

Abstract
Background:
Actin remodeling is a key regulator of mast cell (MC) migration and secretion. However, the precise mechanism underlying the coordination of these processes has remained obscure.
Objective:
We sought to characterize the actin rearrangements that occur during MC secretion or chemotactic migration and identify the underlying mechanism of their coordination.
Methods:
Using high-resolution microscopy, we analyzed the dynamics of actin rearrangements in MCs triggered to migration by IL-8 or prostaglandin E2 or to FcεRI-stimulated secretion.
Results:
We show that a major feature of the actin skeleton in MCs stimulated to migration is the buildup of pericentral actin clusters that prevent cell flattening and converge the secretory granules (SGs) in the cell center. This migratory phenotype is replaced on encounter of an IgE cross-linking antigen that stimulates secretion through a secretory phenotype characterized by cell flattening, reduction of actin mesh density, ruffling of cortical actin, and mobilization of SGs. Furthermore, we show that knockdown of mammalian diaphanous-related formin 1 (mDia1) inhibits chemotactic migration and its typical actin rearrangements, whereas expression of an active mDia1 mutant recapitulates the migratory actin phenotype and enhances cell migration while inhibiting FcεRI-triggered secretion. However, mice deficient in mDia1 appear to have normal numbers of MCs in various organs at baseline.
Conclusion:
Our results demonstrate a unique role of actin rearrangements in clustering the SGs and inhibiting their secretion during MC migration. We identify mDia1 as a novel regulator of MC response that coordinates MC chemotaxis and secretion through its actin-nucleating activity.
Keywords: Mast cells, actin, mammalian diaphanous-related for-min 1, chemotaxis, exocytosis
GRAPHICAL ABSTRACT
Mast cells (MCs) are hematopoietic derived cells of the immune system with undifferentiated precursors that migrate in the blood and invade tissues. The cells then complete their maturation and differentiation under the influence of local growth factors and cytokines.1 Although best known for their involvement in maladaptive acquired immune responses, such as allergy and asthma, MCs also play important protective roles in innate host defense.2,3 Mature MCs contain cytoplasmic secretory granules (SGs) in which inflammatory mediators are stored.4–6 When activated by allergen/antigen-stimulated cross-linking of cell-bound allergen-specific IgE class antibodies or other stimuli, such as neuropeptides or pathogens, MCs degranulate, releasing their SG content by using exocytosis.7–9 MC numbers increase in inflamed tissues by proliferation, migration, and survival.1,10–12 Proliferation and survival factors, such as stem cell factor (SCF) or TGF-β, as well as agonists of G protein-coupled receptors, such as prostaglandin E2 (PGE2), which is generated in inflamed tissues, also serve as chemotactic factors.11–14 In addition, MCs also express chemokine receptors that play an important role in progenitor and MC homing to sites of inflammation. Consistent with this notion, the expression profile of chemokine receptors varies depending on the MC origin and their maturation.12 Some receptors, such as CXCR1 and CXCR4, are constitutively expressed on both progenitor and mature MCs, whereas others are expressed only on mature MCs.12,15–17
It was previously noted that MCs do not degranulate during migration,18,19 yet chemotactic factors synergize with secretagogues to potentiate MC degranulation. 13,20–24 The actin cytoskeleton was implicated in restricting secretion during MC migration.13,18,19 However, how precisely these actin rearrangements harmonize these responses has remained largely unknown. Here, using high-resolution microscopy, we characterized the actin structures and dynamics in MCs under the influence of a chemoattractant, secretagogue, or their combination. We demonstrate a tight regulation of SG positioning by chemoattractant-induced rearrangements of the actin cytoskeleton. We also identify mammalian diaphanous-related formin 1 (mDia1), a member of the formin family of actin-polymerizing proteins and effector protein of the small GTPase RhoA,25–28 as a novel regulator of MC responses. mDia1 has been implicated in actin remodeling during migration of immune cells, including T cells, neutrophils, and dendritic cells.29–31 Here we show that mDia1 coordinates MC chemotaxis and secretion through its actin-nucleating activity.
METHODS
Antibodies and reagents
Horseradish peroxidase-conjugated goat anti-mouse (catalog no. 115-035-166) and anti-rabbit (catalog no. 115-035-003) IgG were from Jackson ImmunoResearch Laboratories (West Grove, Pa). Monoclonal mouse anti-mDia1 (catalog no. 610848, 1:2,000 dilution) was from BD Biosciences (San Jose, Calif). Monoclonal rabbit anti-syntaxin 3 (catalog no. ab133750, 1:100 dilution) was from Abcam (Cambridge, Mass). Monoclonal mouse anti-CXCR1 (catalog no. MAB330) was from R&D Systems (McKinley Place, Minn). Monoclonal mouse anti–phosphorylated extracellular signal-regulated kinase (ERK) 1/2 (catalog no. M8159, 1:10,000 dilution) and monoclonal mouse anti–dinitrophenol (DNP) IgE (catalog #D8406, clone SPE-7) were from Sigma-Aldrich (St Louis, Mo), and polyclonal rabbit anti-ERK2 (catalog no. SC-154, 1:1,000 dilution) was from Santa Cruz Biotechnology (Santa Cruz, Calif). Polyclonal rabbit anti-Akt (catalog no. 9272, 1:1,000 dilution) and polyclonal rabbit anti–phosphorylated Akt (Ser473, catalog no. 9271, 1:1,000 dilution) were from Cell Signaling Technology (Beverly, Mass). DNP-conjugated human serum albumin (catalog no. A6661), fibronectin from bovine plasma (catalog no. F1141), polyethylenimine (catalog no. 408727), polybrene (catalog no. TR-1002), poly-D-lysine (catalog no. P6407), and cytochalasin D (CytD; catalog no. C8273) were from Sigma-Aldrich. Zeocin (catalog no. ant-zn) was from InvivoGen (San Diego, Calif). Recombinant human IL-8 (catalog no. 200-08M), murine IL-3 (catalog no. 213-13), human SCF (catalog no. 250-03), and murine SCF (catalog no. 300-07) were from PeproTech (Rocky Hill, NJ). Puromycin (catalog no. 58-58-2) was from Calbiochem (San Diego, Calif). Alexa Fluor 647–phalloidin (catalog no. A22278, 1:200 dilution) was from Invitrogen (Carlsbad, Calif), HiLyte Plus 488–conjugated goat anti-rabbit IgG (catalog no. 61056-H488, 1:200 dilution) was from Anaspec (Fremont, Calif), and PGE2 (catalog no. 14010) was from Cayman Chemicals (Ann Arbor, Mich). Alexa Fluor 700 anti-mouse lineage cocktail (17A2/RB6-8C5/RA3-6B2/Ter-119/M1/70, catalog no. 79923), phycoerythrin anti-mouse IL-33 receptor (IL-33R) α/ST2 (DIH9, catalog no. 145304), allophycocyanin anti-mouse FcεRIα (MAR-1, catalog no. 134316), peridinin-chlorophyll-protein complex/Cy5.5 anti-mouse Ly-6C (HK1.4, catalog no. 128012), phycoerythrin-Cy7 anti-mouse Sca1 (D7, catalog no. 106113), and allophycocyanin-Cy7 anti-mouse/rat/human CD27 (LG.3A10, catalog no. 124226) were from BioLegend (San Diego, Calif). BV421 rat anti-mouse CD117 (2B8, catalog no. 562609) and BV421 rat anti-mouse Integrin (37 (FIB504, catalog no. 564283) were from BD Biosciences.
Plasmids used in this study
Neuropeptide Y (NPY)–mRFP was a gift from Dr U. Ashery (Tel Aviv University, Tel Aviv, Israel). Green fluorescent protein (GFP)–coiled coil 1 (CC1; dominant negative [DN]–dynein) was a gift from Dr E. Perlson (Tel Aviv University). LifeAct-GFP and Myc-mDia1 constructs were gifts from Dr B.-Z. Shilo (Weizmann Institute of Science). mDia1-targeting short hairpin RNAs (shRNAs) were from Sigma-Aldrich (MISSION shRNA Lentiviral Transduction Particles).
After screening 5 different mDia1-targeting shRNAs, clone ID TRCN0000108685 was selected for further analyses. Control scramble pLKO.1-Puro and packaging plasmids pLP1, pLP2, and VSV-G were gifts from Dr C. Luxenburg (Tel Aviv University).
Cell culture
RBL-2H3 (herein referred to as RBL) and RBL-CXCR1 cells were maintained in adherent cultures in low-glucose Dulbecco modified Eagle medium (catalog no. 01-050-1A; Biological Industries, Bet-Haemek, Israel) supplemented with 10% FBS (catalog no. 12657; Gibco, Carlsbad, Calif), 2 mmol/L l-glutamine, 100 μg/mL streptomycin, 100 U/mL penicillin, and 12 U/mL nystatin (Biological Industries). LAD-2 cells (a kind gift from Dr A. S. Kirshenbaum and Dr D. Metcalfe, Laboratory of Allergic Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Md) were cultured in StemPro (catalog no. 10640-019; Gibco, Carlsbad, Calif) supplemented with 100 ng/mL human recombinant SCF, 2 mmol/L l-glutamine, 100 μg/mL streptomycin, and 100 U/mL penicillin. HMC-1 cells (a kind gift from Dr J. Butterfield, Mayo Clinic, Rochester, Minn) were cultured in RPMI-1640 (catalog no. R8758; Sigma-Aldrich) supplemented with 10% FBS, 2 mmol/L l-glutamine, 100 μg/mL streptomycin, 100 U/mL penicillin, and 12 U/mL nystatin.
Bone marrow–derived mast cells (BMMCs) were isolated from 6- to 10-week-old C57/BL6 mice in complete medium consisting of RPMI-1640 supplemented with 10% FBS, 2 mmol/L l-glutamine, 100 ;g/mL streptomycin, 100 U/mL penicillin, 1 mmol/L sodium pyruvate, nonessential amino acids, 10 mmol/L HEPES (pH 7.4), and 6 pmol/L 2-mercaptoethanol. BMMCs were subsequently cultured for 4 to 5 weeks in the presence of 20 ng/mL IL-3. Cell purity (95% to 97%) was confirmed by analyzing expression of c-Kit and FcεRI by using flow cytometry. All cell lines were maintained in a humidified atmosphere of 5% CO2 at 37°C.
Mice
mDia1 knockout mice harboring GFP were as previously described.32 Genotype profiling with the primers described in Table E1 in this article’s Online Repository at www.jacionline.org confirmed that the mice were selectively depleted of mDia1. All experiments were approved by the Institutional Animal Care and Use Committees at Northwestern University.
Cell transfection by means of electroporation
Transient transfection of RBL cells was performed, as previously described.33 Briefly, RBL cells (1.5 × 107) were transfected by means of electroporation at 300 V and a pulse length of 20 ms by using an ECM830 electroporator (BTX, Holliston, Mass). Cells were immediately replated in tissue-culture dishes containing growth medium for the desired time periods. For generation of RBL cells stably expressing CXCR1, the cells were selected with 50 μg/mL zeocin for 2 weeks.
Generation of mDia1 knockdown cells by means of lentivirus infection
For virus preparation, HEK293FT cells were seeded onto poly-D-lysine–precoated 6-well plates (1 × 106 cells/well) and transiently transfected with 0.75 μg of pLKO.1-Puro vector containing either control scrambles shRNA or mDia1 targeting shRNA together with 0.5 μg of pLP1, 0.18 μg of pLP2, and 0.26 μg pLP/VSV-G lentiviral packaging and helper plasmids in the presence of 26 μg/mL polyethylenimine in serum-free D-10 medium supplemented with 1 mmol/L sodium pyruvate and 1% sodium bicarbonate. The next day, the medium was replaced by D-10 medium supplemented with 10% FBS. After 30 hours, lentiviral particle–containing media were collected and either filtered through a 0.2-μm filter, placed in aliquots, and kept at −80°C for infection of RBL-CXCR1 cells or concentrated by means of filtration through a 0.45-μm filter, followed by centrifugation at 100,000g at 4°C and resuspension of the lentiviral particles in 200 μL of BMMC culture media for infection of BMMCs.
For infection, RBL-CXCR1 cells were seeded onto 6-well plates (3 × 105 cells/well) in Dulbecco modified Eagle medium. The next day, the medium was replaced by RBL culture medium containing 8 μg/mL polybrene and 10 μL of viral particles containing medium, and the cells were incubated for a further 18 hours.
For selection, cells were cultivated for 48 hours in medium containing 2 μg/mL puromycin. mDia1 knockdown (KD) was confirmed by means of immunoblotting and quantitative real-time PCR. BMMCs were grown for 10 days in the presence of 30 ng/mL murine SCF to increase their infectability.14 Cells (1 × 107) were then suspended in 2 mL of BMMC culture medium supplemented with 20 ng/mL IL-3, 30 ng/mL SCF, 10 μg/mL polybrene, and 10 μL of concentrated viral particles and centrifuged for 30 minutes at 800g at 37°C, followed by resuspension in 13 mL of the same medium, except for omission of the viral particles. The next day, the medium was replaced by BMMC culture medium containing 20 ng/mL IL-3 and 30 ng/mL SCF. Cells were analyzed within 48 to 72 hours after infection.
