

Abstract
The ATP-grasp superfamily of enzymes shares an atypical nucleotide-binding site known as the ATP-grasp fold. These enzymes are involved in many biological pathways in all domains of life. One ATP-grasp enzyme, d-alanine–d-alanine ligase (Ddl), catalyzes ATP-dependent formation of the d-alanyl–d-alanine dipeptide essential for bacterial cell wall biosynthesis and is therefore an important antibiotic drug target. Ddl is activated by the monovalent cation (MVC) K+, but despite its clinical relevance and decades of research, how this activation occurs has not been elucidated. We demonstrate here that activating MVCs bind adjacent to the active site of Ddl from Thermus thermophilus and used a combined biochemical and structural approach to characterize MVC activation. We found that TtDdl is a type II MVC-activated enzyme, retaining activity in the absence of MVCs. However, the efficiency of TtDdl increased ∼20-fold in the presence of activating MVCs, and it was maximally activated by K+ and Rb+ ions. A strict dependence on ionic radius of the MVC was observed, with Li+ and Na+ providing little to no TtDdl activation. To understand the mechanism of MVC activation, we solved crystal structures of TtDdl representing distinct catalytic stages in complex with K+, Rb+, or Cs+. Comparison of these structures with apo TtDdl revealed no evident conformational change on MVC binding. Of note, the identified MVC binding site is structurally conserved within the ATP-grasp superfamily. We propose that MVCs activate Ddl by altering the charge distribution of its active site. These findings provide insight into the catalytic mechanism of ATP-grasp enzymes.
Keywords: structural biology, enzyme mechanism, enzyme structure, enzyme catalysis, X-ray crystallography, enzyme kinetics, metal ion–protein interaction, ATP-grasp, d-alanine–d-alanine ligase (Ddl), metal activation, monovalent cation
Introduction
The ATP-grasp superfamily includes more than 20 enzymes, with many having been structurally characterized (Table S1). The defining feature of this superfamily is the ATP-grasp fold, which binds ATP between two α+β domains. In addition to ATP, the catalytic mechanism of ATP-grasp enzymes generally utilizes separate carboxylate and nucleophilic substrates, with the reaction considered to proceed through formation of an acylphosphate intermediate (Fig. 1A). Although utilization of ATP is common in members of the superfamily, the composition of additional substrates varies, resulting in a wide range of biomolecular products. These enzymes are therefore involved in a diverse range of biological pathways in all domains of life, such as the oxidative stress response (GSH synthetase), gluconeogenesis, fatty acid synthesis, amino acid catabolism (carboxylases), purine biosynthesis (Pur synthetases), antibiotic biosynthesis (l-amino acid ligases), and cell wall biosynthesis (d-alanine–d-alanine ligase) (Table S1). Therefore, the role of ATP-grasp enzymes in these various pathways has been extensively studied, with many investigated for their promise as drug targets and for molecular biosynthesis.
Figure 1.

Catalytic mechanism of ATP-grasp enzymes. A, the general mechanism of ATP-grasp enzymes proceeds through two half-reactions. i, the carboxylate substrate reacts with ATP to form an acylphosphate intermediate, requiring a divalent metal (Mg2+, Mn2+, Ca2+). ii, the nucleophile substrate attacks the electrophilic acylphosphate intermediate to form the product. B, the proposed catalytic mechanism of Ddl. This proceeds with an ordered ter-ter mechanism (three substrates and three products) through two half-reactions. The first half-reaction (i) involves the phosphorylation of d-Ala1 (1) to form the acylphosphate intermediate d-Ala–phosphate (2). In the second half-reaction (ii), d-Ala–phosphate is attacked by d-Ala2 (3), resulting in formation of a tetrahedral intermediate, d-Ala–d-Ala–phosphate (4). This second intermediate then collapses within the active site to form d-Ala–d-Ala (5) and Pi (6). It has been determined that K+ is also required for optimal activity; however, the mechanism by which this occurs has been elusive.
d-alanine–d-alanine ligase (Ddl), one of the first characterized ATP-grasp enzymes, catalyzes the energy-dependent conjugation of two d-alanine molecules to form the d-alanyl–d-alanine dipeptide and is essential for cell wall biosynthesis. Because of this important role, Ddl has been primarily investigated as an antibiotic drug target and has been identified as the primary target of the second-line anti-tuberculosis agent d-cycloserine (DCS). Ddl has also been investigated for in vitro biosynthesis of d-amino acid dipeptides (1, 2). The Ddl enzyme from Thermus thermophilus (TtDdl) has been used previously as a model to investigate the reaction mechanism of these enzymes and was employed in this study (3). The enzymatic activity of TtDdl requires formation of a dimer, with a single monomer consisting of an N-terminal domain (Met1–Gly104), a central domain (Ala105–Leu192), and a C-terminal domain (Ser193–Thr319) (Fig. 2). Each subunit possesses a single ATP-binding site, formed by the ATP-grasp fold, and two d-Ala–binding sites. ATP and the two d-Ala substrates bind in adjacent sites at the center of the Ddl monomer to facilitate formation of the d-alanyl–d-alanine dipeptide (d-Ala–d-Ala). Studies of Ddl from other bacterial species have identified that the first d-Ala site has higher affinity for d-Ala than the second (4–7). The catalytic mechanism of Ddl is also accompanied by cumulative conformational changes induced by substrate binding. For TtDdl, this involves three loop regions: loop 1 (aa 155–161, the P-loop), loop 2 (aa 217–234, the Ω -loop), and loop 3 (aa 289–293) (Fig. 2).
Figure 2.
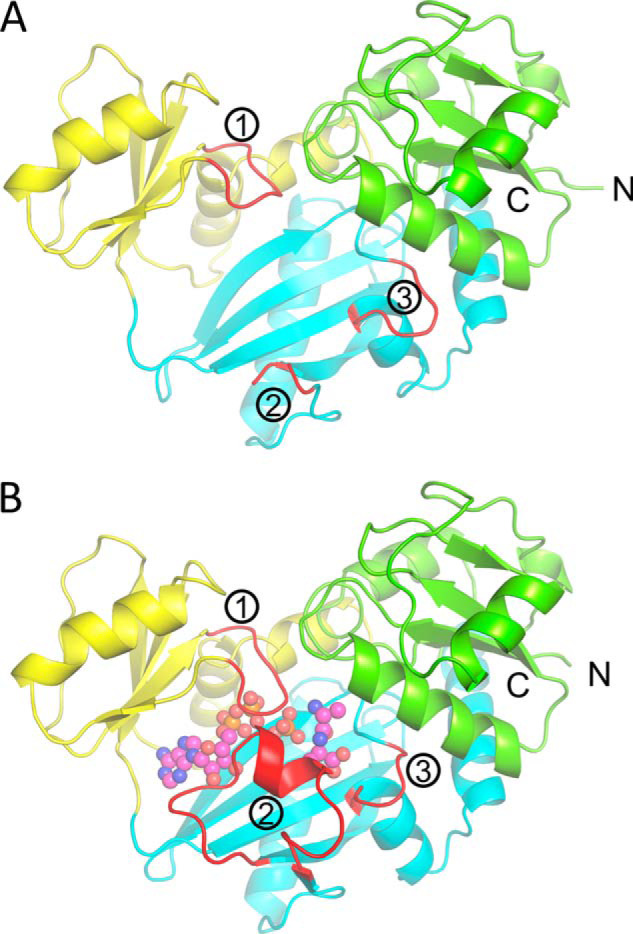
Overall fold of TtDdl. A, apo form of a single TtDdl monomer (PDB code 6U1C). The N-terminal domain, central domain, and C-terminal domain of the TtDdl monomer are colored green, yellow, and cyan, respectively. Loops 1–3 are labeled and shown in red. For apo TtDdl, all loops exist in the open conformation, with the Ω-loop (2) disordered and not observed for aa 217–234. B, the product complex (ADP, Pi, and d-Ala–d-Ala–bound) of a single TtDdl monomer (PDB code 6U1J). All loops are closed, with the Ω-loop becoming ordered and covering the substrate binding pocket. ADP, Pi, and d-Ala–d-Ala are shown as pink spheres.
