

Abstract
Core fucose is an N-glycan structure synthesized by α1,6-fucosyltransferase 8 (FUT8) localized to the Golgi apparatus and critically regulates the functions of various glycoproteins. However, how FUT8 activity is regulated in cells remains largely unclear. At the luminal side and uncommon for Golgi proteins, FUT8 has an Src homology 3 (SH3) domain, which is usually found in cytosolic signal transduction molecules and generally mediates protein-protein interactions in the cytosol. However, the SH3 domain has not been identified in other glycosyltransferases, suggesting that FUT8's functions are selectively regulated by this domain. In this study, using truncated FUT8 constructs, immunofluorescence staining, FACS analysis, cell-surface biotinylation, proteomics, and LC-electrospray ionization MS analyses, we reveal that the SH3 domain is essential for FUT8 activity both in cells and in vitro and identified His-535 in the SH3 domain as the critical residue for enzymatic activity of FUT8. Furthermore, we found that although FUT8 is mainly localized to the Golgi, it also partially localizes to the cell surface in an SH3-dependent manner, indicating that the SH3 domain is also involved in FUT8 trafficking. Finally, we identified ribophorin I (RPN1), a subunit of the oligosaccharyltransferase complex, as an SH3-dependent binding protein of FUT8. RPN1 knockdown decreased both FUT8 activity and core fucose levels, indicating that RPN1 stimulates FUT8 activity. Our findings indicate that the SH3 domain critically controls FUT8 catalytic activity and localization and is required for binding by RPN1, which promotes FUT8 activity and core fucosylation.
Keywords: Src homology 3 domain (SH3 domain); glycosyltransferase; N-linked glycosylation; glycosylation; fucosyltransferase; α1,6-fucosyltransferase; core fucosylation; ribophorin I; posttranslational modification; protein-protein interaction; glycan
Introduction
Protein functions are dynamically regulated by posttranslational modifications including glycosylation, the most common form of protein modification in mammals (1). Glycans are composed of an enormous number of structures and are roughly divided into N- and O-glycans, depending on their core structures (1, 2). N-Glycans, the focus of this study, are highly conserved posttranslational modifications occurring in the secretory pathway. More than 7,000 proteins are known to be N-glycosylated in humans (3), and these N-glycans are critically involved in various physiological phenomena (4). Mechanistically, the alteration of an N-glycan structure on a certain protein brings about changes in protein functions, including folding, activity, and localization as well as modulation in cell properties, including cell adhesion, cell-cell interaction, and signaling (5). Furthermore, N-glycan alterations are also involved in disease development and aggravation (6). Consistent with this, knockout of an N-glycan–synthesizing enzyme in mice often results in spontaneous development or improvement of various disease-like phenotypes, such as cancer (7), chronic obstructive pulmonary disease (8), and Alzheimer's disease (9). Therefore, to understand and regulate the physiological and pathological functions of N-glycans, it is important to elucidate the mechanisms of how N-glycan expression is regulated in cells and dysregulated in disease states.
N-Glycans are biosynthesized in the endoplasmic reticulum (ER) and the Golgi apparatus by the stepwise actions of glycosyltransferases (4). Initiation of N-glycosylation involves an attachment of the common triglucosylated glycan (Glc3Man9GlcNAc2) to an asparagine in the consensus sequence (Asn-X-Thr/Ser) by oligosaccharyltransferase (OST) (10). The transferred glycan is further modified by various glycosyltransferases through the secretory pathway. The early steps of N-glycan synthesis in the ER produce common oligomannosidic structures, whereas the late steps in the Golgi produce a number of N-glycan structures that vary in cell type–, protein–, and even glycosylation site–specific manners, leading to the expression of complex N-glycans. Golgi-localized glycosyltransferases involved in the late steps are typically type-II membrane proteins consisting of an N-terminal cytosolic tail, a transmembrane domain, a stem region, and a large C-terminal catalytic domain (11). Almost all human glycosyltransferase genes (∼180) have been identified, which allows us to propose the enzymes responsible for the most major N-glycan biosynthetic pathways. However, it remains largely unclear how cellular activity and trafficking of each glycosyltransferase are regulated.
Among the various glycosyltransferases involved in N-glycan biosynthesis, we focused on α1,6-fucosyltransferase (FUT8), which transfers a fucose to the innermost GlcNAc residue to produce core fucose (Fig. 1A) (12, 13). Studies using Fut8-knockout mice revealed the various physiological functions of core fucose, including regulation of epidermal growth factor receptor signaling (14), T cell receptor signaling (15), and synaptic plasticity (16). Furthermore, core fucosylation is involved in diseases. For example, Fut8-deficient mice spontaneously develop emphysema-like changes in the lung (8), and a recent report showed that core fucose is a driver of melanoma metastasis (17). Additionally, removal of core fucose from IgG1 drastically enhances the antibody-dependent cellular cytotoxicity (18). These studies highlight the importance of regulating the levels of core fucose and FUT8 activity for therapeutic applications.
Figure 1.

Core fucosylation activity of FUT8 WT and FUT8ΔSH3. A, schematic model of core fucosylation by FUT8. B, the three-dimensional structure of human FUT8 luminal domain (left) and domain structures of FUT8 WT, FUT8ΔSH3, and soluble forms of FUT8. C, plasmid constructs used in this study. D, FUT8 WT or FUT8ΔSH3 was expressed in HEK293 FUT8KO cells. The proteins in cell lysates were separated by SDS-PAGE and then analyzed by Western blotting with anti-FUT8 (sheep Ab) and GAPDH Abs or lectin blotting with PhoSL. E, soluble FUT8 WT and ΔSH3 were expressed in COS7 cells and purified from the media using a Ni2+ column. An arrow indicates the positions of purified FUT8 WT and ΔSH3, which were visualized by CBB staining. F, purified FUT8 WT and ΔSH3 were reacted with the oligosaccharide acceptor (GnGn-bi-Asn-PNSNB) and GDP-Fuc, and the acceptor substrate and the product were separated using HPLC. G, the specific activity of FUT8, calculated by the peak area in F (n = 3, mean ± S.D. (error bars)). The measurements were taken from triplicates of one sample.
FUT8 is considered to be localized in the Golgi, having a typical type II topology (19). However, a previous structural study revealed that the FUT8 luminal domain has a unique C-terminal Src homology 3 (SH3) domain (Fig. 1B) (20). The SH3 domain is usually found in signal transduction molecules in the cytosol and mediates protein-protein interactions by recognizing a proline-rich peptide (21). No other glycosyltransferase has been found to be equipped with this domain, suggesting that the SH3 domain in FUT8 specifically regulates FUT8 functions, such as core fucosylation activity, subcellular localization, and/or interaction with other molecules.
In this study, by using mutant FUT8 lacking the SH3 domain, we show that the SH3 domain is essential for FUT8 catalytic activity and subcellular trafficking. We also identified a specific binding protein, ribophorin I (RPN1), which interacts with FUT8 in an SH3 domain–dependent fashion and positively regulates FUT8 activity. Together, these findings advance our understanding of the regulation mechanisms of FUT8 activity and localization, providing new insights into how glycosyltransferase functions are regulated in cells.
Results
SH3 domain is essential for FUT8 activity
To determine whether the SH3 domain is required for FUT8 activity, we generated constructs of human FUT8 mutants that lack the SH3 domain (FUT8ΔSH3). Because the distance between the N and C termini of the SH3 domain (Asn503–Glu563) is ∼9.6 Å (right box in Fig. 1B), we replaced the SH3 domain with a linker ∼10 Å in size consisting of 4 glycine or 6 glycine units (4Gly or 6Gly) (Fig. 1C). To examine the activity of the FUT8ΔSH3 mutants in living cells, WT and mutant FUT8 were expressed in HEK293 FUT8KO cells (16), and their product glycans were detected by lectin blots with the core fucose-specific lectin PhoSL (Pholiota squarrosa lectin) (22). Compared with the faint signals in the mock-treated sample, expression of FUT8 WT drastically increased the signals for the fucosylated proteins (Fig. 1D, first and second lanes). This indicated that PhoSL specifically recognized core fucose synthesized by FUT8 in cells. In contrast, the samples from FUT8ΔSH3 barely recovered expression of core fucose (Fig. 1D, third and fourth lanes), showing that the cellular activity of FUT8ΔSH3 was much lower than that of FUT8 WT. To directly examine whether FUT8ΔSH3 mutants lack catalytic activity itself, we expressed and purified recombinant soluble FUT8 and its ΔSH3 mutants (Fig. 1, C and E). To measure the activity of purified soluble FUT8 and its mutants, these enzymes were incubated with a fluorescently labeled acceptor N-glycan (GnGnbiAsn-PNS), and the reaction mixtures were analyzed by reverse-phase HPLC, which allows separation of the acceptor substrate and the product (Fig. 1, F and G). The FUT8 activity was calculated based on the peak area of the product (Fig. 1G). Recombinant FUT8 WT efficiently transferred a fucose, whereas no product peak was observed for the FUT8ΔSH3 mutants (Fig. 1F and Fig. S1), demonstrating that FUT8ΔSH3 is catalytically inactive in vitro. These findings clearly show that SH3 domain of FUT8 is essential for FUT8 activity.
