

Abstract
Mitochondrial dysfunction is implicated in sporadic and familial Parkinson's disease (PD). However, the mechanisms that impair homeostatic responses to mitochondrial dysfunction remain unclear. Previously, we found that chronic, low-dose administration of the mitochondrial complex I inhibitor 1-methyl-4-phenylpyridinium (MPP+) dysregulates mitochondrial fission–fusion, mitophagy, and mitochondrial biogenesis. Given that PTEN-induced kinase 1 (PINK1) regulates mitochondrial function, dynamics, and turnover, we hypothesized that alterations in endogenous PINK1 levels contribute to depletion of mitochondria during chronic complex I injury. Here we found that chronic MPP+ treatment of differentiated SH-SY5Y neuronal cells significantly decreases PINK1 expression prior to reductions in other mitochondrial components. Furthermore, Bcl2-associated athanogene 6 (BAG6, BAT3, or Scythe), a protein involved in protein quality control and degradation, was highly up-regulated during the chronic MPP+ treatment. BAG6 interacted with PINK1, and BAG6 overexpression decreased the half-life of PINK1. Conversely, siRNA-mediated BAG6 knockdown prevented chronic MPP+ stress-induced loss of PINK1, reversed MPP+-provoked mitochondrial changes, increased cell viability, and prevented MPP+-induced dendrite shrinkage in primary neurons. These results indicate that BAG6 up-regulation during chronic complex I inhibition contributes to mitochondrial pathology by decreasing the levels of endogenous PINK1. Given that recessive mutations in PINK1 cause familial PD, the finding of accelerated PINK1 degradation in the chronic MPP+ model suggests that PINK1 loss of function represents a point of convergence between the neurotoxic and genetic causes of PD.
Keywords: PTEN-induced putative kinase 1 (PINK1), mitochondria, Parkinson disease, neurodegeneration, protein degradation, BAG6, mitochondrial complex I inhibition, mitochondrial dysfunction, MPP+, neuron injury
Introduction
Parkinson's disease (PD) is a chronic, progressive movement disorder, characterized by degeneration of nigrostriatal dopaminergic neurons. Cells and brain tissue from PD patients exhibit reduced mitochondrial complex I activity (1), and exposure to complex I inhibitors such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and the pesticide rotenone cause parkinsonian neurodegeneration and may contribute to risk of PD (2, 3). Although the majority of PD cases are sporadic, ∼10% show a familial etiology. Genetic models based upon mutations in α-synuclein, Parkin, PINK1, LRRK2, and DJ-1 have further emphasized the role of mitochondrial dyshomeostasis in the pathogenesis of PD (1, 4, 5). Hence, understanding the mechanisms by which genetic and environmental susceptibilities may interact would provide valuable insights into PD pathogenesis.
The PTEN-induced putative kinase 1 (PINK1) is a ubiquitous serine-threonine kinase, localized in both cytosol and mitochondria (6–9). A normal mitochondrial membrane potential allows full-length PINK1 to be imported into mitochondria, where it is cleaved by several mitochondrial proteases, including mitochondrial inner membrane presenilin-associated rhomboid-like protein (PARL), matrix-processing peptidase (MPP), or ATP-dependent Clp protease ATP-binding subunit (ClpX) (10, 11). Processed PINK1 may regulate mitochondrial respiration, or it is exported to the cytosol (12), where its stability is regulated by complexing with chaperone proteins (7). PINK1 is implicated in maintaining mitochondrial function, reducing mitochondrial oxidative stress, regulating mitochondrial transport, fission–fusion, and autophagy (10, 13–16). Loss of PINK1 function alters mitochondrial clearance, increases the production of reactive oxygen species (14, 17, 18), and renders cells more susceptible to neurotoxicity (19, 20). Recently, PINK1 has been shown to regulate mitochondrial bioenergetics by phosphorylating a subunit of complex I, providing a possible link between the complex I dysfunction observed in PD and loss of PINK1 function (21). In addition to these mitochondrial functions, the cytosolic pool of PINK1 regulates neurite outgrowth and dendritic complexity (15) and is sufficient for protecting neurons against MPTP intoxication (8, 15).
The BAG (Bcl-2–associated athanogene) proteins, BAG1–6, were initially identified because of their ability to interact with Bcl-2 (22). In recent years, it has been shown that BAG family members regulate not only cell death but also protein quality control by functioning as co-chaperones. BAG family members can interact with various chaperones including CHIP and the Hsp70/Hsc70 complex (23) through conserved C-terminal BAG domains. Members of the BAG family have been implicated in PD (24–27), and overexpression of BAG2 (25, 28) and BAG5 act to stabilize PINK1 (27). Other members of the BAG family act to negatively regulate HSP70 function (29, 30).
Although it has been shown that engineered knockdown of PINK1 increases sensitivity to acute MPP+ and rotenone elicited apoptosis (8, 18), chronic, low-dose treatment with complex I inhibitors elicit different mechanisms of injury (31). Here, we show that chronic MPP+ treatment elicits decreased expression of full-length and processed forms of endogenous PINK1 in both cytosolic and mitochondrial fractions. Mechanistically, there was a marked up-regulation of BAG6, which acts to accelerate PINK1 degradation. Furthermore, either restoration of PINK1 or knockdown of BAG6 conferred neuroprotection, reversing the pathological effects of MPP+ on cell viability, mitochondrial morphology, and neurite shortening/dendritic retraction.
Results
Chronic treatment with MPP+ in neuronally differentiated SH-SY5Y cells causes selective depletion of endogenous PINK1
In contrast to the mm doses of MPP+ commonly used to elicit acute injury in SH-SY5Y cells (32, 33), we previously found that chronic, low-dose exposure reduces mitochondrial content through suppression of mitochondrial biogenesis in addition to mitophagy (31). Given that the low level complex I deficiency observed in Parkinson's patient cells is not associated with high levels of acute cell death, we selected a dose of MPP+ that was sublethal at 1 week of treatment with ∼20–25% toxicity at 2 weeks (31) for further study.
Chronic treatment of differentiated SH-SY5Y cells with 250 μm MPP+ resulted in reduced expression of both full-length (FL) and processed (dN) forms of PINK1 normalized to GAPDH (Fig. 1, A and B, and Table 1). Loss of PINK1 was not due simply to mitophagy, because it occurred prior to the loss of other mitochondrial proteins (Fig. 1, B and C). Because PINK1 localizes to both mitochondria and cytosol, we further studied its alteration in these subcellular compartments. MPP+ elicited decreased PINK1 levels in both fractions, with a more striking percentage of decrease observed in the cytosol (Fig. 1D). There were no significant decreases in PINK1 mRNA levels, suggesting that the loss of PINK1 is due to enhanced degradation (Fig. 1E).
Figure 1.
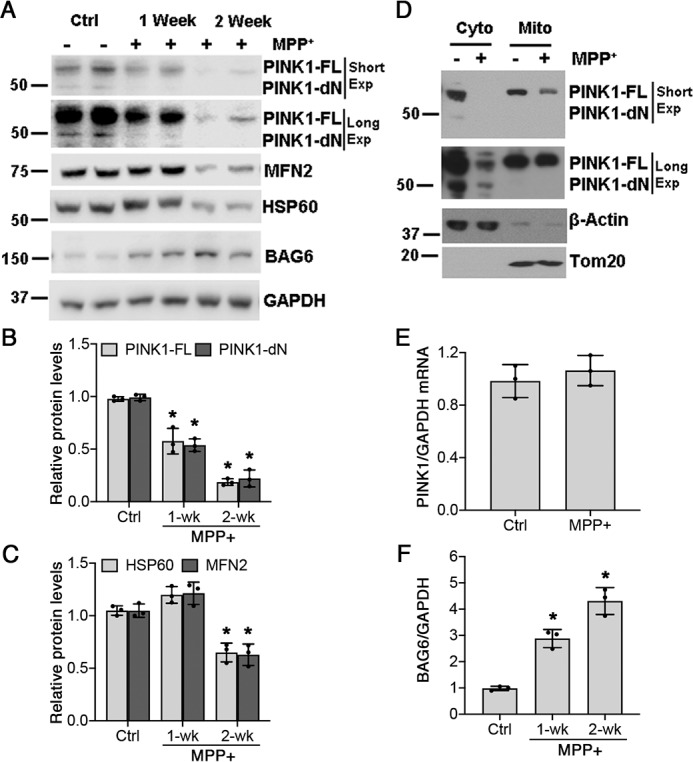
Endogenous PINK1 is depleted in chronic MPP+-treated cells. A, levels of endogenous PINK1 (FL and dN forms) are decreased by chronic MPP+ exposure (Exp, 250 μm). B, quantification of PINK1-FL and PINK1-dN bands normalized to GAPDH. For PINK1-FL, p = 0.0016 for 1-week (wk) MPP+ versus Ctrl and p < 0.0001 for 2-week MPP+ versus Ctrl. For PINK1-dN, p = 0.0003 for 1-week MPP+ versus Ctrl and p < 0.0001 for 2-week MPP+ versus Ctrl (*). C, quantification of HSP60 and MFN2 bands normalized to GAPDH. For HSP60, p = 0.0017 for 2-week MPP+ versus Ctrl (*). For MFN2, p = 0.0044 for 2-week MPP+ versus Ctrl (*). D, chronic MPP+ treatment depletes PINK1 levels in both cytosolic and mitochondrial fractions. E, chronic MPP+ treatment does not affect PINK1 mRNA levels. F, quantification of BAG6 expression in chronic MPP+-treated SH-SY5Y cells normalized to GAPDH. p = 0.0013 for 1-week MPP+ versus Ctrl and p < 0.0001 for 2-week MPP+ versus Ctrl (*). Graphs represent means ± S.D.; n = 3 independent experiments. The data were analyzed by one-way ANOVA (see Table 1 for details) followed by post hoc Bonferroni corrected t-tests.
