

Abstract
There is a growing body of clinical and experimental evidence that neurodegenerative diseases and epileptogenesis after an acquired brain insult may share common etiological mechanisms. Acquired epilepsy commonly develops as a comorbid condition in patients with neurodegenerative diseases such as Alzheimer's disease, although it is likely much under diagnosed in practice. Progressive neurodegeneration has also been described after traumatic brain injury, stroke, and other forms of brain insults. Moreover, recent evidence has shown that acquired epilepsy is often a progressive disorder that is associated with the development of drug resistance, cognitive decline, and worsening of other neuropsychiatric comorbidities. Therefore, new pharmacological therapies that target neurobiological pathways that underpin neurodegenerative diseases have potential to have both an anti‐epileptogenic and disease‐modifying effect on the seizures in patients with acquired epilepsy, and also mitigate the progressive neurocognitive and neuropsychiatric comorbidities. Here, we review the neurodegenerative pathways that are plausible targets for the development of novel therapies that could prevent the development or modify the progression of acquired epilepsy, and the supporting published experimental and clinical evidence.
Keywords: AMPA, amyloid‐β, glutamate, mTOR, neuroinflammation, tau
Key points.
Neurodegenerative diseases and epileptogenesis after an acquired brain insult may share common etiological mechanisms
Targeting neurodegenerative pathways have the potential to have both anti‐epileptogenic and disease‐modifying effects in acquired epilepsy
Modification of tau, amyloid‐β, neuroinflammation, mTOR, and AMPA pathways are plausible targets for the development of epilepsy therapies
1. INTRODUCTION
Epilepsy is one of the most common and disabling neurological disorders worldwide. The etiologies of acquired epilepsy are diverse, but a causative epileptogenic brain injury, such as stroke, status epilepticus, traumatic brain injury (TBI), or infection, can be identified in a proportion of patients.1 There is increasing evidence that acquired epilepsy can be a progressive disorder, associated with cognitive decline and worsening of other neuropsychiatric comorbidities and the development of pharmacoresistance.2, 3, 4, 5, 6, 7 Clinical and experimental evidence has shown an association of epilepsy with different neurodegenerative pathways such as tau, amyloid‐β‐related, the mammalian target of rapamycin (mTOR).8
Neurodegeneration is a broad term defined as the progressive alterations of neuronal function, which often involves neuronal death, and has been described in a wide variety of brain conditions such as stroke, traumatic brain injury, multiple sclerosis, Alzheimer disease, amyotrophic lateral sclerosis, Parkinson's disease, Huntington disease, and acquired epilepsy.9, 10, 11, 12 Observations in experimental models of acquired epilepsy in animals and in vitro are providing a better understanding of different neurodegenerative pathways that may contribute to excitotoxicity, cell death, neurogenesis, and axonal sprouting, which could provide possible pharmacological targets for the development of anti‐epileptogenic or disease‐modifying therapies (Figure 1). A clear etiological link between the neurodegeneration with the development of epileptic seizures has not yet been proven, and it remains possible that the neurodegeneration observed in patients and animal models with acquired epilepsy is an incidental result of the injury or a secondary effect of the repeated epileptic seizures.12, 13, 14 Nevertheless, studies targeting neurodegenerative mechanisms have reported protective effects against epileptogenesis following an acquired brain insult and thus provide evidence linking these and promise for the future development of this approach clinically.15, 16, 17, 18, 19, 20, 21, 22, 23
Figure 1.
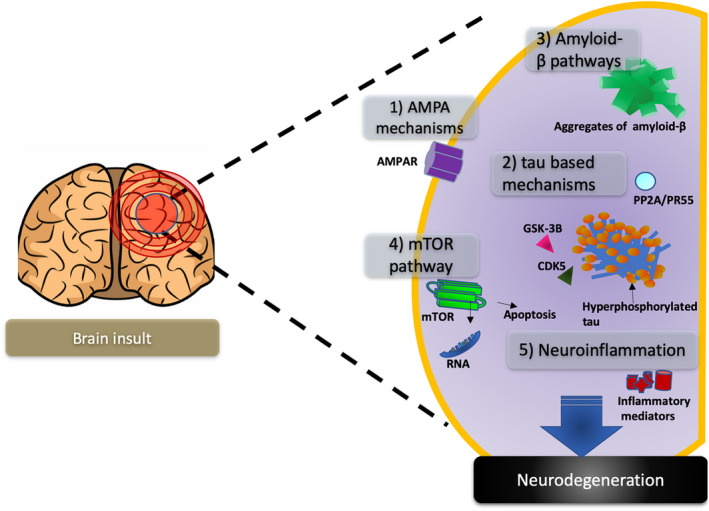
Neurodegenerative pathways in acquired epilepsy. A brain insult triggers a cascade of mechanisms that may be involved in the development of acquired epilepsy, and five neurodegenerative pathways implicated in the development of acquired epilepsy, (1) AMPA mechanisms, (2) tau‐based mechanisms, (3) amyloid‐β pathways, (4) mTOR pathway, and (5) neuroinflammatory mediators, are reviewed in this manuscript as they represent potential targets for drug development
This review focuses on some of the neurodegenerative mediators and pathways, such as AMPA receptors, tau, amyloid, mTOR, and neuroinflammation that represent potential targets to prevent or modify acquired epilepsy, and the published experimental literature supporting this approach.
2. NEURODEGENERATIVE MECHANISMS RELEVANT TO ACQUIRED EPILEPSIES
The acquired epilepsies comprise a heterogeneous group in which a structural abnormality or metabolic condition secondary to a brain injury has been attributed to play a major role in the risk of developing epilepsy.24 TLE is the most common form of acquired epilepsy that is often resistant to drug treatment, where seizures continue to occur despite anti‐epileptic drug treatment.25 Despite decades of study of TLE, and more than 15 new anti‐epileptic drugs that have been introduced into clinical practice, at least 30% of the patients are resistant to medical treatment.25, 26 A variety of different brain insults can be the trigger of the acquired epileptogenic, such as status epilepticus (SE), febrile seizures, TBI, infection, prenatal or perinatal injuries, congenital abnormalities, brain tumors, autoimmune, or genetic disorders associated with brain malformations, with the chance of the development of epilepsy likely enhanced by genetic determinants.1, 26, 27, 28, 29, 30, 31 Epileptogenesis is a cascade of molecular, functional, and structural processes that are triggered by a brain insult and are capable of generating spontaneous seizures. During epileptogenesis, the limbic structures manifest a variety of neurodegenerative changes that may contribute to the development of acquired epilepsy. The initial insult is often followed by a latent period that comprises a cascade of molecular, morphological, functional, and structural changes.32 This latent period is variable from months to years in humans 33 and continues to create a hyperexcitable network prone to develop spontaneous seizures.4, 34, 35, 36, 37, 38, 39, 40, 41, 42
Moreover, acquired epilepsy, specifically affecting the temporal lobe, is associated with an increased incidence of neuropsychiatric disturbances, including anxiety, depression, memory, and learning disabilities.43, 44, 45, 46, 47 These associated neuropsychiatric comorbidities often worsen over time, resembling in some cases a neurodegenerative condition.12 The underlying pathogenic mechanisms may relate to the progressive nature of epilepsy and its impact on the function of the different brain regions involved in cognition and the cumulative effects of therapies and epigenetic factors. Learning, cognition, verbal, and long‐term memory are often affected in acquired epilepsy, since the most common focus is located in the limbic system.48, 49, 50, 51 The duration of epilepsy has been correlated with the degree of hippocampal sclerosis, cortical atrophy, and reduced psychometric intelligence in some studies,48, 49, 50, 51 but not others.52, 53