The catalytic mechanism of Ddl is proposed to proceed with an ordered ter-ter mechanism through two half-reactions and involves formation of two reaction intermediates: an acylphosphate intermediate and a tetrahedral intermediate (Fig. 1B). In the first half-reaction, ATP and two Mg2+ bind and facilitate closure of the P-loop (3, 8). Subsequently, the first d-Ala (d-Ala1) binds adjacent to ATP, inducing closure of the Ω-loop. Here, the carboxylate of d-Ala1 attacks the γ-phosphate of ATP (1), leading to formation of the acylphosphate intermediate d-Ala–phosphate (2). In the second half-reaction, a second d-Ala (d-Ala2) then enters the active site. The primary amine of d-Ala2 must be in its deprotonated form (NH2) to react with d-Ala–phosphate. The mechanism by which this deprotonation occurs remains unclear for Ddl (8, 9). The deprotonated d-Ala2 then attacks the phosphorylated carbonyl carbon of d-Ala1 (3), resulting in formation of a tetrahedral intermediate, d-Ala–d-Ala–phosphate (4). This coincides with closure of loop 3 and formation of a “fully closed” complex (3, 10). The tetrahedral intermediate then collapses to form the d-Ala–d-Ala dipeptide and Pi (5). Following collapse, Pi is released first from TtDdl, followed by d-Ala–d-Ala and ADP (8, 11).
It has also been determined that Ddl is activated by the monovalent cation (MVC) potassium (K+) (12). For over 60 years, K+ has been included for the kinetic analysis of Ddl with little insight into how MVCs activate the enzyme. As a result, the mechanism by which this activation occurs remains unknown (Fig. 1B). Activation of enzymes by MVCs is categorized into one of two general mechanisms, referred to as type I or type II activation (13, 14). Type I activation involves direct interaction of the MVC with the substrate, and so the MVC is required for enzyme activity. For type II activation, the MVC is not essential for enzyme activity. In this case, the MVC binds at an allosteric site, causing an increase in substrate binding affinity and/or enzyme activity. Ddl has been determined previously to be a type II activated enzyme. The pioneering kinetic study of Ddl by Neuhaus (12) revealed that the MVC K+ was required for optimal activity of Enterococcus faecalis Ddl, characterized by a 1.4-fold increase in kcat, and a 3-fold decrease in the Km for the d-Ala substrate in the presence of 10 mm KCl. NH4+ has also been shown to be a less potent activator than K+, whereas Na+ did not affect enzyme activity. Similar observations of K+ activation were later reported by Prosser and De Carvalho (6) for Mycobacterium tuberculosis Ddl. Kinetic studies of other ATP-grasp enzymes have also reported activation by MVCs (Table S1). Similar to Ddl, the mechanism for MVC activation of other ATP-grasp enzymes is poorly understood, largely because the location of MVC binding remains unknown. Therefore, it has yet to be determined whether this superfamily shares the same mechanism for MVC activation or whether unique mechanisms exist.
Historically, a kinetics approach has been used to identify activation of enzymes by MVCs; however, this does not provide full details of the activation mechanism. Recent advances in structural and spectroscopic techniques have allowed a more complete understanding of these mechanisms (13, 15). To this end, X-ray crystallography has proven to be an especially powerful technique. By exploiting the unique anomalous scattering properties of heavy metal MVCs, such as Rb+ and Cs+, MVC binding sites can be identified through cocrystallization with the enzyme of interest. This also allows direct comparison of enzyme structure in the absence and presence of MVCs, providing insight into the mechanism of activation. In this study we have investigated the MVC activation of TtDdl using a combination of steady-state kinetic analysis and X-ray crystallography. This combined approach revealed that TtDdl is activated by the larger MVCs K+, NH4+, Rb+, and Cs+ and allowed the identification of an MVC binding site within the active site of TtDdl. Based on these findings, we proposed a mechanism for MVC activation of Ddl. Comparison with members of the ATP-grasp superfamily revealed that this previously unidentified site is structurally conserved in many ATP-grasp enzymes. The broader significance of this discovery with respect to MVC activation of other enzymes in the ATP-grasp superfamily is also addressed.
Results
TtDdl is selectively activated by MVCs
A kinetics analysis of TtDdl was completed to determine the effect of K+ and other MVCs on catalysis. Although K+ has been shown previously shown to activate Ddl from other species, this had not been determined for TtDdl. For this purpose, a colorimetric antimony–phosphomolybdate assay was used to monitor production of Pi from the enzymatic activity of TtDdl. The kinetic parameters kcat and Km were determined for the ATP and d-Ala1/d-Ala2 substrates in a low-MVC reaction buffer (Table 1, see “Experimental procedures”). These were determined as 16.2 μm, 1250 μm, and 4020 μm, respectively. To investigate the effect of K+ on TtDdl activity, the kinetic parameters were also determined in the presence of 10, 50, and 100 mm KCl (Table 1). For Km ATP and kcat, a slight decrease was observed in the presence of 50 mm and 100 mm KCl. In contrast, Km d-Ala2 decreased 10-fold in the presence of 10 mm KCl and up to 20-fold for 50 and 100 mm KCl. A similar decrease was also reflected in Km d-Ala1. This reduction in d-Ala Km is consistent with previous observations (5, 6). To compare the effect of these MVCs on TtDdl activity, kcat/Km d-ala2 was used as a measure of enzyme efficiency. As addition of KCl showed little to no effect on kcat, this translated to a ∼20-fold more efficient enzyme at 50 mm KCl. This indicates that TtDdl is a more efficient enzyme in the presence of K+ and, therefore, that it activated by this MVC.
Table 1.
Effect of KCl on TtDdl catalysis
| Conditiona | Km ATP | Km d-Ala2 | Km d-Ala1 | kcat | kcat/Km d-Ala2 |
|---|---|---|---|---|---|
| μm | μm | μm | s−1 | s−1 m−1 | |
| Low MVCb | 16.2 ± 1.1 | 4,020 ± 630c | 1,250 ± 490c | 1.26 ± 0.04c | 315 ± 50c |
| 10 mm KCl | 17.3 ± 1.0 | 315 ± 19 | <55d | 1.48 ± 0.02 | 4,690 ± 290 |
| 50 mm KCl | 12.4 ± 0.5 | 150 ± 15 | <50d | 1.07 ± 0.02 | 71,40 ± 710 |
| 100 mm KCl | 12.1 ± 0.7 | 190 ± 24 | <50d | 1.04 ± 0.03 | 54,60 ± 700 |
a All kinetic parameters reported were determined from two experiments, each with three technical replicates unless stated otherwise. Error represents ± S.D.
b <1 mm Na+ present with ATP.
c Determined from five experiments, each with three technical replicates.
d Lowest concentration of d-Ala assayed.
In the case of MVC-activated enzymes, a preference for Na+ or K+ is commonly observed, with MVCs of similar ionic radii often able to substitute with a comparable effect. Therefore, the effects of LiCl, NaCl, NH4Cl, RbCl, and CsCl salts on TtDdl activity were also determined at an intermediate concentration of 50 mm (Fig. 3 and Table S2). The smaller MVCs Li+ and Na+ produced a small effect, with an apparent 1.5-fold increase in TtDdl efficiency compared with the low-MVC condition. In contrast, larger MVCs showed a 10- to 20-fold increase in efficiency. This confirms that activation of TtDdl is not the result of an ionic strength effect. Interestingly, substrate inhibition by d-Ala was observed in the presence of the activating MVCs K+, NH4+, Rb+, and Cs+ (Fig. S2). This has not been reported previously for Ddl. We predict that this may be due to nonproductive binding of d-Ala2, a result of the low Km d-Ala2, which was determined to be decreased ∼10-fold compared with Ddl from other species. The order of MVC activation was thus determined as K+≈ Rb+ > NH4+≈ Cs+ > Na+ ≈ Li+ ≈ low MVC. Therefore, this kinetics analysis demonstrated that K+ and the larger MVCs NH4+, Rb+, and Cs+ activate TtDdl.
Figure 3.
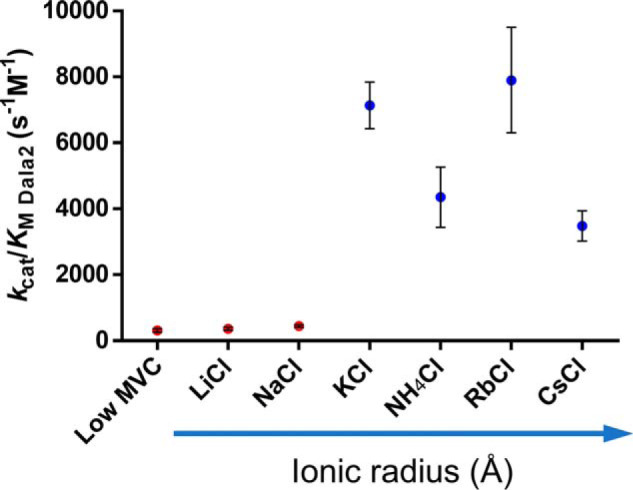
TtDdl is activated by MVCs. Addition of 50 mm KCl, NH4Cl, RbCl, and CsCl (blue) resulted in increased TtDdl efficiency, indicated by an increase in kcat/Km d-Ala2. This effect was in contrast to the low-MVC, 50 mm LiCl, and 50 mm NaCl conditions (red), showing that this was not due to a change in Cl− concentration or ionic strength. Data show kcat/Km d-Ala2 calculated from two experiments, except for the low-MVC condition (n = 5 experiments). Error bars represent ± S.D.; calculated as detailed under “Experimental procedures.”