Table 1.
One-way ANOVA analysis
| Figure | F(DFn, DFd) | p value |
|---|---|---|
| 1B, PINK1-FL | F (2,6) = 86.84 | p < 0.0001 |
| 1B, PINK1-dN | F (2,6) = 122.9 | p < 0.0001 |
| 1C, HSP60 | F (2,6) = 44.20 | p = 0.0003 |
| 1C, MFN2 | F (2,6) = 31.77 | p = 0.0006 |
| 1F | F (2,6) = 64.98 | p < 0.0001 |
| 7B | F (2,6) = 15.33 | p = 0.0044 |
| 7C | F (2,6) = 10.60 | p = 0.0107 |
Restoration of PINK1 expression rescues MPP+-induced neurodegenerative phenotypes
To determine whether loss of PINK1 played a pathogenic role in the chronic MPP+ model, we transfected neuronally differentiated SH-SY5Y cells with plasmids expressing either GFP or GFP-tagged WT PINK1. The cells were probed for the human mitochondrial p60 protein after treatment with 250 μm MPP+ or vehicle for 2 weeks, and mitochondrial morphology was studied as previously described (14). As expected, chronic MPP+ treatment elicited mitochondrial fission with loss of interconnectivity (Fig. 2, B and C, and Table 2) and reduced mitochondrial content (Fig. 2D). Overexpression of PINK1 significantly restored each of these indices of mitochondrial injury (Fig. 2, A–D) and protected against chronic MPP+-induced cell death (Fig. 2E).
Figure 2.

Restoration of PINK1 rescues mitochondrial fragmentation, cell death, and neurite shortening in chronic MPP+-treated SH-SY5Y cells. Differentiated SH-SY5Y cells were treated with either vehicle (Veh) or 250 μm MPP+ for 2 weeks. There is a significant interaction between MPP+ treatment and PINK1 overexpression for all dependent variables analyzed (see Table 2). A–C, PINK1-GFP prevents mitochondrial fragmentation induced by chronic MPP+ treatment, as analyzed by mitochondrial elongation (*, p < 0.0001 for GFP (MPP+ versus vehicle); †, p < 0.0001 for MPP+ (PINK1-GFP versus GFP); B) and mitochondrial connectivity (*, p = 0.0007 for GFP (MPP+ versus vehicle); †, p = 0.0028 for MPP+ (PINK1-GFP versus GFP); C). D, PINK1-GFP rescues the decreased mitochondrial content (percentage of the area occupied by mitochondria per cell) caused by chronic MPP+ treatment. *, p = 0.0002 for GFP (MPP+ versus vehicle); †, p = 0.0003 for MPP+ (PINK1-GFP versus GFP). E, PINK1-GFP prevents chronic MPP+-induced cell death. *, p < 0.0001 for GFP (MPP+ versus vehicle); †, p = 0.0002 for MPP+ (PINK1-GFP versus GFP). F and G, PINK-GFP prevents chronic MPP+-induced neurite shortening. *. p = 0.001 for GFP (MPP+ versus vehicle); †. p = 0.0021 for MPP+ (PINK1-GFP versus GFP. All graphs represent means ± S.D.; n = 3 independent experiments for B–D and G; n = 4 independent experiments for E. The data were analyzed by two-way ANOVA with post hoc t tests (see Table 2 for analysis of main factors and interactions). Scale bars, 20 μm in A and 50 μm in F.
Table 2.
Two-way ANOVA analysis
| Figure | Analysis | F(DFn, DFd) | p |
|---|---|---|---|
| 2B | Factor A: Plasmid | F (1,8) = 75.65 | p < 0.0001 |
| Factor B: MPP+ vs. vehicle | F (1,8) = 110.9 | p < 0.0001 | |
| A * B: Interaction | F (1,8) = 28.93 | p = 0.0007 | |
| 2C | Factor A: Plasmid | F (1,8) = 30.80 | p = 0.0005 |
| Factor B: MPP+ vs. vehicle | F (1,8) = 55.69 | p < 0.0001 | |
| A * B: Interaction | F (1,8) = 6.053 | p = 0.0393 | |
| 2D | Factor A: Plasmid | F (1,8) = 57.73 | p < 0.0001 |
| Factor B: MPP+ vs. vehicle | F (1,8) = 60.80 | p < 0.0001 | |
| A * B: Interaction | F (1,8) = 13.69 | p = 0.0060 | |
| 2E | Factor A: Plasmid | F (1,8) = 30.68 | p = 0.0001 |
| Factor B: MPP+ vs. vehicle | F (1,8) = 44.95 | p < 0.0001 | |
| A * B: Interaction | F (1,8) = 14.02 | p = 0.0028 | |
| 2G | Factor A: Plasmid | F (1,8) = 20.69 | p = 0.0019 |
| Factor B: MPP+ vs. vehicle | F (1,8) = 30.57 | p = 0.0006 | |
| A * B: Interaction | F (1,8) = 14.65 | p = 0.0050 | |
| 4B | Factor A: siRNA | F (1,8) = 6.336 | p = 0.0360 |
| Factor B: MPP+ vs. vehicle | F (1,8) = 29.16 | p = 0.0006 | |
| A * B: Interaction | F (1,8) = 15.23 | p = 0.0045 | |
| 4C | Factor A: siRNA | F (1,8) = 167.0 | p < 0.0001 |
| Factor B: MPP+ vs. vehicle | F (1,8) = 523.5 | p < 0.0001 | |
| A * B: Interaction | F (1,8) = 75.21 | p < 0.0001 | |
| 5B | Factor A: siRNA | F (2,12) = 4.832 | p = 0.0289 |
| Factor B: MPP+ vs. vehicle | F (1,12) = 57.78 | p < 0.0001 | |
| A * B: Interaction | F (2,12) = 5.021 | p = 0.0260 | |
| 5C | Factor A: siRNA | F (2,12) = 8.956 | p = 0.0042 |
| Factor B: MPP+ vs. vehicle | F (1,12) = 267.0 | p < 0.0001 | |
| A * B: Interaction | F (2,12) = 10.19 | p = 0.0026 | |
| 5D | Factor A: siRNA | F (2,12) = 13.86 | p = 0.0008 |
| Factor B: MPP+ vs. vehicle | F (1,12) = 104.8 | p < 0.0001 | |
| A * B: Interaction | F (2,12) = 10.53 | p = 0.0023 | |
| 5E | Factor A: siRNA | F (2,12) = 7.495 | p = 0.0043 |
| Factor B: MPP+ vs. vehicle | F (1,12) = 160.5 | p < 0.0001 | |
| A * B: Interaction | F (2,12) = 14.03 | p = 0.0002 | |
| 5F | Factor A: siRNA | F (2,12) = 6.483 | p = 0.0123 |
| Factor B: MPP+ vs. vehicle | F (1,12) = 38.76 | p < 0.0001 | |
| A * B: Interaction | F (2,12) = 3.362 | p = 0.0693 | |
| 7E | Factor A: siRNA | F (3,16) = 4.763 | p = 0.0147 |
| Factor B: MPP+ vs. vehicle | F (1,16) = 100.6 | p < 0.0001 | |
| A * B: Interaction | F (3,16) = 4.531 | p = 0.0175 |
In addition to mitochondrial injury, MPP+ is known to cause neurite retraction (34). Restoration of PINK1 expression reversed the effects of MPP+ on neurite shortening (Fig. 2, F and G). These data suggest that the loss of PINK1 plays a pathogenic role in chronic MPP+-induced neurodegeneration.
BCL2-associated athanogene (BAG6) is pathologically up-regulated during chronic MPP+ injury
PINK1 is known to be stabilized by the heat shock protein 90 chaperone system (9, 35). However, we found no changes in the expression of HSP90, CDC37, or CHIP (Fig. S1). There were small decreases in BAG5 and BAG2 protein levels (Fig. 3, A–C), recently implicated in stabilizing PINK1 levels (25). However, the most striking change elicited by chronic MPP+ was a large up-regulation of BAG6 protein expression (Fig. 3, A and D), which was negatively correlated with PINK1 protein levels (Figs. 1, B, and F, and 3, A and E). The increase in BAG6 protein expression was linked to a small but statistically significant increase in mRNA expression (Fig. 3F).