During epileptogenesis, a wide spectrum of potentially pro‐epileptogenic neurodegenerative changes is seen in limbic structures including mossy fiber sprouting4, 34, 35, 36, 37, 38, 39, 40, 41, 42; neuronal reorganization‐synaptic remodeling54, 55, 56, 57; neurogenesis58; blood‐brain barrier disruption, γ‐aminobutyric acid (GABA) receptor, and GABAergic neurons changes59, 60, 61, 62, 63; alterations in peptide and brain‐derived neurotrophic factor (BDNF) expression36, 64, 65; neuroinflammation23, 66; changes in ion channels67; alterations in axonal transport, amyloid‐β peptide, tau, and PP2A pathology15, 68; and other cellular and functional changes.54, 56, 57, 69, 70, 71, 72, 73 These neuropathological changes are not specific of epilepsy; however, they are similar to those of neurodegenerative disorders such as Alzheimer's disease,12, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86 even when there is no clear history of epilepsy.12 Neurodegeneration is a progressive process that evolves during acquired epileptogenesis87; however, some studies suggest that the neurodegeneration may not directly result in the epileptogenesis, but it may induce other processes that do.88, 89 Hippocampal sclerosis is the pathological landmark of chronic drug‐resistant mesial TLE and is characterized by neuronal loss in the hippocampus, reactive gliosis, and reorganization of the synaptic connections. However, not all with TLE have hippocampal sclerosis.90, 91, 92 Neuronal cell loss is commonly seen in patients with acquired epilepsy and in different animal models, including the post‐traumatic and post‐status epilepticus models.15, 22 Neuronal death causes a cascade of changes that includes massive release of intracellular Ca2+, oxidative stress, and activation of apoptotic pathways such as caspase, P53 and Bcl,70, 71, 75, 78, 84 and others.93, 94, 95, 96, 97, 98, 99, 100, 101, 102 In response to this, there is an activation and increased presence of astrocytes and microglia, a process known as gliosis. Glial cells and microglia release pro‐inflammatory cytokines, such as interleukin 1ß (IL‐1ß) and tumor necrosis factor α (TNF‐α), which promote gliosis103, 104 and perpetuate a chronic inflammatory state that further influences the hyperexcitability and promotes aberrant neurogenesis and tissue remodeling, which further enhance epileptogenesis.103, 105, 106
Mossy fiber sprouting is another pathological landmark seen in the hippocampal formation of acquired chronic epilepsy patients, in particular those with mesial TLE, and in animal models that represents aberrant synaptic remodeling and hyperexcitable neuronal network formation.105, 107, 108, 109 The increase in interictal epileptiform spike frequency in the chronic epileptic phase in animal models of TLE correlates with the development of spontaneous seizures, neuronal loss, and mossy fiber sprouting.91, 110 The imbalance between inhibitory and excitatory mechanisms plays a role in the development of epilepsy, but also in the initiation and maintenance of spontaneous seizures.71, 72 N‐methyl‐D‐aspartate (NMDA) receptor activation might play a role for inducing the trans‐synaptic alterations that underlie TLE epileptogenesis.33 In fact, repeated seizures may lead to loss of GABAergic inhibitory interneurons in the hippocampus.111, 112, 113 In addition, regulation of inhibitory and excitatory receptors can be influenced by neuropeptides such as brain BDNF.64, 114, 115 However, the mechanisms of GABA, NMDA, BDNF, and other neuropeptide‐related neurodegeneration are out of the scope of this review and will not be discussed in the current manuscript.
This review will focus on the discussion of the AMPA, tau, amyloid‐β, mTOR, and neuroinflammatory pathways as potential targets for drug development.
3. AMPA RECEPTOR NEURODEGENERATIVE MECHANISMS
The majority of fast excitatory synaptic neurotransmission in the central nervous system is mediated via glutamate activation of α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazole propionic acid (AMPA) receptors.115, 116 This neurotransmitter system not only drives abnormal hyperexcitable circuitry during an epileptic seizure but also could initiate neurodegenerative processes by excessive calcium uptake and pushing the cells toward apoptotic cell death.117
The AMPARs primarily mediate fast neurotransmission by serving as a glutamate‐gated cation channel. In addition to initiating neuronal firing, AMPARs also underlie aspects of synaptic plasticity116 such as long‐term potentiation, learning, and memory.118 Glutamate chronic neuronal excitotoxicity is a newer concept, but has linked glutamate excitotoxicity to neurodegenerative processes in Huntington's disease, Parkinson's disease, and Alzheimer's dementia.9
Similarly, excessive glutamate receptor activation has been linked epilepsy.119 In acquired epilepsy, particularly in TLE, increases in glutamate excitotoxicity have been described as an important initial mechanism for neuronal injury that leads to neuronal cell death and neurodegenerative processes that may lead to overall hyperexcitable tissue reorganization by mechanisms described earlier.54, 73 These events could in turn promote increased burst firing in reticular neurons enhancing epileptogenic circuit synchrony, promoting the development of seizures.120, 121, 122, 123, 124
The hippocampus has been identified as the seizure initiating zone in many TLE patients as well as in different animal models.125 GABAergic neurons are found primarily in the basket cells located in deep portions of the granule cell layer in the dentate gyrus.26, 126 However, repeated seizures and excitotoxicity lead to death of these interneurons that are critical to maintain the balance between excitation and inhibition in the hippocampus.105, 113, 127
Glutamate is the major excitatory neurotransmitter in the hippocampal formation.73 The mossy fiber pathway runs from the dentate gyrus granule cells to the pyramidal cells of the CA3; after excitotoxicity and neuronal cell death due to the initial brain insult or to repeated seizures, the mossy fibers are reorganized and sprout into the inner molecular layers of the dentate gyrus to form aberrant synaptic terminals with dendrites of GABAergic interneuron basket cells and with granule cells.113 This abnormal reorganization renders the hippocampus hyperexcitable and prone to the development of spontaneous seizures.107
Therefore, strategies that can inhibit the AMPA receptor activity have the potential to reduce excessive excitatory responses that could to neurodegenerative changes and may be promising targets for the development of anti‐epileptogenic and disease‐modifying drugs.128, 129
Perampanel is a non‐competitive and highly selective AMPA receptor antagonist that has recently completed phase III of clinical trials and has been approved as an adjunctive treatment for drug‐resistant partial‐onset seizures.130, 131, 132, 133 Perampanel also decreases intracellular Ca2+ concentration induced by AMPA receptor activation that would have the net effect of decreasing excitability.134, 135 Perampanel has shown to reduce neuronal cell death in the hippocampus and the piriform cortex in the lithium‐pilocarpine post‐SE model,136 but does not have anti‐epileptogenic properties.137 Similar neuroprotective effects have been described with other AMPA antagonists.138
4. TAU‐BASED MECHANISMS IN EPILEPSY
There is building clinical and experimental evidence linking tau‐based neurodegenerative mechanisms with the epilepsy development,15, 68 suggesting for a neurodegenerative basis for acquired epilepsies. Tau, a microtubule‐associated protein, performs important physiological functions in neurons, including providing stabilization to microtubules as well as contributing to axonal transport. The binding of tau to microtubules and thereby its physiological functions are regulated by a balance in phosphorylated and non‐phosphorylated forms of tau (as reviewed by Zheng et al139 for details on tauopathies). A partially phosphorylated tau is needed for the physiological functioning, whereas a hyperphosphorylated tau can aggregate and lead to impairment of normal functions and cessation of cell survival mechanisms and contribute to neurodegeneration.140, 141
Abnormalities in the expression and phosphorylation of tau have also been described in stroke,142 epilepsy, Alzheimer's disease, frontotemporal dementia, and chronic traumatic encephalopathies.143 In clinical situations, pathological deposition of hyperphosphorylated tau has been observed in brain samples obtained from surgical resection of epileptic tissue from patients with drug‐resistant chronic TLE.144, 145, 146, 147 Similarly, brain tissue from other drug‐resistant epileptic conditions such as focal cortical dysplasia also displays hyperphosphorylated tau.148 Moreover, tau‐based pathologies are also reported acutely after traumatic brain injury, a major component for secondary injury, and is associated with neurological symptoms and cognitive decline,68, 145 as also reported in animal models of TBI.68, 149 Elevated levels of tau and hyperphosphorylated tau have also been observed in cerebrospinal fluid from epilepsy patients with partial and convulsive seizures, as well as following status epilepticus.150, 151, 152 Such changes were determinants of poor prognosis and higher risk to develop epilepsy.150 These findings in human patients suggest a possible role in targeting tau pathologies as disease‐modifying treatment, but also provide a possible biomarker for predicting epilepsy development after a brain insult that should be further investigated and validated.
