed by centrifugation to remove the Au-NPs. The SN38-oligonucleotide was then quantified with the Quan-iT OliGreen ssDNA Assay (ThermoFisher; Figure S3). The concentration of Au-NP was determined by measuring the absorbance at 520 nm.
The following oligonucleotide sequences were used: Survivin thiol; 5′-CCCAGCCTTCCAGCTCCTTGAAAAAAAAAAA-3′. Survivin alkyne; 5′-TCAAGGAGCTGG-3′. Scramble thiol; 5′-ACCATTAACCGATCATCCAAGAAAAAAAA-3′. Scramble Alkyne; 5′-CTTGGATGATCGGTT-3′. EWS-FLI1 thiol; 5′-TAAGAAGGGTTCTGCTGCCCGAAAAA-3′. EWS-FLI1 alkyne; 5′-GCAGCAGAACCCTTC-3′.
Generation of Inducible EWS-FLI1 mRNA Cell Line.
A donor plasmid was generated to insert a doxycycline-inducible EWS-FLI1 transgene into the AAVS1 locus using a CRISPR/Cas9 approach. Gene synthesis (IDT) was used to construct a plasmid with AAVS1 homology arms (derived from the AAV-CAGGS-EGFP Addgene Plasmid #22212) flanking a multiple cloning site (MCS). We then PCR amplified, using Q5 High-Fidelity Polymerase (New England Biolabs), a portion of tetracycline-inducible pLVX-TetOne-Puro vector (Clontech) and used In-Fusion (Clontech) cloning to insert the fragment into the MCS of the AAVS1-homology arm plasmid. The EWS-FLI1 transgene was then PCR amplified from a codon-optimized, synthetic EWS-FLI1 gene (GenScript) and transferred into the MCS site of the TetOne-Puro vector fragment using In-Fusion (Clontech) cloning. The complete plasmid was then transfected into HEK-293T cells, along with gRNA_AAVS1-T1 (Addgene Plasmid #41817) and pCas9_GFP (Addgene Plasmid #44719), according to the FuGENE 6 (Roche) protocol. Two days after the transfection, puromycin (1 μg/mL) was added and the cells were selected with antibiotics for 7 days.
Viability and Apoptosis Assays.
Ewing sarcoma cells were plated in 96 well plates and treated with Au-NPs. After 48 h, viability was assessed with the CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer’s instructions. All experiments were performed in triplicate with at least 3 wells per condition. Annexin-V staining was used to measure apoptosis. For these experiments, Ewing sarcoma cells were treated with Au-NPs for 48 h, stained with annexin-V (eBioscience) and Fixable Viability Dye (eBioscience), and analyzed on a BD FACS Canto flow cytometer. All viability and apoptosis experiments were performed in triplicate with at least 3 wells per condition.
Clonogenic Assay.
Ewing sarcoma cells were pretreated for 4h with scrambled Au-NP, SN38-scrambled Au-NP, SN38-Survivin Au-NP, SN38-EWS-FLI1 Au-NP 3 nM, or untreated. Next, 500 cells from each condition were seeded in 6 well plates in triplicate in complete media. Cultures were incubated for 10–14 days. Colonies were fixed with 4% PFA in PBS for 15 min at room temperature then washed 4 times in PBS and counted using a Zeiss TIRF microscope.
Xenograft Studies.
NSG (NOD.Cg-Prkdcscid, Il2rgtm1Wjl/SzJ; Jackson Laboratories) mice were maintained under aseptic conditions and received autoclaved supplies and irradiated food. Mouse care was in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Minnesota.
GFP expressing TC71 cells were prepared in 70% IMDM/30% Matrigel (Corning) and injected subcutaneously into both flanks of NSG mice. When tumors became palpable, Survivin Au-NP, SN38-Survivin Au-NP, SN38-EWS/FLI1 Au-NP, or PBS was injected into the tumor. For each mouse one tumor was injected with Au-NP and the other PBS. 48 h after treatment, the mice were euthanized and the tumors were removed. Tumors were manually dissociated by forcing through a 40 μm mesh filter. Apoptosis and viability were assessed by flow cytometry as described above after gating on GFP positive TC71 cells.
Au-NP Uptake and Competition Assay.