We next sought to identify the amino acids in the SH3 domain that are required for FUT8 activity. Structural comparison with plant FUT1 in complexes with a donor analog (GDP) and acceptor xyloglucan (Protein Data Bank code 5KOR) (23) suggested that the FUT8 SH3 domain is located in the vicinity of the acceptor substrate (Fig. 2A). Therefore, we selected several candidate residues (Asp519, His535, Lys541, Val543, Arg545, Thr550, and Leu552) that are expected to interact with acceptor glycans or glycoproteins (Fig. 2A), and we replaced the targeted amino acids with alanine. The point mutants were expressed and purified as a soluble form (Fig. 2B), and their specific activity was measured (Fig. 2, C and D). H535A, H535A/K541A, and T550A/L552A showed dramatic reduction in activity (Fig. 2D). T550A and L552A single mutants showed almost the same and moderately reduced activity compared with WT, respectively (Fig. S2). These findings indicate that His535 in SH3 domain is critically important for the FUT8 activity.
Figure 2.

Identification of the essential amino acids in FUT8 SH3 domain. A, the structure of the FUT8 SH3 domain as a ribbon model. A donor analog (GDP) and acceptor xyloglucan (acceptor for plant FUT1) were modeled based on the FUT1 structure (Protein Data Bank code 5KOR). Light blue, SH3 domain; orange, mutated amino acids. B, soluble forms of FUT8 point mutants were expressed and purified from the COS7 culture media and then subjected to CBB staining. C, purified point mutants were mixed with the FUT8 substrates, and the reaction mixtures were separated by HPLC. D, the activity relative to that of FUT8 WT was calculated by the peak areas in C and is shown as the mean ± S.D. (error bars) (n = 3, triplicates from one sample; ***, p < 0.001, Tukey–Kramer test).
FUT8 SH3 domain influences the cell-surface localization of FUT8
FUT8 transfers a fucose in the Golgi apparatus and was reported to be localized in the Golgi (19). As described above, FUT8ΔSH3 did not have catalytic activity, which might lead to overall misfolding of FUT8ΔSH3 and its entrapment in the ER. Therefore, we examined whether FUT8ΔSH3 maintained its Golgi localization. Immunofluorescence staining of FUT8 WT or FUT8ΔSH3 expressed in HEK293 FUT8KO cells showed that FUT8 WT and ΔSH3 both largely overlapped with the Golgi marker GM130 (Fig. 3A), indicating that these proteins are mainly localized in the Golgi. This excludes the possibility that deletion of the SH3 domain caused an overall misfolding. Surprisingly, we found that FUT8 WT was also partially localized at the cell surface (Fig. 3A), and cell-surface localization was barely observed for the FUT8ΔSH3 mutants. Because cell-surface localization of glycosyltransferases, including FUT8, has rarely been reported, the SH3 domain of FUT8 may be uniquely involved in subcellular trafficking of FUT8. To confirm the unusual cell-surface localization of FUT8, we carried out two other experiments. Cell-surface proteins from FUT8 WT– or FUT8ΔSH3–expressing cells were biotinylated and subsequently purified using streptavidin beads. FUT8 WT was found to be biotinylated, whereas the ΔSH3 mutants were more weakly labeled (Fig. 3B, left panels), consistent with the findings observed from immunofluorescence staining. Additionally, biotinylation of endogenous FUT8 was also confirmed using untransfected HEK293 WT and FUT8KO cells (Fig. 3B, right panels), suggesting that endogenous FUT8 is also partially localized at the cell surface. The antibody specificities for detection of endogenous FUT8 were validated in Fig. S3. We next quantitated the levels of cell-surface FUT8, ΔSH3, or point mutants using FACS analysis (Fig. 3C). We successfully detected FUT8 WT without permeabilization, further confirming that FUT8 is partially localized at the cell surface. We also found that the quantity of cell-surface FUT8ΔSH3 was ∼50% of that of FUT8 WT (Fig. 3D), again confirming the decreased cell-surface localization of FUT8ΔSH3. Collectively, these data demonstrated that FUT8 is partially localized at the cell surface depending on its SH3 domain.
Figure 3.

Subcellular localization of FUT8 WT and ΔSH3. A, HEK293 FUT8KO cells transfected with the plasmids for FUT8 WT or ΔSH3 were stained with anti-GM130 (green) and anti-FUT8 (sheep Ab) (red) antibodies and DAPI (blue). Scale bar, 10 μm. B, cell-surface proteins were biotinylated, and the biotinylated proteins were purified from cell lysates by streptavidin-conjugated beads and analyzed by Western blotting with anti-FUT8 (sheep Ab (left) and mouse Ab (right)) and anti-N-cadherin Abs. Left, HEK293 FUT8KO transfectants; right, untransfected HEK293 WT and FUT8KO cells. C, surface proteins of HEK293 FUT8KO cells expressing FUT8 WT or ΔSH3 were stained with anti-FUT8 sheep antibody and analyzed by FACS. Red line, mock-transfected cells; blue line, FUT8 WT- or ΔSH3-expressing cells. Bars, percentage of FUT8-positive cells. D, expression levels of the cell-surface FUT8 relative to that of the FUT8 WT in FACS analysis. The graph shows the mean ± S.D. (error bars) (n = 3).
SH3 domain of FUT8 is required for binding with RPN1
The above data revealed that the SH3 domain of FUT8 is critically involved in activity and trafficking of FUT8. As the SH3 domain is generally involved in protein-protein interactions (21), we hypothesized that the regulation of FUT8 functions by the SH3 domain is also mediated by interaction of the SH3 domain with other proteins. To identify the protein that specifically binds to the SH3 domain of FUT8, we carried out immunoprecipitation of expressed FUT8 WT or FUT8ΔSH3 from the HEK293 FUT8KO cell lysates. Three protein bands that were co-immunoprecipitated with FUT8 WT but weakly with FUT8ΔSH3 (Fig. 4A, arrows) were excised and then identified by proteomics (Fig. 4A and Table 1). RPN1 was unambiguously identified in the immunoprecipitates. RPN1 is a subunit of the mammalian OST and involved in interaction with ribosomes through its cytosolic domain (24–26), but its functions are largely unclear. Western blot analyses of the immunoprecipitates with the anti-FUT8 antibody confirmed that RPN1 binds with FUT8 WT but not with FUT8ΔSH3 (Fig. 4B). These data demonstrate that FUT8 binds with the OST subunit RPN1 and that this binding depends on the SH3 domain of FUT8.
Figure 4.
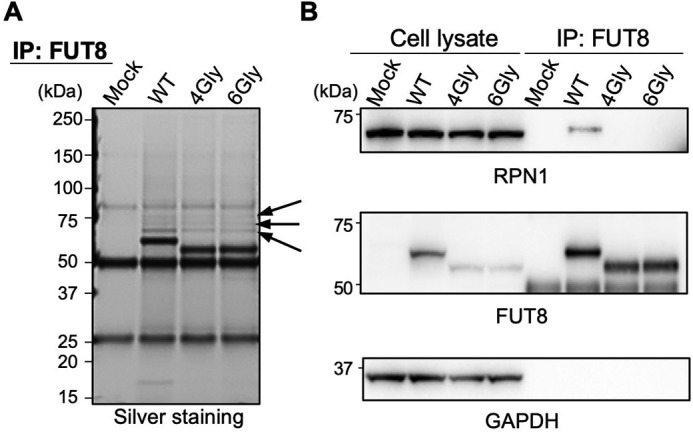
RPN1 was identified as the protein that binds to FUT8 in an SH3 domain–dependent manner. A, FUT8 WT and ΔSH3 expressed in HEK293 FUT8KO cells were immunoprecipitated (IP), and the precipitated proteins were subjected to silver staining. Arrows, co-immunoprecipitated proteins with FUT8 that were identified by proteomics. B, HEK293 FUT8KO cells were transfected with the plasmids for FUT8 WT and ΔSH3, lysed, and subjected to immunoprecipitation with anti-FUT8 (sheep Ab) antibodies. The immunoprecipitated proteins were analyzed by Western blotting with anti-FUT8 (sheep Ab), anti-RPN1, and anti-GAPDH Abs.
Table 1.