The involvement of tau‐based mechanisms in epilepsy has been further supported by neuropathological findings from both genetic153, 154 and acquired models of epilepsy.15 Furthermore, there is evidence that genetic155, 156 or pharmacological15, 157, 158 manipulations of tau phosphorylation alter seizure induction or epilepsy development after an epileptogenic brain insult. In addition to the impact of hyperphosphorylated tau, total tau has been implicated in altering excitation/inhibition balance,159 Roberson et al, who showed that mice with tau genetic knockout display reduced seizure severity and latency to chemoconvulsant‐induced seizures.159
There is also building evidence that tau‐based mechanisms enhance neuronal excitability. An unstable microtubule assembly at axonal segments dysregulates the resting membrane potential and thereby generations of action potentials.160 Accordingly, not only pharmacological regulation or genetic manipulation of tau phosphorylation but also inhibiting total tau has shown protection against induced seizures.156, 157, 161, 162 Interestingly, tau knockout mice that underwent experimental stroke using a middle cerebral artery occlusion, developed less pentylenetetrazol evoked seizures, were protected from excitotoxic brain damage neurological deficits following stroke by site‐specific inhibition of glutamate‐induced and Ras/ERK‐mediated toxicity.162 Contrary to this, recent work has also suggested a role of tau phosphorylation in diminishing neuronal activity and attenuated seizure activity to 4‐aminopyridine induction.163, 164
These findings support a role for tauopathies in neuronal excitability, which may be dependent on the stage of the disease and may determine a compensatory mechanism to combat disease progression. Nevertheless, tau phosphorylation is evidenced to affect neuronal excitability and may alter excitation/inhibition balance providing conditions for epileptic seizures to occur. Whether these effects are relevant during the early phases of epileptogenesis after an epileptogenic insult is an important question. The activity of protein phosphatase 2A (PP2A), the main dephosphorylating enzyme, is increased, and glycogen synthase kinase‐3B (GSK‐3B) and cyclin‐dependent kinase 5 (CDK5), the primary phosphorylating enzymes, decreased, within few hours after the kainic acid‐induced status epilepticus165 as well as following a traumatic brain injury.68 This tips the balance between tau phosphorylation and dephosphorylation in favor of the former, resulting in an accumulation in the brain of hyperphosphorylated tau. This is prevented by the treatment of rats following a variety of epileptogenic brain insults with the oxidized selenium salt and sodium selenate, which specifically increases the activity and expression of the specific PP2A subunit, PR55, that is responsible for dephosphorylating hyperphosphorylated tau.15, 166, 167 Although the exact mechanism of how increased phosphorylation induces epileptogenesis is yet to be identified, it provides for an exciting target for the development of disease‐modifying therapies in established epilepsies as well as anti‐epileptogenesis treatment for inhibiting epilepsy after an epileptogenic insult.15, 21, 167
In the amygdala kindling, post‐SE and post‐TBI rat models of acquired epilepsy, treatment with sodium selenate inhibited the development of limbic epileptogenesis as well as cognitive and sensorimotor impairments.68 Moreover, sodium selenate treatment given acutely after the epileptogenic insult in these models prevented the decrease in PP2A activity and PR55 levels seen following these brain insults, as well as reducing the accumulation of hyperphosphorylated tau, mitigating neurodegenerative changes in the brain, and reducing the number of spontaneous seizures in these animal models.15, 68 Sodium selenate is currently being evaluated in human clinical trials for adult patients with prostate cancer and Alzheimer's disease.168, 169 Given all this pre‐clinical evidence, it seems plausible that targeting the tau neurodegenerative pathways with sodium selenate could be an anti‐epileptogenic therapy in human TLE.
5. AMYLOID‐β PATHWAY IN ACQUIRED EPILEPSY
The deleterious effects of overexpression and reduced clearance of the amyloid precursor protein (APP) and its proteolytic product, the amyloid‐β peptide, in Alzheimer's disease have been widely documented in the literature.170, 171, 172 Amyloid‐β can be found in different aggregates forms in the brain, as soluble monomers, oligomers, or protofibrils before aggregating into insoluble fibrils.173, 174 The amyloid‐β plaques are usually formed outside the cell and exert neurodegenerative effects influence by different pathological mechanisms.171 Patients with Alzheimer's disease have an 8‐ to 10‐fold risk of developing spontaneous seizures than the general population.175, 176, 177, 178, 179 Recently, attention has shifted to the importance of these two proteins and the mechanisms in the development of epilepsy.180
Dysregulation of APP after brain injury has been demonstrated in a number of human and animal studies; however, the effects of this APP regulation are still under debate.8
Numerous lines of evidence in both humans and in pre‐clinical transgenic models that express APP have demonstrated that an epileptogenic brain injury triggers overexpression of APP.8, 180, 181, 182, 183, 184, 185 An increase in APP immunoreactivity has been shown in neurons and astrocytes in the weight drop model of TBI.186, 187 APP protein expression is increased post‐fluid percussion injury in the cortex and hippocampus acutely 1‐h injury.184, 185 Similar results have been reported in the kainic acid‐induced post‐SE model of acquired epilepsy.188 Similarly, in patients APP can readily be detected within hours up to 2.5 years after TBI.181, 182, 183
Evidence shows that increased amyloid production and deposition contribute to the development of acquired epilepsy,189 and Tg2576 mice that overexpress human APP has been shown to be susceptible to the development of seizures in the amygdala kindling model of acquired epilepsy.190 Expression levels of APP and amyloid‐β protein significantly increased in cortex and the hippocampus of the patients with temporal lobe epilepsy refractory to medical treatment.191 Furthermore, amyloid‐β deposits start to accumulate around 10 years before the onset of clinical signs and symptoms of dementia.192 Amyloid‐β deposits have shown to increase neuronal excitability and induce hippocampal network reorganization and hyperactivation processes that have been described as epileptogenic.193, 194 Interestingly, in Alzheimer's patients, hippocampal hyperactivation only occurs in the initial stages of the disease. However, epileptiform activity and seizures can occur throughout the whole course of disease.192, 193, 195, 196
Consistently with the aforementioned evidence, different mice models that overexpress APP also show hyperexcitation in individual neurons, interictal spikes, and spontaneous seizures in cortical and hippocampal networks.83, 197, 198, 199, 200, 201, 202, 203, 204, 205, 206, 207 One of them, the APdE9 transgenic mouse model, generated by crossing transgenic mice expressing the APP human protein and the human PS1‐dE9 (deletion of exon 9),208 has been reported to have spontaneous seizures and increased neuronal excitability.204, 205, 206, 207 Similarly, another study analyzed four different lines of transgenic mice expressing familial mutant or wild‐type human APP and reported aberrant synchronous activity in cortical and hippocampal networks, as well as spontaneous seizures in the mutant mice.63 Moreover, these transgenic mice were more susceptible to develop seizures after pentylenetetrazol, pilocarpine, or kainic acid administration.63 It has been hypothesized that this aberrant excitatory neuronal activity induced by human APP and increased Amyloid‐β production could trigger compensatory inhibitory mechanisms constraining the capacity for synaptic plasticity and contributing to network dysfunction.63, 205 Other mechanisms of epileptogenicity described for amyloid‐β deposits have been related to the excessive dopamine release and activation of the dopamine 1 receptor.209, 210, 211 Excessive activation of dopamine 1 receptors disrupts the GABAergic inhibitory input by reducing GABA release from fast‐spiking interneurons.210 This leads to an excitatory/inhibitory imbalance and consequently hyperexcitability of pyramidal cells. The hyperexcitability of pyramidal cells further increases the amyloid‐β deposits creating a vicious cycle.212, 213 Furthermore, amyloid‐ß accumulation induces microglia activation and release of pro‐inflammatory mediators, which can promote the development of seizures and epilepsy.8
On the other hand, some authors have hypothesized that injured neuronal cells upregulate APP production as an attempt to repair the damage caused by injury.8, 214, 215, 216 Severe experimental TBI has shown to increase the expression of genes encoding proteins involved in amyloid‐β clearance and reduced amyloid‐β plaques in the mouse APP/PS1 model, which overexpresses APP.217 Similarly, in the PDAPP mice model, which exhibits high human APP expression, amyloid‐β deposits were reduced even 8 months after TBI, while sham animals displayed increasing amyloid burden.218, 219 Interestingly, mice lacking APP (APP‐KO) showed increased vulnerability to mild TBI compared with the wild‐type controls (WT) after a mild CCI.220 In addition, intracerebroventricular administration of recombinant secreted fragment APPα (sAPPα) in APP‐KO mice reduced the functional deficits observed after moderate TBI.221 This neuroprotective aspect of APP upregulation is based on the hypothesis that the sAPPα may be neuroprotective and the secreted fragment APP ß (sAPPß) is not.222, 223 However, the exact mechanism by which APP and amyloid‐β can be epileptogenic needs to be elucidated.