Figure 3.
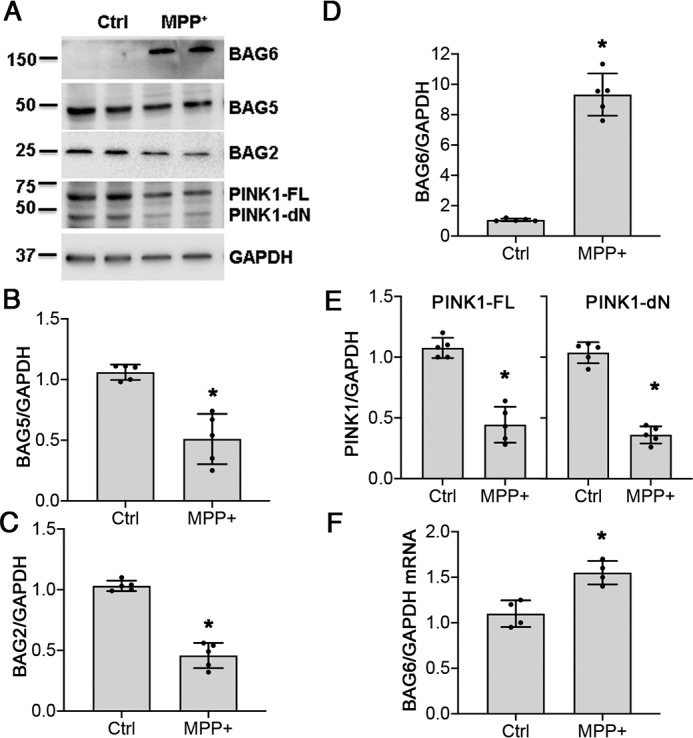
Chronic MPP+ treatment induces BAG6 expression. Differentiated SH-SY5Y cells were either untreated (Ctrl) or treated with 250 μm of MPP+ for 2 weeks, and protein expression was analyzed. A, chronic MPP+ treatment induces BAG6 expression and decreases BAG2, BAG5, and PINK1 levels. B, BAG5 protein quantification. *, p = 0.0005. C, BAG2 protein quantification. *, p < 0.0001. D, BAG6 protein quantification. *, p < 0.0001; see also Fig. 1F. E, PINK1-FL. *, p < 0.0001 and PINK1-dN protein quantification, *, p < 0.0001. F, BAG6 mRNA expression normalized to GAPDH, p = 0.0037. Graphs represent means ± S.D.; n = 5 independent experiments for B–E; n = 4 independent experiments for F; Student's two-tailed t test.
To examine whether BAG6 up-regulation played a causal role in mediating PINK1 depletion during MPP+ toxicity, we applied siRNA targeting BAG6 2 days after initiating MPP+ intoxication and every 4 days until the end of the experiment. This procedure elicited a stable reduction in BAG6 expression (Fig. S2), which persisted in the presence of MPP+ (Fig. 4A). RNAi knockdown of BAG6 significantly reversed the changes in PINK1 expression elicited by 2 weeks of chronic MPP+ toxicity (Fig. 4, A–C). These results suggest that BAG6 plays an important role in down-regulating PINK1 expression under chronic stress.
Figure 4.
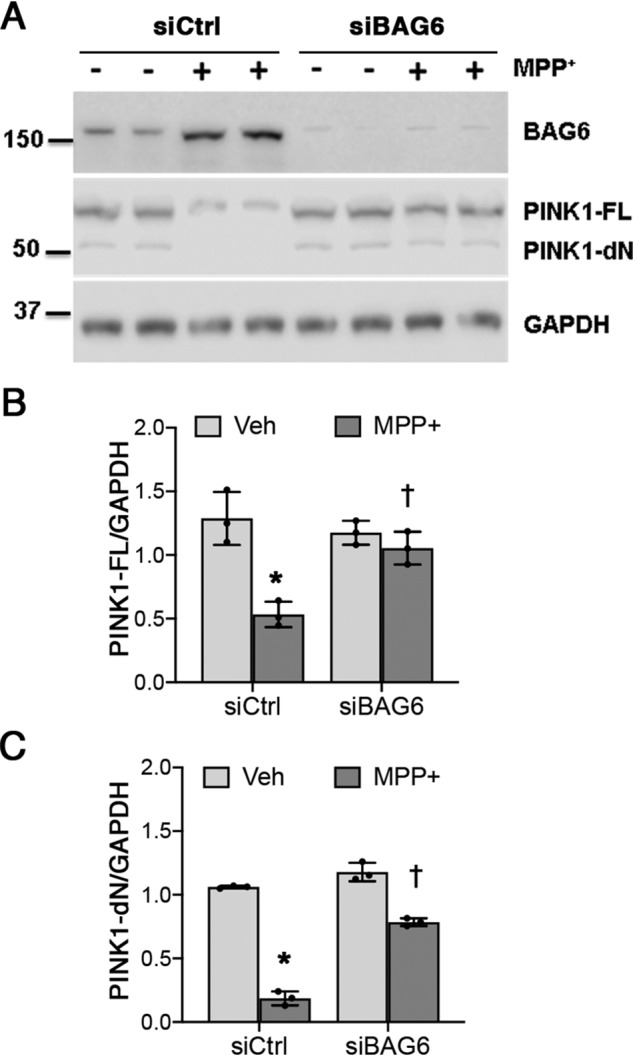
BAG6 knockdown rescues the loss of PINK1 in chronic MPP+-treated SH-SY5Y cells. A, control siRNA (siCtrl) and siBAG6 transfected cells were treated with 250 μm MPP+ for 2 weeks as described under “Experimental procedures.” B, PINK1-FL quantification. *, p = 0.001 for siCtrl (MPP+ versus vehicle (Veh)); †, p = 0.0113 for MPP+ (siBAG6 versus siCtrl). C, PINK1-dN quantification. *, p < 0.001 for siCtrl (MPP+ versus vehicle); †, p < 0.001 for MPP+ (siBAG6 versus siCtrl). Graphs represent means ± S.D.; n = 3 independent experiments; two-way ANOVA with post hoc t tests (see Table 2 for analysis of main factors and interactions).
Blocking the MPP+-induced BAG6 up-regulation by siRNA also reversed indices of MPP+-induced pathology (Fig. 5, A–D) and protected against cell death (Fig. 5E). Although knockdown of BAG6 by itself had little basal effect on neurite length, possibly because BAG6 expression is very low under basal conditions (Fig. 3A), BAG6 knockdown in MPP+-treated cells protected against neurite shortening (Fig. 5, F and G). Use of a second siRNA sequence against BAG6 showed similar protection (Fig. S3).
Figure 5.

Knockdown of BAG6 protein prevents neurite shortening and reduces MPP+-induced mitochondrial fragmentation and cell death. Differentiated SH-SY5Y cells were treated with siRNA followed by MPP+ as in Fig. 4. A–D, siRNA-mediated knockdown of BAG6 prevents chronic MPP+ treatment–induced mitochondrial changes. A, mitochondria were visualized by immunostaining for mitochondrial antigen 60 kDa (Mito P60), and nuclei were visualized using 4′,6′-diamino-2-phenylindole. Scale bars, 20 μm. B and C, knockdown of BAG6 with siRNA rescues chronic MPP+-induced changes in mitochondrial elongation (*, p = 0.0009 for Ctrl (MPP+ versus vehicle (Veh)); *, p = 0.0027 for control siRNA (siCtrl, MPP+ versus vehicle); †, p = 0.045 for MPP+ (siBAG6 versus siCtrl); B) and mitochondrial connectivity index (*, p < 0.0001 for Ctrl (MPP+ versus vehicle); *, p < 0.0001 for siCtrl (MPP+ versus vehicle); †, p = 0.0034 for MPP+ (siBAG6 versus siCtrl); C). D, knockdown of BAG6 rescues chronic MPP+-induced loss of mitochondrial content (percentage of cellular area occupied by mitochondria). *, p < 0.0001 for Ctrl (MPP+ versus vehicle); *, p < 0.0001 for siCtrl (MPP+ versus vehicle); †, p = 0.0011 for MPP+ (siBAG6 versus siCtrl). E, chronic MPP+ treatment–induced cell death is partially rescued by down-regulation of BAG6 expression using siRNA. *, p < 0.0001 for Ctrl (MPP+ versus vehicle); *, p < 0.0001 for siCtrl (MPP+ versus vehicle); †, p = 0.0001 for MPP+ (siBAG6 versus siCtrl). F and G, chronic MPP+ treatment–induced neurite shortening in differentiated SH-SY5Y cells is rescued by siRNA mediated knockdown of BAG6. *, p = 0.0181 for Ctrl (MPP+ versus vehicle); *, p = 0.0043 for siCtrl (MPP+ versus vehicle); †, p = 0.0214 for MPP+ (siBAG6 versus siCtrl). Scale bar, 50 μm. Graphs represent means ± S.D.; n = 3 independent experiments for B–D and F; n = 4 independent experiments for E; two-way ANOVA with post hoc Bonferroni corrected t test (see Table 2 for analysis of main factors and interaction).