Proteomic identification of the proteins that co-precipitated with FUT8
| Sample no. | Protein | Gene | Mr | Score | Peptide | Coverage | Accession no. | Note | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | Endoplasmic reticulum chaperone BiP | HSPA5, GRP78 | 72,333 | 328 | 18 | 32 | P11021 | NP_005338.1 |
| 2 | 1 | Heat shock cognate 71-kDa protein | HSPA8, HSC70 | 70,898 | 162 | 13 | 21 | P11142 | NP_006588.1 |
| 2 | Heat shock 70-kDa protein 1A heat shock 70-kDa protein 1B | HSPA1A HSPA1B | 70,052 | 78 | 7 | 13 | P0DMV8 P0DMV9 | BAG58351.1 | |
| 3 | Dolichyl-diphosphooligosaccharide–protein-glycosyltransferase subunit 1 | RPN1 | 68,569 | 73 | 7 | 14 | P04843 | NP_002941.1 | |
| 4 | α-(1,6)-Fucosyltransferase | FUT8 | 66,516 | 46 | 4 | 6 | Q9BYC5 | NP_835368.1 | |
| 3 | 1 | Dolichyl-diphosphooligosaccharide–protein-glycosyltransferase subunit 1 | RPN1 | 68,569 | 275 | 13 | 26 | P04843 | NP_002941.1 |
| 2 | α-(1,6)-fucosyltransferase | FUT8 | 66,516 | 78 | 3 | 5 | Q9BYC5 | NP_835368.1 |
RPN1 positively regulates FUT8 activity
To examine how RPN1 is involved in regulation of FUT8 functions, HEK293 WT cells were treated with siRNA targeting RPN1 (siRPN1), and the levels of FUT8 mRNA, protein, and enzyme activity were measured. Western blotting results showed that treatment with siRPN1 effectively depleted RPN1 protein (Fig. 5A), confirming that the siRNA works well. Endogenous FUT8 protein was comparably expressed between the control and the RPN1-knocked down cells (Fig. 5A). Strikingly, the enzymatic activity of FUT8 in the siRPN1-treated cells was significantly reduced compared with that in the control cells (Fig. 5 (B and C) and Fig. S4). In contrast, the mRNA levels of FUT8 in the siRPN1-treated cells were only slightly reduced, compared with the siControl-treated cells (Fig. 5D). These data strongly suggest that RPN1 positively regulates cellular FUT8 activity at the protein level.
Figure 5.

RPN1 positively regulates FUT8 activity. A, HEK293 WT cells were treated with siRPN1 or siControl for 48 h. The cell lysates were analyzed by Western blotting with anti-FUT8 (mouse Ab), anti-RPN1, and anti-GAPDH Abs. B, the lysates of HEK293 WT cells treated with siRPN1 or siControl were incubated with the FUT8 acceptor substrate and GDP-Fuc, and the acceptor substrates and products were separated by HPLC. C, the FUT8-specific activities were calculated by the peak areas in B and shown as the mean ± S.D. (error bars) (n = 3). D, the mRNA expression levels of FUT8, MGAT3, and MGAT5 in HEK293 WT cells treated with siRPN1 were quantified by real-time PCR and shown as the values relative to those in cells treated with siControl (n = 3, mean ± S.D.). The mRNA levels were normalized to the GAPDH levels. *, p < 0.05, Mann–Whitney U test.
N-Glycomic analysis of RPN1-knocked down cells
We finally investigated by N-glycomic analysis whether RPN1 also regulates FUT8 activity in cells. HEK293 WT cells were treated with siControl or siRPN1 for 96 h, and the total N-glycans were released with peptide:N-glycosidase F (PNGase F), labeled with aminoxy TMT0, and analyzed by LC-ESI MS (Fig. 6 and Fig. S5). Although RPN1 is a subunit of OST and might be critically involved in N-glycosylation by OST, the N-glycan profiles in siRPN1-treated cells did not show drastic changes compared with siControl cells (Fig. 6A), consistent with a previous finding that RPN1 is not essential for N-glycosylation activity of the OST complex (27, 28). We detected 89 N-glycan isomers (Table S1), and the presence of core fucose in many of these glycans was confirmed by an unambiguous diagnostic ion in the MS/MS spectra (Fig. S5A). The glycans confirmed to be core-fucosylated are highlighted in Table S1 (colored in yellow), and the total amount of the core fucosylated glycans was found to be reduced in the RPN1-knocked down cells (Fig. 6B). When we compared the three major pairs of core-fucosylated and nonfucosylated glycans (Fig. 6C and Fig. S5B), the signal intensities of the core fucosylated glycans in the siRPN1-treated cells were markedly lower than in control cells, whereas the intensity of disialyl nonfucosylated glycan (No. 50) in the siRPN1-treated cells was comparable with that in control cells, and the intensity of trisialyl nonfucosylated glycan (No. 73) in the knockdown cells was even higher than in control cells. This suggests that core fucosylation of these glycans is decreased by RPN1 depletion. Together, these findings indicate that RPN1 positively regulates FUT8 activity in cells.
Figure 6.

LC-MS analysis of N-glycans in HEK293 cells treated with siRPN1. A, base peak chromatogram (BPC) from the LC-ESI MS analysis of N-glycans from HEK293 WT cells treated with siControl or siRPN1. N-Glycans were labeled with aminoxy tandem mass tag 0 (aminoxyTMT0). The deduced structures of the major N-glycans are shown. Pink, glycans increased in siRPN1; blue, glycans decreased in siRPN1; gray, glycans unchanged between the siControl and siRPN1. B, sum of the signal intensities of nonfucosylated, core-fucosylated glycans, and fucosylated glycans with unknown linkage from LC-MS analysis. C, LC-MS signal intensities of major pairs of core-fucosylated and nonfucosylated N-glycans derived from siControl- and siRPN1-treated cells.
Discussion
As described herein, the unique SH3 domain found in FUT8 is essential for the core fucosylation activity and cell-surface trafficking of FUT8. We also discovered that the OST subunit RPN1 specifically interacts with FUT8 to positively regulate FUT8 activity at the protein level. To our knowledge, a specific interacting partner of FUT8 has not been identified, and thus RPN1 is a novel and unique regulator of core fucosylation.
In general, the SH3 domain binds to proteins by recognizing proline-rich sequences. Our in vitro enzyme assays showed that FUT8 WT is fully active toward the oligosaccharide-type substrate (Fig. 1F), whereas no activity was found for FUT8ΔSH3, suggesting that the SH3 domain of FUT8 is required for the binding to acceptor glycans rather than the polypeptide part of the glycoprotein substrates. We also found that His535 in the SH3 domain is critical for the FUT8 reaction, which is consistent with a previous structural model in which His535 is located in the vicinity of the α1,3-mannose arm of the acceptor glycan (29). Therefore, the functions of the SH3 domain of FUT8 are different from the usual SH3 domain found in other molecules in the cytosol and are required for the fucose transfer reaction either directly or indirectly. One possibility is that SH3 directly interacts with a part of acceptor glycan. Another possibility is that the SH3 domain assists FUT8 folding through interaction with a chaperone-like binding partner and that ΔSH3 might lack the active fold without the aid of the SH3 domain.
At almost the same time as the initial submission of this manuscript, two papers were reported that revealed the crystal structures of mammalian FUT8 complex with donor and acceptor substrates (30, 31). In a crystal structure, the SH3 domain was shown to be involved in the interaction with the neighboring FUT8 molecule in the dimer (31), which was also supported by the recent biochemical study (32). The crystal structure paper also suggests that this interaction restricts movement of the SH3 domain. In addition, in both crystal structures, His535 interacted with the GlcNAc residue on the α1,3-mannose arm (30, 31). The presence of the GlcNAc residue in acceptor glycan was shown to be a basically prerequisite for the FUT8 action (33), which is consistent with both our mutagenesis data on His535 and the recently published crystal structure data. Intriguingly, it should be noted that some specific glycoproteins can be core-fucosylated by FUT8 even without the GlcNAc (34). How FUT8 recognizes an acceptor substrate without His535-GlcNAc interaction is an important next question to be solved.
We revealed that RPN1 binding to FUT8 depends on the SH3 domain and positively regulates FUT8 activity. Although the functions of RPN1 remain to be understood, several proteins were already reported to interact with RPN1. For example, a complex structure of yeast OST, Sec61, and a ribosome showed that cytosolic regions of RPN1 were located near the ribosome (35), suggesting that RPN1 physically interacts with the ribosome and connects the OST complex and the ribosome (36). RPN1 also binds to a membrane-anchored ER-resident lectin, malectin, which specifically recognizes Glc2Man9GlcNAc2 (G2Man9). This interaction was found to trap misfolded proteins in the ER for degradation (37, 38). Additionally, RPN1 was shown to selectively interact with a misfolded form of a model protein, suggesting that RPN1 acts as a chaperone (37). It is possible that, in the case of FUT8, RPN1 also enhances folding of FUT8 in the ER as a chaperone-like molecule. As RPN1 is a subunit of a large OST complex, it will be important to clarify whether regulation of FUT8 activity is accomplished intrinsically by RPN1 or in coordination with other OST subunits. Further investigation of whether FUT8 also interacts with other OST subunits will be needed in the future.