Nevertheless, the current evidence in the pre‐clinical models and in patients suggests that the modulation of APP and amyloid‐β production, the prevention of amyloid‐β aggregation, or promotion of its clearance could be potential therapeutic targets to prevent the development of epilepsy.224 The modulation of APP secretases, proteins that cleaves and prevents APP aggregation, has been explored as a therapeutic approach with conflicting evidence. Loane and colleagues show that pharmacological inhibition of γ‐secretase decreased APP and amyloid‐β production and reduced neurodegeneration and improved motor and cognitive recovery after controlled cortical impact‐induced TBI.225 In contrast, γ‐secretase inhibition failed to hypersynchronous oscillatory activity and spontaneous seizures in a APP transgenic mice model of Alzheimer's disease.212 Intracerebroventricular administration of APP96‐110, a peptide that interacts with the D1 heparin binding site, following controlled cortical impact in mice showed promising neuroprotective effects.226, 227 Furthermore, APP96‐110 administration in Sprague Dawley rats after a diffuse TBI improved cognitive outcomes and reduced axonal injury.227
Reduction of inhibition of the c‐Jun N‐terminal kinase (JNK) pathway is another potential therapeutic strategy to prevent the development of epilepsy.228 Aberrant activation of JNK intracellular signaling cascade has been reported in Alzheimer's disease patients and in mouse models, suggesting that it might be involved in a number of neurodegenerative mechanisms associated with the disease.228, 229, 230 SP600125, a specific JNK inhibitor, has been described to reduce APP expression levels and amyloid‐ß production, inhibition of inflammatory responses, and apoptotic neurodegeneration among other pathological features of after TBI. Remarkably, treatment with SP600125 also seems to redirect APP processing from the amyloidogenic to the non‐amyloidogenic pathway, without affecting amyloid‐ß clearance but suppressing its production.228
Valproic acid, one of the most commonly prescribed antiseizure drugs, has shown to affect the production of amyloid231 and to reduced epileptiform activity in mice models that overproduce amyloid‐ß; however, the effects are not sustained after treatment discontinuation.232 Similar effects have been shown with lamotrigine233 and bexarotene.234 Huperzine A, an acetylcholinesterase inhibitor, has shown promising anticonvulsant,235 reducing amyloid accumulations and synaptic deficits.236, 237 Levetiracetam reduces abnormal spike‐wave activity, and in chronic use (12 days), reverses hippocampal remodeling and cognitive deficits in mice model that overexpresses human APP.238
Although these approaches have been widely explored in the TBI and Alzheimer's disease research field, the findings can promote insights and bolster the rationale for developing an APP and amyloid target therapy as disease‐modifying or anti‐epileptogenic therapy in acquired epilepsy.8
6. MECHANISTIC TARGET OF MAMMALIAN RAPAMYCIN PATHWAY
The mammalian target of rapamycin (mTOR) is another pathway that can contribute to neurodegeneration in epilepsy as well established to be involved in neurodegenerative conditions such as Alzheimer's and Parkinsonism.239 mTOR is an intracellular signaling protein that belongs to the phosphatidylinositol 3‐kinase (PI3K)‐related kinase family, whose activity is mediated through a serine‐threonine protein kinase and has been recognized as one of the pivotal cellular signal pathway to control cell survival and proliferation.240 mTOR exists in two multiprotein complexes, namely mTORC1 that is rapamycin‐sensitive and mTORC2 that is rapamycin insensitive. mTOR signaling is regulated by signals from growth factors and nutrients that bind to receptors at the membrane to activate intracellular signaling mechanisms such as PI3K, and generates PIP3 to activate mTOR, which induces protein kinase B (Akt) signaling. This inhibits negative regulators of mTOR such as tuberous sclerosis complex (TSC1/2) and induces mTOR signaling to lead protein translation of key proteins involved in synaptic plasticity, learning, and memory apart from the proteins involved in cell growth mechanisms. In the brain, mTORC1 regulates neuronal excitability, memory formation, and learning, and on the other hand, mTORC2 is found to be involved in cytoskeletal integrity and cell migration.241 For a detailed review of mTOR signaling, readers are referred to Perluigi et al239
With regard to the role of mTOR pathway in neurodegeneration, a major focus of research attention has been mechanisms related to autophagy,242, 243 a self‐consuming mechanism that plays a key role in cell survival by removing toxic proteins and defunct organelles.244 Similarly, mitochondria‐mediated mechanisms of inducing apoptosis are also associated with this pathway.245 Accumulation or aggregation of pathological proteins is a common mechanism among neurodegenerative conditions and is negatively regulated by autophagy.246 mTOR pathway–mediated regulation of autophagy has been increasingly investigated and rapamycin led inhibition of this pathway has exhibited strong effects in inhibiting aggregation of the pathological misfolded proteins.247, 248 Indeed, rapamycin‐mediated inhibition of mTOR against neurodegeneration has been described to be neuroprotective in Parkinson's, Alzheimer's, and Huntington diseases.248, 249, 250
Considering neurodegenerative pathology in epilepsy, it is indeed relevant that mTOR pathway is involved in epileptogenesis and seizure‐inducing mechanisms.241 Abnormal mTOR activation has been reported in both genetic and acquired epilepsies. Mutations and genetic polymorphisms of TSC1 or TSC2 proteins that are intrinsic inhibitors of mTOR pathway leading to its over‐activation have been associated with the development of epilepsy in humans.251, 252 Similar outcomes have also been reported in transgenic TSC1 and 2 mouse models, with rapamycin showing antiseizure and anti‐epileptogenesis effects.253, 254, 255 Accordingly, other intracellular signaling mechanisms of mTOR such as phosphatase and tensin homolog deleted on chromosome ten (PTEN), Akt, and DEPDC5 have all been associated with the development of epilepsy.256, 257, 258, 259 Similarly, dysregulation of the mTOR signaling has also been observed in acquired epilepsies, including increased expression of phosphor‐mTOR patients with mesial temporal lobe epilepsy.260, 261 Further, the mTOR pathway is reported to be dysregulated after experimental epileptogenic insults in animal models, including SE and traumatic brain injury.262, 263 Inhibiting mTOR signaling by rapamycin has been reported to provide anti‐epileptogenesis effects in animal models following an epileptogenic insult,264, 265 as well as in animal models following the establishment of pharmacoresistant epileptic seizures.261
While considering the mechanisms by which mTOR pathway contributes to epilepsy, it is important to recognize the myriad of the cellular process involved and how a dysregulation of those could affect neuronal excitability and contribute to neuronal circuit reorganization to promote epileptic seizures. Apart from neurodegeneration, mTOR pathways are involved in neurogenesis, newborn cell survival and migration, axonal sprouting, neuronal plasticity, and altered expression of ion channels and receptors, all of these mechanisms have been considered to be characteristic of epileptogenesis and may contribute to an excitable brain network.266
Rapamycin inhibits mTORC1 and has been shown to have antiseizure effects.17, 267 Treatment with rapamycin during or after the initial epileptogenic injury has shown to reduce the percentage of mice that developed post‐traumatic epilepsy and the frequency of spontaneous seizures, as well as neuronal degeneration.268, 269 Similar results have also been shown in the post‐SE18, 20 and neonatal hypoxia models of acquired epilepsy.19, 270 However, these results have not been replicated in the amygdala kindling271 and in the pilocarpine‐induced post‐SE mice model of TLE16 where rapamycin treatment was unable to reduce the occurrence of spontaneous seizures. Similarly, rapamycin did not persistently prevent mossy fiber sprouting and was unable to reduce granule cell proliferation, hilar neuron loss, or generation of ectopic granule cells.16, 17, 19, 268, 272
7. NEUROINFLAMMATION
Neuroinflammation plays a substantial role in promoting neurodegenerative changes in acquired epilepsies.273, 274 On the other hand, neuroinflammation could be promoted by cells undergoing death by the release of damage‐associated molecular patterns (DAMPs) such as high‐mobility group box 1 proteins, purine metabolites, or proteins released from damaged extracellular matrix due to the dying of neurons.275 These events lead to a cycle of pathways that lead to enhanced neuroinflammation and promoting further cell loss. Such events could contribute both to the pathological and functional outcomes of an epileptogenic insult. A detailed discussion of the inflammatory molecules involved in human and experimental acquired epilepsies and how such molecules may be involved in promoting seizures and epileptogenesis is discussed in another review in this special issue.276 Pharmacological target of neuroinflammation, particularly modulating interleukins, cyclooxygenase‐2, prostanoid pathways, and several chemokines are promising anti‐epileptogenic and disease‐modifying targets.277, 278 Of particular interest has been the involvement of interleukin‐1β (IL‐1β) in the pathogenesis of acquired epilepsies, with consistent reports of either its receptor blockers or inhibitors of biosynthesis providing neuroprotective, anticonvulsant, or anti‐epileptic effects as well as disease‐modifying effects in epilepsy models.279, 280, 281, 282, 283 Interestingly, an increased inflammation has also been reported in models of genetic epilepsies,284, 285 with inhibition of IL‐1β synthesis providing a seizure‐suppressant effect.285 Furthermore, blockage of purinergic receptor P2X7 receptor involved in the release of pro‐inflammatory cytokines has been reported to reduce epileptic seizures or overall frequency of spontaneous recurrent seizures.286, 287
High‐mobility group box 1 protein (HMGB‐1) is another molecule that has been increasingly investigated recently, in terms of anti‐epileptic effects as well as promising findings of it providing a potential biomarker of epilepsy.288 HMGB‐1 is a DAMP and is secreted by neurons and glial cells following the formation of inflammasome and leads to the release of pro‐inflammatory cytokines after activation of its receptors—Toll‐like receptor 4 (TLR‐4) and receptor for advanced glycation end products (RAGE).275 Along with the potential of HMGB‐1 in suppressing seizures289, 290 along with reported disease‐modifying effects,291 the pathologic disulfide isoforms of HMGB‐1 have been reported to be a prospective biomarker for experimental acquired epilepsies292 as well as in human childhood and adult epilepsies.288, 292 Notably, it has also been associated with a decline in the cognitive functions,293 a major neuropsychiatric comorbidity associated with acquired epilepsies.