Ewing sarcoma cells were incubated with DNA-Au-NPs backfilled with Cy5-PEG-Thiol and then assessed by flow cytometry at 3, 5, and 20 h. The percent of cells positive for Cy5 and the median fluorescence intensities (MFI) were quantified. For the competition assays, murine bone marrow cells (red cell lysed) were mixed at specified ratios with Ewing sarcoma cells stained with Cell-Trace Violet (Invitrogen). DNA-Au-NPs backfilled with Cy5-PEG-Thiol (Nanocs) were then added to the cells and incubated overnight. The cells were then analyzed by flow cytometry. Within the violet gate (Ewing sarcoma cells), the percent of Cy5 positive cells was measured.
Western Blot.
A673, EW8, TC32, TC71, and U2OS cells were harvested and lysed with RIPA buffer. Protein concentrations were determined with the Pierce BCA Protein Assay Kit (Thermo). Samples were subjected to SDS-PAGE and transferred to nitrocellulose membranes using the iBlot Dry Blotting System (Invitrogen). Proteins were probed for β-Actin (Thermo Scientific) or survivin (Cell Signaling Technologies) and detected using Luminata Forte chemiluminescence substrate (Millipore) and imaged on HyBlot CL Autoradiography Film (Denville Scientific).
Statistical Analysis.
Experiments were performed in at least triplicate and data are shown as the mean plus or minus the SEM. GraphPad Prism 7 software (GraphPad Software, La Jolla, CA) was used to perform Student’s t tests or ANOVA tests. Data were considered statistically significant when P < 0.05.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the Children’s Cancer Research Fund (PMG). This work also utilized the University of Minnesota Masonic Cancer Center shared Flow Cytometry Facility, which is supported in part by NIH P30 CA77598. We thank members of the Gordon lab for helpful discussions.
ABBREVIATIONS
- Au-NP
gold nanoparticle
- SN-38
7-Ethyl-10-hydroxy-camptothecin
- EFS
Event free survival
- RMS
rhabdomyosarcoma
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.7b00774.
Figures S1–S6 and additional characterization of the SN38-oligonucleotides (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Gossai NP, Naumann JA, Li N-S, Zamora EA, Gordon DJ, Piccirilli JA, and Gordon PM (2016) Drug conjugated nanoparticles activated by cancer cell specific mRNA. Oncotarget 7, 38243–38256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Li N-S, Gossai N, Naumann JA, Gordon PM, and Piccirilli JA (2016) Efficient Synthetic Approach to Linear Dasatinib-DNA Conjugates by Click Chemistry. Bioconjugate Chem. 27, 2575–2579. [DOI] [PubMed] [Google Scholar]
- (3).Bala V, Rao S, Boyd BJ, and Prestidge CA (2013) Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J. Controlled Release 172, 48–61. [DOI] [PubMed] [Google Scholar]
- (4).Zhang H, Wang J, Mao W, Huang J, Wu X, Shen Y, and Sui M (2013) Novel SN38 conjugate-forming nanoparticles as anticancer prodrug: In vitro and in vivo studies. J. Controlled Release 166, 147–158. [DOI] [PubMed] [Google Scholar]
- (5).Iyer R, Croucher JL, Chorny M, Mangino JL, Alferiev IS, Levy RJ, Kolla V, and Brodeur GM (2015) Nanoparticle delivery of an SN38 conjugate is more effective than irinotecan in a mouse model of neuroblastoma. Cancer Lett. 360, 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Alferiev IS, Iyer R, Croucher JL, Adamo RF, Zhang K, Mangino JL, Kolla V, Fishbein I, Brodeur GM, Levy RJ, and Chorny M (2015) Nanoparticle-mediated delivery of a rapidly activatable prodrug of SN-38 for neuroblastoma therapy. Biomaterials 51, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Norris RE, Shusterman S, Gore L, Muscal JA, Macy ME, Fox E, Berkowitz N, Buchbinder A, and Bagatell R (2014) Phase 1 evaluation of EZN-2208, a polyethylene glycol conjugate of SN38, in children adolescents and young adults with relapsed or refractory solid tumors. Pediatr Blood Cancer 61, 1792–1797. [DOI] [PubMed] [Google Scholar]