Surprisingly, we found that FUT8 is also partially localized at the cell surface, and its level at the cell surface is reduced by deleting the SH3 domain. Typically, glycosyltransferases are localized in the Golgi, depending on their cytoplasmic tail, transmembrane domain, and a stem region (39). The early studies demonstrated that Golgi-resident glycosyltransferases have shorter transmembrane domains compared with the plasma membrane proteins (40–42). In contrast, our findings indicate that FUT8 localization is governed by distinct mechanisms, independent of its transmembrane domain. To date, cell-surface localization of glycosyltransferases has not been studied well, and only several cases were reported, including B4GALT1, ST8SIA1, and a fucosyltransferase (43–45). B4GALT1, a major galactosyltransferase involved in the biosynthesis of lactosamine structure and lactose (46), was extensively studied with regard to its cell-surface localization. Of two B4GALT1 isoforms that differ in the length of their cytosolic tail, only the longer isoform was found to be localized to the cell surface and interact with matrix proteins, including laminin and ZP3 for cell adhesion and acrosome reaction (47, 48), whereas the shorter isoform showed Golgi-restricted localization. Considering that the mutant mice lacking only the longer form showed comparable B4GALT1 activity with WT mice (49), the surface enzyme seems to have functions different from the Golgi enzyme. Phosphorylation of the cytosolic tail was suggested to be involved in the cell-surface trafficking of B4GALT1 (50), but the detailed mechanism largely remains unclear. The trafficking mechanism of FUT8 to the cell surface is likely different from B4GALT1, because the luminal SH3 domain mediates the surface localization of FUT8. It is possible that an unknown cargo receptor recognizes the FUT8 SH3 domain to transport it to the cell surface. To date, there are only few examples of cargo receptors involved in the trafficking from the Golgi to the cell surface even in the case of other secretory or membrane proteins. Identification of a FUT8 SH3-specific cargo receptor is of importance to further our knowledge of how proteins are sorted to the post-Golgi compartments. Note that FUT8 also has several isoforms and that one transcript corresponds to a short form lacking the cytosolic tail and the transmembrane domain (NM_004480.4). This is an intriguing topic for future work to investigate where this nonmembrane type FUT8 is localized and what functions it has.
In general, regulation mechanisms of glycosyltransferase activity and localization in cells have not yet been fully understood. Considering our findings of the cell-surface localization of FUT8, Golgi localization of typical glycosyltranferases could be more dynamic than previously thought, and other enzymes could also be transiently localized at the cell surface or in endosomal compartments. Furthermore, although formation of homo- and heteroenzyme complex of Golgi glycosyltransferases were already found in many cases (51–55), interaction of Golgi glycosyltransferases with other classes of molecules is largely unclear. Our finding of the association between FUT8 and an OST subunit could lead to elucidation of a novel mode of regulation of glycosylation. By using CRISPR techniques, endogenous glycosyltransferases can be knocked out or genetically modified. This could further accelerate future research and help to clarify the detailed mechanisms of glycosyltransferase localization and interaction with other molecules. Additionally, to understand how glycosyltransferases act in the Golgi, structural studies of glycosyltransferases will also be needed (56). Moreover, it remains largely unclear how glycosyltransferases act on specific target glycoproteins, although these enzymes including FUT8 modify their substrates in a protein-specific manner (34). As fucosylated glycans have various physiological and pathological roles (57), unraveling the detailed regulation mechanisms of fucosyltransferases, including FUT8, could be useful for clinical applications as well as advancement of our knowledge of the roles of fucosylated glycans.
Experimental procedures
Reagents
The following antibodies were used: sheep anti-FUT8 (R&D Systems, AF5768), mouse anti-FUT8 (Fujirebio, clone 15C6), rabbit anti-RPN1 (Abcam, ab198508), mouse anti-GAPDH (Merck Millipore, MAB374), mouse anti-N-cadherin (BD Biosciences, 610920), rabbit anti-GM130 (Cell Signaling Technology, 12480), HRP-conjugated anti-mouse IgG (GE Healthcare, NA931V), HRP-conjugated anti-sheep IgG (Abcam, ab6900), HRP-conjugated anti-rabbit IgG (GE Healthcare, NA934V), Alexa546-conjugated anti-sheep IgG (Invitrogen, A21098), and Alexa488-conjugated anti-rabbit IgG (Invitrogen, A21206). Biotinylated PhoSL (22) was kindly provided by J-Oil Mils.
Structure representation
The three-dimensional structural inspection of the FUT8 SH3 domain was carried out using PyMOL (PyMOL Molecular Graphics System, version 2.0, Schrödinger, LLC, New York). The structural comparison with FUT1 was performed using SUPERPOSE (58).
Plasmid construction
pcDNA 6.2/humanFUT8 (a kind gift from Dr. Hiroaki Korekane (RIKEN)) was used as the template for PCR amplification of the FUT8ΔSH3 and FUT8 point mutants along with the primers listed in Table S2. For construction of FUT8ΔSH3, the SH3 domain (Ala504–Ile562) was replaced by a glycine linker (GGGG or GGGGGG). The large fragment encoding the N terminus to Asn503 was amplified by PCR, digested with BamHI, and phosphorylated by T4PNK. The oligonucleotides encoding Glu563 to the C terminus were annealed to produce a 3′-EcoRI-sticky end. These two fragments were then ligated to the BamHI-EcoRI sites of pcDNA6/myc-His A. The plasmids for the full-length FUT8 point mutants were constructed using pcDNA 6.2/hFUT8 as a template and the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer's protocol. The plasmid for the His6-tagged soluble FUT8 (pcDNA-IH/hFUT8) was constructed as described previously (59). For the soluble mutant enzymes, the DNA fragments encoding the catalytic region (Arg68 to C terminus) were amplified by PCR using pcDNA 6.2/hFUT8 mutants as templates and ligated to the EcoRI-NotI sites of pcDNA-IH (60).
Cell culture
HEK293, HEK293/FUT8KO (16), and COS7 cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 50 μg/ml kanamycin under 5% CO2 conditions at 37 °C. HEK293/FUT8KO cells, which were generated by zinc-finger technology, were kindly provided by Dr. Jianguo Gu (Tohoku Medical and Pharmaceutical University). The protein concentrations of cell lysates were measured using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific), and the same amounts of proteins were loaded to each lane in SDS-PAGE.
Plasmid transfection
The expression and empty plasmids were transfected into cells plated in a 10- or 6-cm dish at 70–80% confluence using Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific), according to the manufacturer's protocol. For expression of recombinant soluble FUT8, polyethyleneimine MAX (Polyscience) was used as described below.
Western and lectin blotting, silver and CBB staining
Proteins were resolved by 5–20% SDS-PAGE. For CBB staining, proteins separated in the gel were incubated with GelCode Blue Safe Protein Stain (Thermo Fisher Scientific) and visualized using FUSION-SOLO 7s EDGE (Vilber Lourmat). For silver staining, separated proteins were stained using Silver Stain II Kit Wako (Wako) according to the manufacturer's protocol. For Western blotting, proteins separated in the gel were transferred to nitrocellulose membranes. The membranes were blocked with PBS containing 5% skim milk and 0.1% Tween 20 and incubated with primary antibody followed by HRP-conjugated secondary antibody. For lectin blotting, the membranes were blocked with PBS containing 0.1% Tween 20 and were incubated with biotinylated lectin diluted in TBS containing 0.1% Tween 20 followed by incubation with HRP-streptavidin (VECTASTAIN ABC Standard Kit). Protein bands were detected with Western Lightning Plus-ECL (PerkinElmer Life Sciences) or SuperSignal West Femto Maximum Sensitivity substrate (Thermo Fisher Scientific) using FUSION-SOLO 7s EDGE (Vilber Lourmat). Representative images of three or more experiments are shown in the figures. The raw data for Western blots are shown in Fig. S6.
Purification of recombinant FUT8
Recombinant soluble FUT8 and its mutants were purified as described previously (59). Briefly, the expression plasmids were transfected into COS7 cells on 15-cm dishes at 70–80% confluence using polyethyleneimine MAX. After 6 h, the medium was replaced with Opti-MEM I followed by further incubation for 72 h. The His6-tagged FUT8 and its mutants were purified from the medium using a Ni2+ column, followed by desalting using a NAP-5 gel filtration column (GE Healthcare).