Cell activation
RBL or RBL-CXCR1 cells were either seeded onto 24-well plates at 5 × 105 cells/well for secretion assays or at 1 × 105 cells/wells on plates containing glass coverslips for confocal microscopy assays. For signaling assays and Western blot analyses, cells were seeded onto 6-well plates at 1 × 106 cells/well. Cells were sensitized overnight with 0.25 μg/mL mouse anti-DNP–specific monoclonal IgE. After 3 washes in Tyrode buffer (10 mmol/L HEPES [pH 7.4], 130 mmol/L NaCl, 5 mmol/L KCl, 1.8 mmol/L CaCl2, 1 mmol/L MgCl2, 5.6 mmol/L glucose, and 0.1% BSA), cells were triggered in Tyrode buffer at 37°C with 50 ng/mL DNP-HSA (antigen) or 50 ng/mL IL-8 or IL-8 followed by DNP-HSA for the desired time periods.
For activation of BMMCs, cells were grown at 1 × 106 cells/mL overnight with anti-DNP-specific IgE. The next day, cells were washed, resuspended in Tyrode buffer at 1.25 × 106 cells/mL, and activated as above for secretion assays or seeded at 6 × 105 cells/mL onto 24-well plates containing glass coverslips that were coated overnight with 100 μg/mL fibronectin in PBS and washed 3 times with Tyrode buffer. Cells were activated in the same buffer at a final volume of 200 μL. For details, see Fig E1 in this article’s Online Repository at www.jacionline.org.
Secretion of β-hexosaminidase
Activity of the SG-associated enzyme β-hexosaminidase was determined, as previously described.34 Briefly, 20-μL aliquots of supernatants and cell lysates derived from cells activated as described above were incubated for 90 minutes at 37°C with 50 μL of substrate solution consisting of 1.3 mg/mL p-nitrophenyl-N-acetyl-β-d-glucosaminide in 0.1 mol/L citrate (pH 4.5). Reactions were stopped by addition of 180 μL of 0.2 mol/L glycine (pH 10.7). OD was measured at 405 nm by using an absorbance microplate reader (Infinite F50; Tecan, Männedorf, Switzerland). Secretion is expressed as a percentage of total cellular β-hexosaminidase activity.
Secretion of NPY-mRFP
Secretion of NPY-mRFP was measured, as previously described.33 Briefly, fluorescence of cell supernatants and cell lysates (200 μL) derived from cells that were transiently transfected with NPY-mRFP and activated as described above was measured by using a fluorescence microplate reader (Infinite 200; Tecan) with a 590-nm, 20-nm bandwidth excitation filter and a 635-nm, 35-nm bandwidth emission filter. Autofluorescence of nontransfected RBL cells was set as reference. The amount of secretion is presented as a percentage of total cellular NPY-mRFP.
Determination of tissue histamine levels and MC numbers in mice
Age-, sex-, and strain-matched wild-type (WT) and mDia1 knockout mice (n = 6-7) were used to assess the role of mDia1 in constitutive homeostatic MC accumulation and persistence in tissues. Tissues were harvested from ears and spleens; bone marrow and peritoneal lavage cells were also collected. Ears and spleens were homogenized in PBS containing protease inhibitor, whereas peritoneal lavage cells were disrupted in RIPA buffer containing protease inhibitor. Total histamine liberated was quantified by means of ELISA (ab213975; Abcam) and then corrected per microgram of total protein (BCA Protein Assay; Thermo Scientific). For bone marrow analysis, progenitor MCs were counted by using flow cytometry (LSRII) with a gating strategy that included lineage-negative, CD117+, Sca1−, Ly-6C−, FcεRIα−, CD27−, integrin β7+, IL-33Rα/ST2+ cells. A minimum of 1 million events was collected (sources included those listed above).
Immunostaining and confocal analyses
RBL-CXCR1 cells grown on 12-mm round glass coverslips (thick #1; Thermo Scientific) and activated as described above were washed 3 times with ice-cold PBS and fixed for 20 minutes at room temperature with 4% paraformaldehyde (catalog no. 15710; ElectronMicroscopy Sciences, Hatfield, Pa) in PBS. For BMMCs, medium was replaced with ice-cold 4% paraformaldehyde, and cells were incubated on ice for 5 minutes, followed by 15 minutes of incubation at room temperature. Cells (RBL-CXCR1 or BMMCs) were then permeabilized for 15 minutes at room temperature with 0.1%Triton X-100, 5% FBS, and 2% BSA diluted in PBS. Cells were subsequently incubated for 1 hour at room temperature with the primary antibodies, followed by 3 washes and a 1-hour incubation with the appropriate secondary antibody. After washing, cells were mounted (catalog no. E18–18; Golden Bridge Life Science, Mukilteo City, Wash) and analyzed with a Leica SP5 laser scanning confocal microscope (Leica, Wetzlar, Germany) equipped with a HyD detector by using a ×63 oil/1.4NA objective for imaging of RBL-CXCR1 cells or ×100 oil/1.4 NA for imaging of BMMCs. Three-dimensional reconstructions were performed by using Imaris software (Bitplane, Zurich, Switzerland).
Time-lapse and total internal reflection fluorescence microscopy
RBL-CXCR1 cells transiently cotransfected with NPY-mRFP and LifeAct-GFP were seeded at 2 × 105 cells per chamber in 8-well chamber borosilicate coverglass systems (Ibidi μ-slide 8-well glass bottom, catalog no. 80827). The next day, cells were activated under the microscope, and images were acquired withaZeiss510 (Zeiss, Oberkochen, Germany) or Leica SP5 microscope equipped with a HyD detector by using a ×63 oil/1.4 NA objective equipped with a top-stage incubator (Okolab, Ottaviano, Italy) set to 37°C and 5% CO2.
For total internal reflection fluorescence (TIRF) microscopy, images were acquired with a TILL Photonics iMIC TIRF microscope (FEI, Munich, Germany) using an Olympus 100× NA 1.49 TIRF objective. Images were acquired with an Andor iXon 897 EMCCD camera (Andor, Belfast, United Kingdom), and the imaging protocol was controlled by using TILL Photonics Live Acquisition software. TIRF angle was set to provide minimal penetration of the evanescent wave while still producing measurable signal from the LifeAct and NPY structures. We used the 360° TIRF feature, which azimuthally spins the laser beam on the circumference of the objective back focal plane, creating homogenous TIRF illumination across the field. Data were analyzed by using ImageJ software (National Institutes of Health, Bethesda, Md).
RNA purification and quantitative real-time PCR
Total cellular RNA was extracted with the GENEzol TriRNA Pure Kit (catalog no. GZXD200; Genaid, New Taipei, Taiwan), according to the manufacturer’s protocol. cDNA was generated by using 2 mg of total RNA and high-capacity reverse transcriptase (Applied Biosystems, Foster City, Calif) in a total volume of 20 μL. cDNA was assessed by using real-time PCR (StepOne; Applied Biosystems) with Power SYBR Green PCR Master Mix (catalog no. 4367659; Applied Biosystems), as previously described,35 and analyzed by using StepOne V.2.3 software. The following primers were used: mDia1, sense 5′ GCTGTCTGCCCTCTGTATCC 3′ and antisense 5′ AGCCTCTAAAACTCGTTCATTCA 3′; mDia2, sense 5′ TGTTCTCAAAGTTTGTAACCATGAA 3′ and antisense 5′ TTAAGCTCCTTGGCGACTG 3′; and mDia3, sense 5′ GCGTGAGATGGTTGTCCAG 3′ and antisense 5′ AGGGTGAACTCATGTTTGTTGTT 3′.
Migration assays
RBL-CXCR1 cells were transiently transfected with 10 μg of LifeAct-GFP. The next day, cells were washed 3 times with RBL culture medium supplemented with 0.1% BSA and 10 mmol/L HEPES (pH 7.4) and gently scraped and resuspended to a concentration of 1 × 106 cells/mL in the same medium. BMMCs were washed 3 times with BMMC culture medium supplemented with 0.1% BSA and 10 mmol/L HEPES (pH 7.4). Cells (1.5 × 105; RBL-CXCR1 or BMMCs) in a total volume of 150 μL were seeded in the upper chamber of a transwell with an 8-μm pore size polycarbonate membrane insert (catalog no. 3422; Corning, Corning, NY) that was precoated with 100 μg/mL fibronectin for 24 hours at 4°C. Cells were incubated for 2 hours in a 5% CO2 humidified atmosphere at 37°C, followed by addition of either 50 ng/mL IL-8 (RBL-CXCR1) or 100 nmol/L PGE2 (BMMCs) in 350 μL to the bottom chamber. Cells were allowed to migrate for 4 hours in a humidified atmosphere of 5% CO2 at 37°C. Numbers of RBL-CXCR1 cells migrating through the filter was quantified by replacing the medium in the bottom chamber with PBS supplemented with 4% paraformaldehyde and fixing the cells for 20 minutes. The upper chamber was then gently wiped with a cotton swab, and the inserts were mounted on a glass coverslip (24 × 60 mm, thickness #1, 0.13-0.11 mm; catalog no. E18-18; Golden Bridge Life Science). Numbers migrating cells were quantified by imaging 7 fields of each chamber with aLeicaSP5 laser scanning confocal microscope with an open pinhole and ×20/0.1 numeric aperture objective and counting the LifeAct-GFP-expressing cells in each area by using the multipoint tool (ImageJ software).
Media from the bottom chamber were collected, and the migrated cells were palleted by means of centrifugation at 300g to quantify BMMC migration. Cell pellets were resuspended in 50 μL of PBS, and cells were immediately counted with a hemocytometer.
Western blot analyses
Cell lysates were prepared by means of addition of a lysis buffer consisting of 150 mmol/L sucrose, 80 mmol/L β-glycerophosphate, 2 mmol/L EDTA, 2 mmol/L ethyleneglycol-bis-(β-aminoethylether)-N,N,N′,N′-tetraacetic acid, 2 mmol/L NaVO3, 10 mmol/L sodium pyrophosphate, and 1% Triton X-100. Samples normalized according to protein content or cell number were separated by using SDS-PAGE with 7.5–12% polyacrylamide gels and electrophoretically transferred to nitrocellulose membranes. Blots were blocked for 1 hour in TBST (10 mmol/LTris–HCl [pH 8.0], 150 mmol/L NaCl, and 0.05% Tween 20) containing 5% skim milk, followed by overnight incubation at 40C with the desired primary antibodies. Blots were washed 3 times with TBST and incubated for 1 hour at room temperature with horseradish peroxidase–conjugated secondary antibodies. Immunoreactive bands were visualized by using enhanced chemiluminescence, according to the manufacturer’s instructions.
Statistical analysis
Data are expressed as means ± SEMs. P values were determined by using an unpaired 2-tailed Student t test.