Identification of a catalytic MVC binding site
After determining that TtDdl is activated by MVCs we aimed to identify catalytically relevant binding sites by X-ray crystallography. An approach to identify such sites is crystallization of the target protein in the presence of MVCs (16). For this purpose, the larger electron density and anomalous scattering properties of Rb+ and Cs+ were exploited. Cocrystals of TtDdl complexed with Mg2+-ATP, d-Ala–d-Ala, and Rb+ or Mg2+-ATP, d-Ala–d-Ala, and Cs+ were grown as described under “Experimental procedures.” Datasets for both conditions were collected at 13,000 eV at the Australian Synchrotron, and the structures were solved to 1.90 Å and 2.30 Å (17). Additional datasets were collected at 15,250 eV and 8,500 eV for the Rb+- and Cs+-bound structures, respectively, to obtain a strong anomalous signal. Anomalous difference Fourier maps were used to identify MVC binding sites. In total, two separate MVC sites were identified per TtDdl monomer for Rb+ and Cs+ (Fig. 4A). Anomalous peak heights and atomic parameters for each site are detailed in Table S4. Site 1 is located at the edge of the dimerization interface, coordinated by Trp133 and solvent (Fig. S3). This is distant from the active site and unlikely to be responsible for the activating effect. This conclusion is supported by the low occupancies for Rb+ modeled at this site, despite a high concentration of 150 mm RbCl in the crystallization condition.
Figure 4.

MVC binding sites of TtDdl. A, anomalous difference maps for TtDdl-Rb+anom (15,250 eV, blue mesh, top panel) and TtDdl-Cs+anom (8,500 eV, purple mesh, bottom panel) contoured to 6σ and 5σ, respectively. The corresponding models are shown as yellow and green ribbons, respectively, with ATP and d-Ala–d-Ala shown as sticks. B, Rb+ (blue sphere) and Cs+ (cyan sphere) binding at site 2. Site 2 (the K+ cleft) is in close proximity to the active site. Electron density for simulated annealing composite omit maps (2Fo-Fc, 1.5σ) is shown for the ligands of Rb+-bound (blue mesh) and Cs+-bound (purple mesh) TtDdl. Bound Mg2+ is shown as green spheres (Rb+) and dark green spheres (Cs+). The three loop regions of TtDdl (red; P-loop, Ω-loop, and loop 3) are in the closed conformation. C, the coordination network of Rb+ (blue spheres) and Cs+ (cyan spheres) at the K+ cleft extends to Mg2+ and ATP. The coordination spheres of Rb+ and Cs+ are shown as blue and cyan dashes, respectively. Coordinating waters are shown as red spheres (Rb+) and pink spheres (Cs+), with the water bridging the MVC and Mg2+ labeled (H2OB). Coordinating residues and the residues forming the K+ cleft are shown as yellow sticks (Rb+) and green sticks (Cs+). The coordination spheres of bound Mg2+, green spheres (Rb+) and dark green spheres (Cs+), are shown as green and dark green dashes, respectively. The P-loop and Ω-loop are omitted for clarity. D, electron density for feature-enhanced maps (2Fo-Fc, 2σ) of TtDdl-Rb+ (left panel, blue mesh) and TtDdl-Cs+ (right panel, purple mesh) site 2 coordination spheres.
In contrast, site 2, referred to here as the K+ cleft, is in close proximity to the active site (Fig. 4B). The K+ cleft is defined by the side chains of Glu282, Asn284, Met114, Lys116, and Glu87 and the backbone of Leu283 and Cys113. Rb+ and Cs+ share the same coordination sphere of seven coordinating interactions (Fig. 4, C and D). This includes the carboxylate of Glu282, the amide of Asn284, the backbone carbonyl O of Cys113 and Leu283, and three ordered waters, arranged in a capped trigonal prismatic geometry. Notably Glu282 also coordinates to both Mg2+ that bridge the triphosphate group of ATP. This suggests an important role of Glu282 in Ddl catalysis, supported by a previous site-directed mutagenesis study of Escherichia coli Ddl that reported a ∼500-fold decrease in activity for an equivalent Glu282→Gln mutant (9). Additionally, multiple sequence alignments and structural superposition of TtDdl to other Ddl enzymes also reveals that the residues forming the K+ cleft are highly conserved (Fig. S4). Together, this provides strong evidence that MVC binding at the K+ cleft is responsible for activation of TtDdl.
K+ binding at the K+ cleft does not induce conformational change
Having identified the MVC binding site of TtDdl, we further investigated the influence of K+ at separate stages of the Ddl reaction mechanism. The mechanism of MVC activation for type II activated enzymes commonly involves conformational change in the vicinity of the MVC binding site. As TtDdl is a type II activated enzyme, K+-induced conformational change was investigated by capturing TtDdl at different stages of the catalytic cycle. By completing crystallization experiments (see “Experimental procedures”), five structures of TtDdl representing four different stages of Ddl catalysis were solved: Apo TtDdl (PDB code 6U1C); TtDdl in complex with ADP, Pi, and K+ (PDB code 6U1H); TtDdl in complex with ADP, the phosphorylated form of d-cycloserine (DCSP), and K+ (PDB code 6U1I); TtDdl in complex with ADP, Pi, d-Ala–d-Ala, and K+ (PDB code 6U1J); and TtDdl in complex with ADP, CO32−, d-Ala–d-Ala, and K+ (PDB code 6U1K) (Fig. S5). The high resolution (1.67 Å) of the latter structure allowed clear identification of K+ bound at the K+ cleft. The mean K+-O bond length of 2.91 Å across all of these structures is consistent with a reported value of 2.91 ± 0.179 Å for [7]K+ (18). A complete summary of these atomic parameters for each modeled cation is reported in Table S5. In each of these structures, K+ shared the same coordination sphere described for Rb+ and Cs+ (Fig. 5A and Fig. S6). Flexibility in coordinating residues of the K+ cleft was also observed, being dependent on the ionic radius of the bound MVC. Accommodation of the bulkier Cs+ produced the greatest effect, possibly explaining the reduced potency compared with activation by K+ and Rb+. As the activating effect of these MVCs was associated with the d-Ala substrate, the possibility of conformational change at the K+ cleft or within the d-Ala pocket was considered. To identify whether K+ binding at the K+ cleft induces conformational change, the captured states of K+-bound TtDdl were superposed with apo TtDdl (Fig. 5B). Overall, minimal conformational change was observed for the coordinating residues of the K+ cleft. The only observable difference was rotation of Glu282 from the apo to K+ bound enzyme, which is consistent with ordering of the side chain following binding of ATP and Mg2+. Therefore, this suggests that MVC activation of TtDdl is not elicited through conformational change at the K+ cleft. As the activating effect of K+ was associated with the d-Ala substrates, the d-Ala1 binding pocket was similarly compared (Fig. 5C). Again, no conformational change was observed in the residues forming the d-Ala1 binding pocket. Notably, the key residues Glu13 and Arg268, which facilitate hydrogen bond–mediated substrate recognition, remain in the same conformation in the apo and ligand-bound forms of TtDdl. The only considerable difference is in the conformation of the P-loop; however, this is accounted for by the absence of a nucleotide substrate in the structure of apo TtDdl. This structural analysis shows that K+ binding at the K+ cleft does not result in rearrangement of active-site residues and suggests that activation of TtDdl occurs through a mechanism that does not involve significant conformational change.
Figure 5.