BAG6 interacts with PINK1, increasing PINK1 turnover
To determine whether PINK1 and BAG6 physically interact, we co-expressed PINK1-GFP with V5-tagged BAG6 in HEK293 cells. Cell extracts were immunoprecipitated using anti-GFP–conjugated microbeads to pulldown PINK1, resulting in co-immunoprecipitation of BAG6 (Fig. 6A). Reciprocal pulldown experiment revealed that BAG6 pulled down both the full-length and processed forms of PINK1 (Fig. 6B).
Figure 6.

BAG6–PINK1 interaction and PINK1 turnover. A, co-immunoprecipitation of BAG6-V5 in PINK-GFP pulldowns from transfected HEK293 cells. B, co-immunoprecipitation of PINK1 by V5-tagged BAG6 in transfected HEK293 cells. C and D, overexpression of BAG6-V5 accelerated the loss of co-transfected FLAG-tagged PINK1 in cycloheximide-treated HEK293 cells compared with cells co-overexpressing GFP and FLAG-tagged PINK1. E, the half-life of PINK1 isoforms was estimated by fitting a one-phase exponential decay curve for each independent experiment using Prism 8 software (GraphPad). For PINK1-FL, *, p = 0.0084 for BAG6-V5 versus GFP. For PINK1-dN, *, p = 0.0430 for BAG6-V5 versus GFP. F and G, steady-state levels of endogenous PINK1 was determined by transfecting SH-SY5Y cells with 2 μg/well of V5-tagged BAG6 plasmid or vector control. GFP transfection was used to monitor transfection efficiency and normalize densitometry ratios. For PINK1-FL, *, p = 0.0066 for BAG6-V5 versus Ctrl. For PINK1-dN, *, p = 0.0104 for BAG6-V5 versus Ctrl. All graphs represent means ± S.D., n = 3 independent experiments, two-tailed Student's t test.
To determine whether BAG6 modulates PINK1 degradation, we co-expressed BAG6 or GFP with FLAG-tagged PINK1 in HEK293 cells and examined PINK1-FLAG levels after cycloheximide treatment to block new protein synthesis. Cells expressing BAG6 enhanced the degradation of both FL and dN forms of PINK1 compared with GFP (Fig. 6, C and D), causing significant reductions in the half-lives (t½) of PINK1-FL and PINK1-dN (Fig. 6E). Elevating BAG6 expression also decreased steady-state levels of PINK1 in the absence of MPP+ (Fig. 6, F and G).
Increased BAG6 expression is associated with decreased PINK1 levels and dendritic shortening in primary mouse neurons
We next studied the effects of chronic, low-dose MPP+ exposure in primary mouse cortical neurons. Primary neurons are more sensitive to MPP+, with an LD50 of ∼30 μm (36). Under conditions of acute toxicity (25–50 μm), BAG6 levels were decreased at 48 h (Fig. S4). Given that we were able to create a chronic model in SH-SY5Y cells by dropping to ∼10% of the LD50, we studied the effects of chronic, repetitive treatment of primary neurons with 2.5–5 μm of MPP+. In contrast to acute toxicity, chronic low-dose exposure to MPP+ increased endogenous BAG6 expression accompanied by significantly decreased PINK1 expression (Fig. 7, A–C). As previously reported by others (37), antibodies that recognize mouse PINK1 are not robust, and we could only detect the stronger full-length band.
Figure 7.

Chronic, low-dose MPP+ elevates BAG6 and reduces PINK1 levels in primary neurons, and siBAG6 protects against neuronal injury. A–C, mouse primary cortical neurons were plated at 1 × 106 cells/well in 6-well plates. At DIV7, the cells were treated with 2.5 and 5 μm of MPP+ for an additional 7 days, with half medium changes containing fresh toxin on DIV9 and DIV12. The cells were harvested and lysed for Western blotting analysis at DIV14. For BAG6 densitometry, *, p = 0.01854 for 2.5 μm MPP+ versus vehicle (Veh); *, p = 0.0065 for 5.0 μm MPP+ versus vehicle. For PINK1 densitometry, *, p = 0.0184 for 2.5 μm MPP+ versus vehicle; *, p = 0.0287 for 5.0 μm MPP+ versus vehicle. The data were analyzed by one-way ANOVA followed by Bonferroni's multiple comparison test (see Table 1 for details). D, mouse primary cortical neurons were either transfected with control siRNA (siCtrl) or 25 nm of siBAG6#1 or siBAG6#2 at DIV7. A second transfection with the same amount of siRNA was done at DIV11. The cells were harvested and lysed at DIV14 for Western blotting analysis (left panel) and densitometry (right panel) of Bag6 knockdown in mouse primary cortical neurons. *, p = 0.01442 for siBAG6#1 versus siCtrl; *, p = 0.0485 for siBAG6#2 versus siCtrl; Bonferroni-corrected two-tailed t test. E, mouse primary neurons plated on LabTek chamber slides were transfected with 25 nm of the indicated siRNA as described for D. The neurons were treated with 5 μm of MPP+ as described for A. The dendritic arbor of transfected neurons was visualized by co-transfection with GFP (left panels). Summated dendrite length per neuron was quantified (right panel) as described under “Experimental procedures.” *, p < 0.0001 for Ctrl (MPP+ versus vehicle); *, p = 0.0002 for siCtrl (MPP+ versus vehicle); †, p = 0.0170 for MPP+ (siBAG6#1 versus siCtrl); †, p = 0.0354 for MPP+ (siBAG6#2 versus siCtrl). Scale bar, 100 μm. The data were analyzed by two-way ANOVA with post hoc Bonferroni-corrected t test (see Table 2 for analysis of main factors and interactions). All graphs represent means ± S.D. from three independent experiments.
Using two distinct siRNA sequences (Fig. 7D), we studied the effects of mouse Bag6 knockdown on MPP+-induced neuron injury. Both sequences conferred significant protection against MPP+ in primary neurons (Fig. 7E).
Discussion
Although acute treatment of cells with mitochondrial uncouplers results in accumulation of full-length PINK1 (1, 4, 16, 31, 38), the effect of less severe, chronic mitochondrial stress has not been previously delineated. MPP+ is the active metabolite of MPTP, which is recognized as an environmental cause of human parkinsonian injury, and these compounds are widely used in cell culture and rodent models of PD (31, 39). Early studies using isolated brain mitochondria demonstrated that MPP+ binds specifically to complex I, inhibiting activity without eliciting irreversible damage (42). It is commonly accepted that MPP+ toxicity results from its ability to inhibit mitochondrial complex I activity, as evidenced by the resistance of rho zero (ρ0) cells to MPP+-mediated cell death (32). Nevertheless, cytotoxic mechanisms that appear to be independent of complex I inhibitory effects have also been reported for MPP+, rotenone, and paraquat (40, 41). Indeed, metabolomic, transcriptomic, and proteomic studies demonstrate multiple effects of MPP+ treatment that may result directly or indirectly from complex I inhibition or occur independently of electron transport dysfunction (43, 44). Although these studies confirm primarily mitochondrial or oxidative changes, other mechanisms may also contribute to MPP+ toxicity in acute or chronic settings.
Using a low-dose MPP+ model that elicits no cell death at 1 week and ∼20% cell death at 2 weeks (31), we found that chronic complex I inhibition resulted in a significant decrease of endogenous PINK1 from both cytosolic and mitochondrial fractions of differentiated neuronal cells, with a more robust depletion noted in the cytosol. We also discovered that chronic MPP+ treatment robustly increased the expression of BAG6 protein. Similar effects of low-dose, chronic treatment were also observed in primary mouse neurons. Knockdown of BAG6 not only prevented PINK1 loss but also reduced the neurite shortening, mitochondrial fragmentation, and cell death induced by chronic MPP+ treatment, conferring protection against MPP+-induced injury in primary neurons. Additionally, we found that BAG6 physically interacts with PINK1 and regulates PINK1 stability by increasing its turnover.