RESULTS
Actin structure and dynamics in MCs triggered to migration or secretion
To be able to capture the dynamics of actin rearrangements during MC migration or secretion in real time, we introduced LifeAct-GFP, an actin marker that allows tracking of actin in live cells by using high-resolution imaging, into RBL cells. RBL cells were chosen for this task because they share similar exocytic machineries with primary MCs and display similar patterns of degranulation36 while offering the important advantage of increased transfectability. This latter property enables functional genomics analyses of these processes33 and therefore has marked RBL cells as an attractive model for studies on MC exocytosis.36
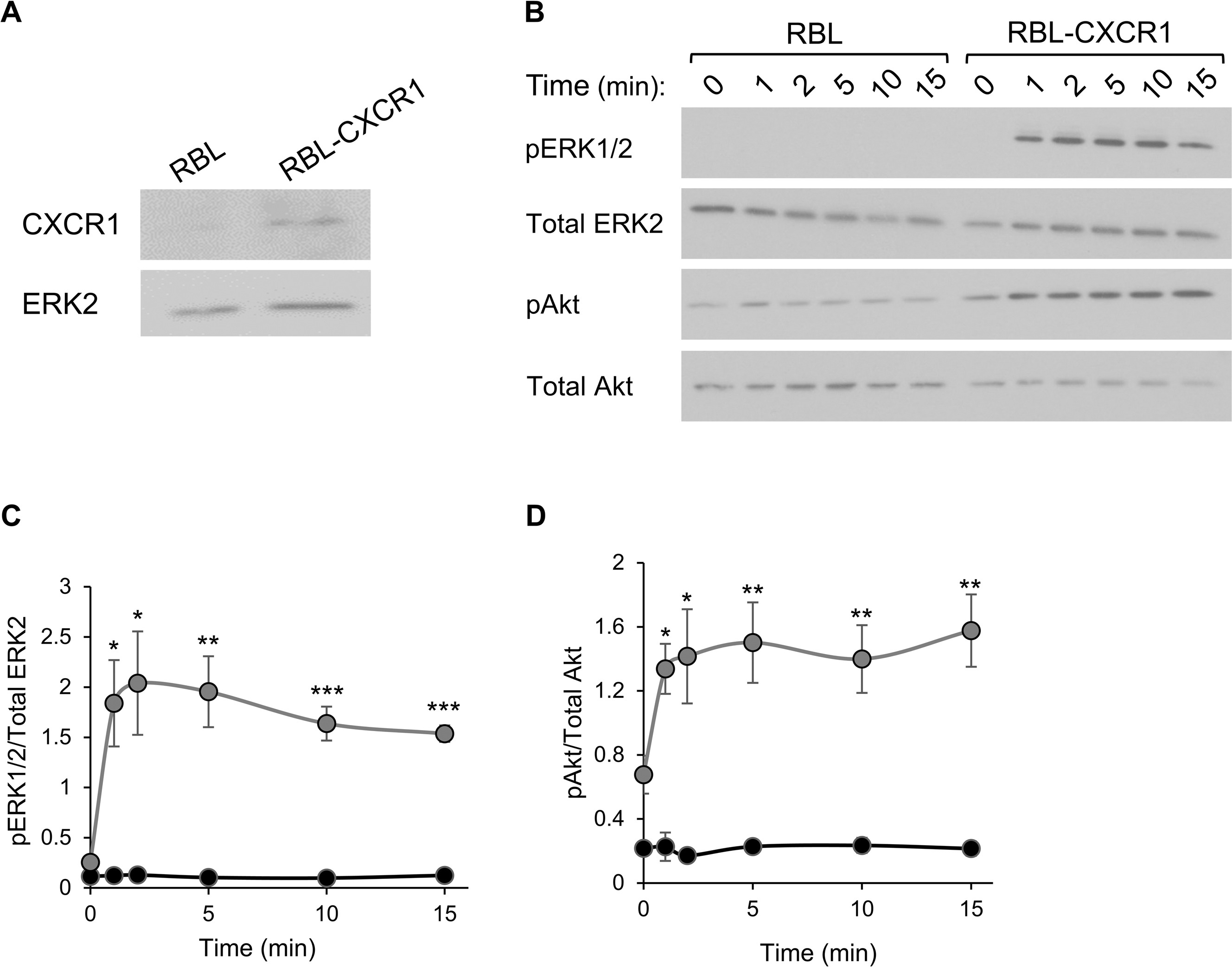
To generate a model that will allow exclusive monitoring of chemotactic responses, we adopted a previously described paradigm for MC chemotactic migration that was based on the stable expression of CXCR1, the human receptor for IL-8, in RBL cells (see Fig E2, A, in this article’s Online Repository at www.jacionline.org).37–43 We confirmed the functionality of this model in our hands by analyzing the ability of IL-8 to stimulate signaling in our stably transfected cells and promote their chemotactic migration. Indeed, IL-8 stimulated phosphorylation of the ERK1/2 and Akt kinases in RBL-CXCR1 cells but not in their parental counterpart (see Fig E2, B–D). IL-8 also significantly stimulated their chemotactic migration (Fig 1, A), whereas it had no effect on naive RBL cells (data not shown). In contrast, cell triggering by IgE/antigen had no effect on cell migration (Fig 1, A), whereas at the same range of concentrations, it significantly stimulated secretion of the SG-associated enzyme β-hexosaminidase by either naive or CXCR1-expressing cells (Fig 1, B–D). IL-8 did not stimulate secretion at concentrations that effectively stimulated migration (Fig 1, B). However, prior exposure to IL-8 significantly increased IgE/antigen-stimulated release (Fig 1, B), whereas the presence of antigen inhibited IL-8–stimulated migration (Fig 1, A). Hence this model recapitulated the crosstalk that exists in MCs between chemokine receptors and FcεRI signaling.23
FIG 1.
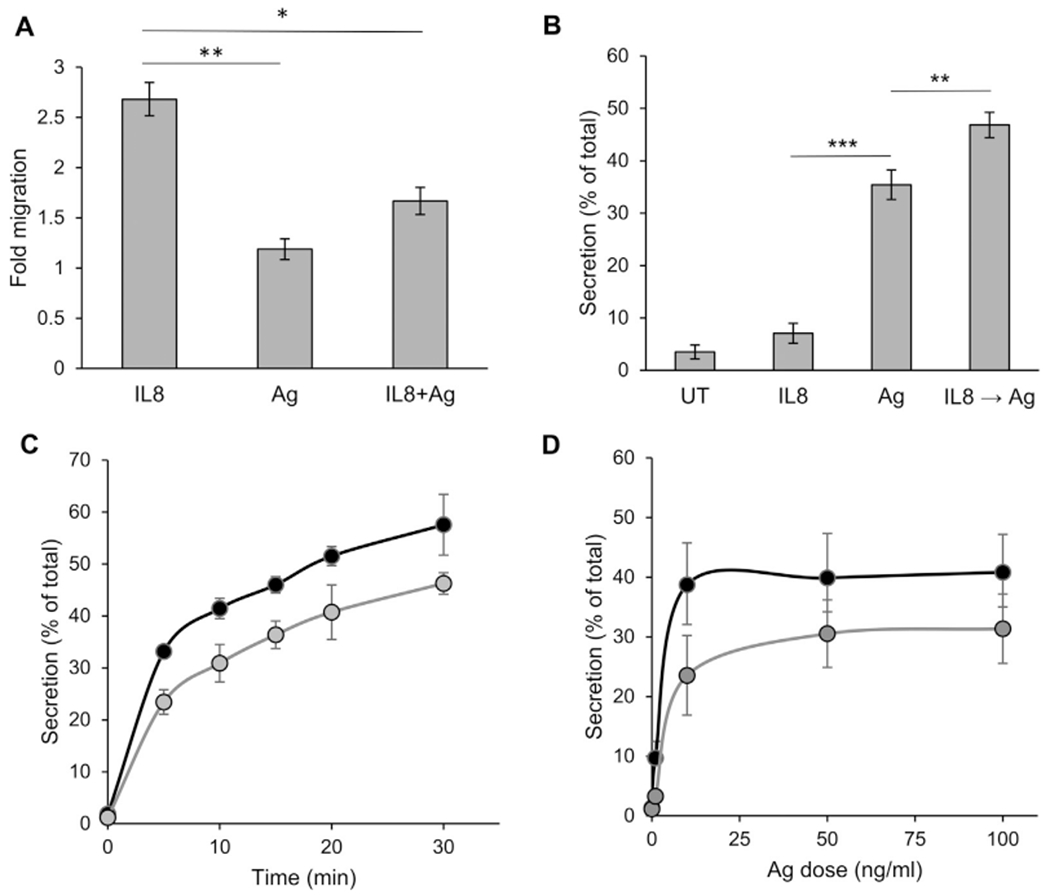
RBL-CXCR1 cells migrate in response to IL-8 and degranulate in response to IgE/antigen (IgE/Ag). A, Chemotactic migration of RBL-CXCR1 cells sensitized with 0.25 μg/mL DNP-specific IgE was assayed, as described in the Methods section, in response to 50 ng/mL IL-8, 50 ng/mL DNP-HSA (Ag), or both, as indicated. Stimulated migration was calculated relative to migration of untreated cells. Results are means ± SEMs of 5 separate experiments. *P = 2.89E-2 and **P = 5.84E-3. B, RBL-CXCR1 cells sensitized with 0.25 μg/mL DNP-specific IgE were either left untreated (UT) or triggered for 30 minutes with 50 ng/mL IL-8,50 ng/mL DNP-HSA (Ag), or 50 ng/mL IL-8,followed by a further 30 minutes of incubation with 50 ng/mL DNP-HSA (IL8 → Ag), as indicated. β-Hexosaminidase secretion was determined as described in the Methods section and is presented as a percentage of total cellular activity. Results are means ± SEMs of 8 separate experiments. **P = 8E-3 and ***P = 8.38E-7. C and D, RBL (black circles) and RBL-CXCR1 (gray circles) cells were sensitized with 0.25 μg/mL IgE and activated with 50 ng/mL DNP-HSA for the indicated time periods or for 30 minutes with the indicated concentrations of DNP-HSA. Results are means ± SEMs of 3 separate experiments.
Finally, we confirmed that our model agrees well with previous reports on the opposing requirements of actin rearrangements of migration and secretion.44 Consistent with the reported requirement of actin polymerization for cell migration,44 treatment of RBL-CXCR1 cells with CytD, a drug that inhibits actin polymerization, inhibited IL-8–stimulated migration by 80% (Fig 2, A), whereas this treatment significantly enhanced IgE/antigen-induced secretion (Fig 2, B), which stands in agreement with the proposed role of actin as a barrier to exocytosis.12,45 Indeed, analysis of the dynamics of cortical actin near the plasma membrane during exocytosis by using TIRF microscopy of cells that coexpressed LifeAct-GFP and the SG reporter NPY-mRFP33 revealed actin depolymerization that preceded SG fusion with the plasma membrane (Fig 2, C and D, see Movie E1 in this article’s Online Repository at www.jacionline.org), implying formation of actin-free exit spots in the plasma membrane, through which the SGs discharge their contents.46
FIG 2.
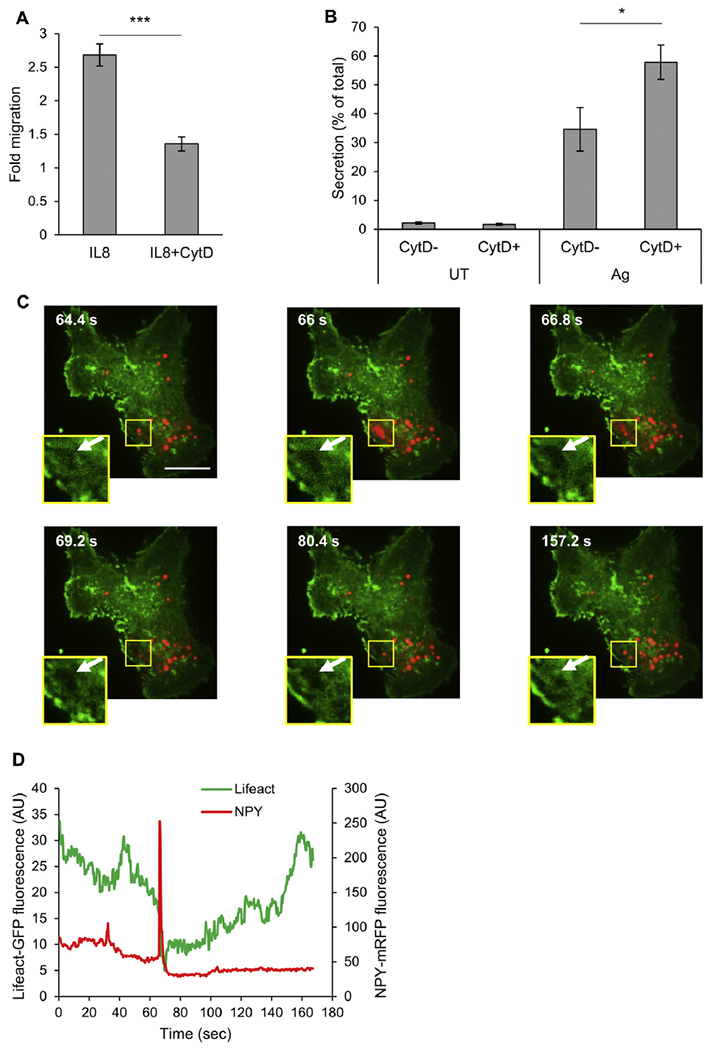
Actin depolymerization inhibits migration but stimulates secretion. A, Chemotactic migration of RBL-CXCR1 cells was assayed in response to 50 ng/mL IL-8 in the absence or presence of 10 μmol/L CytD, as indicated. Results are means ± SEMs of 5 separate experiments. ***P = 9.57E-3. B, RBL-CXCR1 cells were sensitized with 0.25 μg/mL IgE and activated by 50 ng/mL DNP-HSA (Ag) in the absence or presence of 10 μmol/L CytD, as indicated. β-Hexosaminidase secretion was measured. Results are means ± SEMs of 6 separate experiments. *P = 3.6E-2. C, RBL-CXCR1 cells were cotransfected with 15 μg of LifeAct-GFP (green) and 15 μg of NPY-mRFP (red), sensitized with 0.25 μg/mL IgE, and activated by 50 ng/mL DNP-HSA. Cells were imaged in real time by means of TIRF microscopy, as described in the Methods section. Insets are magnifications of the boxed areas. Arrows point to the “exit spot” at which actin is depolymerized to allow SG exocytosis. D, Quantification of fluorescent signals in the boxed areas as a function of time of trigger.
Having established our MC model, at high resolution, we next characterized the structure of the actin meshwork during CXCR1 or FcεRI signaling. In this regard confocal images of resting RBL-CXCR1 cells or cells triggered by either IL-8 or IgE/antigen were taken. The results, which were presented also as heat maps of actin density, revealed marked and trigger-type dependent differences in actin rearrangements. In resting cells actin mainly concentrated at the cell periphery (Fig 3, A). However, IL-8 induced robust spreading of cell and actin redistribution to form dense clusters at the cell center (Fig 3, A, and see Movie E2 in this article’s Online Repository at www.jacionline.org). In sharp contrast, in IgE/antigen-triggered cells strokes of dense actin formed membrane ruffles, whereas the overall density of actin in the cell cortex significantly decreased (Fig 3, A, and see Movie E3 in this article’s Online Repository at www.jacionline.org).
FIG 3.