FUT8 activity assay
FUT8 activity was measured as described previously (60, 61). In brief, a cell lysate or purified FUT8 was incubated in 10 μl of the reaction buffer (100 mm MES-NaOH, pH 7.0, 200 mm GlcNAc, 0.5% Triton X-100, 1 mg/ml BSA), 1 mm GDP-fucose, and 10 μm fluorescently labeled acceptor substrate (GnGn-bi-Asn-PNSNB (N-(2-(2-pyridylamino)ethyl)succinamic acid 5-norbornene-2,3-dicarboxyimide ester)). The FUT8 reaction was stopped by boiling at 95 °C for 5 min, and 40 μl of water was added to the mixture. After centrifugation at 15,000 × g for 3 min, 10 μl of the supernatant was injected to an HPLC equipped with an ODS column (Tosoh; 4.6 × 150 mm).
Immunofluorescence staining
Cells cultured on an 8-well glass chamber slide were fixed with 4% PFA/PBS. The cells were washed with PBS and then permeabilized with PBS containing 0.1% Triton X-100 and 1% BSA. After washing with PBS, cells were incubated with primary antibodies followed by with Alexa488- or Alexa546-conjugated secondary antibodies and DAPI. Fluorescence was visualized using a BZ-X800 all-in-one fluorescence microscope (KEYENCE).
FACS analysis
Cells cultured on 6-cm dishes were fixed in 4% PFA/PBS. The cells were washed with PBS and suspended in FACS buffer (1% BSA in PBS) that is used for blocking and antibody dilution. The cells were incubated with sheep anti-FUT8 followed by Alexa546-conjugated anti-sheep IgG. Fluorescence signals were detected using a BD FACSMelody cell sorter (BD Bioscience), and the data were analyzed using FlowJo (FlowJo LLC).
Cell-surface biotinylation
Cells on 6-cm dishes were washed with PBS and incubated with 1 mg/ml EZ-Link Sulfo-NHS-SS-Biotin (Thermo Fisher Scientific) in PBS at 4 °C for 30 min. The biotinylation reaction was quenched by adding 100 mm glycine in PBS. After washing with PBS containing 100 mm glycine, the cells were harvested and lysed with TBS containing 1% Triton X-100, followed by ultracentrifugation at 100,000 × g for 20 min. Streptavidin-conjugated magnetic beads (Tamagawa Seiki) were added to the supernatants, followed by gentle rotation at 4 °C for 2 h or overnight. The beads were washed three times with TBS containing 0.1% Triton X-100, and the biotinylated proteins bound to the beads were eluted by boiling in 2× SDS sample buffer containing 2 mm biotin.
Immunoprecipitation
Cells were lysed with 1% Triton X-100 in PBS, and the lysates were ultracentrifuged at 100,000 × g for 20 min. The supernatants were precleared with Dynabeads protein G (Thermo Fisher Scientific) followed by rotation at 4 °C for 30 min. Then the precleared lysates were rotated with Dynabeads protein G and antibodies at 4 °C for 2 h or overnight. The beads were washed three times with 0.1% Triton X-100 in PBS and then boiled with SDS sample buffer to elute the bound proteins.
Proteomics
FUT8 WT and FUT8ΔSH3 mutants were overexpressed in HEK293 FUT8KO cells and immunoprecipitated from the cell lysates with anti-FUT8 antibody. The immunoprecipitates were subjected to SDS-PAGE and CBB staining. Three bands (around 75 kDa) from FUT8 WT-expressing cell samples, which corresponded to proteins specifically co-precipitated with FUT8, were excised. Subsequent proteomic identification was carried out at JPROteomics (Sendai, Japan). The gel pieces were destained with 50 mm NH4HCO3 in 50% CH3CN, dehydrated with CH3CN, and evaporated. The samples were reduced by incubation in 100 mm DTT in 100 mm NH4HCO3 at 56 °C for 30 min. After removing the supernatant, 100 mm iodoacetamide in 100 mm NH4HCO3 was added, and then the sample was incubated at 37 °C for 30 min for alkylation. The supernatant was removed, and the samples were washed first with 100 mm NH4HCO3 and then with CH3CN and evaporated. The proteins were digested in the gel with trypsin in 50 mm Tris-HCl, pH 7.5, at 35 °C for 17 h. The samples were desalted using a ZipTipC18 column (Millipore), and the peptides were eluted with 3 μl of 70% CH3CN containing 0.1% TFA. The samples were diluted with 20 μl of 2% CH3CN containing 0.1% formic acid and analyzed using a Bio NanoLC (KYA Technologies) linked to a QSTAR XL qTOF mass spectrometer (Applied Biosystems). The peptides were separated using an HiQ sil C18W-3 column (0.1 × 50 mm, KYA Technologies) and a four-step linear gradient (10 min at 2% CH3CN, 0.1% formic acid; 30 min at 2–41% CH3CN, 0.1% formic acid; 10 min at 41–80% CH3CN, 0.1% formic acid; 20 min at 80% CH3CN, 0.1% formic acid) at a flow rate of 200 nl/min. The MS spectra were obtained in the positive ion mode (ion spray voltage: 1.8 kV). The assignment of the MS/MS data to tryptic peptides was performed using ABSciex Analyst QS software version 1.1 (Applied Biosystems/SCIEX). All MS/MS data were searched using MASCOT MS/MS Ions Search engine (Matrix Science, April 24, 2019) against the NCBIprot 20180429 database (number of entries: 152,462,470 sequences, 55,858,910,152 residues; taxonomy: Homo sapiens). Mass tolerance for both precursor ions and fragment ions was ±0.2 Da. Cysteine carbamidomethylation and methionine oxidation were included as variable modifications. The significance threshold for MASCOT MS/MS Ions Search was set at p < 0.05, and one missed trypsin cleavage was allowed.
siRNA treatment
Cells at 30–40% confluence on 6-cm dishes were transfected with 80 pmol of siRNA using 20 μl of Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific). After 48 h, the cells were harvested for SDS-PAGE. For LC-MS analysis of N-glycans, the cells on 6-cm dishes were transfected with 80 pmol of siRNA using 20 μl of Lipofectamine RNAiMAX. After 48 h, the cells were cultured on 10-cm dishes and then transfected with 160 pmol of siRNA using 40 μl of Lipofectamine RNAiMAX, followed by a 48-h incubation. The following siRNAs were purchased from Qiagen: SI00706874 and SI03175074 for RPN1 and 1027281 for the mock treatment.
RNA extraction and quantitative PCR
The total RNA was extracted from cultured cells on a 6-cm dish with TRI Reagent (Molecular Research Center) and reverse-transcribed using the SuperScript IV First-Strand Synthesis System (Thermo Fisher Scientific) with random hexamers. The target cDNAs were amplified using a TaqMan Gene Expression Master Mix (Applied Biosystems) and the primers and probes listed below, and the amplified cDNAs were detected using a CFX Connect Real-Time PCR Detection System (Bio-Rad). The primers and probes were purchased from Applied Biosystems as follows: Hs00189535_m1 for FUT8, Hs02379589_s1 for MGAT3, Hs00159136_m1 for MGAT5, and Hs99999905_m1 for GAPDH. The mRNA levels of FUT8, MGAT3, and MGAT5 were normalized to that of the GAPDH level.