The biological effects of PINK1 are associated with its protein level, kinase activity, localization, and protein–protein interactions. In mitochondria, PINK1 may regulate the phosphorylation of TRAP1, OMM-anchored Parkin, NCLX, or the mitochondrial protease HtrA2 to regulate oxidative stress–induced apoptosis, mitophagy, mitochondrial calcium efflux, or mitochondrial dynamics (5–7, 45–48). Although full-length mitochondrial PINK1 triggers Parkin-dependent mitophagy of depolarized mitochondria (8, 9, 49–51), recent studies indicate that cytosolic PINK1 may have distinct functions compared with outer mitochondrial membrane-stabilized PINK1 (15). Indeed, N-terminally truncated, cytosolic PINK1 has been reported to suppress autophagy/mitophagy (14), to confer neuroprotection (8), and to promote dendrite outgrowth and neuron differentiation (15, 52). These effects may be mediated through activation of Akt (53) or through activation of PKA (52), thereby promoting PKA phosphorylation of p47 (52), DRP1 (54), or LC3 (34, 55) to affect neuronal growth or differentiation (52) mitochondrial fission (14) or LC3-mediated cargo targeting (56), respectively. Alternatively, PINK1 may directly bind parkin in the cytosol, inhibiting its ability to translocate to the outer mitochondrial membrane for initiating carbonyl cyanide p-trifluoromethoxyphenylhydrazone–dependent mitophagy (57). Taken together, these studies indicate that PINK1 plays multiple roles in promoting neuronal health and function. The MPP+-induced decreases of both processed and full-length PINK1 would be predicted to impair both its progrowth and mitochondrial quality control functions to impact both synaptodendritic and mitochondrial aspects of neuronal health.
In healthy cells, PINK1 is continuously turned over with a half-life that varies from 0.45 to 2.3 h depending on the cell type (7, 26, 35, 58–60). Uncoupling the mitochondria prevents its import, resulting in stabilization of full-length PINK1 at the mitochondrial surface; upon washout of CCCP, accumulated FL-PINK1 reverts to its original half-life (49). The variability in PINK1 half-lives may relate to cell type– and context-dependent differences in expression of chaperone proteins, because PINK1 mutants that are unable to bind HSP90 or CDC37 show a 2–4-fold increase in turnover (35, 60).
Indeed, the PINK1 protein is known to be stabilized by chaperone proteins such as HSP90α, CDC37, and TRAPs (9, 35), as well as BAG2 and BAG5 (25, 27, 28). Interestingly, there was also a mild decrease in expression of both BAG2 and BAG5. It is possible that loss of these chaperones also contributed to the loss of PINK1 in the chronic MPP+ model. The most striking change elicited by chronic, low-level complex I impairment was a large up-regulation of BAG6 expression. Given the effects on both full-length and processed PINK1, it is unclear whether BAG6 interacts with PINK1 at the mitochondrial surface or in the cytosol, because equilibrium changes in one compartment are likely to affect the other.
BAG6, also known as Bat3/Scythe, is a multifunctional protein involved in gene regulation, cell cycle, apoptosis, and protein quality control (61). Bag6 acts as a co-chaperone, negatively regulating HSP70's protein folding capacity (29), in opposition to BAG2 and BAG5. BAG6 acts to target misfolded proteins for proteasomal degradation (62), regulates the distribution of damaged mitochondria (63), and stabilizes the apoptosis-inducing factor (64), which has been implicated in MPP+ toxicity (36). BAG6 knockout neurons are resistant to ER stress–induced cell death (66). Our current data demonstrate that accelerated degradation of the neuroprotective protein PINK1 also contributes to the detrimental effects of BAG6.
PINK1 plays an important role in the regulation of mitochondrial dynamics and function (14, 67–70). There was increased mitochondrial fragmentation and swelling caused by chronic MPP+ treatment, accompanied by reduced mitochondrial content. Interestingly, siBAG6 transfection not only suppressed BAG6 up-regulation but also rescued the loss of PINK1 and prevented each of these chronic MPP+-induced mitochondrial changes. These data suggest that the up-regulation of BAG6 expression plays a central role in chronic MPP+-induced mitochondrial pathology.
Neuritic shortening is a common pathological change in neurodegenerative diseases and has been reported in multiple PD models to include 6-hydroxydopamine, PINK1 knockdown, and mutant LRRK2 (15, 30, 71–73). Here, we found that overexpression of PINK1 reversed the MPP+-induced neurite shortening, consistent with the previously reported role of PINK1 in promoting dendrite outgrowth (15, 52). These prodifferentiation effects of PINK1 may be mediated through indirect activation of Akt (53) or by the ability of PINK1 to phosphorylate and activate PKA (52). In addition, PINK1 may influence neurite outgrowth through its effects on mitochondrial trafficking (15), because the ability to deliver mitochondria plays a limiting role in synaptogenesis (65, 74). Interestingly, siRNA against BAG6 not only prevented MPP+-induced loss of endogenous PINK1 but also protected against neurite shortening in differentiated SH-SY5Y cells and dendritic shrinkage in primary mouse neurons.
In conclusion, our data indicate a novel role for BAG6 in regulating PINK1 levels under stress conditions. Under basal conditions, BAG6 is maintained at low levels. However, upon chronic MPP+-induced stress, BAG6 levels are elevated, resulting in accelerated PINK1 degradation. Restoration of PINK1 levels through either overexpression or via BAG6 RNAi protected against chronic MPP+-induced changes in mitochondrial structure, neurite shortening, and cell death. These data suggest a possible point of mechanistic convergence between neurotoxic/environmental and genetic causes of PD, which centers on the loss of PINK1 function.
Experimental procedures
Cell lines, primary neuron culture, and treatments
SH-SY5Y cells (ATCC, Manassas, VA) were maintained in antibiotic-free Advanced Dulbecco's modified Eagle's medium with 5% heat-inactivated fetal calf serum (BioWhittaker, Walkersville, MD), 2 mm glutamine, and 10 mm HEPES. HEK293 cells (ATCC) were maintained in antibiotic free DMEM (BioWhittaker) supplemented with 10% fetal bovine serum (Mediatech), 2 mm l-glutamine, and 10 mm HEPES in a humidified incubator at 37 °C and 5% CO2. Timed pregnant female C57BL/6 mice were purchased from Charles Rivers Laboratories. All procedures for derivation of primary neuron cultures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Primary cortical neurons from embryonic day 14–16 male or female pups were isolated from cerebral cortices as described previously (15, 30). Neurons were plated at 150,000 cells/cm2 in LabTek II coverglass chamber slides coated with poly-l-lysine (0.1 mg/ml). They were maintained in antibiotic-free1 Neurobasal medium supplemented with 2% B27 and 2 mm GlutaMAX (Gibco, Bethesda, MD). Half of the medium was replaced with fresh medium every other day.
SH-SY5Y cells were plated in 6-well plates or LabTek II coverglass chamber slides (Thermo Fisher) and neuronally differentiated with 10 μm retinoic acid (Sigma–Aldrich) for 72 h prior to and during each experiment. SH-SY5Y cells were treated with 250 μm MPP+ three times per week for up to 2 weeks. Mouse primary cortical neurons were treated with 2.5 or 5 μm MPP+ from DIV7 for 1 week with half medium change containing fresh toxin every 2 days. For cycloheximide experiments, HEK293 cells were transfected with the indicated plasmids for 48 h. Transfected cells were treated with 100 μg/ml cycloheximide (Sigma–Aldrich) diluted in fresh DMEM from a 100 mg/ml stock. Cells were treated at staggered intervals every 30 min, and at all time points were quickly harvested and lysed after 2 h (30 min after the last set of cells was treated). Untreated transfected cells were used as the zero time point.
RNAi and DNA transfection
siRNAs targeting different portions of the BAG6 mRNA sequence were employed: assay HSS111844 (siBAG6#1), which targets human, mouse, and rat sequences, and assay s15467 (siBAG6#2), which targets human and mouse sequences (Thermo Fisher). SH-SY5Y cells were transfected with the siRNAs 2 days after the first dose of MPP treatment and again every 4 days, which results in persistent knockdown (Fig. S2). For DNA transfection in differentiated SH-SY5Y cells, the cells were transfected with 750 ng/well of either GFP-tagged WT PINK1, or control vector (Genecopoeia, Rockville, MD) 2 days before MPP+ treatment. HEK293 cells were co-transfected with either 2 μg/well PINK1-FLAG and GFP or PINK1-FLAG and 1 μg/well BAG6-V5 (HsCD00442162, DNASU plasmid repository) in 6-well plates at 2 days prior to cycloheximide chase experiments. For co-immunoprecipitation experiments, the cells were transfected with 2 μg/well of PINK1-GFP or control vector and 1 μg/well BAG6-V5 in a 6-well plate for 48 h. For immunoblotting experiments, mouse cortical neurons were transfected with 25 nm of the two siRNAs targeting different regions of Bag6 mRNA at 1 day after the start of MPP+ treatment and again at DIV 11. Mouse primary cortical neurons were transfected with 500 ng/well GFP-expressing plasmid 48 h before fixing the cells for total dendrite length analysis.
Immunoprecipitation
48 h after plasmid transfection, HEK293 cells were lysed in buffer containing 1% Triton X-100 and protease/phosphatase inhibitors. Protein concentration was determined by Coomassie Blue protein assay (Pierce). Equal amounts of protein (1 mg of total protein) were used for IP. For IP of GFP-tagged PINK1, the uMACS GFP isolation kit (catalog no. 130-091-125, Miltenyi Biotec, Gaithersburg, MD) was used according to the manufacturer-recommended protocol. For reverse IP, protein lysates were incubated with 2 μg of mouse anti-V5 antibody (Santa Cruz), 50 μl of protein G–agarose beads (catalog no. 16-266, EMD Millipore) overnight at 4 °C. The beads were pelleted at 5000 × g for 5 min, the supernatant was removed, and the beads were washed three times with wash buffer 1 and twice with wash buffer 2 from the GFP isolation kit. IPed complexes were eluted by boiling the beads in 50 μl of elution buffer followed by centrifugation at 10,000 × g for 5 min. The supernatant was used for SDS-PAGE.