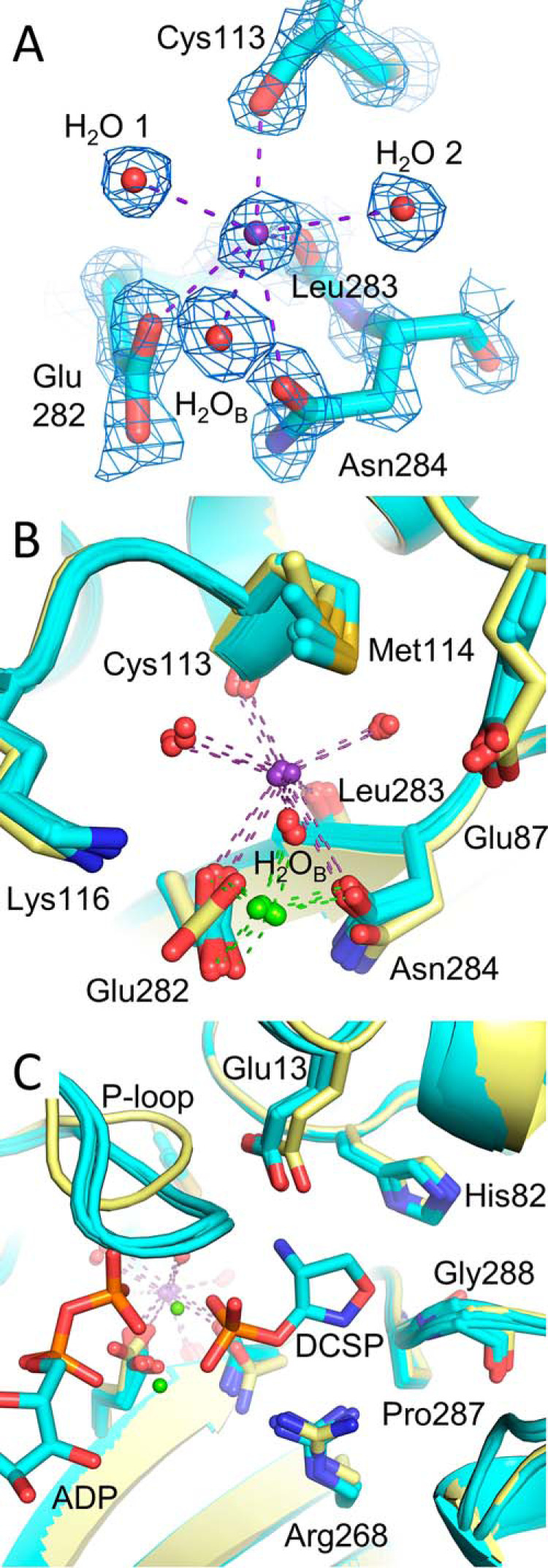
K+ binding at the K+ cleft does not induce conformational change. A, electron density for feature enhanced map (2Fo-Fc, 2σ) of TtDdl-K+ (PDB code 6U1K) at the K+ cleft. The coordination sphere of K+ (purple spheres, purple dashes) is analogous to that observed for Rb+ and Cs+ (Fig. 4, C and D). B, superposition of apo (yellow) and K+-bound (cyan) TtDdl structures (Table S3). Minimal conformational change is evident on K+ binding at the K+ cleft throughout the catalytic cycle. Bound Mg2+ is shown as green spheres, with coordinating bonds shown as green dashes. Coordinating waters are shown as red spheres. Bound ligands were omitted for clarity. C, superposition of apo (yellow) and K+-bound (cyan) TtDdl structures shows no change in conformation of residues forming the d-Ala binding sites. ADP and DCSP (PDB code 6U1I) are shown as cyan sticks, with the DCS moiety indicating the position of the d-Ala1 pocket. Mg2+ is shown as green spheres. The P-loop is seen to be in the open conformation for apo TtDdl, as no nucleotide is bound. The Ω-loop is omitted for clarity.
The K+ cleft is structurally conserved between members of the ATP-grasp superfamily
Enzymes of the ATP-grasp superfamily share high conservation in their nucleotide binding sites and in the first step of catalysis, which involves formation of an acylphosphate intermediate (Fig. 1A). As summarized in Table S1, some ATP-grasp enzymes have been determined to be activated by MVCs. Therefore, we predicted that MVCs may also activate other members of the ATP-grasp superfamily by a similar mechanism. Structures have been solved by X-ray crystallography for many of these enzymes, allowing direct comparison with the K+ cleft of TtDdl. Initially, examples of ATP-grasp enzymes with an MVC bound at this position were considered. In all, two cases were identified, with the first of these being carbamoyl phosphate synthetase (CPS) (19). Superposition of the carboxyphosphate domain of E. coli CPS with TtDdl revealed that the K+ cleft of TtDdl overlays with a bound K+ (K+3) of CPS (Fig. 6, A and B). Closer inspection of this site reveals high conservation between the two enzymes, with the only major difference being substitution of Met114 in TtDdl to Glu126 of CPS, resulting in an additional interaction with the bound K+. The second case identified was for the biotin carboxylase (BC) domain of E. coli acetyl-CoA carboxylase (20). A previous study investigating BC catalysis required CsCl in the crystallization condition for an Arg16→Glu16 mutant. In this structure, Cs+ is clearly bound at the same position as the K+ cleft of TtDdl (Fig. 6C). Compared with TtDdl, Met114 is substituted for Gly in BC. Additionally, structural comparison with WT E. coli BC shows that the Arg16→Glu16 mutation does not affect the conformation of this site (Fig. S7). Together, these examples demonstrate that MVC binding at this position is also possible for ATP-grasp enzymes other than Ddl. Following this, the K+ cleft of TtDdl was compared with the analogous position in other ATP-grasp enzymes. A total of 25 different ATP-grasp enzymes were compared by structural superposition to TtDdl (PDB code 6U1H) (Figs. S8 and S9 and Table S6).
Figure 6.

MVC binding sites of ATP-grasp enzymes. A, superposition of the carboxyphosphate domain of E. coli carbamoyl phosphate synthetase (EcCPS; blue, PDB code 1JDB) with TtDdl in complex with ADP and Pi (yellow, PDB code 6U1H). In the ATP-grasp superfamily, CPS is unique in that it has a type I mechanism of activation, with K+ essential for its activity. The three separate K+ binding sites (K+1, K+2, and K+3; gray spheres) of the EcCPS carboxyphosphate domain are labeled. MVC activation of CPS has been attributed to K+1 and K+2, whereas the role of K+3 remains unknown. Superposition with TtDdl reveals that K+3 binds at the same position as the K+ cleft. Bound Mg2+ (TtDdl) and Mn2+ (EcCPS) are shown as green and dark green spheres, respectively. K+ bound to TtDdl is shown as a purple sphere. Bound ADP and Pi are shown as sticks. B, comparison of K+3 of EcCPS (gray sphere, gray dashes) with K+ at the K+ cleft of TtDdl (purple sphere, purple dashes). The overall structure of the site is highly conserved between the two enzymes, with the key difference being an additional contact from Glu126 of CPS at the equivalent position of Met114 of TtDdl. Residues forming the K+ cleft are shown as sticks. C, superposition of E. coli biotin carboxylase (EcBC; light blue, PDB code 3RV4) with TtDdl (PDB code 6U1H). For EcBC, Cs+ (cyan sphere, cyan dashes) was modeled at the same position as K+ (purple sphere, purple dashes) bound at the K+ cleft of TtDdl. Coordinating waters are shown as red spheres (K+) and pink spheres (Cs+). Residues forming the K+ cleft are shown as sticks. Bound Mg2+ is shown as green spheres (TtDdl) and dark green spheres (EcBC). The P-loop is omitted for clarity.
This revealed that the K+ cleft is structurally conserved for most of these ATP-grasp enzymes and for the d-amino acid ligases VanA and VanG, complete conservation of the K+ cleft was observed. Overall, the Glu282 and Asn284 positions showed high conservation, being conserved in 80% and 56% of the ATP-grasp enzymes analyzed, respectively. Although the Met114 residue was poorly conserved, most substitutions at this position project away from the K+ cleft and do not block the MVC binding site. Of all the enzymes analyzed, kinase and thiokinase members showed the least conservation in the primary and secondary structure of the K+ cleft (Fig. S8 and Table S6). One plausible explanation for this is differences in the catalytic mechanism for these enzymes compared with Ddl, which rely on phosphorylation of a catalytic histidine residue rather than a carboxylate substrate. Overall, the K+ cleft of Ddl was observed to be structurally conserved in ATP-grasp enzymes, particularly for Glu282, with most of these enzymes catalyzing reactions that proceed through formation of an acylphosphate intermediate. This suggests that other enzymes of the ATP-grasp superfamily may share the same type II mechanism for MVC activation as proposed for Ddl.