- (8).Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, Le Deley M-C, Kovar H, Grimer R, Whelan J, Claude L, Delattre O, Paulussen M, Picci P, Sundby Hall K, van den Berg H, Ladenstein R, Michon J, Hjorth L, Judson I, Luksch R, Bernstein ML, Marec-Bérard P, Brennan B, Craft AW, Womer RB, Juergens H, and Oberlin O. (2015) Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J. Clin. Oncol. 33, 3036–3046. [DOI] [PubMed] [Google Scholar]
- (9).Kurucu N, Sari N, and Ilhan IE (2015) Irinotecan and temozolamide treatment for relapsed Ewing sarcoma: a single-center experience and review of the literature. Pediatr. Hematol. Oncol. 32, 50–59. [DOI] [PubMed] [Google Scholar]
- (10).Cheung CHA, Huang C-C, Tsai F-Y, Lee JY-C, Cheng SM, Chang Y-C, Huang Y-C, Chen S-H, and Chang J-Y (2013) Survivin - biology and potential as a therapeutic target in oncology. OncoTargets Ther. 6, 1453–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tirado OM, Mateo-Lozano S, and Notario V (2005) Roscovitine is an effective inducer of apoptosis of Ewing’s sarcoma family tumor cells in vitro and in vivo. Cancer Res. 65, 9320–9327. [DOI] [PubMed] [Google Scholar]
- (12).Greve B, Sheikh-Mounessi F, Kemper B, Ernst I, Götte M, and Eich HT. (2012) Survivin, a target to modulate the radiosensitivity of Ewing’s sarcoma. Strahlenther. Onkol. 188, 1038–1047. [DOI] [PubMed] [Google Scholar]
- (13).Fröhling S, and Döhner H. (2008) Chromosomal abnormalities in cancer. N. Engl. J. Med. 359, 722–734. [DOI] [PubMed] [Google Scholar]
- (14).Bala V, Rao S, Bateman E, Keefe D, Wang S, and Prestidge CA (2016) Enabling Oral SN38-Based Chemotherapy with a Combined Lipophilic Prodrug and Self-Microemulsifying Drug Delivery System. Mol. Pharmaceutics 13, 3518–3525. [DOI] [PubMed] [Google Scholar]
- (15).Bala V, Rao S, Li P, Wang S, and Prestidge CA (2016) Lipophilic Prodrugs of SN38: Synthesis and in Vitro Characterization toward Oral Chemotherapy. Mol. Pharmaceutics 13, 287–294. [DOI] [PubMed] [Google Scholar]
- (16).Barnaby SN, Sita TL, Petrosko SH, Stegh AH, and Mirkin CA (2015) Therapeutic applications of spherical nucleic acids. Cancer Treat. Res. 166, 23–50. [DOI] [PubMed] [Google Scholar]
- (17).Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC, and Mirkin CA (2010) Gold nanoparticles for biology and medicine. Angew. Chem., Int. Ed. 49, 3280–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Prigodich AE, Seferos DS, Massich MD, Giljohann DA, Lane BC, and Mirkin CA (2009) Nano-flares for mRNA regulation and detection. ACS Nano 3, 2147–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Prigodich AE, Randeria PS, Briley WE, Kim NJ, Daniel WL, Giljohann DA, and Mirkin CA (2012) Multiplexed nanoflares: mRNA detection in live cells. Anal. Chem. 84, 2062–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, Patidar R, Hurd L, Chen L, Shern JF, Liao H, Wen X, Gerard J, Kim J-S, Lopez Guerrero JA, Machado I, Wai DH, Picci P, Triche T, Horvai AE, Miettinen M, Wei JS, Catchpool D, Llombart-Bosch A, Waldman T, and Khan J (2014) The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 10, e1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).May WA, Grigoryan RS, Keshelava N, Cabral DJ, Christensen LL, Jenabi J, Ji L, Triche TJ, Lawlor ER, and Reynolds CP (2013) Characterization and drug resistance patterns of Ewing’s sarcoma family tumor cell lines. PLoS One 8, e80060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Smith MA, Morton CL, Phelps D, Girtman K, Neale G, and Houghton PJ (2008) SK-NEP-1 and Rh1 are Ewing family tumor lines. Pediatr. Blood Cancer 50, 703–706. [DOI] [PubMed] [Google Scholar]