Quantitative RT-PCR
Total RNA was extracted using RNeasy kit (Qiagen). A total of 1 μg of RNA was used for reverse transcription reaction using the SuperScript IV one-step RT-PCR system (Life Technologies, Inc.). The cDNA was quantified by Q-PCR using PINK1 TaqMan probes (Hs00260868_m1, Life Technologies, Inc.) and BAG6 (Hs00190383_m1, Life technologies) and normalized to GAPDH mRNA (4333764F).
Western blotting analysis and densitometry
SH-SY5Y, mouse cortical neurons, and HEK293 cells were lysed in buffer containing the following: 150 mm NaCl, 5 mm EDTA, 25 mm HEPES, 10% glycerol, and 1% Triton X-100 supplemented with protease inhibitors as described previously (33). Equal amounts of protein as determined by Coomassie Plus protein assay (Thermo Scientific) were resolved on a 10% gel by SDS-PAGE and transferred to polyvinylidene difluoride membrane. The membranes were blocked with 5% nonfat milk and probed with antibodies as listed in Table 3 overnight with gentle agitation at 4 °C. Immunoreactive bands were detected using anti-mouse or anti-rabbit horseradish peroxidase–conjugated secondary antibodies (GE Healthcare) followed by exposure to ECL solution. The images were acquired using the Odyssey Fc imaging system (Li-Cor) for densitometry using Image Studio software (Li-Cor). Densitometry data were normalized to loading control, and the half-lives of PINK1 isoforms were estimated by fitting a one-phase exponential decay curve using Prism 8 software (GraphPad, San Diego, CA).
Table 3.
List of antibodies used in the study
| Antibody | Species | Catalog number | Source | Dilution |
|---|---|---|---|---|
| PINK1 | Rabbit | BC100-494 | Novus Biological | 1:1000 |
| TOM20 | Rabbit | Sc11415 | Santa Cruz | 1:10,000 |
| β-ACTIN | Mouse | A5316 | Sigma | 1:2000 |
| HSP60 | Mouse | 611563 | BD Transduction | 1:1000 |
| BAG6 | Mouse | Sc365928 | Santa Cruz | 1:1000 |
| BAG5 | Rabbit | NB100-56091 | Novus Biological | 1:1000 |
| BAG2 | Mouse | Sc101216 | Santa Cruz | 1:1000 |
| HSP90 | Rabbit | 4877 | Cell Signaling | 1:1000 |
| CDC37 | Rabbit | 3604 | Cell Signaling | 1:1000 |
| CHIP | Rabbit | Sc66830 | Santa Cruz | 1:1000 |
| GAPDH | Rabbit | Ab37168 | Abcam | 1:10,000 |
| GFP | Rabbit | A6455 | Invitrogen | 1:10,000 |
| V5 | Mouse | Sc271944 | Santa Cruz | 1:1000 |
| FLAG | Rabbit | F7425 | Sigma | 1:1000 |
| β-Tubulin | Rabbit | Ab6046 | Abcam | 1:5000 |
Fluorescence microscopy
SH-SY5Y cells and mouse cortical neurons were fixed in 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in PBS. The cells were blocked with Superblock buffer (catalog no. 37515, Thermo Scientific). GFP or PINK1-GFP cells were stained with mouse anti-human mitochondrial antigen 60KD (1:400) and rabbit anti-human GFP (1:1000) for mitochondrial morphology analysis or GFP for neurite length or total dendrite length analysis as previously described (71). The cells were then incubated with Alexa 488 donkey anti-rabbit (Molecular Probes, Eugene, CA) or Cy3-donkey anti-mouse secondary antibodies (1:500; Jackson ImmunoResearch Laboratories, West Grove, PA). Labeled cells were imaged using an IDX71 Olympus fluorescence microscope (Olympus America Inc., Melville, NY; excitation/emission filter, 490/520 nm; 541/572 nm) or an inverted Fluoview 1000 laser-scanning confocal microscope (Olympus America).
Quantification of mitochondrial morphology and neurite length
The parameters of mitochondrial morphology including mitochondrial area, perimeter, and mitochondrial connectivity were analyzed using ImageJ software (National Institutes of Health). Mitochondrial connectivity was calculated as the square root of the perimeter/circularity ratio normalized by the minor elliptical axis of mitochondria as described previously with slight modification (12). The inverse circularity was used as a measure of mitochondrial elongation. Differentiated SH-SY5Y cells typically exhibit a small cell body and one major neurite projecting from the cell body. The longest neurite from the cell body was measured using the intensity-tracing algorithm in ImageJ (34). For neurite length and total dendritic length analysis, color images were extracted to 8-bit grayscale images for quantification using ImageJ supplemented with the NeuronJ plug-in.
Cell death assay and quantification
After 2 weeks of chronic MPP+ treatment, SH-SY5Y cells were gently washed once with warm DMEM and replaced with DMEM containing Hoechst (Thermo Scientific; 1:1000) and propidium iodide (Molecular Probes, Eugene, OR; 1:1000) and incubated for 20 min at 37 °C. After that, the medium was replaced with dye free medium and incubated for 30 min at 37 °C to remove extra dye. Before imaging, the cells were washed once with warm DMEM and imaged at room temperature using Olympus IX71 microscope using 10× objective. Seven to nine random fields were imaged per condition. For quantification, the color images were background subtracted and converted to grayscale using ImageJ. After thresholding, the particle counting plugin was used to count total cells (Hoechst) and propidium iodide–positive cells. Percent dead cells was determined using the formula [(total propidium iodide − positive cells)/(total number of cells)] × 100.
Statistics
All graphed data represent means ± S.D. from independent, replicate experiments. Two group data were analyzed using two-tailed Student's t test. Multigroup data were analyzed using one-way or two-way ANOVA. If the null hypothesis was rejected, post hoc comparisons using Bonferroni-corrected two-tailed t tests were performed to test specific hypotheses in the context of each experiment. Values of p < 0.05 were considered significant.
Data availability
All data described are contained within the manuscript.
Supplementary Material
This article contains supporting information.
Author contributions—M. V. and J. Z. formal analysis; M. V., J. Z., and K. Z. Q. W. investigation; M. V., J. Z., and C. T. C. writing-original draft; M. V., J. Z., K. Z. Q. W., and C. T. C. writing-review and editing; J. Z. and C. T. C. conceptualization; C. T. C. supervision; C. T. C. funding acquisition; C. T. C. project administration.
Funding and additional information—This work was supported in part by National Institutes of Health Grants R01-AG026389, R01-NS065789, and R01-NS101628 (co-funded by NINDS/NIA). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- PD
- Parkinson's disease
- ANOVA
- analysis of variance
- MPP+
- 1-methyl-4-phenylpyridinium
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- FL
- full-length
- dN
- processed
- DIV
- day(s) in vitro
- IP
- immunoprecipitation
- DMEM
- Dulbecco's modified Eagle's medium
- Ctrl
- control.