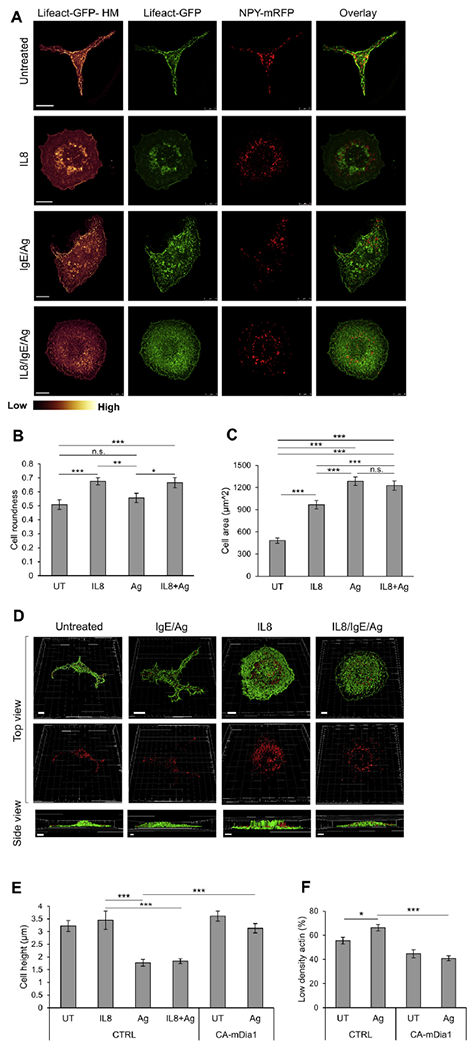
Characterization of the actin meshwork, cell height, and SG distribution under migratory and secretory conditions. A, RBL-CXCR1 cells were transiently cotransfected with 10 μg of LifeAct-GFP (green) and 15 μg of NPY-mRFP (red) sensitized with 0.25 μg/mL IgE and either left untreated (UT) or triggered for 30 minutes with 50 ng/mL IL-8, 50 ng/mL DNP-HSA (IgE/Ag), or IL-8 followed by DNP-HSA (IL8/IgE/Ag), as indicated. Cells were processed for microscopy, as described in the Methods section, and imaged with a Leica SP5 confocal microscope. LifeAct-GFP fluorescence is also presented as heat maps (Lifeact-HM). Scale bars = 10 μm. B, Cell roundness (range, 0-1) was calculated by tracing the cells’ footprints and using the roundness measurement parameter in ImageJ software. *P = .034, **P = .007, and ***P ≤ 3E-3 (n ≥ 20 for each treatment from at least 3 different experiments). C, Cell area was calculated by tracing the cells’ footprints and using the area measurement parameter in ImageJ software. ***P≤3E-3 (n ≥ 20 for each treatment from at least 3 different experiments). D, Confocal images were 3-dimensionally reconstructed by using Imaris software. Scale bars = 5 μm. E, Quantification of the average cell height derived from 3-dimensional images of cells transfected with 30 mg of either empty vector (CTRL) or CA mDia1, as indicated. Cells were either left untreated (UT) or triggered for 30 minutes with IL-8, IgE/antigen, or IL-8 followed by antigen. ***P< 8.4E-4 (n = 15 cells for each treatment). F, Quantification of LifeAct-GFP fluorescence in RBL-CXCR1 cells cotransfected with LifeAct-GFP and either empty vector (CTRL) or CA mDia1, as in Fig 3, E. Cells were IgE sensitized with 0.25 μg/mL IgE and either left untreated (UT) or activated for 30 minutes by 50 ng/mL DNP-HSA (Ag), as indicated. Cells were imaged at the footprint with a Leica SP5 confocal microscope. The LifeAct-GFP signal was classified as low or high according to a uniform threshold and quantified as the percentage of cell area with low/high-density actin per cell. Quantification is based on 9 (UT) to 25 cells derived from 3 separate experiments. *P = 1.23E-2 and ***P = 3.23E-7. n.s., Not significant.
The 2 types of trigger also differently affected cell morphology. IL-8–triggered cells became rounder (Fig 3, B), and their footprint area (ie, the focal plane of the cell cortex at the junction with the coverslip) significantly increased (Fig 3, C). In contrast, IgE/ antigen-triggered cells only significantly increased in their footprint area (Fig 3, B and C). Three-dimensional reconstruction of z-stacks of the confocal images demonstrated that IgE/antigen-triggered cells have flattened, whereas IL-8–triggered cells retained a high center, most likely because of the architecture of the ring-shaped dense actin wall that formed under these conditions (Fig 3, D). Quantitative analysis of the results indicated that the average height of resting cells of 3.2 ± 0.2 mm did not change after an IL-8 trigger and maintained an average of 3.4 ± 0.3 μm (Fig 3, E), whereas the average height of IgE/ antigen-triggered cells was reduced to 1.7 ± 0.1 μm (Fig 3, E).
Quantification of the cortical actin meshwork, measured as a percentage of low-density actin area at a focal plane at the base of the cell (close to the coverslip), confirmed that IgE/antigen-induced flattening affected the density of the cortical actin meshwork, impeding a porous geometry of the actin bundles, as reflected in a significant increase in low-density actin area (Fig 3, F).
Cellular distribution of SGs in MCs triggered to migration or secretion
We envisioned that the distinct geometric actin remodeling imposed by a chemokine, as opposed to a secretagogue, can dictate unique spatiotemporal features of the SGs. To investigate this possibility, we analyzed the SG position depicted by labeling with NPY-mRFP under each condition. Close inspection of the images and their 3-dimensional reconstructions demonstrated that in both resting and IgE/antigen-triggered cells, SGs were scattered throughout the cell (Fig 3, A and D, and see Movies E4 and E5 in this article’s Online Repository at www.jacionline.org). In sharp contrast, in IL-8–triggered cells the majority of SGs converged at the cell center in close proximity to the central actin clusters that formed in these cells (Fig 3, A and D, and see Movie E6 in this article’s Online Repository at www.jacionline.org).
To understand the dynamics of the SGs, we also monitored the differently triggered cells by using time-lapse microscopy. Consistent with previous observations,47,48 the SGs were highly mobile in IgE/antigen-triggered cells (see Movie E3). However, opposite to the IgE/antigen trigger, the SGs remained static and clustered at the cell center in the IL-8–triggered cells (see Movie E2).
Sequential activation by IgE/antigen of IL-8–triggered cells relieves central clustering of the SGs
After their chemotactic migration, MCs encounter the secretagogue and undergo degranulation. Therefore it is anticipated that secretory signals would override the migratory secretion-restricting actin signal. To test this notion, we analyzed the actin structure and dynamics in cells that were sequentially activated, first by IL-8 and then by IgE/antigen. Indeed, analyses of confocal images and their 3-dimensional reconstructed images confirmed that the cells acquired a secretory actin phenotype. Specifically, the cells have flattened, reducing their average height to 1.8 ± 0.09 μm (Fig 3, E); their central actin became considerably less dense; and their SGs dispersed throughout the entire cell (Fig 3, A and D, and see Movie E7 in this article’s Online Repository at www.jacionline.org).
In this context it is noteworthy that similar phenotypic shifts were also conferred on IgE/antigen-triggered cells as a function of the antigen concentration. Hence, unlike the significant increase in cell area accompanied by a significant reduction in cell height, which is induced by antigen concentrations that stimulate secretion (ie, 50 ng/mL DNP-HSA; Fig 3 and see Fig E3, A, C, and D, in this article’s Online Repository at www.jacionline.org), exposure of cells to 10 pg/mL DNP-HSA, which is in agreement with previous results,49 stimulated cell migration but not secretion (see Fig E3, A and B) and significantly increased the cell area, but this change was not accompanied by a reduction in cell height (see Fig E3, C and D). Furthermore, in a similar manner to IL-8, cell triggering with IgE/antigen under conditions that induced cell migration was associated with an increase in cell roundness (see Fig E3, E) and SG clustering (see Fig E3, F).
SG clustering in PGE2-triggered MCs and its relief by means of sequential activation by IgE/antigen
Our high-resolution analyses of the actin rearrangements that occur in RBL-CXCR1 cells triggered by a secretagogue, chemoattractant, or their combination are highly consistent with previous observations that noted increased actin polymerization in PGE2-triggered BMMCs that was reversed in cells triggered by both IgE/antigen and PGE2.44 Because RBL cells do not respond to PGE2 (data not shown), to investigate whether increased actin polymerization in PGE2-triggered MCs is also linked to SG clustering, we visualized BMMCs triggered by either PGE2, IgE/antigen, or their combination by means of high-resolution confocal imaging. Staining with Alexa Fluor 647–labeled phalloidin, which stains F-actin, was used to visualize the actin cytoskeleton, whereas the SGs were visualized by immunostaining of syntaxin 3, an SG membrane–resident SNARE protein.50 Three-dimensional reconstruction of confocal images revealed that in a similar manner to RBL-CXCR1 cells, IgE/antigen-activated BMMCs, under conditions that cause secretion (see Fig E4 in this article’s Online Repository at www.jacionline.org), have flattened, displaying a 40% reduction in their average cell height that was accompanied by a decrease in the density of the actin meshwork and an increase in peripheral SGs (Fig 4, A and B, and see Movies E8 and E9 in this article’s Online Repository at www.jacionline.org). In contrast, in cells activated by PGE2, the cell center remained high, the actin meshwork was dense, and the SGs clearly clustered in the cell center (Fig 4, A and B, and see Movie E10 in this article’s Online Repository at www.jacionline.org). Furthermore, sequential exposure to PGE2 followed by IgE/antigen, which in agreement with previous results24 led to potentiation of secretion (see Fig E4), completely suppressed the “migratory phenotype,” allowing the “secretory phenotype” to dominate, as reflected in flattening of the cells, reduction in actin meshwork density, and peripheral distribution of the SGs (Fig 4, A and B, and see Movie E11 in this article’s Online Repository at www.jacionline.org).
FIG 4.
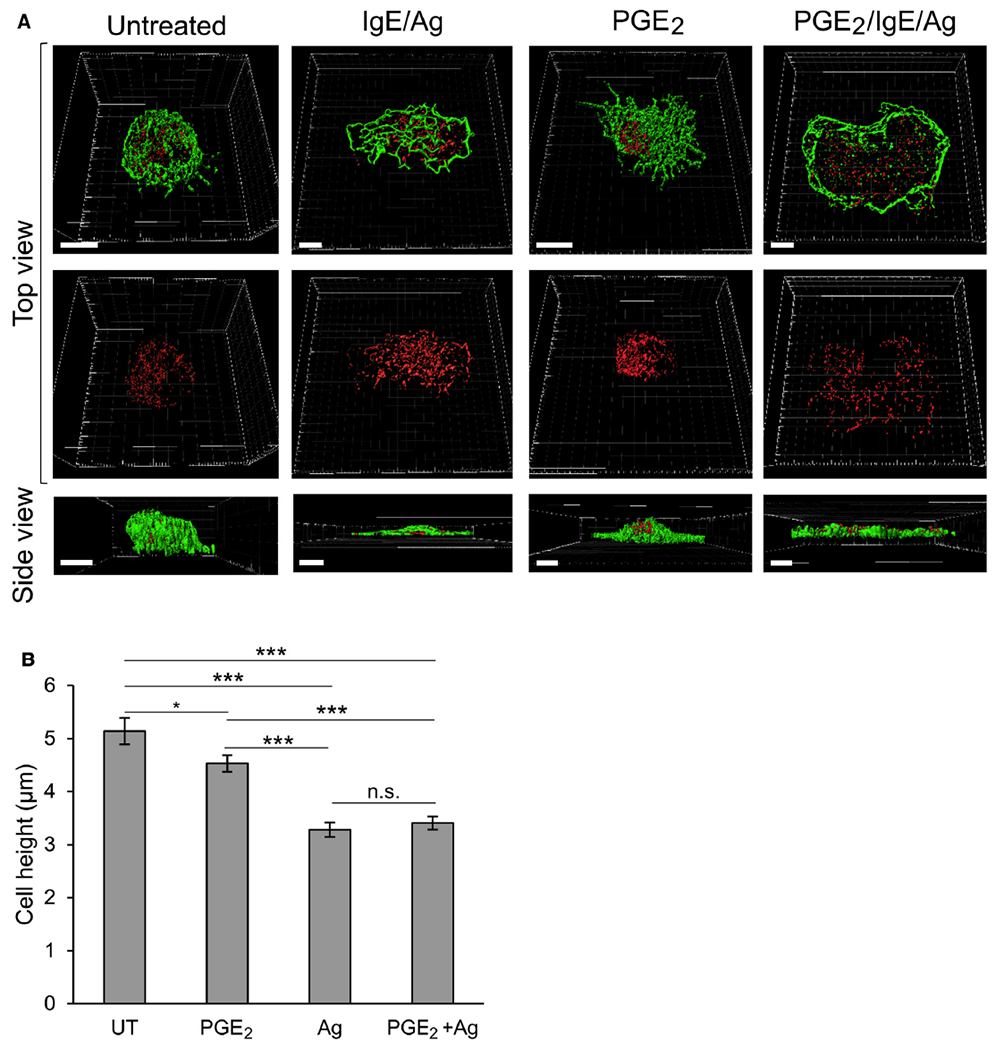
Three-dimensional characterization of BMMCs, cell height, and SG distribution under migratory and secretory conditions. A, BMMCs were sensitized with 0.25 μg/mL IgE and either left untreated (UT) or triggered for 30 minutes with 100 nmol/L PGE2, 50 ng/mL DNP-HSA (IgE/Ag), or PGE2, followed by DNP-HSA, as indicated. Cells were stained with anti-syntaxin 3 antibodies and Alexa Fluor 647 phalloidin and processed for microscopy, as described in the Methods section. Images were taken with a Leica SP5 confocal microscope and 3-dimensionally reconstructed by using Imaris software. Scale bars = 5 μm. B, Quantification o average cell height derived from the 3-dimensional images of BMMCs that were either left untreated (UT) or triggered for 30 minutes with PGE2, IgE/antigen, or PGE2 followed by antigen. *P< .04 and ***P < 1.57E-7 (n ≥46 cells for each treatment). n.s., Not significant.