In addition to the cytokines, chemokines are other molecules contributing to the inflammatory response by mobilizing the immune cells to the site of injury. Chemokines and its receptor systems such as fractalkine/CX3CR1 and CCL2/CCR5 systems have been reported to be involved in neurodegenerative mechanisms following an epileptogenic insult.273, 294, 295, 296 Despite reports of inhibiting the signaling of these chemokines displaying neuroprotective and antiseizure effects, a disease‐modifying or anti‐epileptic effects of them are yet to be established.
Finally, the role of promoting anti‐inflammatory cytokines to induce neuroinflammation toward an M2 phenotype has been increasingly investigated in epilepsy models.274 This is important considering the fact that inflammation is a fairly heterogeneous process and is also involved in repair of damaged tissue. A disturbed balance in pro‐ and anti‐inflammatory response has been shown in models of acquired and genetic epilepsies.297, 298 Though, directly attempting to release anti‐inflammatory cytokines at the epileptogenic focus did not provide protective effects against epileptogenesis or neuroprotection.299 Overall, future studies are warranted to investigate the potential of other strategies modulating neuroinflammation in this manner.
8. CONCLUSIONS
There are significant neuropathological and neurobiological parallels between neurodegenerative diseases and acquired epileptogenesis, and both are commonly comorbid in patients and chronic animal models. All current therapies just systematically suppress seizures, but have no sustained effect to prevent the development of epilepsy, and do not mitigate its progression, or to reverse once it is established. Novel therapies targeting neurodegenerative pathways, such as tau, amyloid‐β, mTOR, and neuroinflammation, have potentially to be anti‐epileptogenic and/or disease‐modifying therapies for patients with acquired epilepsy. These therapies may not only have beneficial effects on the epilepsy itself, but also on the associated neurocognitive and neuropsychiatric comorbidities. There is promising evidence from animal models of acquired epilepsy that compounds targeting these neurodegenerative pathways may have such anti‐epileptogenic effects. However, further research is needed to validate these findings before proceeding into clinical trials.
DISCLOSURES
Neither of the authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
ACKNOWLEDGMENTS
PMCE and IA were funded by Melbourne University Early Career Research Grant. This work was supported by the National Institute of Neurological Disorders and Stroke (NINDS) Centers without Walls [grant number U54 NS100064], a NHMRC Program Grant to TOB (#APP1091593), and The NHMRC Peter Doherty Early Career Fellowship to PMCE (#APP1166170).
Casillas‐Espinosa PM, Ali I, O'Brien TJ. Neurodegenerative pathways as targets for acquired epilepsy therapy development. Epilepsia Open. 2020;5:138–154. 10.1002/epi4.12386
REFERENCES
- 1. Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde BW, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–85. [DOI] [PubMed] [Google Scholar]
- 2. Leonardi M, Ustun TB. The global burden of epilepsy. Epilepsia. 2002;43(Suppl 6):21–5. [DOI] [PubMed] [Google Scholar]
- 3. de Boer HM, Mula M, Sander JW. The global burden and stigma of epilepsy. Epilepsy Behav. 2008;12:540–6. [DOI] [PubMed] [Google Scholar]
- 4. Pitkänen A, Sutula T. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal‐lobe epilepsy. Lancet Neurol. 2002;1:173–81. [DOI] [PubMed] [Google Scholar]
- 5. Kwan P, Brodie MJ. Refractory epilepsy: a progressive, intractable but preventable condition? Seizure. 2002;11:77–84. [DOI] [PubMed] [Google Scholar]
- 6. Coan AC, Cendes F. Epilepsy as progressive disorders: what is the evidence that can guide our clinical decisions and how can neuroimaging help? Epilepsy Behav. 2013;26:313–21. [DOI] [PubMed] [Google Scholar]
- 7. Cole AJ. Is epilepsy a progressive disease? The neurobiological consequences of epilepsy. Epilepsia. 2000;41(Suppl 2):S13–22. [DOI] [PubMed] [Google Scholar]
- 8. Ali I, Silva J, Liu S, Shultz S, Kwan P, Jones N, et al. Targeting neurodegeneration to prevent post‐traumatic epilepsy. Neurobiol Dis. 2019;123:100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–42. [DOI] [PubMed] [Google Scholar]
- 10. Bourdenx M, Koulakiotis NS, Sanoudou D, Bezard E, Dehay B, Tsarbopoulos A. Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: examples of amyloidopathies, tauopathies and synucleinopathies. Prog Neurobiol. 2017;155:171–93. [DOI] [PubMed] [Google Scholar]
- 11. Marriott AL, Rojas‐Mancilla E, Morales P, Herrera‐Marschitz M, Tasker RA. Models of progressive neurological dysfunction originating early in life. Prog Neurobiol. 2017;155:2–20. [DOI] [PubMed] [Google Scholar]
- 12. Ono T, Galanopoulou A. Epilepsy and epileptic syndrome. Adv Exp Med Biol. 2012;724:99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bertoglio D, Amhaoul H, Van Eetveldt A, Houbrechts R, Van De Vijver S, Ali I, et al. Kainic Acid‐induced post‐status epilepticus models of temporal lobe epilepsy with diverging seizure phenotype and neuropathology. Front Neurol. 2017;8:588–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pitkänen A, Kharatishvili I, Narkilahti S, Lukasiuk K, Nissinen J. Administration of diazepam during status epilepticus reduces development and severity of epilepsy in rat. Epilepsy Res. 2005;63:27–42. [DOI] [PubMed] [Google Scholar]
- 15. Liu S‐J, Zheng P, Wright D, Dezsi G, Braine E, Nguyen T, et al. Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2A and hyperphosphorylated tau. Brain. 2016;139:1919–38. [DOI] [PubMed] [Google Scholar]
- 16. Buckmaster P, Lew F. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J Neurosci. 2011;31:2337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heng K, Haney M, Buckmaster P. High‐dose rapamycin blocks mossy fiber sprouting but not seizures in a mouse model of temporal lobe epilepsy. Epilepsia. 2013;54:1535–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang X, Zhang H, Yang J, Wu J, McMahon J, Lin Y, et al. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol Dis. 2010;40:193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Vliet E, Forte G, Holtman L, den Burger JCG, Sinjewel A, de Vries H, et al. Inhibition of mammalian target of rapamycin reduces epileptogenesis and blood‐brain barrier leakage but not microglia activation. Epilepsia. 2012;53:1254–63. [DOI] [PubMed] [Google Scholar]
- 20. Zeng L‐H, Rensing N, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jones N, Nguyen T, Corcoran N, Velakoulis D, Chen T, Grundy R, et al. Targeting hyperphosphorylated tau with sodium selenate suppresses seizures in rodent models. Neurobiol Dis. 2012;45:897–901. [DOI] [PubMed] [Google Scholar]
- 22. Shultz S, Wright D, Zheng P, Stuchbery R, Liu S‐J, Sashindranath M, et al. Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain. 2015;138:1297–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Semple B, O'Brien T, Gimlin K, Wright D, Kim S, Casillas Espinosa P, et al. Interleukin‐1 receptor in seizure susceptibility after traumatic injury to the pediatric brain. J Neurosci. 2017;37:7864–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scheffer I, French J, Hirsch E, Jain S, Mathern G, Moshé S, et al. Classification of the epilepsies: new concepts for discussion and debate‐Special report of the ILAE Classification Task Force of the Commission for Classification and Terminology. Epilepsia Open. 2016;1:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kwan P, Schachter SC, Brodie MJ. Drug‐resistant epilepsy. N Engl J Med. 2011;365:919–26. [DOI] [PubMed] [Google Scholar]