Discussion
The structural and kinetic evaluation of TtDdl with MVCs has provided important insight into the mechanism of MVC activation. These experiments support a type II mechanism of activation. A marked reduction was observed in the Km for both d-Ala substrates in the presence of the larger MVCs K+, NH4+, Rb+, and Cs+; however, no significant conformational change at the active site of TtDdl is evident. Although the major effect involves the d-Ala substrates, MVCs were identified to bind closer in proximity to the nucleotide binding pocket, forming an extensive coordination network with the bound Mg2+ and ATP. Enzyme catalyzed phosphoryl transfer reactions require a divalent metal as an essential cofactor (21). For TtDdl, this is facilitated by two Mg2+ that act as Lewis acids, balancing the buildup of negative charge on the triphosphate leaving group of ATP. This promotes nucleophilic attack by d-Ala1 to form the acylphosphate intermediate d-Ala–phosphate. The proposed Mg2+–ATP complex for TtDdl in the absence of an activating MVC is depicted in Fig. 7. Mg1 coordinates the carboxylate side chains of Asp270 and Glu282 and the oxyanions of the α/γ Pi. The carboxylate side chain of Glu282 also acts as a bidentate ligand for Mg2, which coordinates the oxyanions of the β/γ Pi (Fig. 7A). We propose that, when an activating MVC is bound at the K+ cleft, the charge distribution of this coordination network is altered (Fig. 7B). This may occur through the MVC attenuating the negative charge of the Glu282 carboxylate anion balanced by bidentate coordination of Mg2. This would increase the Lewis acidity of Mg2, allowing it to stabilize additional negative charge from the β/γ Pi. This reduces electrostatic repulsion with the carboxylate of d-Ala1, increasing the binding affinity of d-Ala1 and resulting in an increased rate of d-Ala–phosphate formation. The complex of TtDdl with DCSP shown in Fig. 7, C and E, mimics the binding mode of d-Ala–phosphate and demonstrates how MVC binding at the K+ cleft could also modulate the Lewis acidity of Mg1 and Mg2 prior to nucleophilic attack by d-Ala2. This model is consistent with the ordered nature of the Ddl catalytic mechanism. If the binding affinity of d-Ala1 is increased, then this would result in faster d-Ala–phosphate formation. As d-Ala–phosphate formation is increased, d-Ala2 can more readily react with d-Ala–phosphate to form the tetrahedral intermediate and, upon collapse, d-Ala–d-Ala. This explains the observed decrease in Km for both d-Ala substrates. The fact that a similar effect is not observed in kcat supports product release being the rate-limiting step, consistent with previous isotope exchange experiments utilizing d-Ala–d-Ala and d-Ala–d-X ligases (11, 22). The high conservation observed in the K+ cleft suggests that this mechanism of MVC activation applies generally for Ddl enzymes (Fig. S4). Furthermore, the structural conservation of the K+ cleft and the key coordinating Glu residue between the ATP-grasp enzymes suggests that this mechanism of activation may apply more broadly across the superfamily.
Figure 7.

Proposed mechanism of MVC activation for TtDdl. A, proposed Mg2+–ATP complex in the absence of an activating MVC. Mg1 and Mg2 (green spheres, green dashes) act to stabilize the negative charge of the triphosphate moiety of ATP, facilitating nucleophilic attack by d-Ala1. The model was prepared by superimposing the structures of TtDdl in complex with RbCl (PDB code 6U1D, yellow) and TtDdl in complex with ADP and d-Ala (PDB code 2ZDH, cyan). Coordinating atoms and waters are shown as red spheres. B, Mg2+–ATP complex in the presence of an activating MVC. The bound MVC (purple sphere, purple dashes) balances the negative charge of the Glu282 carboxylate anion, allowing Mg2 to withdraw additional electron density from the β/γ Pi. This reduces the electrostatic repulsion between the γ Pi of ATP and the nucleophilic carboxylate anion of d-Ala1, increasing the binding affinity of the d-Ala1 substrate. The model was prepared as described in Fig. 6A. C, reaction intermediate complex following phosphoryl transfer. TtDdl in complex with Mg2+–ADP and DCSP mimics the acylphosphate intermediate. Upon transfer of the γ-Pi to DCS, Mg1 shifts to stabilize the negative charge of all three phosphate groups, replacing the interaction with Asp270 by coordination of an additional water. The model represents the structure of TtDdl in complex with DCSP (PDB code 6U1I). D, chemical representation of the Mg2+–ATP complex in the presence of an activating MVC. Dashed lines represent coordinating bonds, with distances labeled in angstroms (PDB code 6U1D). The arrow adjacent to Glu282 represents the shift of charge for the carboxylate group on MVC binding. An asterisk indicates that the bond projects out of the page. E, chemical representation of the Mg2+–ADP–DCSP complex in the presence of an activating MVC. Dashed lines represent coordinating bonds, with distances labeled in angstroms (PDB code 6U1I. An asterisk indicates that the bond projects out of the page.
This discovery of structural conservation at the K+ cleft within the functionally diverse members of the ATP-grasp superfamily provides a basis for further investigation of MVC activation. Several ATP-grasp enzymes require MVCs for maximal activity; however, for many, the effect of MVCs has yet to be elucidated (Table S1). As the functions of ATP-grasp enzymes are so diverse, understanding the role of MVCs would provide insight into many catalytic schemes. This is particularly relevant for ATP-grasp enzymes that show promise as drug targets and for enzyme-catalyzed chemical synthesis. Members of the ATP-grasp superfamily that have been investigated as drug targets include Ddl, biotin carboxylase, tubulin tyrosine ligase, and the purine biosynthesis enzyme PurK (23–27). In this study, we demonstrate that the presence of activating MVCs has a substantial effect on the efficiency of Ddl, likely by altering the charge distribution at the active site. If this is found to apply to other ATP-grasp enzymes, we predict that using K+ concentrations that more closely reflect physiological conditions will allow more accurate characterization of inhibitors in vitro. More recently, l-amino acid ligases (LALs) have also been investigated for the industrial production of a range of products, including antibiotics and peptides for use in healthcare and consumable products (28–31). Despite many ATP-grasp enzymes having high similarity to Ddl, to our knowledge, activation of LALs by MVCs has not been investigated. Investigating activation by MVCs may lead to improvements in activity and broaden substrate specificity of LALs. Such examples demonstrate the importance of further characterizing MVC activation for the ATP-grasp superfamily.
Conclusion
Through structural investigation of T. thermophilus Ddl, we identified the site responsible for MVC activation, the K+ cleft, providing the first mechanistic insight into K+ activation of Ddl. Type II MVC activation of TtDdl was also confirmed, with activation being dependent on the ionic radius. For the group I MVCs Li+–Cs+, the smaller Li+ and Na+ showed little to no activation. In contrast, maximal activation was observed for the larger K+ and Rb+. Crystal structures of TtDdl captured at distinct stages of the catalytic mechanism revealed minimal conformational change in the presence of K+, suggesting that a conformational change is not responsible for the activating effect. We propose that activation occurs through K+ altering the charge distribution within the active site, increasing the binding affinity for the carboxylate substrate d-Ala1 and promoting formation of the acylphosphate intermediate. Structural comparison of TtDdl with other ATP-grasp enzymes revealed that the K+ cleft is structurally conserved, suggesting that the same mechanism of activation may be applicable to other ATP-grasp enzymes. Therefore, we recommend that future studies of ATP-grasp enzymes consider activation by MVCs.
Experimental procedures
Materials
For the enzyme activity assay, potassium antimonyl tartrate trihydrate and ammonium molybdate tetrahydrate were purchased from Sigma-Aldrich (St. Louis, MO, USA). Ascorbate and all chloride salts were of analytical grade or higher. KH2PO4 standards were prepared from a 10 mm KH2PO4 stock solution. For crystallization experiments, 50% PEG 3350, magnesium formate dihydrate (1 m) and BisTris (pH 6.5) (1 m) were purchased from Hampton Research (Aliso Viejo, CA, USA). A stock solution of BisTris (pH 6.8) was prepared by dropwise addition of 2 m NaOH. ATP, ADP, d-Ala, DCS, and d-Ala–d-Ala were purchased from Sigma-Aldrich.