- (23).Jensen SA, Day ES, Ko CH, Hurley LA, Luciano JP, Kouri FM, Merkel TJ, Luthi AJ, Patel PC, Cutler JI, Daniel WL, Scott AW, Rotz MW, Meade TJ, Giljohann DA, Mirkin CA, and Stegh AH (2013) Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci. Transl. Med. 5, 209ra152–209ra152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bao C, Conde J, Curtin J, Artzi N, Tian F, and Cui D (2015) Bioresponsive antisense DNA gold nanobeacons as a hybrid in vivo theranostics platform for the inhibition of cancer cells and metastasis. Sci. Rep. 5, 12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Fukuda S, and Pelus LM (2006) Survivin, a cancer target with an emerging role in normal adult tissues. Mol. Cancer Ther. 5, 1087–1098. [DOI] [PubMed] [Google Scholar]
- (26).Wang C (2012) Childhood rhabdomyosarcoma: recent advances and prospective views. J. Dent. Res. 91, 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Weigel BJ, Lyden E, Anderson JR, Meyer WH, Parham DM, Rodeberg DA, Michalski JM, Hawkins DS, and Arndt CAS (2016) Intensive Multiagent Therapy, Including Dose-Compressed Cycles of Ifosfamide/Etoposide and Vincristine/Doxorubicin/Cyclophosphamide, Irinotecan, and Radiation, in Patients With High-Risk Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J. Clin. Oncol. 34, 117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Pappo AS, Lyden E, Breitfeld P, Donaldson SS, Wiener E, Parham D, Crews KR, Houghton P, and Meyer WH (2007) Two consecutive phase II window trials of irinotecan alone or in combination with vincristine for the treatment of metastatic rhabdomyosarcoma: the Children’s Oncology Group. J. Clin. Oncol. 25, 362–369. [DOI] [PubMed] [Google Scholar]
- (29).Mascarenhas L, Lyden ER, Breitfeld PP, Walterhouse DO, Donaldson SS, Paidas CN, Parham DM, Anderson JR, Meyer WH, and Hawkins DS (2010) Randomized phase II window trial of two schedules of irinotecan with vincristine in patients with first relapse or progression of rhabdomyosarcoma: a report from the Children’s Oncology Group. J. Clin. Oncol. 28, 4658–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lewis TB, Coffin CM, and Bernard PS (2007) Differentiating Ewing’s sarcoma from other round blue cell tumors using a RT-PCR translocation panel on formalin-fixed paraf inembedded tissues. Mod. Pathol. 20, 397–404. [DOI] [PubMed] [Google Scholar]
- (31).Giovannini M, Biegel JA, Serra M, Wang JY, Wei YH, Nycum L, Emanuel BS, and Evans GA (1994) EWS-erg and EWS-Fli1 fusion transcripts in Ewing’s sarcoma and primitive neuroectodermal tumors with variant translocations. J. Clin. Invest. 94, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).de Alava E, Panizo A, Antonescu CR, Huvos AG, Pardo-Mindaán FJ, Barr FG, and Ladanyi M (2000) Association of EWSFLI1 type 1 fusion with lower proliferative rate in Ewing’s sarcoma. Am. J. Pathol. 156, 849–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lane LA, Qian X, Smith AM, and Nie S (2015) Physical chemistry of nanomedicine: understanding the complex behaviors of nanoparticles in vivo. Annu. Rev. Phys. Chem. 66, 521–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Zhao H, Rubio B, Sapra P, Wu D, Reddy P, Sai P, Martinez A, Gao Y, Lozanguiez Y, Longley C, Greenberger LM, and Horak ID (2008) Novel prodrugs of SN38 using multiarm poly(ethylene glycol) linkers. Bioconjugate Chem. 19, 849–859. [DOI] [PubMed] [Google Scholar]
- (35).Taton TA (2002) Preparation of gold nanoparticle-DNA conjugates. Current Protocols in Nucleic Acid Chemistry, Chapter 12, Unit 12.2.1-12.2.12 DOI: 10.1002/0471142700.nc1202s09 [DOI] [PubMed] [Google Scholar]
- (36).Barnaby SN, Lee A, and Mirkin CA (2014) Probing the inherent stability of siRNA immobilized on nanoparticle constructs. Proc. Natl. Acad. Sci. U. S. A. 111, 9739–9744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.