- 26. Sharma AK, Reams RY, Jordan WH, Miller MA, Thacker HL, Snyder PW. Mesial temporal lobe epilepsy: pathogenesis, induced rodent models and lesions. Toxicol Pathol. 2007;35:984–999. Review. [DOI] [PubMed] [Google Scholar]
- 27. Engel J Jr. Introduction to temporal lobe epilepsy. Epilepsy Res. 1996;26:141–150. Review. [DOI] [PubMed] [Google Scholar]
- 28. McNamara JO. Emerging insights into the genesis of epilepsy. Nature. 1999;399:A15–22. [DOI] [PubMed] [Google Scholar]
- 29. Herman S. Epilepsy after brain insult: targeting epileptogenesis. Neurology. 2002;59:S21–6. [DOI] [PubMed] [Google Scholar]
- 30. Anderson G, Temkin N, Chandler W, Winn HR. Effect of valproate on hemostatic function in patients with traumatic brain injury. Epilepsy Res. 2003;57:111–9. [DOI] [PubMed] [Google Scholar]
- 31. McNamara JO. Cellular and molecular basis of epilepsy. J Neurosci. 1994;14:3413–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Powell K, Lukasiuk K, O'Brien T, Pitkänen A. Are alterations in transmitter receptor and ion channel expression responsible for epilepsies? Adv Exp Med Biol. 2014;813:211–29. [DOI] [PubMed] [Google Scholar]
- 33. Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol. 2004;73:1–60. Review. [DOI] [PubMed] [Google Scholar]
- 34. Upreti C, Otero R, Partida C, Skinner F, Thakker R, Pacheco LF, et al. Altered neurotransmitter release, vesicle recycling and presynaptic structure in the pilocarpine model of temporal lobe epilepsy. Brain. 2012;135:869–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sanabria ER, Su H, Yaari Y. Initiation of network bursts by Ca2+‐dependent intrinsic bursting in the rat pilocarpine model of temporal lobe epilepsy. J Physiol. 2001;532:205–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Binder DK, Croll SD, Gall CM, Scharfman HE. BDNF and epilepsy: too much of a good thing? Trends Neurosci. 2001;24:47–53. Review. [DOI] [PubMed] [Google Scholar]
- 37. Raza M, Blair RE, Sombati S, Carter DS, Deshpande LS, DeLorenzo RJ. Evidence that injury‐induced changes in hippocampal neuronal calcium dynamics during epileptogenesis cause acquired epilepsy. Proc Natl Acad Sci USA. 2004;101:17522–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Blair RE, Sombati S, Lawrence DC, McCay BD, DeLorenzo RJ. Epileptogenesis causes acute and chronic increases in GABAA receptor endocytosis that contributes to the induction and maintenance of seizures in the hippocampal culture model of acquired epilepsy. J Pharmacol Exp Ther. 2004;310:871–80. [DOI] [PubMed] [Google Scholar]
- 39. Tu B, Timofeeva O, Jiao Y, Nadler JV. Spontaneous release of neuropeptide Y tonically inhibits recurrent mossy fiber synaptic transmission in epileptic brain. J Neurosci. 2005;25:1718–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Powell KL, Lukasiuk K, O'Brien TJ, Pitkanen A. Are alterations in transmitter receptor and ion channel expression responsible for epilepsies? Adv Exp Med Biol. 2014;813:211–29. [DOI] [PubMed] [Google Scholar]
- 41. Ben AY. Cell death and synaptic reorganizations produced by seizures. Epilepsia. 2001;42(Suppl 3):5–7. [DOI] [PubMed] [Google Scholar]
- 42. Holmes GL, Sarkisian M, Ben Ari Y, Chevassus‐Au‐Louis N. Mossy fiber sprouting after recurrent seizures during early development in rats. J Comp Neurol. 1999;404:537–53. [DOI] [PubMed] [Google Scholar]
- 43. Stores G. Cognitive function in children with epilepsy. Dev Med Child Neurol. 1971;13:390–3. [DOI] [PubMed] [Google Scholar]
- 44. Stafstrom CE, Chronopoulos A, Thurber S, Thompson JL, Holmes GL. Age‐dependent cognitive and behavioral deficits after kainic acid seizures. Epilepsia. 1993;34:420–32. [DOI] [PubMed] [Google Scholar]
- 45. Kandratavicius L, Lopes Aguiar C, Bueno Júnior L, Romcy Pereira R, Hallak JEC, Leite J. Psychiatric comorbidities in temporal lobe epilepsy: possible relationships between psychotic disorders and involvement of limbic circuits. Revista Brasileira de Psiquiatria. 2012;34:454–66. [DOI] [PubMed] [Google Scholar]
- 46. Tellez Zenteno J, Patten S, Jetté N, Williams J, Wiebe S. Psychiatric comorbidity in epilepsy: a population‐based analysis. Epilepsia. 2007;48:2336–44. [DOI] [PubMed] [Google Scholar]
- 47. Ott D, Siddarth P, Gurbani S, Koh S, Tournay A, Shields WD, et al. Behavioral disorders in pediatric epilepsy: unmet psychiatric need. Epilepsia. 2003;44:591–7. [DOI] [PubMed] [Google Scholar]
- 48. Vazquez B, Devinsky O. Epilepsy and anxiety. Epilepsy Behav. 2003;4(Suppl 4):S20–25. [DOI] [PubMed] [Google Scholar]
- 49. Kimiskidis V, Valeta T. Epilepsy and anxiety: epidemiology, classification, aetiology, and treatment. Epileptic Disord. 2012;14:248–56. [DOI] [PubMed] [Google Scholar]
- 50. Hinnell C, Williams J, Metcalfe A, Patten S, Parker R, Wiebe S, et al. Health status and health‐related behaviors in epilepsy compared to other chronic conditions–a national population‐based study. Epilepsia. 2010;51:853–61. [DOI] [PubMed] [Google Scholar]
- 51. Valente KDR, Busatto FG. Depression and temporal lobe epilepsy represent an epiphenomenon sharing similar neural networks: clinical and brain structural evidences. Arq Neuropsiquiatr. 2013;71:183–90. [DOI] [PubMed] [Google Scholar]
- 52. Motamedi G, Meador K. Epilepsy and cognition. Epilepsy Behav. 2003;4(Suppl 2):S25–S38. [DOI] [PubMed] [Google Scholar]
- 53. O'Leary DS, Lovell MR, Sackellares JC, Berent S, Giordani B, Seidenberg M, et al. Effects of age of onset of partial and generalized seizures on neuropsychological performance in children. J Nerv Ment Dis. 1983;171:624–9. [DOI] [PubMed] [Google Scholar]
- 54. Delorenzo RJ, Sun DA, Deshpande LS. Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintenance of epilepsy. Pharmacol Ther. 2005;105:229–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sanabria ERG, da Silva A, Spreafico R, Cavalheiro E. Damage, reorganization, and abnormal neocortical hyperexcitability in the pilocarpine model of temporal lobe epilepsy. Epilepsia. 2002;43(Suppl 5):96–106. [DOI] [PubMed] [Google Scholar]
- 56. Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–34. [DOI] [PubMed] [Google Scholar]
- 57. Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Coulter DA. Epilepsy‐associated plasticity in gamma‐aminobutyric acid receptor expression, function, and inhibitory synaptic properties. Int Rev Neurobiol. 2001;45:237–52. [DOI] [PubMed] [Google Scholar]
- 60. Gu F, Parada I, Shen F, Li J, Bacci A, Graber K, et al. Structural alterations in fast‐spiking GABAergic interneurons in a model of posttraumatic neocortical epileptogenesis. Neurobiol Dis. 2017;108:100–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Koch KL, Stern RM, Vasey MW, Seaton JF, Demers LM, Harrison TS. Neuroendocrine and gastric myoelectrical responses to illusory self‐motion in humans. Am J Physiol. 1990;258:E304–310. [DOI] [PubMed] [Google Scholar]
- 62. Olsen RW, Avoli M. GABA and epileptogenesis. Epilepsia. 1997;38:399–407. [DOI] [PubMed] [Google Scholar]