Active mDia1 recapitulates IL-8-induced actin remodeling and central clustering of SGs in a dynein-independent fashion
Given the remarkable remodeling of the actin cytoskeleton noted in IL-8-triggered MCs, we speculated that mDia1, a member of the formin family of actin-polymerizing proteins, which was previously shown to control chemotactic responses of immune cells,29–31 might be involved in this process. To address this possibility, we first examined the expression profile of mDia1 in a number of MC models and found that mDia1 was indeed expressed in all of them, including BMMCs, the human MC lines HMC-1 and LAD-2, and RBL cells (Fig 5, A).
FIG 5.
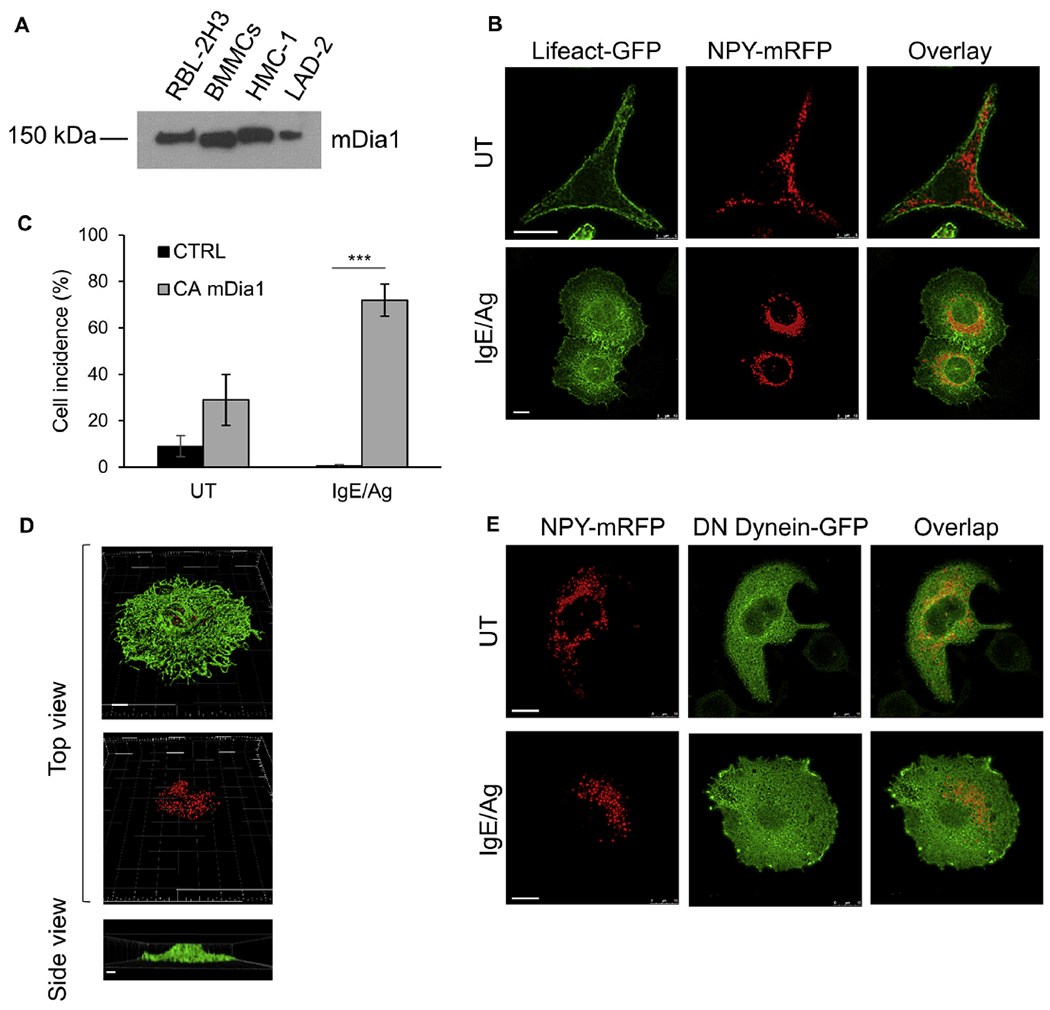
mDia1 enforces a migratory actin phenotype. A, Expression of mDia1 was determined by resolving lysates (100 μg) derived from naive RBL cells, BMMCs, HMC-1 cells, and LAD2 cells by means of SDS-PAGE and immunoblotting with anti-mDia1 antibodies. B, RBL-CXCR1 cells cotransfected with 30 μg of CAmDia1, 10 μg of LifeAct-GFP (green), and 15 μg of NPY-mRFP (red) were IgE sensitized and either left untreated (UT) or activated with 50 ng/mL DNP-HSA (IgE/Ag) for 30 minutes. Cells were processed for confocal microscopy and imaged with a Leica SP5 confocal microscope. Scale bars = 10 μm. C, Incidence of cells displaying converged SGs was calculated based on images of cells that were cotransfected with LifeAct-GFP, NPY-mRFP, and either empty vector (CTRL) or CA mDia1 and either left untreated (UT) or activated with IgE/antigen as in Fig 5, B. Quantification is based on 63 cells for each treatment derived from 3 separate experiments. ***P < .001. D, Images of cells transfected and activated were 3-dimensionally constructed by using Imaris software. Scale bars = 5 μm. E, RBL-CXCR1 cells were cotransfected with 20 μg of CA mDia1, 30 μg of p150glued-CC1-GFP (green), and 10 μg of NPY-mRFP (red) and sensitized with 0.25 μg/mL IgE. Cells were subsequently left untreated (UT) or activated with 50 ng/mL DNP-HSA for 30 minutes (IgE/Ag). Cells were processed for confocal microscopy and imaged with a Leica SP5 confocal microscope. Scale bars = 10 μm.
mDia1 is normally autoinhibited by its inhibitory diaphanous autoregulatory domain (DAD) and requires binding of active RhoA for activation.25–28 Therefore, to investigate the possible role of mDia1 in MCs, we expressed a constitutively active mutant of mDia1, which lacks its autoinhibitory DAD (herein referred to as CA mDia1). We analyzed the effect of CA mDia1 on the actin structure and cellular distribution of the SGs. Surprisingly, although CA mDia1 is supposedly active, its overexpression did not significantly affect the actin skeleton or SG distribution in resting cells (Fig 5, B, compared with Fig 3, A). However, marked changes in both actin distribution and SG location were noted on antigen triggering. Such cells displayed an exaggerated “migratory actin phenotype” despite their FcεRI trigger to secretion (Fig 5, B). Specifically, CA mDia1–expressing and IgE/antigen-triggered cells demonstrated central clustering of the SGs by a dense pericentral ring of actin that formed under these conditions (Fig 5, B). Quantification of the results indicated SG clustering in 70% of the cells (Fig 5, C). Three-dimensional reconstruction of confocal images of these cells revealed that despite their antigen trigger, the cells did not flatten and retained their central height (Figs 3, E, and 5, D, and see Movie E12 in this article’s Online Repository at www.jacionline.org), which is reminiscent of cells triggered by IL-8 (Fig 3, E). In addition, the area of their low-density actin meshwork has not increased in response to the secretory trigger (Fig 3, F).
Because mDia1 was previously shown to bind the heavy chain of the dynein motor protein,51 we investigated whether CA mDia1 enforced central clustering of the SGs by promoting their retrograde transport similar to clustering that we have previously noted in cells expressing a constitutively active mutant of the small GTPase Rab12.48 For this purpose, we coexpressed CA mDia1 with the CC1 domain of the p150Glued subunit of dynactin, which exerts a DN effect on dynein-mediated transport.52 However, unlike its profound inhibitory effect on Rab12-mediated SG clustering,48 DN dynein did not prevent SG clustering caused by CA mDia1 (Fig 5, E).
mDia1 regulates MC migration and secretion
Given the phenotypic shift in the actin phenotype from secretory to migratory inflicted by CA mDia1 on IgE/antigen-triggered cells, we next examined the effect of expression of this active mutant on IgE/antigen-triggered secretion. Results showed that, consistent with its marked effect on actin remodeling, CA mDia1 significantly inhibited secretion by 60% (Fig 6, A). Importantly, this inhibition could be relieved by preventing actin polymerization by CytD (Fig 6, B) but not by expression of DN dynein (Fig 6, C). Therefore these results implicated actin remodeling in mediating CA mDia1 function in SG clustering and inhibition of their exocytosis. Notably, expression of WT mDia1 neither affected actin remodeling in IgE/antigen-triggered cells (data not shown) nor inhibited secretion (Fig 6, A). Therefore relieving the autoinhibitory constraint imposed by the DAD seems to be a prerequisite for full activation of mDia1 by an IgE/antigen trigger.
FIG 6.
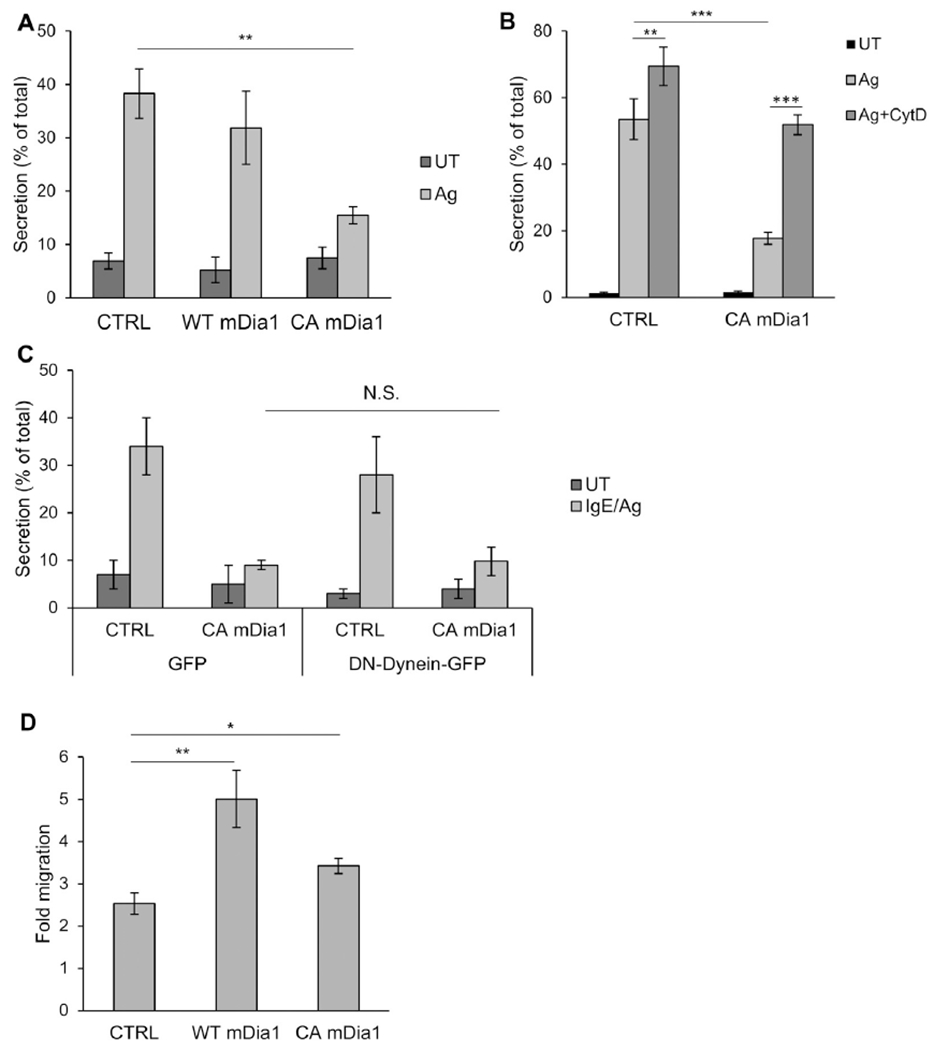
CA mDia1 stimulates migration and inhibits secretion. A, RBL-CXCR1 cells cotransfected with 10 μg of NPY-mRFP and 20 μg of either empty vector (CTRL) or WT or CAmDia1 and sensitized with IgE and were either left untreated (UT) or activated with 50 ng/mLDNP-HSA for 30 minutes (IgE/Ag). NPY-mRFP secretion was measured. Results are means ± SEMs of 4 separate experiments. **P = 2E-3. B, RBL-CXCR1 cells transfected and IgE sensitized as in Fig 6, A, were either left untreated (UT) or pretreated with vehicle or 10 μmol/L CytD for 15 minutes and then activated with 50 ng/mL DNP-HSA for 30 minutes, as indicated. Cells were assayed for NPY-mRFP secretion. Results are means ± SEMs of 3 separate experiments **P = 6.02E-3 and ***P ≤ 6.2E-4. C, RBL-CXCR1 cells were cotransfected with 15 μg of NPY-mRFP and 20 μg of either empty vector and 30 μg of GFP, CA mDia1 and GFP, empty vector and p150glued-CC1-GFP, or CA mDia1 and p150glued-CC1-GFP, as indicated. Cells were IgE sensitized and either left untreated (UT) or activated with 50 ng/ml DNP-HSA for 30 minutes (IgE/Ag). NPY-mRFP secretion was assayed. Results are mean ± SEMs of 3 separate experiments. N.S., Not significant. D, RBL-CXCR1 cells were cotransfected with 10 μg of LifeAct-GFP and 20 μg of either empty vector (CTRL), WT, or CA mDia1, as indicated. Cell migration was assayed in response to 50 ng/mL IL-8. Fold migration was calculated relative to basal migration measured in the absence of IL-8. Results are means ± SEMs of 9 separate experiments. *P = 1.26E-2 and **P = 5E-3.