N-Glycan analysis of cell membrane proteins by LC-ESI MS
The N-glycans from the cell membrane proteins were released (62, 63), labeled with aminoxyTMT0 reagent (Thermo Fisher Scientific) and analyzed by LC-ESI MS according to previous procedures (60, 64) with modifications as follows. HEK293 cells (1 × 107 cells) were homogenized in 2 ml of lysis buffer (50 mm Tris-HCl, pH 7.4, 0.1 m NaCl, 1 mm EDTA, and protease inhibitor mixture (Roche Applied Science)) using a Polytron homogenizer (seven times for 15 s in an ice bath), followed by centrifugation at 2,000 × g for 20 min at 4 °C to remove nuclei and unbroken cells. The supernatant was diluted with 2 ml of Tris buffer (50 mm Tris-HCl, pH 7.4, 0.1 m NaCl) and then ultracentrifuged at 120,000 × g for 80 min at 4 °C. The membrane pellet was suspended in 100 μl of the Tris buffer, followed by the addition of 400 μl of the Tris buffer containing 1% Triton X-114 with pipetting. The lysate was incubated on ice for 10 min and then at 37 °C for 20 min, followed by phase partitioning by centrifugation at 1,940 × g for 2 min. The upper aqueous phase was removed, and the lower detergent phase was mixed with 1 ml of ice-cold acetone and kept at −25 °C overnight. After centrifugation at 1,940 × g for 2 min, the precipitated membrane proteins were dissolved in 10 μl of 8 m urea and spotted (2.5 μl × 4 times) onto an ethanol-pretreated polyvinylidene difluoride membrane without electrophoresis. After drying at room temperature for >4 h, the membrane was washed once with ethanol for 1 min and then three times with water for 1 min. The protein on the membrane was stained for 5 min with Direct Blue 71 (Sigma–Aldrich) (800 μl of solution A (0.1% Direct Blue 71) in 10 ml of solution B (acetic acid/ethanol/water at 1:4:5)). After destaining with solution B for 1 min, the membrane was dried at room temperature for >3 h. The protein spots were excised from the polyvinylidene difluoride membrane and placed into a well of a 96-well plate. The spots were covered with 100 μl of 1% (w/v) poly(vinylpyrrolidone) 40000 in 50% (v/v) methanol, agitated for 20 min, and washed with water (100 μl five times). PNGase F (2 units in 10 μl of 20 mm phosphate buffer, pH 7.3; Roche Applied Science) was added to the well, and the spots were incubated at 37 °C for 15 min followed by the addition of 10 μl of water and then were incubated at 37 °C overnight. The samples were sonicated in the 96-well plate for 10 min, and the released N-glycans (20 μl) were transferred to 1.5-ml polypropylene tubes. The well was washed with water (50 μl twice), and the washings were combined and evaporated. The dried N-glycans were reacted with aminoxyTMT0 reagent (Thermo Fisher Scientific; 0.02 mg in 200 μl of 95% methanol, 0.1% acetic acid solution) by continuous shaking for 15 min at room temperature. After evaporating the reaction solution, 200 μl of 95% methanol was added to the samples, followed by further shaking for 15 min. After evaporating the methanol, 100 μl of 10% acetone solution was added to the samples, followed by incubation at room temperature for 15 min with continuous shaking. The acetone was evaporated, and excess reagent was removed using Sepharose CL4B. The samples were dried, dissolved in 20 μl of 10 mm ammonium bicarbonate, and analyzed by LC-ESI MS and MS/MS. N-Glycans labeled with aminoxyTMT0 were separated on a carbon column (5 μm HyperCarb, 1-mm inner diameter × 100 mm; Thermo Fisher Scientific; column oven: 40 °C) using an Accela HPLC pump (flow rate: 50 μl/min) and a sequence of isocratic and two segmented linear gradients as follows: 10 mm ammonium bicarbonate for 0–8 min; 9–22.5% (v/v) acetonitrile in 10 mm ammonium bicarbonate for 8–38 min; 22.5–51.75% (v/v) acetonitrile in 10 mm ammonium bicarbonate for 38–73 min; and then increasing to 81% (v/v) acetonitrile in 10 mm ammonium bicarbonate for 7 min followed by re-equilibration with 10 mm ammonium bicarbonate for 15 min. The eluate was introduced continuously into an ESI source (LTQ Orbitrap XL, Thermo Fisher Scientific). MS spectra were obtained in the positive ion mode using Orbitrap (mass range: m/z 800–2,000; capillary temperature: 300 °C; source voltage: 4.5 kV; capillary voltage: 18 V; tube lens voltage: 110 V). For MS/MS analysis, the top three precursor ions were fragmented by higher-energy collisional dissociation using a stepped collision energy (normalized collision energy: 35.0; width: 40.0; steps: 3; minimum signal required: 10,000; isolation width: 4.00; activation time: 100) using an Orbitrap. Monoisotopic masses were assigned with possible monosaccharide compositions using the GlycoMod software tool (mass tolerance for precursor ions is ±0.01 Da, RRID:SCR_001602), and the proposed glycan structures were further verified through annotation using a fragmentation mass–matching approach based on the MS/MS data. Xcalibur software version 2.2 (Thermo Fisher Scientific) was used to show the base peak chromatogram and extracted-ion chromatogram and to analyze the MS and MS/MS data. The relative abundances (%) of each glycan structure were calculated by setting the total peak intensities of all detected glycan peaks without oligomannose-type glycans as 100%.
Data availability
Proteomics raw data and search files for protein identification of co-precipitated proteins with FUT8 have been deposited to the ProteomeXchange Consortium (announced ID: PXD018032) via the jPOST partner repository (announced ID: JPST000763). Glycomic raw data for glycan structure analysis have been deposited to GlycoPOST (announced ID: GPST000061). All of the other data are contained in the article.
Supplementary Material
Acknowledgments
We thank Chizuko Yonekawa and Yuko Tokoro (Gifu University) for technical assistance. We also thank Dr. Jianguo Gu (Tohoku Medical and Pharmaceutical University) for kindly providing HEK293 FUT8KO cells. We thank Edanz Group for editing a draft of the manuscript.
This article contains supporting information.
Author contributions—S. T. and Y. K. conceptualization; S. T., M. T., M. Nagae, M. Nakano, and Y. K. investigation; S. T. and Y. K. writing-original draft; T. H., M. Nakano, and Y. K. writing-review and editing; Y. K. supervision; Y. K. funding acquisition; Y. K. project administration.
Funding and additional information—This work was supported in part by Grant-in-Aid for Scientific Research (C) 17K07356 (to Y. K.), a Leading Initiative for Excellent Young Researchers (LEADER) project (to Y. K.) from the Japan Society for the Promotion of Science (JSPS), CREST Grant 18070267 (to Y. K.) from the Japan Science and Technology Agency (JST), grants from the Takeda Science Foundation (to Y. K.), and a grant from the Uehara Memorial Foundation (to Y. K.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- ER
- endoplasmic reticulum
- B4GALT1
- β1,4-galactosyltransferase 1
- FUT8
- fucosyltransferase 8
- OST
- oligosaccharyltransferase
- PhoSL
- Pholiota squarrosa lectin
- PNGase F
- peptide:N-glycosidase F
- PNSNB
- N-(2-(2-pyridylamino)ethyl)succinamic acid 5-norbornene-2,3-dicarboxyimide ester
- RPN1
- ribophorin I
- SH3
- Src homology 3
- TMT0
- tandem mass tag 0
- PFA
- paraformaldehyde
- ESI
- electrospray ionization
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- HRP
- horseradish peroxidase
- CBB
- Coomassie Brilliant Blue
- DAPI
- 4′,6-diamidino-2-phenylindole.
References
- 1. Varki A. (2017) Biological roles of glycans. Glycobiology 27, 3–49 10.1093/glycob/cww086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moremen K. W., Tiemeyer M., and Nairn A. V. (2012) Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462 10.1038/nrm3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sun S., Hu Y., Ao M., Shah P., Chen J., Yang W., Jia X., Tian Y., Thomas S., and Zhang H. (2019) N-GlycositeAtlas: a database resource for mass spectrometry-based human N-linked glycoprotein and glycosylation site mapping. Clin. Proteomics 16, 35 10.1186/s12014-019-9254-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stanley P., and Taniguchi N. A. M. (2017) N-glycans. in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H., eds) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 5. Zhao Y.-Y., Takahashi M., Gu J.-G., Miyoshi E., Matsumoto A., Kitazume S., and Taniguchi N. (2008) Functional roles of N-glycans in cell signaling and cell adhesion in cancer. Cancer Sci. 99, 1304–1310 10.1111/j.1349-7006.2008.00839.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ohtsubo K., and Marth J. D. (2006) Glycosylation in cellular mechanisms of health and disease. Cell 126, 855–867 10.1016/j.cell.2006.08.019 [DOI] [PubMed] [Google Scholar]