- 63. Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien‐Ly N, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Scharfman HE, Goodman JH, Sollas AL. Actions of brain‐derived neurotrophic factor in slices from rats with spontaneous seizures and mossy fiber sprouting in the dentate gyrus. J Neuroscience. 1999;19:5619–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Scharfman H. Does BDNF contribute to temporal lobe epilepsy? Epilepsy Curr. 2002;2:92–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Curia G, Lucchi C, Vinet J, Gualtieri F, Marinelli C, Torsello A, et al. Pathophysiogenesis of mesial temporal lobe epilepsy: is prevention of damage antiepileptogenic? Curr Med Chem. 2014;21:663–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Casillas Espinosa P, Hicks A, Jeffreys A, Snutch T, O'Brien T, Powell K. Z944, a Novel selective T‐type calcium channel antagonist delays the progression of seizures in the Amygdala Kindling Model. PLoS One. 2015;10:e0130012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shultz SR, Wright DK, Zheng P, Stuchbery R, Liu SJ, Sashindranath M, et al. Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain. 2015;138:1297–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Scharfman HE. Blockade of excitation reveals inhibition of dentate spiny hilar neurons recorded in rat hippocampal slices. J Neurophysiol. 1992;68:978–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Scharfman HE. Electrophysiological diversity of pyramidal‐shaped neurons at the granule cell layer/hilus border of the rat dentate gyrus recorded in vitro. Hippocampus. 1995;5:287–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ueda Y, Tsuru N. Simultaneous monitoring of the seizure‐related changes in extracellular glutamate and gamma‐aminobutyric acid concentration in bilateral hippocampi following development of amygdaloid kindling. Epilepsy Res. 1995;20:213–9. [DOI] [PubMed] [Google Scholar]
- 72. During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993;341:1607–10. [DOI] [PubMed] [Google Scholar]
- 73. Casillas‐Espinosa PM, Powell KL, O'Brien TJ. Regulators of synaptic transmission: roles in the pathogenesis and treatment of epilepsy. Epilepsia. 2012;53(Suppl 9):41–58. [DOI] [PubMed] [Google Scholar]
- 74. Chin J, Scharfman HE. Shared cognitive and behavioral impairments in epilepsy and Alzheimer's disease and potential underlying mechanisms. Epilepsy Behav. 2013;26:343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Costa C, Romoli M, Calabresi P. Late onset epilepsy and Alzheimer's disease: exploring the dual pathogenic role of amyloid‐beta. Brain. 2018;141:e60. [DOI] [PubMed] [Google Scholar]
- 76. DiFrancesco JC, Tremolizzo L, Polonia V, Giussani G, Bianchi E, Franchi C, et al. Adult‐onset epilepsy in presymptomatic Alzheimer's disease: a retrospective study. J Alzheimers Dis. 2017;60:1267–74. [DOI] [PubMed] [Google Scholar]
- 77. Dulla CG, Coulter DA, Ziburkus J. From molecular circuit dysfunction to disease: case studies in epilepsy, traumatic brain injury, and Alzheimer's disease. Neuroscientist. 2016;22:295–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Garg N, Joshi R, Medhi B. Cracking novel shared targets between epilepsy and Alzheimer's disease: need of the hour. Rev Neurosci. 2018;29:425–42. [DOI] [PubMed] [Google Scholar]
- 79. Horvath A, Szucs A, Hidasi Z, Csukly G, Barcs G, Kamondi A. Prevalence, semiology, and risk factors of epilepsy in Alzheimer's disease: an ambulatory EEG study. J Alzheimers Dis. 2018;63:1045–54. [DOI] [PubMed] [Google Scholar]
- 80. Jiang XW, Lu HY, Xu Z, Liu TY, Wu Q, Yang Y, et al. Silico analyses for key genes and molecular genetic mechanism in epilepsy and Alzheimer's disease. CNS Neurol Disord Drug Targets. 2018;17:608–17. [DOI] [PubMed] [Google Scholar]
- 81. Lam AD, Deck G, Goldman A, Eskandar EN, Noebels J, Cole AJ. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer's disease. Nat Med. 2017;23:678–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nicastro N, Assal F, Seeck M. From here to epilepsy: the risk of seizure in patients with Alzheimer's disease. Epileptic Disord. 2016;18:1–12. [DOI] [PubMed] [Google Scholar]
- 83. Palop J, Chin J, Roberson E, Wang J, Thwin M, Bien Ly N, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rapoport E, Shimshony A. Health hazards to the small ruminant population of the Middle East posed by the trade of sheep and goat meat. Rev Sci Tech. 1997;16:57–64. [DOI] [PubMed] [Google Scholar]
- 85. Rauramaa T, Saxlin A, Lohvansuu K, Alafuzoff I, Pitkanen A, Soininen H. Epilepsy in neuropathologically verified Alzheimer's disease. Seizure. 2018;58:9–12. [DOI] [PubMed] [Google Scholar]
- 86. Subota A, Pham T, Jette N, Sauro K, Lorenzetti D, Holroyd‐Leduc J. The association between dementia and epilepsy: a systematic review and meta‐analysis. Epilepsia. 2017;58:962–72. [DOI] [PubMed] [Google Scholar]
- 87. Naegele J. Neuroprotective strategies to avert seizure‐induced neurodegeneration in epilepsy. Epilepsia. 2007;48(Suppl 2):107–17. [DOI] [PubMed] [Google Scholar]
- 88. Pitkänen A. Treatment with antiepileptic drugs: Possible neuroprotective effects. Neurology. 1996;47:S12–S16. [DOI] [PubMed] [Google Scholar]
- 89. Nairismägi J, Pitkänen A, Kettunen M, Kauppinen R, Kubova H. Status epilepticus in 12‐day‐old rats leads to temporal lobe neurodegeneration and volume reduction: a histologic and MRI study. Epilepsia. 2006;47:479–88. [DOI] [PubMed] [Google Scholar]
- 90. Mathern GW, Babb TL, Pretorius JK, Melendez M, Levesque MF. The pathophysiologic relationships between lesion pathology, intracranial ictal EEG onsets, and hippocampal neuron losses in temporal lobe epilepsy. Epilepsy Res. 1995;21:133–47. [DOI] [PubMed] [Google Scholar]
- 91. Mathern GW, Cifuentes F, Leite JP, Pretorius JK, Babb TL. Hippocampal EEG excitability and chronic spontaneous seizures are associated with aberrant synaptic reorganization in the rat intrahippocampal kainate model. Electroencephalogr Clin Neurophysiol. 1993;87:326–39. [DOI] [PubMed] [Google Scholar]
- 92. Thom M, Sisodiya S, Beckett A, Martinian L, Lin W‐R, Harkness W, et al. Cytoarchitectural abnormalities in hippocampal sclerosis. J Neuropathol Exp Neurol. 2002;61:510–9. [DOI] [PubMed] [Google Scholar]
- 93. Dragunow M, Yamada N, Bilkey DK, Lawlor P. Induction of immediate‐early gene proteins in dentate granule cells and somatostatin interneurons after hippocampal seizures. Mol Brain Res. 1992;13:119–26. [DOI] [PubMed] [Google Scholar]
- 94. Herdegen T, Sandkühler J, Gass P, Kiessling M, Bravo R, Zimmermann MJUN, et al. and CREB transcription factor proteins in the rat cortex: basal expression and induction by spreading depression and epileptic seizures. J Comp Neurol. 1993;333:271–88. [DOI] [PubMed] [Google Scholar]
- 95. Dragunow M, Young D, Hughes P, MacGibbon G, Lawlor P, Singleton K, et al. Is c‐Jun involved in nerve cell death following status epilepticus and hypoxic‐ischaemic brain injury? Mol Brain Res. 1993;18:347–52. [DOI] [PubMed] [Google Scholar]
- 96. Gass P, Bruehl C, Herdegen T, Kiessling M, Lutzenburg M, Witte OW. Induction of FOS and JUN proteins during focal epilepsy: congruences with and differences to [14C]deoxyglucose metabolism. Mol Brain Res. 1997;46:177–84. [DOI] [PubMed] [Google Scholar]