In sharp contrast to its inhibitory action on secretion and consistent with its induction of a migratory actin phenotype, CA mDia1 significantly stimulated IL-8–induced cell migration (Fig 6, D). Moreover, WT mDia1 effectively and strongly stimulated migration, suggesting that IL-8 is capable of relieving mDia1 from its autoinhibitory constraint and activating its functionality in controlling migration (Fig 6, D).
To obtain direct evidence for the involvement of mDia1 in chemotactic migration to IL-8, we analyzed the effect of mDia1 KD by shRNA. A number of mDia1-targeting shRNAs were tested, and the most effective and specific shRNA, which depleted more than 90% of the protein (Fig 7, A) without affecting expression levels of mDia2 or mDia3, which are also endogenously expressed in RBL cells (Fig 7, B), was chosen to test the effect of mDia1 KD on IL-8-stimulated migration. Indeed, IL-8–stimulated migration was significantly impaired in mDia1 KD cells (Fig 7, C). In contrast, no significant effect was recorded on IgE/antigen-stimulated secretion (Fig 7, D). Furthermore, although RBL-CXCR1 cells expressing control shRNA (SCR) responded to IL-8 similarly to nontransfected cells, displaying SG clustering and buildup of a central actin ring, in IL-8–stimulated and mDia1 KD RBL-CXCR1 cells, the SGs distributed throughout the cell and actin remained cortical despite the chemotactic trigger (Fig 7, E). Quantitative analysis of the results demonstrated that only 46% of the mDia1 KD RBL-CXCR1 cells displayed central actin clusters and convergence of the SGs compared with 79% of RBL-CXCR1 cells that expressed control shRNA (Fig 7, F). Therefore these results implicated mDia1 in actin remodeling and SG clustering downstream of IL-8–activated CXCR1.
FIG 7.
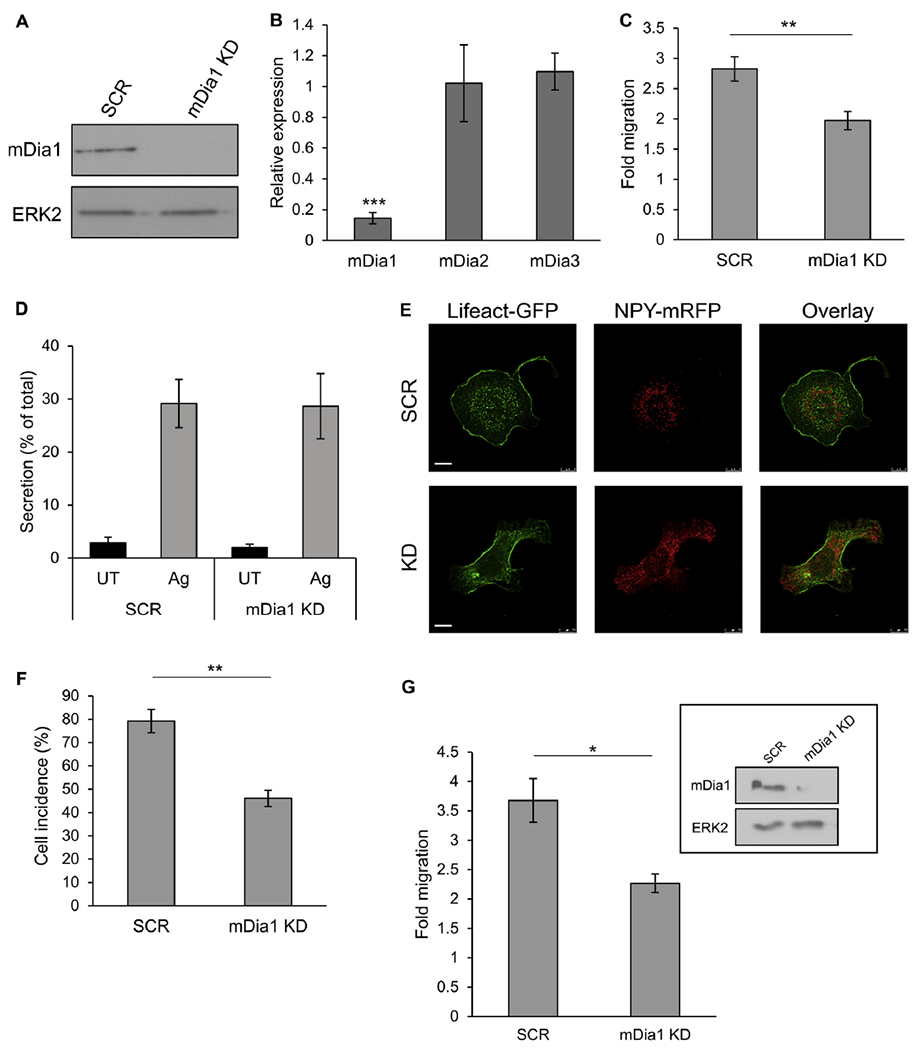
mDia1 KD inhibits chemotactic migration and migratory actin rearrangements. A, Cell lysates (100 μg) derived from RBL-CXCR1 cells stably infected with control scrambled shRNA (SCR) or mDia1-targeting shRNA (mDia1 KD) were resolved by using SDS-PAGE and immunoblotted with anti-mDia1 antibodies. Blots were reprobed with anti-total ERK2 antibodies as protein-loading controls. A representative blot of 3 similar experiments is shown. B, Relative expression of mDia1, mDia2, and mDia3 in control and mDia1 KD RBL-CXCR1 cells was determined by using quantitative RT-PCR. Results are means ± SEMs of 3 separate experiments. ***P = 1.77E-4. C, Migration of control and mDia1 KD RBL-CXCR1 cells was assayed in response to 50 ng/mL IL-8. Fold migration was calculated relative to migration in the absence of IL-8. Results are means ± SEMs of 9 separate experiments. **P = 4E-3. D, IgE-sensitized control and mDia1 KD RBL-CXCR1 cells were either left untreated (UT) or activated with 50 ng/mL DNP-HAS for 30 minutes (Ag). Secretion of β-hexosaminidase was determined. Results are means ± SEMs of 3 separate experiments. E and F, Control and mDia1 KD RBL-CXCR1 cells were cotransfected with 10 μg of LifeAct-GFP (green) and 15 μg of NPY-mRFP (red). Cells were activated by 50 ng/mL IL-8 for 30 minutes, processed for confocal microscopy, and imaged with a Leica SP5 confocal microscope. The incidence of cells featuring central actin and pericentral clustering of the SGs was quantified based on 50 cells for each treatment derived from 3 separate experiments. **P = 7E-3. G, Migration of control and mDia1 KD BMMCs was assayed in response to 100 nmol/L PGE2. Fold migration was calculated relative to migration in the absence of PGE2. Results are means ± SEMs of 3 separate experiments. *P = .047. The inset depicts a representative blot of cell lysates (30 μg) derived from BMMCs infected with either control scrambled shRNA (SCR) or mDia1-targeting shRNA (mDia1 KD) probed with anti-mDia1 antibody and with anti–total ERK2 antibody as a protein-loading control.
Given the similarities in actin rearrangements noted in IL-8–triggered RBL-CXCR1 cells and PGE2-triggered BMMCs, we also analyzed the effect of mDia1 KD on BMMCs chemotaxis in response to PGE2. Indeed, mDia1-targeting shRNA, which depleted more than 90% of the protein (Fig 7, G), significantly inhibited PGE2-induced migration (Fig 7, G). Therefore, taken together, our results strongly implicate mDia1 as a general regulator of MC chemotactic migration.
mDia1 knockout mice have normal MC numbers in tissues
Next, we asked whether depletion of mDia1 affects the normal homing of MC progenitors to the tissues, where they complete their differentiation and maturation. To this goal, we compared the amount of histamine that is primarily attributable to the presence of MCs in mDia1 knockout mice in several tissues and body compartments. However, focusing on the ear, spleen, and peritoneal compartment, no differences in the amount of tissue histamine were detected (Fig 8), suggesting that MC homing remained intact. Consistent with this notion, flow cytometric analyses of bone marrow derived from mDia1 knockout mice revealed no differences in the amount of Lineage−, CD117+, Sca1−, Ly-6C−, FcεRIα−, CD27−, integrin β7+, IL-33Rα/ST2+ cells, which correspond to MC progenitors in the bone marrow (data not shown). Therefore in this regard MCs behave similarly to lymphocytes and platelets, which we have previously shown to maintain their steady-state amounts in peripheral blood of mDia1 knockout mice.53
FIG 8.
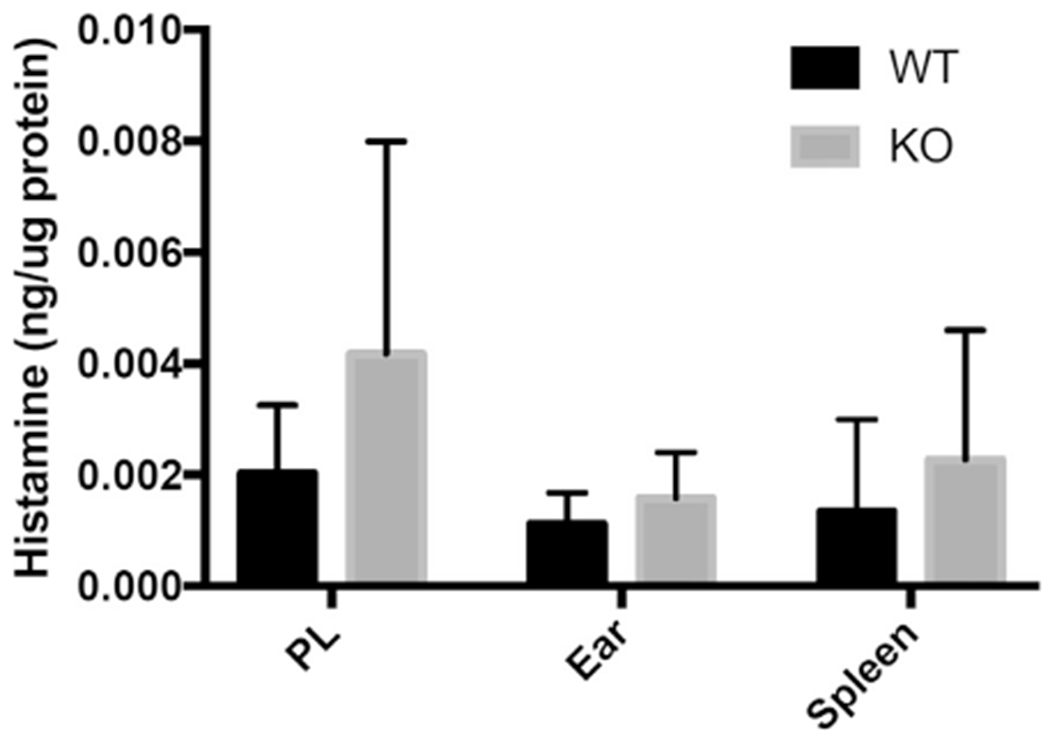
The amount of tissue histamine is unaltered in mDia1 knockout mice: effect of a congenital deficiency of mDia1 on mouse MC numbers in various locations. WT and mDia1 knockout mice (KO) were assessed for histamine content in lysates of cells isolated from peritoneal lavage fluid (PL) and ears and spleens corrected for micrograms of protein. Values represent means ± SDs from 6 to 7 mice.
DISCUSSION
The actin cytoskeleton plays important roles during cell migration and secretion. In the migrating cell, constant remodeling of actin filaments allows continuous rolling of the cell and balance and maintenance of the leading and trailing edges.54 Actin remodeling is also required for recycling of focal adhesion complexes that are imperative for cell migration.55 During secretion, the actin skeleton can facilitate SG transport or expulsion of their content, but it might also negatively regulate secretion by serving as a barrier that prevents SG fusion with the plasma membrane.56,57 Cells of the immune system, such as MCs, respond to both chemoattractants and secretagogues and therefore undergo the actin rearrangements required for each of these processes, which do not necessarily comply.44 However, how these actin rearrangements are coordinated has remained largely unknown.
To address this question, we adopted the RBL-CXCR1 cell line as a reliable MC model and used high-resolution confocal microscopy to image the actin meshwork architecture and SG localization during triggers to migration or secretion. Consistent with previous studies,44 our results clearly demonstrate distinct actin remodeling in response to these signaling types. Furthermore, we show that these distinct patterns of actin are tightly linked with changes in the spatial distribution of the SGs. During secretion, the actin meshwork becomes porous, allowing SG movement and dispersion throughout the cell. This change involves stretching of the actin meshwork because of cell flattening. In addition, disruption of cortical actin contributes to formation of exit spots at the plasma membrane, at which the SGs fuse and release their content. In sharp contrast, the major feature of the migrating cell is the buildup of pericentral actin clusters that prevent cell flattening and thereby keep the actin meshwork dense and the SGs captured in the cell center.