Protein expression and purification
The vector pET16b was purchased from Genscript (Piscataway, NJ, USA) for overexpression of TtDdl with a C-terminal hexahistidine tag. The TtDdl-pET16b vector was transformed into E. coli BL21(DE3). For protein expression, two flasks containing 1 liter of lysogeny broth supplemented with 100 μg/ml ampicillin were inoculated with 20 ml of an overnight culture. Cells were grown at 37 °C to an A600 of 0.5 and induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside. TtDdl was expressed for 16 h at 16 °C. Cells were pelleted by centrifugation at 5,000 × g for 20 min. Cell pellets were resuspended in 15 ml of buffer A (20 mm Tris-HCl (pH 8.0), 300 mm NaCl, 5 mm imidazole, and 5 mm β-mercaptoethanol) and lysed by five rounds of cell disruption. The lysate was clarified by centrifugation at 40,000 × g for 40 min. The supernatant was applied to a Zetasep nickel-nitrilotriacetic acid column (emp Biotech) and eluted with a gradient of buffer B (20 mm Tris-HCl (pH 8.0), 300 mm NaCl, 250 mm imidazole, and 5 mm β-mercaptoethanol). For crystallography, TtDdl was dialyzed against 4 liters of storage buffer overnight at 4 °C (20 mm Tris-HCl (pH 7.5), 50 mm NaCl, and 1 mm DTT). Protein was concentrated at 4 °C using an Amicon Ultra centrifugal filter (10,000 molecular weight cut off, Sigma-Aldrich) to 10 mg/ml and stored at −80 °C. For enzyme assays, TtDdl was then applied to a HiPrep 26/60 Sephacryl S-300 HR gel filtration column (GE Life Sciences, Chicago, IL, USA) equilibrated with 20 mm Tris-HCl (pH 7.5) and 1 mm DTT. Protein was concentrated using an Amicon Ultra centrifugal filter (10,000 molecular weight cut off, Sigma-Aldrich) to 3 mg/ml and stored at −80 °C. Protein concentration was determined by measuring absorbance at 280 nm.
Measurement of TtDdl activity
To measure the activity of purified TtDdl in the presence and absence of various monovalent cations (Li+, Na+, K+, NH4+, Rb+, and Cs+), the antimony–phosphomolybdate colorimetric end point assay described by Bartolommei and Tadini-Buoninsegni (32) was used, with several modifications. For all experiments, a 2× color reagent (CR) (255 mm sulfuric acid, 1.2 mm ammonium molybdate tetrahydrate, 18 mm ascorbic acid, and 72 μm potassium antimonyl tartrate trihydrate) was prepared from stock solutions of each component and diluted in MilliQ H2O. The assay was optimized for use in a 96-well microplate format. Validation of the assay is provided in Fig. S1. Calibration curves of KH2PO4 standards in the absence and presence of ATP and d-Ala showed linearity for the measured concentration range of 1–15 μm Pi in a 150-μl sample. All experiments were performed at 30 °C in a reaction buffer containing 50 mm Tris-HCl (pH 7.3) and 10 mm MgCl2 with varying concentrations of ATP, d-Ala, and MVC. Prior to starting the reaction, TtDdl and the reaction were equilibrated to 30 °C. To start the reaction, 75 μl of TtDdl (50 mm Tris-HCl (pH 7.3) and 10 mm MgCl2) was added to 75 μl of reaction buffer (50 mm Tris-HCl (pH 7.3) and 10 mm MgCl2 and 2× ATP, d-Ala, and MVC) and mixed by pipetting. The reaction was allowed to proceed for exactly 10 min. To stop TtDdl activity and begin color development, 150 μl of 2× CR was added to the 150-μl sample and mixed by pipetting. Color was developed for 10 min. Color development was quenched with 20 μl of a 30% citric acid solution to prevent interference from acid-catalyzed ATP hydrolysis in the presence of CR, giving a final volume of 320 μl. Absorbance was measured at a wavelength of 890 nm (Pherastar FSX microplate reader; BMG Labtech, Ortenberg, Germany) after 5 min. To account for acid-catalyzed ATP hydrolysis during color development, the absorbance of a no-enzyme blank was subtracted from each sample. A separate calibration curve for 1–15 μm KH2PO4 was measured for each individual experiment, with the slope used to calculate the concentration of Pi released by TtDdl-catalyzed ATP hydrolysis.
Determination of kinetic parameters
For determination of kinetic parameters of ATP (Km, kcat) reactions were assayed for eight concentrations ranging from at least 0.5–10× Km ATP, with 50 mm d-Ala and 6.25 nm TtDdl (Fig. S2). To determine the effect of varying KCl concentrations, the experiment was completed in the presence of 0, 10, 50, and 100 mm KCl. Experiments were repeated at least twice, with three technical replicates for each concentration assayed. For determination of kinetic parameters of d-Ala (Km d-Ala1, Km d-Ala2, kcat), reactions were assayed for seven or more concentrations ranging from at least 0.3–5× Km d-Ala2 with 300 μm ATP and 12.5 nm TtDdl (Fig. S2). To determine the effect of various MVCs, the experiment was completed for 0, 10, 50, and 100 mm KCl and 50 mm LiCl, NaCl, NH4Cl, RbCl, and CsCl. Experiments were repeated at least twice, with three technical replicates for each concentration assayed. To determine Km ATP, data were fit with the Michaelis–Menten Equation (Equation 1) for a single binding site by nonlinear regression. To determine kcat, Km d-Ala1 and Km d-Ala2, data were fit with Equation 2 by nonlinear regression, as derived by Neuhaus (5) for the steady-state kinetics of two identical substrates with ordered binding. As Km d-Ala1 was outside of the range of measurable d-Ala concentrations for the assay in the presence of more than 10 mm KCl, Km d-Ala1 was reported as less than the lowest concentration of d-Ala assayed for these conditions (Table 1). The parameters kcat and Km d-Ala2 and their standard deviations (Δkcat and ΔKm) were then used to calculate kcat/Km d-Ala2 values. The standard deviation for kcat/Km d-Ala2 value Δ (kcat/Km d-Ala2) was calculated as Δ(kcat/Km d-Ala2) = (kcat/Km d-Ala2)[(Δkcat/kcat)2 + (ΔKm d-Ala2/Km d-Ala2)2]1/2.
| (Eq. 1) |
| (Eq. 2) |
Crystallization of TtDdl
For crystallization of TtDdl in complex with Rb+, 10 mg/ml TtDdl was incubated with 5 mm ATP, 5 mm MgCl2, 50 mm d-Ala, and 150 mm RbCl for 20 min at 60 °C. Crystals of TtDdl in complex with Cs+ were similarly obtained by incubation of 10 mg/ml TtDdl with 5 mm ATP, 5 mm MgCl2, 50 mm d-Ala, and 80 mm CsCl for 20 min at 60 °C. Crystals of the product complex of TtDdl were obtained without addition of substrates or products, with 10 mg/ml TtDdl kept on ice prior to crystallization. To obtain apo crystals of TtDdl, 10 mg/ml TtDdl was heated at 60 °C for 20 min prior to crystallization. Crystals of TtDdl in complex with ADP, DCSP, and K+ were obtained by incubating 10 mg/ml TtDdl with 37 mm DCS for 30 min at 60 °C. Crystals of TtDdl in complex with ADP and Pi were obtained by incubation of 10 mg/ml TtDdl with 3 mm ATP, 8 mm MgCl2, and 150 mm KCl for 20 min at 60 °C. Crystals of TtDdl in complex with ADP and d-Ala–d-Ala were obtained by incubating TtDdl with 3 mm ADP, 8 mm MgCl2, 40 mm d-Ala, and 150 mm KCl for 20 min at 60 °C. A carbonate anion (CO32−) was modeled to clearly planar electron density at the expected position of the γ-Pi of bound ATP. This has been observed previously in structures of E. coli Ddl (33). The unexpected presence of products in several of these structures suggests that the TtDdl used for crystallography copurified with substrates/products. The incubation step at 60 °C was included as it appeared to promote crystal formation. Crystals were obtained by adding 1 μl of incubated TtDdl to 2 μl of well solution (0.5 ml) containing 15%–18% PEG 3350, 100 mm BisTris (pH 6.8), and 100 mm magnesium formate using the hanging drop vapor diffusion method in 24-well plates (Costar, Corning, NY, USA) stored at 16 °C.
Data collection, structure determination, and refinement
Crystals were transferred to Paratone-N for cryoprotection and flash-frozen in liquid nitrogen. Datasets were collected at the MX1 and MX2 beamlines of the Australian Synchrotron (17). Indexing and integration were completed using XDS (34). Aimless (CCP4) was used for scaling and merging datasets (35). The structures of TtDdl were solved by molecular replacement using Phaser with a previously solved structure of TtDdl (PDB code 2ZDH) as the search model (3, 36). For data collected at 15,250 eV (RbCl, PDB code 6U1E) and 8,500 eV (CsCl, PDB code 6U1D), the corresponding model solved at 13,000 eV was used. Modeled ligands and waters were removed from all search models. Solutions from Phaser were refined in Phenix. These models were subjected to multiple rounds of rebuilding in Coot, followed by B-factor and positional refinement in Phenix until R-factors converged (37, 38). The results, including data processing and refinement statistics, are presented in Table S3. For the final models, more than 95% of modeled residues were favored in the Ramachandran plot. The only outlier, Gly86, fits well in the 2Fo-Fc electron density map. For identification of MVC binding sites, anomalous difference maps were generated to a resolution of 3.5 Å for the TtDdl–ATP–d-Ala–d-Ala-Rb+anom and TtDdl–ATP–d-Ala–d-Ala–Cs+anom datasets using phenix.maps (38). K+ was modeled using a combination of Fo-Fc difference maps, occupancies, B-factors, and bond lengths for validation.