- 7. Granovsky M., Fata J., Pawling J., Muller W. J., Khokha R., and Dennis J. W. (2000) Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat. Med. 6, 306–312 10.1038/73163 [DOI] [PubMed] [Google Scholar]
- 8. Wang X., Inoue S., Gu J., Miyoshi E., Noda K., Li W., Mizuno-Horikawa Y., Nakano M., Asahi M., Takahashi M., Uozumi N., Ihara S., Lee S. H., Ikeda Y., Yamaguchi Y., et al. (2005) Dysregulation of TGF-β1 receptor activation leads to abnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 102, 15791–15796 10.1073/pnas.0507375102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kizuka Y., Kitazume S., Fujinawa R., Saito T., Iwata N., Saido T. C., Nakano M., Yamaguchi Y., Hashimoto Y., Staufenbiel M., Hatsuta H., Murayama S., Manya H., Endo T., and Taniguchi N. (2015) An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer's disease. EMBO Mol. Med. 7, 175–189 10.15252/emmm.201404438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cherepanova N., Shrimal S., and Gilmore R. (2016) N-Linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 41, 57–65 10.1016/j.ceb.2016.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Breton C., Mucha J., and Jeanneau C. (2001) Structural and functional features of glycosyltransferases. Biochimie 83, 713–718 10.1016/S0300-9084(01)01298-6 [DOI] [PubMed] [Google Scholar]
- 12. Uozumi N., Yanagidani S., Miyoshi E., Ihara Y., Sakuma T., Gao C. X., Teshima T., Fujii S., Shiba T., and Taniguchi N. (1996) Purification and cDNA cloning of porcine brain GDP-l-Fuc:N-acetyl-β-d-glucosaminide α1→6fucosyltransferase. J. Biol. Chem. 271, 27810–27817 10.1074/jbc.271.44.27810 [DOI] [PubMed] [Google Scholar]
- 13. Yanagidani S., Uozumi N., Ihara Y., Miyoshi E., Yamaguchi N., and Taniguchi N. (1997) Purification and cDNA cloning of GDP-l-Fuc:N-acetyl-β-d-glucosaminide:α1–6 fucosyltransferase (α1–6 FucT) from human gastric cancer MKN45 cells. J. Biochem. 121, 626–632 10.1093/oxfordjournals.jbchem.a021631 [DOI] [PubMed] [Google Scholar]
- 14. Wang X., Gu J., Ihara H., Miyoshi E., Honke K., and Taniguchi N. (2006) Core fucosylation regulates epidermal growth factor receptor-mediated intracellular signaling. J. Biol. Chem. 281, 2572–2577 10.1074/jbc.M510893200 [DOI] [PubMed] [Google Scholar]
- 15. Fujii H., Shinzaki S., Iijima H., Wakamatsu K., Iwamoto C., Sobajima T., Kuwahara R., Hiyama S., Hayashi Y., Takamatsu S., Uozumi N., Kamada Y., Tsujii M., Taniguchi N., Takehara T., and Miyoshi E. (2016) Core fucosylation on T cells, required for activation of T-cell receptor signaling and induction of colitis in mice, is increased in patients with inflammatory bowel disease. Gastroenterology 150, 1620–1632 10.1053/j.gastro.2016.03.002 [DOI] [PubMed] [Google Scholar]
- 16. Gu W., Fukuda T., Isaji T., Hang Q., Lee H. H., Sakai S., Morise J., Mitoma J., Higashi H., Taniguchi N., Yawo H., Oka S., and Gu J. (2015) Loss of α1,6-fucosyltransferase decreases hippocampal long term potentiation: implications for core fucosylation in the regulation of AMPA receptor heteromerization and cellular signaling. J. Biol. Chem. 290, 17566–17575 10.1074/jbc.M114.579938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Agrawal P., Fontanals-Cirera B., Sokolova E., Jacob S., Vaiana C. A., Argibay D., Davalos V., McDermott M., Nayak S., Darvishian F., Castillo M., Ueberheide B., Osman I., Fenyö D., Mahal L. K., and Hernando E. (2017) A systems biology approach identifies FUT8 as a driver of melanoma metastasis. Cancer Cell 31, 804–819.e7 10.1016/j.ccell.2017.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shinkawa T., Nakamura K., Yamane N., Shoji-Hosaka E., Kanda Y., Sakurada M., Uchida K., Anazawa H., Satoh M., Yamasaki M., Hanai N., and Shitara K. (2003) The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 278, 3466–3473 10.1074/jbc.M210665200 [DOI] [PubMed] [Google Scholar]
- 19. Kawamoto S., Moriwaki K., Nakagawa T., Terao M., Shinzaki S., Yamane-Ohnuki N., Satoh M., Mehta A. S., Block T. M., and Miyoshi E. (2011) Overexpression of α1,6-fucosyltransferase in hepatoma enhances expression of Golgi phosphoprotein 2 in a fucosylation-independent manner. Int. J. Oncol. 39, 203–208 10.3892/ijo.2011.1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ihara H., Ikeda Y., Toma S., Wang X., Suzuki T., Gu J., Miyoshi E., Tsukihara T., Honke K., Matsumoto A., Nakagawa A., and Taniguchi N. (2007) Crystal structure of mammalian α1,6-fucosyltransferase, FUT8. Glycobiology 17, 455–466 10.1093/glycob/cwl079 [DOI] [PubMed] [Google Scholar]
- 21. Zafra-Ruano A., and Luque I. (2012) Interfacial water molecules in SH3 interactions: getting the full picture on polyproline recognition by protein-protein interaction domains. FEBS Lett. 586, 2619–2630 10.1016/j.febslet.2012.04.057 [DOI] [PubMed] [Google Scholar]
- 22. Kobayashi Y., Tateno H., Dohra H., Moriwaki K., Miyoshi E., Hirabayashi J., and Kawagishi H. (2012) A novel core fucose-specific lectin from the mushroom Pholiota squarrosa. J. Biol. Chem. 287, 33973–33982 10.1074/jbc.M111.327692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rocha J., Cicéron F., de Sanctis D., Lelimousin M., Chazalet V., Lerouxel O., and Breton C. (2016) Structure of Arabidopsis thaliana FUT1 reveals a variant of the GT-B class fold and provides insight into xyloglucan fucosylation. Plant Cell. 28, 2352–2364 10.1105/tpc.16.00519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harada Y., Ohkawa Y., Kizuka Y., and Taniguchi N. (2019) Oligosaccharyltransferase: a gatekeeper of health and tumor progression. Int. J. Mol. Sci. 20, E6074 10.3390/ijms20236074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shrimal S., and Gilmore R. (2019) Oligosaccharyltransferase structures provide novel insight into the mechanism of asparagine-linked glycosylation in prokaryotic and eukaryotic cells. Glycobiology 29, 288–297 10.1093/glycob/cwy093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramírez A. S., Kowal J., and Locher K. P. (2019) Cryo-electron microscopy structures of human oligosaccharyltransferase complexes OST-A and OST-B. Science 366, 1372–1375 10.1126/science.aaz3505 [DOI] [PubMed] [Google Scholar]
- 27. Wilson C. M., Roebuck Q., and High S. (2008) Ribophorin I regulates substrate delivery to the oligosaccharyltransferase core. Proc. Natl. Acad. Sci. U.S.A. 105, 9534–9539 10.1073/pnas.0711846105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilson C. M., and High S. (2007) Ribophorin I acts as a substrate-specific facilitator of N-glycosylation. J. Cell Sci. 120, 648–657 10.1242/jcs.000729 [DOI] [PubMed] [Google Scholar]
- 29. Kötzler M. P., Blank S., Bantleon F. I., Wienke M., Spillner E., and Meyer B. (2013) Donor assists acceptor binding and catalysis of human α1,6-fucosyltransferase. ACS Chem. Biol. 8, 1830–1840 10.1021/cb400140u [DOI] [PubMed] [Google Scholar]
- 30. García-García A., Ceballos-Laita L., Serna S., Artschwager R., Reichardt N. C., Corzana F., and Hurtado-Guerrero R. (2020) Structural basis for substrate specificity and catalysis of α1,6-fucosyltransferase. Nat. Commun. 11, 973 10.1038/s41467-020-14794-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Järvå M. A., Dramicanin M., Lingford J. P., Mao R., John A., Jarman K. E., Grinter R., and Goddard-Borger E. D. (2020) Structural basis of substrate recognition and catalysis by fucosyltransferase 8. J. Biol. Chem. 295, 6677–6688 10.1074/jbc.RA120.013291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ihara H., Okada T., Taniguchi N., and Ikeda Y. (2020) Involvement of the α-helical and Src homology 3 domains in the molecular assembly and enzymatic activity of human α1,6-fucosyltransferase, FUT8. Biochim. Biophys. Acta Gen. Subj. 1864, 129596 10.1016/j.bbagen.2020.129596 [DOI] [PubMed] [Google Scholar]
- 33. Wilson J. R., Williams D., and Schachter H. (1976) The control of glycoprotein synthesis: N-acetylgluocosamine linkage to a mannose residue as a signal for the attachment of l-fucose to the asparagine-linked N-acetylglucosamine residue of glycopeptide from α1-acid glycoprotein. Biochem. Biophys. Res. Commun. 72, 909–916 10.1016/S0006-291X(76)80218-5 [DOI] [PubMed] [Google Scholar]