References
- 1. Zhu J., and Chu C. T. (2010) Mitochondrial dysfunction in Parkinson's disease. J. Alzheimers Dis. 20, S325–S334 10.3233/JAD-2010-100363 [DOI] [PubMed] [Google Scholar]
- 2. Langston J. W., Ballard P., Tetrud J. W., and Irwin I. (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980 10.1126/science.6823561 [DOI] [PubMed] [Google Scholar]
- 3. Betarbet R., Sherer T. B., MacKenzie G., Garcia-Osuna M., Panov A. V., and Greenamyre J. T. (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 3, 1301–1306 10.1038/81834 [DOI] [PubMed] [Google Scholar]
- 4. Bogaerts V., Theuns J., and van Broeckhoven C. (2008) Genetic findings in Parkinson's disease and translation into treatment: a leading role for mitochondria? Genes Brain Behav. 7, 129–151 10.1111/j.1601-183X.2007.00342.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trancikova A., Tsika E., and Moore D. J. (2012) Mitochondrial dysfunction in genetic animal models of Parkinson's disease. Antioxid. Redox Signal. 16, 896–919 10.1089/ars.2011.4200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takatori S., Ito G., and Iwatsubo T. (2008) Cytoplasmic localization and proteasomal degradation of N-terminally cleaved form of PINK1. Neurosci. Lett. 430, 13–17 10.1016/j.neulet.2007.10.019 [DOI] [PubMed] [Google Scholar]
- 7. Lin W., and Kang U. J. (2008) Characterization of PINK1 processing, stability, and subcellular localization. J. Neurochem. 106, 464–474 10.1111/j.1471-4159.2008.05398.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haque M. E., Thomas K. J., D'Souza C., Callaghan S., Kitada T., Slack R. S., Fraser P., Cookson M. R., Tandon A., and Park D. S. (2008) Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc. Natl. Acad. Sci. U.S.A. 105, 1716–1721 10.1073/pnas.0705363105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weihofen A., Ostaszewski B., Minami Y., and Selkoe D. J. (2008) Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum. Mol. Genet. 17, 602–616 10.1093/hmg/ddm334 [DOI] [PubMed] [Google Scholar]
- 10. Jin S. M., Lazarou M., Wang C., Kane L. A., Narendra D. P., and Youle R. J. (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942 10.1083/jcb.201008084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Greene A. W., Grenier K., Aguileta M. A., Muise S., Farazifard R., Haque M. E., McBride H. M., Park D. S., and Fon E. A. (2012) Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 13, 378–385 10.1038/embor.2012.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamano K., and Youle R. J. (2013) PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769 10.4161/auto.24633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu S., Sawada T., Lee S., Yu W., Silverio G., Alapatt P., Millan I., Shen A., Saxton W., Kanao T., Takahashi R., Hattori N., Imai Y., and Lu B. (2012) Parkinson's disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 8, e1002537 10.1371/journal.pgen.1002537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dagda R. K., Cherra S. J. 3rd, Kulich S. M., Tandon A., Park D., and Chu C. T. (2009) Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 284, 13843–13855 10.1074/jbc.M808515200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dagda R. K., Pien I., Wang R., Zhu J., Wang K. Z., Callio J., Banerjee T. D., Dagda R. Y., and Chu C. T. (2014) Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J. Neurochem. 128, 864–877 10.1111/jnc.12494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Narendra D. P., Jin S. M., Tanaka A., Suen D. F., Gautier C. A., Shen J., Cookson M. R., and Youle R. J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLos Biol. 8, e1000298 10.1371/journal.pbio.1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gautier C. A., Kitada T., and Shen J. (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 105, 11364–11369 10.1073/pnas.0802076105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deng H., Jankovic J., Guo Y., Xie W., and Le W. (2005) Small interfering RNA targeting the PINK1 induces apoptosis in dopaminergic cells SH-SY5Y. Biochem. Biophys. Res. Commun. 337, 1133–1138 10.1016/j.bbrc.2005.09.178 [DOI] [PubMed] [Google Scholar]
- 19. Chien W. L., Lee T. R., Hung S. Y., Kang K. H., Wu R. M., Lee M. J., and Fu W. M. (2013) Increase of oxidative stress by a novel PINK1 mutation, P209A. Free Radic. Biol. Med. 58, 160–169 10.1016/j.freeradbiomed.2012.12.008 [DOI] [PubMed] [Google Scholar]
- 20. Chien W. L., Lee T. R., Hung S. Y., Kang K. H., Lee M. J., and Fu W. M. (2011) Impairment of oxidative stress-induced heme oxygenase-1 expression by the defect of Parkinson-related gene of PINK1. J. Neurochem. 117, 643–653 [DOI] [PubMed] [Google Scholar]
- 21. Morais V. A., Haddad D., Craessaerts K., De Bock P. J., Swerts J., Vilain S., Aerts L., Overbergh L., Grünewald A., Seibler P., Klein C., Gevaert K., Verstreken P., and De Strooper B. (2014) PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 344, 203–207 10.1126/science.1249161 [DOI] [PubMed] [Google Scholar]
- 22. Kabbage M., and Dickman M. B. (2008) The BAG proteins: a ubiquitous family of chaperone regulators. Cell Mol. Life Sci. 65, 1390–1402 10.1007/s00018-008-7535-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takayama S., and Reed J. C. (2001) Molecular chaperone targeting and regulation by BAG family proteins. Nat. Cell Biol. 3, E237–E241 10.1038/ncb1001-e237 [DOI] [PubMed] [Google Scholar]
- 24. Kermer P., Köhn A., Schnieder M., Lingor P., Bähr M., Liman J., and Dohm C. P. (2015) BAG1 is neuroprotective in in vivo and in vitro models of Parkinson's disease. J. Mol. Neurosci. 55, 587–595 10.1007/s12031-014-0396-2 [DOI] [PubMed] [Google Scholar]
- 25. Qu D., Hage A., Don-Carolis K., Huang E., Joselin A., Safarpour F., Marcogliese P. C., Rousseaux M. W., Hewitt S. J., Huang T., Im D. S., Callaghan S., Dewar-Darch D., Figeys D., Slack R. S., et al. (2015) BAG2 gene-mediated regulation of PINK1 protein is critical for mitochondrial translocation of PARKIN and neuronal survival. J. Biol. Chem. 290, 30441–30452 10.1074/jbc.M115.677815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cao Y. L., Yang Y. P., Mao C. J., Zhang X. Q., Wang C. T., Yang J., Lv D. J., Wang F., Hu L. F., and Liu C. F. (2017) A role of BAG3 in regulating SNCA/α-synuclein clearance via selective macroautophagy. Neurobiol. Aging 60, 104–115 10.1016/j.neurobiolaging.2017.08.023 [DOI] [PubMed] [Google Scholar]
- 27. Wang X., Guo J., Fei E., Mu Y., He S., Che X., Tan J., Xia K., Zhang Z., Wang G., and Tang B. (2014) BAG5 protects against mitochondrial oxidative damage through regulating PINK1 degradation. PLoS One 9, e86276 10.1371/journal.pone.0086276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Che X., Tang B., Wang X., Chen D., Yan X., Jiang H., Shen L., Xu Q., Wang G., and Guo J. (2013) The BAG2 protein stabilises PINK1 by decreasing its ubiquitination. Biochem. Biophys. Res. Commun. 441, 488–492 10.1016/j.bbrc.2013.10.086 [DOI] [PubMed] [Google Scholar]
- 29. Thress K., Song J., Morimoto R. I., and Kornbluth S. (2001) Reversible inhibition of Hsp70 chaperone function by Scythe and Reaper. EMBO J. 20, 1033–1041 10.1093/emboj/20.5.1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cherra S. J. 3rd, Steer E., Gusdon A. M., Kiselyov K., and Chu C. T. (2013) Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am. J. Pathol. 182, 474–484 10.1016/j.ajpath.2012.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhu J. H., Gusdon A. M., Cimen H., Van Houten B., Koc E., and Chu C. T. (2012) Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP+ toxicity: dual roles for ERK1/2. Cell Death Dis. 3, e312 10.1038/cddis.2012.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fall C. P., and Bennett J. P. Jr. (1999) Characterization and time course of MPP+-induced apoptosis in human SH-SY5Y neuroblastoma cells. J. Neurosci. Res. 55, 620–628 [DOI] [PubMed] [Google Scholar]
- 33. Zhu J. H., Horbinski C., Guo F., Watkins S., Uchiyama Y., and Chu C. T. (2007) Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am. J. Pathol. 170, 75–86 10.2353/ajpath.2007.060524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cherra S. J. 3rd, Kulich S. M., Uechi G., Balasubramani M., Mountzouris J., Day B. W., and Chu C. T. (2010) Regulation of the autophagy protein LC3 by phosphorylation. J. Cell Biol. 190, 533–539 10.1083/jcb.201002108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moriwaki Y., Kim Y. J., Ido Y., Misawa H., Kawashima K., Endo S., and Takahashi R. (2008) L347P PINK1 mutant that fails to bind to Hsp90/Cdc37 chaperones is rapidly degraded in a proteasome-dependent manner. Neurosci. Res. 61, 43–48 10.1016/j.neures.2008.01.006 [DOI] [PubMed] [Google Scholar]
- 36. Chu C. T., Zhu J. H., Cao G., Signore A., Wang S., and Chen J. (2005) Apoptosis inducing factor mediates caspase-independent 1-methyl-4-phenylpyridinium toxicity in dopaminergic cells. J. Neurochem. 94, 1685–1695 10.1111/j.1471-4159.2005.03329.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McWilliams T. G., Prescott A. R., Montava-Garriga L., Ball G., Singh F., Barini E., Muqit M. M. K., Brooks S. P., and Ganley I. G. (2018) Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 27, 439–449.e5 10.1016/j.cmet.2017.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Um J. W., Park H. J., Song J., Jeon I., Lee G., Lee P. H., and Chung K. C. (2010) Formation of parkin aggregates and enhanced PINK1 accumulation during the pathogenesis of Parkinson's disease. Biochem. Biophys. Res. Commun. 393, 824–828 10.1016/j.bbrc.2010.02.090 [DOI] [PubMed] [Google Scholar]