Structure comparison and data analysis
Protein secondary structure was assigned using DSSP (39). Structural superposition was completed using secondary structure matching in Coot (37). Figures for visualizing enzyme structure were generated using PyMOL (40). PDB code 2PQR and APBS were used for electrostatic surface calculations (41). Multiple sequence alignment was performed using Clustal Ω (42).
Data availability
All structures have been deposited into the RCSB Protein Data Bank with accession codes 6U1C, 6U1D, 6U1E, 6U1F, 6U1G, 6U1H, 6U1I, 6U1J, 6U1K. All remaining data are contained within the article.
Supplementary Material
Acknowledgments
This research was undertaken in part using the MX1 and MX2 beamlines at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation detector.
This article contains supporting information.
Author contributions—J. L. P. and A. P. T. data curation; J. L. P., A. P. T., and J. B. B. formal analysis; J. L. P., A. P. T., and S. G. B. validation; J. L. P. and A. P. T. investigation; J. L. P. visualization; J. L. P. and A. P. T. methodology; J. L. P. writing-original draft; J. L. P. and J. B. B. project administration; S. G. B. supervision; S. G. B. and J. B. B. writing-review and editing; J. B. B. conceptualization; J. B. B. resources.
Funding and additional information—J. L. P. is a recipient of an Australian Government Research Training Program stipend scholarship.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- Ddl
- d-alanine–d-alanine ligase
- DCS
- d-cycloserine
- aa
- amino acids
- MVC
- monovalent cation
- DCSP
- phosphorylated form of d-cycloserine
- CPS
- carbamoyl phosphate synthetase
- BC
- biotin carboxylase
- LAL
- l-amino acid ligase
- CR
- color reagent.
References
- 1. Sato M., Kirimura K., and Kino K. (2005) d-amino acid dipeptide production utilizing d-alanine-d-alanine ligases with novel substrate specificity. J. Biosci. Bioeng. 99, 623–628 10.1263/jbb.99.623 [DOI] [PubMed] [Google Scholar]
- 2. Sato M., Kirimura K., and Kino K. (2006) Substrate specificity of thermostable d-alanine-d-alanine ligase from Thermotoga maritima ATCC 43589. Biosci. Biotechnol. Biochem. 70, 2790–2792 10.1271/bbb.60307 [DOI] [PubMed] [Google Scholar]
- 3. Kitamura Y., Ebihara A., Agari Y., Shinkai A., Hirotsu K., and Kuramitsu S. (2009) Structure of d-alanine-d-alanine ligase from Thermus thermophilus HB8: cumulative conformational change and enzyme–ligand interactions. Acta Crystallogr. D 65, 1098–1106 10.1107/S0907444909029710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu S., Chang J. S., Herberg J. T., Horng M.-M., Tomich P. K., Lin A. H., and Marotti K. R. (2006) Allosteric inhibition of Staphylococcus aureus d-alanine: d-alanine ligase revealed by crystallographic studies. Proc. Natl. Acad. Sci. U.S.A. 103, 15178–15183 10.1073/pnas.0604905103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neuhaus F. C. (1962) The enzymatic synthesis of d-alanyl-d-alanine ii. kinetic studies on d-alanyl-d-alanine synthetase. J. Biol. Chem. 237, 3128–3135 [PubMed] [Google Scholar]
- 6. Prosser G. A., and de Carvalho L. P. S. (2013) Kinetic mechanism and inhibition of Mycobacterium tuberculosis d-alanine:d-alanine ligase by the antibiotic d-cycloserine. FEBS J. 280, 1150–1166 10.1111/febs.12108 [DOI] [PubMed] [Google Scholar]
- 7. Zawadzke L. E., Bugg T. D., and Walsh C. T. (1991) Existence of two d-alanine:d-alanine ligases in Escherichia coli: cloning and sequencing of the ddlA gene and purification and characterization of the DdlA and DdlB enzymes. Biochemistry 30, 1673–1682 10.1021/bi00220a033 [DOI] [PubMed] [Google Scholar]
- 8. Fan C., Moews P. C., Walsh C. T., and Knox J. R. (1994) Vancomycin resistance: structure of d-alanine: d-alanine ligase at 2.3 Å resolution. Science 266, 439–443 10.1126/science.7939684 [DOI] [PubMed] [Google Scholar]
- 9. Shi Y., and Walsh C. T. (1995) Active-site mapping of Escherichia coli d-Ala-d-Ala ligase by structure-based mutagenesis. Biochemistry 34, 2768–2776 10.1021/bi00009a005 [DOI] [PubMed] [Google Scholar]
- 10. Lee J. H., Na Y., Song H.-E., Kim D., Park B.-H., Rho S.-H., Im Y. J., Kim M.-K., Kang G. B., Lee D.-S., and Eom S. H. (2006) Crystal structure of the apo form of d-alanine:d-alanine ligase (Ddl) from Thermus caldophilus: a basis for the substrate-induced conformational changes. Proteins 64, 1078–1082 10.1002/prot.20927 [DOI] [PubMed] [Google Scholar]
- 11. Mullins L. S., Zawadzke L. E., Walsh C. T., and Raushel F. M. (1990) Kinetic evidence for the formation of d-alanyl phosphate in the mechanism of d-alanyl-d-alanine ligase. J. Biol. Chem. 265, 8993–8998 [PubMed] [Google Scholar]
- 12. Neuhaus F. C. (1962) The enzymatic synthesis of d-alanyl-d-alanine I: purification and properties of d-alanyl-d-alanine synthetase. J. Biol. Chem. 237, 778–786 [PubMed] [Google Scholar]
- 13. Gohara D. W., and Di Cera E. (2016) Molecular mechanisms of enzyme activation by monovalent cations. J. Biol. Chem. 291, 20840–20848 10.1074/jbc.R116.737833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Di Cera E. (2006) A structural perspective on enzymes activated by monovalent cations. J. Biol. Chem. 281, 1305–1308 10.1074/jbc.R500023200 [DOI] [PubMed] [Google Scholar]
- 15. Shisler K. A., Hutcheson R. U., Horitani M., Duschene K. S., Crain A. V., Byer A. S., Shepard E. M., Rasmussen A., Yang J., Broderick W. E., Vey J. L., Drennan C. L., Hoffman B. M., and Broderick J. B. (2017) Monovalent cation activation of the radical SAM enzyme pyruvate formate-lyase activating enzyme. J. Am. Chem. Soc. 139, 11803–11813 10.1021/jacs.7b04883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Doyle D. A., Morais Cabral J., Pfuetzner R. A., Kuo A., Gulbis J. M., Cohen S. L., Chait B. T., and MacKinnon R. (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77 10.1126/science.280.5360.69 [DOI] [PubMed] [Google Scholar]
- 17. McPhillips T. M., McPhillips S. E., Chiu H.-J., Cohen A. E., Deacon A. M., Ellis P. J., Garman E., Gonzalez A., Sauter N. K., Phizackerley R. P., Soltis S. M., and Kuhn P. (2002) Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 9, 401–406 10.1107/S0909049502015170 [DOI] [PubMed] [Google Scholar]
- 18. Gagné O. C., and Hawthorne F. C. (2016) Bond-length distributions for ions bonded to oxygen: alkali and alkaline-earth metals. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 72, 602–625 10.1107/S2052520616008507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thoden J. B., Raushel F. M., Benning M. M., Rayment I., and Holden H. M. (1999) The structure of carbamoyl phosphate synthetase determined to 2.1 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 55, 8–24 10.1107/S0907444998006234 [DOI] [PubMed] [Google Scholar]
- 20. Chou C. Y., and Tong L. (2011) Structural and biochemical studies on the regulation of biotin carboxylase by substrate inhibition and dimerization. J. Biol. Chem. 286, 24417–24425 10.1074/jbc.M111.220517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cowan J. A. (1998) Metal activation of enzymes in nucleic acid biochemistry. Chem. Rev. 98, 1067–1088 10.1021/cr960436q [DOI] [PubMed] [Google Scholar]