- 34. Yang Q., Zhang R., Cai H., and Wang L.-X. (2017) Revisiting the substrate specificity of mammalian α1,6-fucosyltransferase reveals that it catalyzes core fucosylation of N-glycans lacking α1,3-arm GlcNAc. J. Biol. Chem. 292, 14796–14803 10.1074/jbc.M117.804070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Braunger K., Pfeffer S., Shrimal S., Gilmore R., Berninghausen O., Mandon E. C., Becker T., Förster F., and Beckmann R. (2018) Structural basis for coupling protein transport and N-glycosylation at the mammalian endoplasmic reticulum. Science 360, 215–219 10.1126/science.aar7899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harada Y., Li H., Li H., and Lennarz W. J. (2009) Oligosaccharyltransferase directly binds to ribosome at a location near the translocon-binding site. Proc. Natl. Acad. Sci. U.S.A. 106, 6945–6949 10.1073/pnas.0812489106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Qin S.-Y., Hu D., Matsumoto K., Takeda K., Matsumoto N., Yamaguchi Y., and Yamamoto K. (2012) Malectin forms a complex with ribophorin I for enhanced association with misfolded glycoproteins. J. Biol. Chem. 287, 38080–38089 10.1074/jbc.M112.394288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takeda K., Qin S.-Y., Matsumoto N., and Yamamoto K. (2014) Association of malectin with ribophorin I is crucial for attenuation of misfolded glycoprotein secretion. Biochem. Biophys. Res. Commun. 454, 436–440 10.1016/j.bbrc.2014.10.102 [DOI] [PubMed] [Google Scholar]
- 39. Banfield D. K. (2011) Mechanisms of protein retention in the Golgi. Cold Spring Harb. Perspect. Biol. 3, a005264 10.1101/cshperspect.a005264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wong S. H., Low S. H., and Hong W. (1992) The 17-residue transmembrane domain of β-galactoside α2,6-sialyltransferase is sufficient for Golgi retention. J. Cell Biol. 117, 245–258 10.1083/jcb.117.2.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Munro S. (1995) An investigation of the role of transmembrane domains in Golgi protein retention. EMBO J. 14, 4695–4704 10.1002/j.1460-2075.1995.tb00151.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Munro S. (1991) Sequences within and adjacent to the transmembrane segment of α-2,6-sialyltransferase specify Golgi retention. EMBO J. 10, 3577–3588 10.1002/j.1460-2075.1991.tb04924.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Evans S. C., Youakim A., and Shur B. D. (1995) Biological consequences of targeting β1,4-galactosyltransferase to two different subcellular compartments. Bioessays 17, 261–268 10.1002/bies.950170313 [DOI] [PubMed] [Google Scholar]
- 44. Vilcaes A. A., Demichelis V. T., and Daniotti J. L. (2011) Trans-activity of plasma membrane-associated ganglioside sialyltransferase in mammalian cells. J. Biol. Chem. 286, 31437–31446 10.1074/jbc.M111.257196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ram P. A., Cardullo R. A., and Millette C. F. (1989) Expression and topographical localization of cell surface fucosyltransferase activity during epididymal sperm maturation in the mouse. Gamete Res. 22, 321–332 10.1002/mrd.1120220309 [DOI] [PubMed] [Google Scholar]
- 46. Ramakrishnan B., Boeggeman E., Ramasamy V., and Qasba P. K. (2004) Structure and catalytic cycle of β-1,4-galactosyltransferase. Curr. Opin. Struct. Biol. 14, 593–600 10.1016/j.sbi.2004.09.006 [DOI] [PubMed] [Google Scholar]
- 47. Begovac P. C., Hall D. E., and Shur B. D. (1991) Laminin fragment E8 mediates PC12 cell neurite outgrowth by binding to cell surface β1,4-galactosyltransferase. J. Cell Biol. 113, 637–644 10.1083/jcb.113.3.637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miller D. J., Macek M. B., and Shur B. D. (1992) Complementarity between sperm surface β-l,4-galactosyl-transferase and egg-coat ZP3 mediates sperm-egg binding. Nature 357, 589–593 10.1038/357589a0 [DOI] [PubMed] [Google Scholar]
- 49. Lu Q., and Shur B. D. (1997) Sperm from β1,4-galactosyltransferase-null mice are refractory to ZP3-induced acrosome reactions and penetrate the zona pellucida poorly. Development 124, 4121–4131 [DOI] [PubMed] [Google Scholar]
- 50. Hathaway H. J., Evans S. C., Dubois D. H., Foote C. I., Elder B. H., and Shur B. D. (2003) Mutational analysis of the cytoplasmic domain of β1,4-galactosyltransferase I: influence of phosphorylation on cell surface expression. J. Cell Sci. 116, 4319–4330 10.1242/jcs.00720 [DOI] [PubMed] [Google Scholar]
- 51. Khoder-Agha F., Harrus D., Brysbaert G., Lensink M. F., Harduin-Lepers A., Glumoff T., and Kellokumpu S. (2019) Assembly of B4GALT1/ST6GAL1 heteromers in the Golgi membranes involves lateral interactions via highly charged surface domains. J. Biol. Chem. 294, 14383–14393 10.1074/jbc.RA119.009539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hassinen A., Rivinoja A., Kauppila A., and Kellokumpu S. (2010) Golgi N-glycosyltransferases form both homo- and heterodimeric enzyme complexes in live cells. J. Biol. Chem. 285, 17771–17777 10.1074/jbc.M110.103184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kizuka Y., Matsui T., Takematsu H., Kozutsumi Y., Kawasaki T., and Oka S. (2006) Physical and functional association of glucuronyltransferases and sulfotransferase involved in HNK-1 biosynthesis. J. Biol. Chem. 281, 13644–13651 10.1074/jbc.M601453200 [DOI] [PubMed] [Google Scholar]
- 54. Seko A., and Yamashita K. (2005) Characterization of a novel galactose β1,3-N-acetylglucosaminyltransferase (β3Gn-T8): the complex formation of β3Gn-T2 and β3Gn-T8 enhances enzymatic activity. Glycobiology 15, 943–951 10.1093/glycob/cwi082 [DOI] [PubMed] [Google Scholar]
- 55. McCormick C., Duncan G., Goutsos K. T., and Tufaro F. (2000) The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc. Natl. Acad. Sci. U.S.A. 97, 668–673 10.1073/pnas.97.2.668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Moremen K. W., and Haltiwanger R. S. (2019) Emerging structural insights into glycosyltransferase-mediated synthesis of glycans. Nat. Chem. Biol. 15, 853–864 10.1038/s41589-019-0350-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schneider M., Al-Shareffi E., and Haltiwanger R. S. (2017) Biological functions of fucose in mammals. Glycobiology 27, 601–618 10.1093/glycob/cwx034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 10.1107/S0907444904026460 [DOI] [PubMed] [Google Scholar]
- 59. Kizuka Y., Nakano M., Yamaguchi Y., Nakajima K., Oka R., Sato K., Ren C.-T., Hsu T.-L., Wong C.-H., and Taniguchi N. (2017) An alkynyl-fucose halts hepatoma cell migration and invasion by inhibiting GDP-fucose-synthesizing enzyme FX, TSTA3. Cell Chem. Biol. 24, 1467–1478.e5 10.1016/j.chembiol.2017.08.023 [DOI] [PubMed] [Google Scholar]
- 60. Kizuka Y., Funayama S., Shogomori H., Nakano M., Nakajima K., Oka R., Kitazume S., Yamaguchi Y., Sano M., Korekane H., Hsu T.-L., Lee H.-Y., Wong C.-H., and Taniguchi N. (2016) High-sensitivity and low-toxicity fucose probe for glycan imaging and biomarker discovery. Cell Chem. Biol. 23, 782–792 10.1016/j.chembiol.2016.06.010 [DOI] [PubMed] [Google Scholar]
- 61. Uozumi N., Teshima T., Yamamoto T., Nishikawa A., Gao Y. E., Miyoshi E., Gao C. X., Noda K., Islam K. N., Ihara Y., Fujii S., Shiba T., and Taniguchi N. (1996) A fluorescent assay method for GDP-l-Fuc:N-acetyl-β-d-glucosaminide α1–6fucosyltransferase activity, involving high performance liquid chromatography. J. Biochem. 120, 385–392 10.1093/oxfordjournals.jbchem.a021424 [DOI] [PubMed] [Google Scholar]
- 62. Nakano M., Saldanha R., Göbel A., Kavallaris M., and Packer N. H. (2011) Identification of glycan structure alterations on cell membrane proteins in desoxyepothilone B resistant leukemia cells. Mol. Cell. Proteomics 10, M111.009001 10.1074/mcp.M111.009001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kizuka Y., Nakano M., Miura Y., and Taniguchi N. (2016) Epigenetic regulation of neural N-glycomics. Proteomics 16, 2854–2863 10.1002/pmic.201600053 [DOI] [PubMed] [Google Scholar]
- 64. Nakano M., Mishra S. K., Tokoro Y., Sato K., Nakajima K., Yamaguchi Y., Taniguchi N., and Kizuka Y. (2019) Bisecting GlcNAc is a general suppressor of terminal modification of N-glycan. Mol. Cell. Proteomics 18, 2044–2057 10.1074/mcp.RA119.001534 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomics raw data and search files for protein identification of co-precipitated proteins with FUT8 have been deposited to the ProteomeXchange Consortium (announced ID: PXD018032) via the jPOST partner repository (announced ID: JPST000763). Glycomic raw data for glycan structure analysis have been deposited to GlycoPOST (announced ID: GPST000061). All of the other data are contained in the article.