- 39. Stephans S. E., Miller G. W., Levey A. I., and Greenamyre J. T. (2002) Acute mitochondrial and chronic toxicological effects of 1-methyl-4-phenylpyridinium in human neuroblastoma cells. Neurotoxicology 23, 569–580 10.1016/S0161-813X(02)00060-8 [DOI] [PubMed] [Google Scholar]
- 40. Espino A., Tortosa A., Bendahan G., Bartrons R., Calopa M., Ferrer I., and Ambrosio S. (1994) Stereotaxic administration of 1-methyl-4-phenylpyridinium ion (MPP+) decreases striatal fructose 2,6-bisphosphate in rats. J. Neurochem. 62, 1913–1920 [DOI] [PubMed] [Google Scholar]
- 41. Choi W. S., Kruse S. E., Palmiter R. D., and Xia Z. (2008) Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc. Natl. Acad. Sci. U.S.A. 105, 15136–15141 10.1073/pnas.0807581105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bates T. E., Heales S. J., Davies S. E., Boakye P., and Clark J. B. (1994) Effects of 1-methyl-4-phenylpyridinium on isolated rat brain mitochondria: evidence for a primary involvement of energy depletion. J. Neurochem. 63, 640–648 [DOI] [PubMed] [Google Scholar]
- 43. Krug A. K., Gutbier S., Zhao L., Pöltl D., Kullmann C., Ivanova V., Förster S., Jagtap S., Meiser J., Leparc G., Schildknecht S., Adam M., Hiller K., Farhan H., Brunner T., et al. (2014) Transcriptional and metabolic adaptation of human neurons to the mitochondrial toxicant MPP+. Cell Death Dis. 5, e1222 10.1038/cddis.2014.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Choi J. W., Song M. Y., and Park K. S. (2014) Quantitative proteomic analysis reveals mitochondrial protein changes in MPP+-induced neuronal cells. Mol. Biosyst. 10, 1940–1947 10.1039/c4mb00026a [DOI] [PubMed] [Google Scholar]
- 45. Plun-Favreau H., Klupsch K., Moisoi N., Gandhi S., Kjaer S., Frith D., Harvey K., Deas E., Harvey R. J., McDonald N., Wood N. W., Martins L. M., and Downward J. (2007) The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat. Cell Biol. 9, 1243–1252 10.1038/ncb1644 [DOI] [PubMed] [Google Scholar]
- 46. Pridgeon J. W., Olzmann J. A., Chin L. S., and Li L. (2007) PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 5, e172 10.1371/journal.pbio.0050172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang Y., Ouyang Y., Yang L., Beal M. F., McQuibban A., Vogel H., and Lu B. (2008) Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl. Acad. Sci. U.S.A. 105, 7070–7075 10.1073/pnas.0711845105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kostic M., Ludtmann M. H., Bading H., Hershfinkel M., Steer E., Chu C. T., Abramov A. Y., and Sekler I. (2015) PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Rep. 13, 376–386 10.1016/j.celrep.2015.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C. A., Sou Y. S., Saiki S., Kawajiri S., Sato F., Kimura M., Komatsu M., Hattori N., and Tanaka K. (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221 10.1083/jcb.200910140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vives-Bauza C., and Przedborski S. (2010) PINK1 points Parkin to mitochondria. Autophagy 6, 674–675 10.4161/auto.6.5.12068 [DOI] [PubMed] [Google Scholar]
- 51. Jin S. M., and Youle R. J. (2013) The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 9, 1750–1757 10.4161/auto.26122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang K. Z. Q., Steer E., Otero P. A., Bateman N. W., Cheng M. H., Scott A. L., Wu C., Bahar I., Shih Y. T., Hsueh Y. P., and Chu C. T. (2018) PINK1 interacts with VCP/p97 and activates PKA to promote NSFL1C/p47 phosphorylation and dendritic arborization in neurons. eNeuro 5, ENEURO.0466–18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Murata H., Sakaguchi M., Jin Y., Sakaguchi Y., Futami J., Yamada H., Kataoka K., and Huh N. H. (2011) A new cytosolic pathway from a Parkinson disease-associated kinase, BRPK/PINK1: activation of AKT via mTORC2. J. Biol. Chem. 286, 7182–7189 10.1074/jbc.M110.179390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sandebring A., Thomas K. J., Beilina A., van der Brug M., Cleland M. M., Ahmad R., Miller D. W., Zambrano I., Cowburn R. F., Behbahani H., Cedazo-Mínguez A., and Cookson M. R. (2009) Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS One 4, e5701 10.1371/journal.pone.0005701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dagda R. K., Gusdon A. M., Pien I., Strack S., Green S., Li C., Van Houten B., Cherra S. J. 3rd, Chu C. T. (2011) Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson's disease. Cell Death Differ. 18, 1914–1923 10.1038/cdd.2011.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chu C. T., Ji J., Dagda R. K., Jiang J. F., Tyurina Y. Y., Kapralov A. A., Tyurin V. A., Yanamala N., Shrivastava I. H., Mohammadyani D., Wang K. Z. Q., Zhu J., Klein-Seetharaman J., Balasubramanian K., Amoscato A. A., et al. (2013) Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205 10.1038/ncb2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fedorowicz M. A., de Vries-Schneider R. L., Rüb C., Becker D., Huang Y., Zhou C., Alessi Wolken D. M., Voos W., Liu Y., and Przedborski S. (2014) Cytosolic cleaved PINK1 represses Parkin translocation to mitochondria and mitophagy. EMBO Rep. 15, 86–93 10.1002/embr.201337294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ando M., Fiesel F. C., Hudec R., Caulfield T. R., Ogaki K., Górka-Skoczylas P., Koziorowski D., Friedman A., Chen L., Dawson V. L., Dawson T. M., Bu G., Ross O. A., Wszolek Z. K., and Springer W. (2017) The PINK1 p.I368N mutation affects protein stability and ubiquitin kinase activity. Mol. Neurodegener. 12, 32 10.1186/s13024-017-0174-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guo J. F., Yao L. Y., Sun Q. Y., Cui Y. T., Yang Y., Xu Q., Yan X. X., and Tang B. S. (2017) Identification of Ser465 as a novel PINK1 autophosphorylation site. Transl. Neurodegener. 6, 34 10.1186/s40035-017-0103-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Beilina A., Van Der Brug M., Ahmad R., Kesavapany S., Miller D. W., Petsko G. A., and Cookson M. R. (2005) Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc. Natl. Acad. Sci. U.S.A. 102, 5703–5708 10.1073/pnas.0500617102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Binici J., and Koch J. (2014) BAG-6, a jack of all trades in health and disease. Cell Mol. Life Sci. 71, 1829–1837 10.1007/s00018-013-1522-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Minami R., Hayakawa A., Kagawa H., Yanagi Y., Yokosawa H., and Kawahara H. (2010) BAG-6 is essential for selective elimination of defective proteasomal substrates. J. Cell Biol. 190, 637–650 10.1083/jcb.200908092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hayashishita M., Kawahara H., and Yokota N. (2019) BAG6 deficiency induces mis-distribution of mitochondrial clusters under depolarization. FEBS Open Bio. 9, 1281–1291 10.1002/2211-5463.12677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Desmots F., Russell H. R., Michel D., and McKinnon P. J. (2008) Scythe regulates apoptosis-inducing factor stability during endoplasmic reticulum stress-induced apoptosis. J. Biol. Chem. 283, 3264–3271 10.1074/jbc.M706419200 [DOI] [PubMed] [Google Scholar]
- 65. Li Z., Okamoto K., Hayashi Y., and Sheng M. (2004) The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887 10.1016/j.cell.2004.11.003 [DOI] [PubMed] [Google Scholar]
- 66. Desmots F., Russell H. R., Lee Y., Boyd K., and McKinnon P. J. (2005) The reaper-binding protein scythe modulates apoptosis and proliferation during mammalian development. Mol. Cell. Biol. 25, 10329–10337 10.1128/MCB.25.23.10329-10337.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Poole A. C., Thomas R. E., Yu S., Vincow E. S., and Pallanck L. (2010) The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One 5, e10054 10.1371/journal.pone.0010054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen Y., and Dorn G. W. 2nd. (2013) PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475 10.1126/science.1231031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Liu W., Acín-Peréz R., Geghman K. D., Manfredi G., Lu B., and Li C. (2011) Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc. Natl. Acad. Sci. U.S.A. 108, 12920–12924 10.1073/pnas.1107332108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lee S., Sterky F. H., Mourier A., Terzioglu M., Cullheim S., Olson L., and Larsson N. G. (2012) Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 21, 4827–4835 10.1093/hmg/dds352 [DOI] [PubMed] [Google Scholar]
- 71. Verma M., Callio J., Otero P. A., Sekler I., Wills Z. P., and Chu C. T. (2017) Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD-associated LRRK2 mutants. J. Neurosci. 37, 11151–11165 10.1523/JNEUROSCI.3791-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Plowey E. D., Cherra S. J. 3rd, Liu Y. J., and Chu C. T. (2008) Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 105, 1048–1056 10.1111/j.1471-4159.2008.05217.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. MacLeod D., Dowman J., Hammond R., Leete T., Inoue K., and Abeliovich A. (2006) The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52, 587–593 10.1016/j.neuron.2006.10.008 [DOI] [PubMed] [Google Scholar]
- 74. Mattson M. P., Gleichmann M., and Cheng A. (2008) Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766 10.1016/j.neuron.2008.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data described are contained within the manuscript.