- 22. Healy V. L., Mullins L. S., Li X., Hall S. E., Raushel F. M., and Walsh C. T. (2000) d-Ala–d-X ligases: evaluation of d-alanyl phosphate intermediate by MIX, PIX and rapid quench studies. Chem. Biol. 7, 505–514 10.1016/S1074-5521(00)00135-6 [DOI] [PubMed] [Google Scholar]
- 23. Neuhaus F. C., and Lynch J. L. (1962) Studies on the inhibition of d-alanyl-d-alanine synthetase by the antibiotic d-cycloserine. Biochem. Biophys. Res. Commun. 8, 377–382 10.1016/0006-291X(62)90011-6 [DOI] [PubMed] [Google Scholar]
- 24. Miller J. R., Dunham S., Mochalkin I., Banotai C., Bowman M., Buist S., Dunkle B., Hanna D., Harwood H. J., Huband M. D., Karnovsky A., Kuhn M., Limberakis C., Liu J. Y., Mehrens S., et al. (2009) A class of selective antibacterials derived from a protein kinase inhibitor pharmacophore. Proc. Natl. Acad. Sci. U.S.A. 106, 1737–1742 10.1073/pnas.0811275106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zeczycki T. N., Maurice M. S., and Attwood P. V. (2010) Inhibitors of pyruvate carboxylase. Open Enzym. Inhib. J. 3, 8–26 10.2174/1874940201003010008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dal Piaz F., Vassallo A., Lepore L., Tosco A., Bader A., and De Tommasi N. (2009) Sesterterpenes as tubulin tyrosine ligase inhibitors: first insight of structure-activity relationships and discovery of new lead. J. Med. Chem. 52, 3814–3828 10.1021/jm801637f [DOI] [PubMed] [Google Scholar]
- 27. Firestine S. M., Paritala H., McDonnell J. E., Thoden J. B., and Holden H. M. (2009) Identification of inhibitors of N5-carboxyaminoimidazole ribonucleotide synthetase by high-throughput screening. Bioorg. Med. Chem. 17, 3317–3323 10.1016/j.bmc.2009.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tabata K., Ikeda H., and Hashimoto S. (2005) ywfE in Bacillus subtilis codes for a novel enzyme, l-amino acid ligase. J. Bacteriol. 187, 5195–5202 10.1128/JB.187.15.5195-5202.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kino K., Noguchi A., Nakazawa Y., and Yagasaki M. (2008) A novel l-amino acid ligase from Bacillus licheniformis. J. Biosci. Bioeng. 106, 313–315 10.1263/jbb.106.313 [DOI] [PubMed] [Google Scholar]
- 30. Kino H., Nakajima S., Arai T., and Kino K. (2016) Effective production of Pro–Gly by mutagenesis of l-amino acid ligase. J. Biosci. Bioeng. 122, 155–159 10.1016/j.jbiosc.2016.01.014 [DOI] [PubMed] [Google Scholar]
- 31. Kino K., Kotanaka Y., Arai T., and Yagasaki M. (2009) A novel l-amino acid ligase from Bacillus subtilis NBRC3134, a microorganism producing peptide-antibiotic rhizocticin. Biosci. Biotechnol. Biochem. 73, 901–907 10.1271/bbb.80842 [DOI] [PubMed] [Google Scholar]
- 32. Bartolommei G., and Tadini-Buoninsegni F. (2016) Antimony-phosphomolybdate ATPase assay. Methods Mol. Biol. 1377, 111–120. 10.1007/978-1-4939-3179-8_12 [DOI] [PubMed] [Google Scholar]
- 33. Batson S., de Chiara C., Majce V., Lloyd A. J., Gobec S., Rea D., Fülöp V., Thoroughgood C. W., Simmons K. J., Dowson C. G., Fishwick C. W. G., de Carvalho L. P. S., and Roper D. I. (2017) Inhibition of d-Ala:d-Ala ligase through a phosphorylated form of the antibiotic d-cycloserine. Nat. Commun. 10.1038/s41467-017-02118-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kabsch W. (2010) XDS. Acta Crystallogr. D 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 38. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kabsch W., and Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
- 40. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.1, Schroedinger, LLC, New York [Google Scholar]
- 41. Jurrus E., Engel D., Star K., Monson K., Brandi J., Felberg L. E., Brookes D. H., Wilson L., Chen J., Liles K., Chun M., Li P., Gohara D. W., Dolinsky T., Konecny R., et al. (2018) Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 10.1002/pro.3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Prosser G. A., and de Carvalho L. P. (2013) Metabolomics reveal d-alanine:d-alanine ligase as the target of d-cycloserine in Mycobacterium tuberculosis. ACS Med. Chem. Lett. 4, 1233–1237 10.1021/ml400349n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Meister A. (1985) Glutathione synthetase from rat kidney. Methods Enzymol. 113, 393–399 10.1016/S0076-6879(85)13052-1 [DOI] [PubMed] [Google Scholar]
- 45. Rathbun W. B., Sethna S. S., and Van Buskirk G. (1977) Purification and properties of glutathione synthetase from bovine lens. Exp. Eye Res. 24, 145–158 10.1016/0014-4835(77)90255-X [DOI] [PubMed] [Google Scholar]
- 46. Snoke J. E. (1955) Isolation and properties of yeast glutathione synthetase. J. Biol. Chem. 213, 813–824 [PubMed] [Google Scholar]
- 47. McClure W. R., Lardy H. A., and Kneifel H. P. (1971) Rat liver pyruvate carboxylase I: preparation, properties, and cation specificity. J. Biol. Chem. 246, 3569–3578 [PubMed] [Google Scholar]
- 48. Ruiz-Amil M., De Torrontegui G., Palacián E., Catalina L., and Losada M. (1965) Properties and function of yeast pyruvate carboxylase. J. Biol. Chem. 240, 3485–3492 [PubMed] [Google Scholar]
- 49. Warren G. B., and Tipton K. F. (1974) Pig liver pyruvate carboxylase: purification, properties and cation specificity. Biochem. J. 139, 297–310 10.1042/bj1390297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Edwards J. B., and Keech D. B. (1968) Activation of pig heart propionyl-CoA carboxylase by potassium ions. Biochim. Biophys. Acta 159, 167–175 10.1016/0005-2744(68)90255-6 [DOI] [PubMed] [Google Scholar]
- 51. Giorgio A. J., and Plaut G. W. (1967) The effect of univalent cations on activities catalyzed bovine-liver propionyl-CoA carboxylase. Biochim. Biophys. Acta 139, 487–501 10.1016/0005-2744(67)90052-6 [DOI] [PubMed] [Google Scholar]
- 52. Nielsen N. C., and Stumpf P. K. (1976) Activation of wheat germ acetyl CoA carboxylase by potassium and rubidium. Biochem. Biophys. Res. Commun. 68, 205–210 10.1016/0006-291X(76)90030-9 [DOI] [PubMed] [Google Scholar]
- 53. Roon R. J., and Levenberg B. (1972) Urea amidolyase I: properties of the enzyme from Candida utilix. J. Biol. Chem. 247, 4107–4113 [PubMed] [Google Scholar]
- 54. Argaraña C. E., Barra H. S., and Caputto R. (1978) Release of [14C]tyrosine from tubulinyl-[14C]tyrosine by brain extract: separation of a carboxypeptidase from tubulin-tyrosine ligase. Mol. Cell. Biochem. 19, 17–21 [DOI] [PubMed] [Google Scholar]
- 55. Sidey V. (2016) On the effective ionic radii for ammonium. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 72, 626–633 10.1107/S2052520616008064 [DOI] [PubMed] [Google Scholar]
- 56. Evans H. J., and Wood H. G. (1971) Purification and properties of pyruvate phosphate dikinase from propionic acid bacteria. Biochemistry 10, 721–729 10.1021/bi00781a001 [DOI] [PubMed] [Google Scholar]
- 57. Shashi K., Bachhawat A. K., and Joseph R. (1990) ATP: citrate lyase of Rhodotorula gracilis: purification and properties. Biochim. Biophys. Acta 1033, 23–30 10.1016/0304-4165(90)90189-4 [DOI] [PubMed] [Google Scholar]
- 58. Shannon R. D. (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 32, 751–767 10.1107/S0567739476001551 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All structures have been deposited into the RCSB Protein Data Bank with accession codes 6U1C, 6U1D, 6U1E, 6U1F, 6U1G, 6U1H, 6U1I, 6U1J, 6U1K. All remaining data are contained within the article.