Notably, in natural killer cells and cytotoxic T lymphocytes, convergence of their lytic granules serves to avoid bystander killing and thereby increase the efficiency of target directed killing.58 Hence it is tempting to propose an analogous mechanism in MCs whereby convergence of the SGs would prevent their accidental fusion with the plasma membrane during cell migration. Indeed, given the extensive turnover and constant remodeling of the plasma membrane that take place in a migrating cell and the fact that a fraction of SGs are fusion competent already in resting MCs, with cortical actin serving the only barrier for secretion,59 such a mechanism would ensure retainment of the inflammatory mediators during cell migration and thereby increase the efficacy of secretagogue-induced secretion at the site of inflammation. In particular, in view of the fact that MCs can undergo compound exocytosis,60–62 in which fused granules serve as hooks for the attachment of subsequent fusing SGs, it might be critical for the MC to develop an inhibitory mechanism for restricting SG fusion during migration. Indeed, we show that this constraint is relieved on encounter with a secretagogue, conditions under which the “migratory” actin phenotype is replaced by a “secretory” actin phenotype. These interchangeable actin phenotypes are independent of the MC or chemoattractant type and were noted in CXCR1-expressing RBL cells in response to IL-8 and in BMMCs in response to PGE2.
Moreover, the phenotypic switches between a migratory, secretion-restricting, and secretory phenotype are not confined only to the sequential trigger by a chemokine, followed by a secretagogue. We show that a similar shift takes place when MCs migrate toward an antigen, whereby exposure to low concentrations imposes a “migratory” actin phenotype that supports migration while restricting degranulation, whereas greater concentrations, which are expected to be present at the peak of the chemotaxis gradient, stimulate a switch in the actin phenotype, allowing cell degranulation. Therefore encounter with a high concentration of antigen does not only relieve the secretory barrier imposed by chemokines, such as IL-8 or PGE2, but also relieves its own constraint.
Actin polymerization relies on branching of new filaments mediated by the Arp2/3 complex and elongation of pre-existing filaments, which is mainly mediated by the formin family of actin-polymerizing proteins.63 Although the Arp2/3 complex comprises a permanent composition, the formin family includes multiple actin polymerizers.55,64 Therefore we anticipated that the interchangeable actin architectures would be regulated by the more flexible formin family of actin-polymerizing proteins. In particular, mDia1, a member of this family, was already implicated in control of migration of immune cells.29–31
Therefore we chose to focus on this formin, postulating that mDia1 cycling between active and inactive conformations might govern actin phenotype switching. Indeed, a number of observations support this notion. First, expression of CA mDia1, which is devoid of its autoinhibitory DAD, enforces a migratory actin phenotype on cells triggered to FcεRI-mediated secretion, and accordingly, their degranulation is inhibited. Second, CytD, which prevents actin polymerization, relieves this inhibition, thus indicating that inhibition of secretion by CA mDia1 is coupled to actin remodeling. Third, both WT and CA mDia1 significantly increase IL-8–stimulated migration. Finally, mDia1 KD inhibits IL-8– and PGE2-stimulated migration and also relieves the migratory actin phenotype.
It is interesting to note that the CA mDia1 mutant, which lacks the DAD motif, requires cell activation to be able to impose a migratory actin phenotype. We interpret these results as implying that activation/inactivation cycles of mDia1 require the apposition and removal of 2 sequential signals (see the model in Fig 9). The first signal involves the removal of the autoinhibitory constraint imposed by the DAD or by the binding of chemoattractant-activated RhoA27 or deletion of the DAD, as is the case in the CA mDia1 mutant. The second yet unidentified signal is also elicited by the chemoattractant but can also be transmitted by activated FcεRI (Fig 9). Our model gains support from our findings that overexpressed WT mDia1 increases IL-8-induced cell migration, but unlike CA mDia1, the WT autoinhibited protein cannot switch the secretory actin phenotype into a migratory one nor does it inhibit antigen-induced secretion. This model is compatible with previous results that demonstrated the involvement of RhoA in MC migration,23 as opposed to inhibition of RhoA activity by IgE/antigen18 and its dispensability for antigen-induced secretion.65
FIG 9.
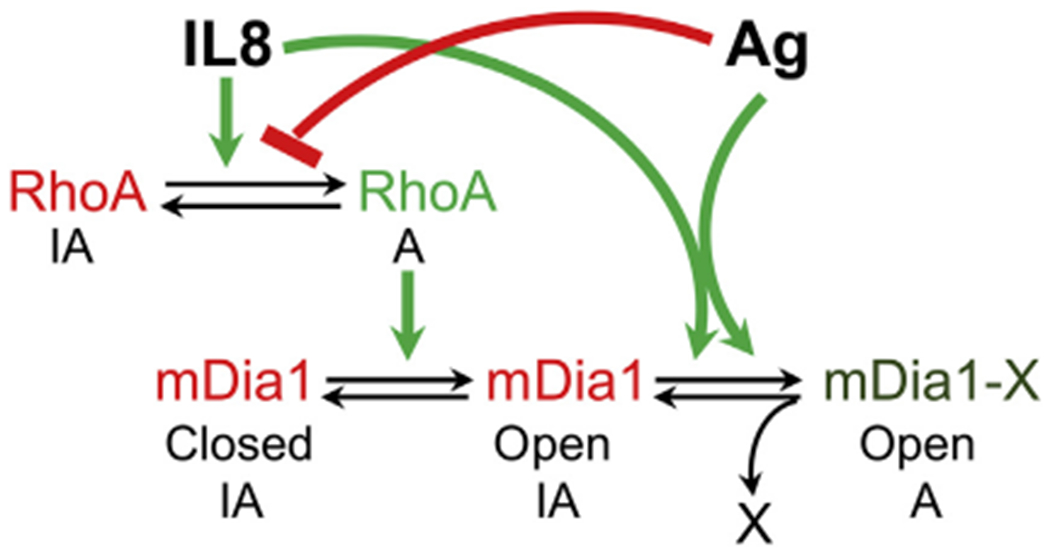
Model for the role of mDia1 in coordinating MC migration and secretion. According to our proposed model, MC activation by IL-8 involves activation of RhoA, which in turn binds to and relieves mDia1 from its autoinhibited state (Closed IA converted to Open IA). Another yet unknown modification is required that can be transduced by either IL-8 or FcεRI signaling to achieve full activation of mDia1 (Open IA converted to mDia1-X Open A). Through its actin-polymerizing activity, mDia1 then builds up the pericentral actin clusters that prevent cell flattening and converge the SGs, thereby inhibiting their release. When IL-8-stimulated cells are exposed to FcεRI signaling, the latter inhibits RhoA activation, thereby reverting mDia1 to its autoinhibited state. Therefore the secretory signal overrides the migratory signal, which is elicited by the chemokine receptor.
The nature of the putative second activating signal is presently unknown. Phosphorylation is an attractive candidate based on its involvement in coactivation of mDia3, another member of the formin family.66 It is interesting to note that mammalian target of rapamycin complex 2 (mTORC2) was implicated in controlling PGE2-triggered chemotaxis by regulating actin polymerization.14 Given the role of both mTORC2 and mDia1 in mechanically induced cytoskeletal reorganization,67,68 it is tempting to speculate that mTORC2 can regulate mDia1 activity downstream of RhoA.
We evaluated the role of mDia1 in MC homing under homeostatic conditions and detected no difference in the amount of histamine in tissues derived from WT versus mDia1 knockout mice. Although we analyzed histamine levels only in the ear, spleen, and peritoneum, the fact that we also detected no differences in the amount of MC progenitors in the bone marrow suggests that homing to other tissues is most likely also unaffected.
Two not necessarily mutually exclusive reasons might account for this observation. First, mDia1 might play a role only in the mobilization of mature MCs in tissues, under conditions of inflammation, whereas it might play no role in the homing of MC progenitors.
Alternatively, loss of mDia1 might be compensated by other members of the formin family. This latter view is supported by a number of independent observations. First, although our in vitro studies demonstrate significant inhibition of cell migration in mDia1 KD cells, they also show that this inhibition is partial and therefore unlikely to affect steady-state distribution. Second, although T-cell migration is regulated by mDia1,69 we have previously shown that lymphocyte numbers in peripheral blood do not change in mDia1 knockout mice.53 This finding is consistent with studies by others that have shown that some of the functions of mDia1 are compensated in mDia1 knockout mice by mDia2.30
Finally, we have previously shown that the reduction in neutrophil numbers in the peripheral blood of mDia1 knockout mice is not due to their defect in migration, which is known to be regulated by mDia1, but rather is the result of impaired endocytosis of CD11b, which increases neutrophil retention in the bone marrow.53 MC progenitors do not express CD11b and therefore might be exempt from such a regulatory constraint. Future studies on mDia1/mDia2 double-knockout mice will aim to resolve the role of formins in vivo.
In conclusion, we have characterized the phenotypic responses of the actin meshwork in MCs activated by a migratory versus secretory signal. We demonstrate the negative regulation imposed by the migratory actin skeleton on spatiotemporal features of the SGs to prevent premature secretion. Finally, we identify mDia1 as a novel regulator of MC responses that coordinates enhanced migration with inhibition of secretion through its actin-nucleating activity.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Key messages.
The actin skeleton significantly differs in MCs stimulated to migration by IL-8 or PGE2 or to secretion by FcεRI.
The secretory actin phenotype overrides the migratory phenotype when chemoattractant-activated cells encounter a secretagogue.
The actin-nucleating formin mDia1 mediates buildup of the migratory actin phenotype and thereby coordinates MC chemotaxis and secretion.
Acknowledgments
We thank Drs Uri Ashery, Adit Ben Baruch, Chen Luxenburg, Eran Perlson, and Ben Zion-Shilo for their generous gifts of cDNAs. We thank Drs Gilad Mass, Leonide Mittleman, Miriam Shaharbani, Yael Zilberstein, and Alexandra Lichtenstein from the Sackler Cellular & Molecular Imaging Center for their invaluable assistance with microscopy and image analyses. We thank Drs Eran Perlson and Eitan Erez Zahavi for their invaluable assistance with the TIRF experiments and Dr Eran Perlson for stimulating discussions and for critical reading of this manuscript.
Supported by grant 933/15 from the Israel Science Foundation founded by the Israel Academy for Sciences (to R.S.-E.) and by the United States-Israel Binational Science Foundation (grant 2013263 to R.S.-E.). This work was supported in part by a travel grant from the Constantiner Institute (to O.K.) and a scholarship from the Ministry of Aliyah and Integration (to P.L.-H.). P.J. is a scholar of the Leukemia and Lymphoma Society. This workis partially supported by the Department of Defense (CA140119; to P.J.).
Abbreviations used
- BMMC
Bone marrow–derived mast cell
- CC1
Coiled coil 1
- CytD
Cytochalasin D
- DAD
Diaphanous autoregulatory domain
- DN
Dominant negative
- DNP
Dinitrophenyl
- ERK
Extracellular signal-regulated kinase
- GFP
Green fluorescent protein
- IL-33R
IL-33 receptor
- KD
Knockdown
- MC
Mast cell
- mDia1
Mammalian diaphanous-related formin 1
- mTORC2
Mammalian target of rapamycin complex 2
- NPY
Neuropeptide Y
- PGE2
Prostaglandin E2
- SCF
Stem cell factor
- SG
Secretory granule
- shRNA
Short hairpin RNA
- TIRF
Total internal reflection fluorescence
- WT
Wild-type
Footnotes
Disclosure of potential conflict of interest: The authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Okayama Y, Kawakami T. Development, migration, and survival of mast cells. Immunol Res 2006;34:97–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mukai K, Tsai M, Starkl P, Marichal T, Galli SJ. IgE and mast cells in host defense against parasites and venoms. Semin Immunopathol 2016;38:581–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan CY, St John AL, Abraham SN. Plasticity in mast cell responses during bacterial infections. Curr Opin Microbiol 2012;15:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mukai K, Tsai M, Saito H, Galli SJ. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol Rev 2018;282:121–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol 2014;14:478–94. [DOI] [PubMed] [Google Scholar]
- 6.Moon TC, Befus AD, Kulka M. Mast cell mediators: their differential release and the secretory pathways involved. Front Immunol 2014;5:569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benhamou M, Blank U. Stimulus-secretion coupling by high-affinity IgE receptor: new developments. FEBS Lett 2010;584:4941–8. [DOI] [PubMed] [Google Scholar]
- 8.Redegeld FA, Yu Y, Kumari S, Charles N, Blank U. Non-IgE mediated mast cell activation. Immunol Rev 2018;282:87–113. [DOI] [PubMed] [Google Scholar]
- 9.Blank U The mechanisms of exocytosis in mast cells. Adv Exp Med Biol 2011; 716:107–22. [DOI] [PubMed] [Google Scholar]
- 10.Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast cell infiltration of airway smooth muscle in asthma. N Engl J Med 2002;346: 1699–705. [DOI] [PubMed] [Google Scholar]
- 11.Ramirez-Valadez KA, Vazquez-Victorio G, Macias-Silva M, Gonzalez-Espinosa C. Fyn kinase mediates cortical actin ring depolymerization required for mast cell migration in response to TGF-β in mice. Eur J Immunol 2017;47:1305–16. [DOI] [PubMed] [Google Scholar]
- 12.Halova I, Draberova L, Draber P. Mast cell chemotaxis—chemoattractants and signaling pathways. Front Immunol 2012;3:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smrž D, Bandara G, Beaven MA, Metcalfe DD, Gilfillan AM. Prevention of F-actin assembly switches the response to SCF from chemotaxis to degranulation in human mast cells. Eur J Immunol 2013;43:1873–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuehn HS, Jung M-Y, Beaven MA, Metcalfe DD, Gilfillan AM. Prostaglandin E2 activates and utilizes mTORC2 as a central signaling locus for the regulation of mast cell chemotaxis and mediator release. J Biol Chem 2011;286:391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juremalm M, Nilsson G. Chemokine receptor expression by mast cells. In: Mast cells in allergic diseases. Basel: Karger; 2005. pp. 130–44. [DOI] [PubMed] [Google Scholar]
- 16.Lippert U, Artuc M, Grützkau A, Möller A, Kenderessy-Szabo A, Schadendorf D, et al. Expression and functional activity of the IL-8 receptor type CXCR1 and CXCR2 on human mast cells. J Immunol 1998;161:2600–8. [PubMed] [Google Scholar]
- 17.Brightling CE, Kaur D, Berger P, Morgan AJ, Wardlaw AJ, Bradding P. Differential expression of CCR3 and CXCR3 by human lung and bone marrow-derived mast cells: implications for tissue mast cell migration. J Leukoc Biol 2005;77:759–66. [DOI] [PubMed] [Google Scholar]
- 18.Tůmová M, Koffer A, Šimíček M, Dráberová L, Dráber P. The transmembrane adaptor protein NTAL signals to mast cell cytoskeleton via the small GTPase Rho. Eur J Immunol 2010;40:3235–45. [DOI] [PubMed] [Google Scholar]
- 19.Taub D, Dastych J, Inamura N, Upton J, Kelvin D, Metcalfe D, et al. Bone marrow-derived murine mast cells migrate, but do not degranulate, in response to chemokines. J Immunol 1995;154:2393–402. [PubMed] [Google Scholar]
- 20.Bischoff SC, Dahinden CA. c-kit ligand: a unique potentiator of mediator release by human lung mast cells. J Exp Med 1992;175:237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vosseller K, Stella G, Yee NS, Besmer P. c-kit receptor signaling through its phosphatidylinositide-3’-kinase-binding site and protein kinase C: role in mast cell enhancement of degranulation, adhesion, and membrane ruffling. Mol Biol Cell 1997;8:909–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laffargue M, Calvez R, Finan P, Trifilieff A, Barbier M, Altruda F, et al. Phosphoinositide 3-kinase gamma is an essential amplifier of mast cell function. Immunity 2002;16:441–51. [DOI] [PubMed] [Google Scholar]
- 23.Toda M, Dawson M, Nakamura T, Munro PMG, Richardson RM, Bailly M, et al. Impact of engagement of FcεRI and CC chemokine receptor 1 on mast cell activation and motility. J Biol Chem 2004;279:48443–8. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen M, Solle M, Audoly LP, Tilley SL, Stock JL, McNeish JD, et al. Receptors and signaling mechanisms required for prostaglandin E2-mediated regulation of mast cell degranulation and IL-6 production. J Immunol 2002;169:4586–93. [DOI] [PubMed] [Google Scholar]
- 25.Kühn S, Geyer M. Formins as effector proteins of Rho GTPases. Small GTPases 2014;5:e983876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe N, Madaule P, Reid T, Ishizaki T, Watanabe G, Kakizuka A, et al. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J 1997;16:3044–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lammers M, Rose R, Scrima A, Wittinghofer A. The regulation of mDia1 by autoinhibition and its release by Rho*GTP. EMBO J 2005;24:4176–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rose R, Weyand M, Lammers M, Ishizaki T, Ahmadian MR, Wittinghofer A. Structural and mechanistic insights into the interaction between Rho and mammalian Dia. Nature 2005;435:513–8. [DOI] [PubMed] [Google Scholar]
- 29.Shi Y, Zhang J, Mullin M, Dong B, Alberts AS, Siminovitch KA. The mDial for-min is required for neutrophil polarization, migration, and activation of the LARG/ RhoA/ROCK signaling axis during chemotaxis. J Immunol 2009;182:3837–45. [DOI] [PubMed] [Google Scholar]
- 30.Tanizaki H, Egawa G, Inaba K, Honda T, Nakajima S, Moniaga CS, et al. Rho-mDia1 pathway is required for adhesion, migration, and T-cell stimulation in dendritic cells. Blood 2010;116:5875–84. [DOI] [PubMed] [Google Scholar]
- 31.Vicente-Manzanares M, Rey M, Perez-Martinez M, Yanez-Mo M, Sancho D, Cabrero JR, et al. The RhoA effector mDia is induced during T cell activation and regulates actin polymerization and cell migration in T lymphocytes. J Immunol 2003; 171:1023–34. [DOI] [PubMed] [Google Scholar]
- 32.Keerthivasan G, Mei Y, Zhao B, Zhang L, Harris CE, Gao J, et al. Aberrant overexpression of CD14 on granulocytes sensitizes the innate immune response in mDia.1 heterozygous del(5q) MDS. Blood 2014;124:780–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Azouz NP, Matsui T, Fukuda M, Sagi-Eisenberg R. Decoding the regulation of mast cell exocytosis by networks of Rab GTPases. J Immunol 2012;189: 2169–80. [DOI] [PubMed] [Google Scholar]
- 34.Baram D, Adachi R, Medalia O, Tuvim M, Dickey BF, Mekori YA, et al. Synap-totagmin II negatively regulates Ca2+-triggered exocytosis of lysosomes in mast cells. J Exp Med 1999;189:1649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azouz NP, Zur N, Efergan A, Ohbayashi N, Fukuda M, Amihai D, et al. Rab5 is a novel regulator of mast cell secretory granules: impact on size, cargo, and exocytosis. J Immunol 2014;192:4043–53. [DOI] [PubMed] [Google Scholar]
- 36.Falcone FH, Wan D, Barwary N, Sagi-Eisenberg R. RBL cells as models for in vitro studies of mast cells and basophils. Immunol Rev 2018;282:47–57. [DOI] [PubMed] [Google Scholar]
- 37.Nasser MW, Raghuwanshi SK, Grant DJ, Jala VR, Rajarathnam K, Richardson RM. Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J Immunol 2009;183:3425–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh V, Raghuwanshi SK, Smith N, Rivers EJ, Richardson RM. G Protein-coupled receptor kinase-6 interacts with activator of G protein signaling-3 to regulate CXCR2-mediated cellular functions. J Immunol 2014;192:2186–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raghuwanshi SK, Su Y, Singh V, Haynes K, Richmond A, Richardson RM. The chemokine receptors CXCR1 and CXCR2 couple to distinct G protein-coupled receptor kinases to mediate and regulate leukocyte functions. J Immunol 2012;189:2824–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barlic J, Khandaker MH, Mahon E, Andrews J, DeVries ME, Mitchell GB, et al. beta-arrestins regulate interleukin-8-induced CXCR1 internalization. J Biol Chem 1999;274:16287–94. [DOI] [PubMed] [Google Scholar]
- 41.Matityahu E, Feniger-Barish R, Meshel T, Zaslaver A, Ben-Baruch A. Intracellular trafficking of human CXCR1 and CXCR2: regulation by receptor domains and actin-related kinases. Eur J Immunol 2002;32:3525–35. [DOI] [PubMed] [Google Scholar]
- 42.Feniger-Barish R, Yron I, Meshel T, Matityahu E, Ben-Baruch A. IL-8-induced migratory responses through CXCR1 and CXCR2: Association with phosphorylation and cellular redistribution of focal adhesion kinase †. Biochemistry 2003;42: 2874–86. [DOI] [PubMed] [Google Scholar]
- 43.Richardson RM, Marjoram RJ, Barak LS, Snyderman R. Role of the cytoplasmic tails of CXCR1 and CXCR2 in mediating leukocyte migration, activation, and regulation. J Immunol 2003;170:2904–11. [DOI] [PubMed] [Google Scholar]
- 44.Kuehn HS, Radinger M, Brown JM, Ali K, Vanhaesebroeck B, Beaven MA, et al. Btk-dependent Rac activation and actin rearrangement following Fc RI aggregation promotes enhanced chemotactic responses of mast cells. J Cell Sci 2010;123: 2576–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frigeri L, Apgar JR. The role of actin microfilaments in the down-regulation of the degranulation response in RBL-2H3 mast cells. J Immunol 1999;162: 2243–50. [PubMed] [Google Scholar]
- 46.Wilson JD, Shelby SA, Holowka D, Baird B. Rab11 regulates the mast cell exo-cytic response. Traffic 2016;17:1027–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith AJ, Pfeiffer JR, Zhang J, Martinez AM, Griffiths GM, Wilson BS. Microtubule-dependent transport of secretory vesicles in RBL-2H3 cells. Traffic 2003;4: 302–12. [DOI] [PubMed] [Google Scholar]
- 48.Efergan A, Azouz NP, Klein O, Noguchi K, Rothenberg ME, Fukuda M, et al. Rab12 regulates retrograde transport of mast cell secretory granules by interacting with the RILP-dynein complex. J Immunol 2016;196:1091–101. [DOI] [PubMed] [Google Scholar]
- 49.Jung ID, Lee H- S, Lee HY, Choi OH. FcεRI-mediated mast cell migration: signaling pathways and dependence on cytosolic free Ca2+ concentration. Cell Signal 2009;21:1698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Munoz I, Danelli L, Claver J, Goudin N, Kurowska M, Madera-Salcedo IK, et al. Kinesin-1 controls mast cell degranulation and anaphylaxis through PI3K-dependent recruitment to the granular Slp3/Rab27b complex. J Cell Biol 2016; 215:203–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daou P, Hasan S, Breitsprecher D, Baudelet E, Camoin L, Audebert S, et al. Essential and nonredundant roles for Diaphanous formins in cortical microtubule capture and directed cell migration. Mol Biol Cell 2014;25:658–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Towns WL, Tauhata SBF, Vaughan PS, Vaughan KT. Transfection-induced defects in dynein-driven transport: evidence that ICs mediate cargo-binding. Cell Motil Cytoskeleton 2009;66:80–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mei Y, Feng G, Rahimi N, Zhao B, Zhang J, Cao L, et al. Loss of mDia1 causes neutropenia via attenuated CD11b endocytosis and increased neutrophil adhesion to the endothelium. Blood Adv 2017;1:1650–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science 2003;302:1704–9. [DOI] [PubMed] [Google Scholar]
- 55.Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J. Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev 2014;94:235–63. [DOI] [PubMed] [Google Scholar]
- 56.Porat-Shliom N, Milberg O, Masedunskas A, Weigert R. Multiple roles for the actin cytoskeleton during regulated exocytosis. Cell Mol Life Sci 2013;70: 2099–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Malacombe M, Bader M-F, Gasman S. Exocytosis in neuroendocrine cells: new tasks for actin. Biochim Biophys Acta 2006;1763:1175–83. [DOI] [PubMed] [Google Scholar]
- 58.Hsu H-T, Mace EM, Carisey AF, Viswanath DI, Christakou AE, Wiklund M, et al. NK cells converge lytic granules to promote cytotoxicity and prevent bystander killing. J Cell Biol 2016;215:875–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Higashio H, Satoh Y, Saino T. Mast cell degranulation is negatively regulated by the Munc13–4-binding small-guanosine triphosphatase Rab37. Sci Rep 2016;6: 22539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alvarez de Toledo G, Fernandez JM. Compound versus multigranular exocytosis in peritoneal mast cells. J Gen Physiol 1990;95:397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gaudenzio N, Sibilano R, Marichal T, Starkl P, Reber LL, Cenac N, et al. Different activation signals induce distinct mast cell degranulation strategies. J Clin Invest 2016;126:3981–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klein O, Roded A, Zur N, Azouz NP, Pasternak O, Hirschberg K, et al. Rab5 is critical for SNAP23 regulated granule-granule fusion during compound exocytosis. Sci Rep 2017;7:15315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pollard TD. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu Rev Biophys Biomol Struct 2007;36:451–77. [DOI] [PubMed] [Google Scholar]
- 64.Schonichen A, Geyer M. Fifteen formins for an actin filament: a molecular view on the regulation of human formins. Biochim Biophys Acta 2010;1803:152–63. [DOI] [PubMed] [Google Scholar]
- 65.Sheshachalam A, Baier A, Eitzen G. The effect of Rho drugs on mast cell activation and degranulation. J Leukoc Biol 2017;102:71–81. [DOI] [PubMed] [Google Scholar]
- 66.Cheng L, Zhang J, Ahmad S, Rozier L, Yu H, Deng H, et al. Aurora B regulates formin mDia3 in achieving metaphase chromosome alignment. Dev Cell 2011; 20:342–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu M, Yuan X, Lu C, Le S, Kawamura R, Efremov AK, et al. mDia1 senses both force and torque during F-actin filament polymerization. Nat Commun 2017;8: 1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sen B, Xie Z, Case N, Thompson WR, Uzer G, Styner M, et al. mTORC2 regulates mechanically induced cytoskeletal reorganization and lineage selection in marrow-derived mesenchymal stem cells. J Bone Miner Res 2014;29:78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sakata D, Taniguchi H, Yasuda S, Adachi-Morishima A, Hamazaki Y, Nakayama R, et al. Impaired T lymphocyte trafficking in mice deficient in an actin-nucleating protein, mDia1. J Exp Med 2007;204:2031–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.