- 97. Engel T, Sanz Rodgriguez A, Jimenez Mateos E, Concannon C, Jimenez Pacheco A, Moran C, et al. CHOP regulates the p53‐MDM2 axis and is required for neuronal survival after seizures. Brain. 2013;136:577–92. [DOI] [PubMed] [Google Scholar]
- 98. Jewett K, Christian C, Bacos J, Lee K, Zhu J, Tsai N‐P. Feedback modulation of neural network synchrony and seizure susceptibility by Mdm2‐p53‐Nedd4‐2 signaling. Mol Brain. 2016;9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kilany A, Raouf ERA, Gaber A, Aloush T, Aref H, Anwar M, et al. Elevated serum Bcl‐2 in children with temporal lobe epilepsy. Seizure (London, England). 2012;21:250–3. [DOI] [PubMed] [Google Scholar]
- 100. El Hodhod MA, Tomoum HY. Abd Al‐Aziz MM, Samaan SM. Serum Fas and Bcl‐2 in patients with epilepsy. Acta Neurol Scand. 2006;113:315–21. [DOI] [PubMed] [Google Scholar]
- 101. Tzeng T‐T, Tsay H‐J, Chang L, Hsu C‐L, Lai T‐H, Huang F‐L, et al. Caspase 3 involves in neuroplasticity, microglial activation and neurogenesis in the mice hippocampus after intracerebral injection of kainic acid. J Biomed Sci. 2013;20:90–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Narkilahti S, Pitkänen A. Caspase 6 expression in the rat hippocampus during epileptogenesis and epilepsy. Neuroscience. 2005;131:887–97. [DOI] [PubMed] [Google Scholar]
- 103. Shapiro L, Wang L, Ribak C. Rapid astrocyte and microglial activation following pilocarpine‐induced seizures in rats. Epilepsia. 2008;49(Suppl 2):33–41. [DOI] [PubMed] [Google Scholar]
- 104. Vezzani A, Ravizza T, Balosso S, Aronica E. Glia as a source of cytokines: implications for neuronal excitability and survival. Epilepsia. 2008;49(Suppl 2):24–32. [DOI] [PubMed] [Google Scholar]
- 105. Cavazos J, Cross D. The role of synaptic reorganization in mesial temporal lobe epilepsy. Epilepsy Behav. 2006;8:483–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Foresti M, Arisi G, Shapiro L. Role of glia in epilepsy‐associated neuropathology, neuroinflammation and neurogenesis. Brain Res Rev. 2011;66:115–22. [DOI] [PubMed] [Google Scholar]
- 107. Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Annals Neurol. 1989;26:321–30. [DOI] [PubMed] [Google Scholar]
- 108. Cavazos JE, Sutula TP. Progressive neuronal loss induced by kindling: a possible mechanism for mossy fiber synaptic reorganization and hippocampal sclerosis. Brain Res. 1990;527:1–6. [DOI] [PubMed] [Google Scholar]
- 109. Buckmaster PS, Dudek FE. In vivo intracellular analysis of granule cell axon reorganization in epileptic rats. J Neurophysiol. 1999;81:712–21. [DOI] [PubMed] [Google Scholar]
- 110. Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid‐treated rats. J Neurosci. 1985;5:1016–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Bouilleret V, Loup F, Kiener T, Marescaux C. Fritschy JM. Early loss of interneurons and delayed subunit‐specific changes in GABA(A)‐receptor expression in a mouse model of mesial temporal lobe epilepsy. Hippocampus. 2000;10:305–24. [DOI] [PubMed] [Google Scholar]
- 112. Loup F, Wieser HG, Yonekawa Y, Aguzzi A, Fritschy JM. Selective alterations in GABAA receptor subtypes in human temporal lobe epilepsy. J Neurosci. 2000;20:5401–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Cavazos JE, Jones SM, Cross DJ. Sprouting and synaptic reorganization in the subiculum and CA1 region of the hippocampus in acute and chronic models of partial‐onset epilepsy. Neuroscience. 2004;126:677–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Brooks Kayal A, Raol Y, Russek S. Alteration of epileptogenesis genes. Neurotherapeutics. 2009;6:312–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Casillas Espinosa P, Powell K, O'Brien T. Regulators of synaptic transmission: roles in the pathogenesis and treatment of epilepsy. Epilepsia. 2012;53(Suppl 9):41–58. [DOI] [PubMed] [Google Scholar]
- 116. Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 117. Nagarkatti N, Deshpande LS, DeLorenzo RJ. Development of the calcium plateau following status epilepticus: role of calcium in epileptogenesis. Expert Rev Neurother. 2009;9:813–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Selcher JC, Xu W, Hanson JE, Malenka RC, Madison DV. Glutamate receptor subunit GluA1 is necessary for long‐term potentiation and synapse unsilencing, but not long‐term depression in mouse hippocampus. Brain Res. 2012;1435:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007;51:333–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Meeren HK, Pijn JP, Van Luijtelaar EL, Coenen AM, Lopes da Silva FH. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci. 2002;22:1480–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Tanaka H, Grooms SY, Bennett MV. Zukin RS. The AMPAR subunit GluR2: still front and center‐stage. Brain Res. 2000;886:190–207. [DOI] [PubMed] [Google Scholar]
- 122. McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE. 2006;2006:re12. [DOI] [PubMed] [Google Scholar]
- 123. Krestel HE, Shimshek DR, Jensen V, Nevian T, Kim J, Geng Y, et al. A genetic switch for epilepsy in adult mice. J Neurosci. 2004;24:10568–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Sanchez RM, Koh S, Rio C, Wang C, Lamperti ED, Sharma D, et al. Decreased glutamate receptor 2 expression and enhanced epileptogenesis in immature rat hippocampus after perinatal hypoxia‐induced seizures. J Neurosci. 2001;21:8154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Sierra A, Gröhn O, Pitkänen A. Imaging microstructural damage and plasticity in the hippocampus during epileptogenesis. Neuroscience. 2015;309:162–72. [DOI] [PubMed] [Google Scholar]
- 126. Goldensohn ES, Salazar AM. Temporal and spatial distribution of intracellular potentials during generation and spread of epileptogenic discharges. Adv Neurol. 1986;44:559–82. [PubMed] [Google Scholar]
- 127. Cavazos JE, Golarai G, Sutula TP. Mossy fiber synaptic reorganization induced by kindling: time course of development, progression, and permanence. J Neurosci. 1991;11:2795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurotherapeutics. 2007;4:18–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Rogawski MA. Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Curr. 2011;11:56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Krauss GL, Bar M, Biton V, Klapper JA, Rektor I, Vaiciene‐Magistris N, et al. Tolerability and safety of perampanel: two randomized dose‐escalation studies. Acta Neurol Scand. 2012;125:8–15. [DOI] [PubMed] [Google Scholar]
- 131. Trinka E, Steinhoff BJ, Nikanorova M, Brodie MJ. Perampanel for focal epilepsy: insights from early clinical experience. Acta Neurol Scand. 2016;133:160–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Brodie M. Practical use of newer antiepileptic drugs as adjunctive therapy in focal epilepsy. CNS Drugs. 2015;29:893–904. [DOI] [PubMed] [Google Scholar]
- 133. Glauser T, Laurenza A, Yang H, Williams B, Ma T, Fain R. Efficacy and tolerability of adjunct perampanel based on number of antiepileptic drugs at baseline and baseline predictors of efficacy: A phase III post‐hoc analysis. Epilepsy Res. 2016;119:34–40. [DOI] [PubMed] [Google Scholar]
- 134. Hanada T, Hashizume Y, Tokuhara N, Takenaka O, Kohmura N, Ogasawara A, et al. Perampanel: a novel, orally active, noncompetitive AMPA‐receptor antagonist that reduces seizure activity in rodent models of epilepsy. Epilepsia. 2011;52:1331–40. [DOI] [PubMed] [Google Scholar]
- 135. Ceolin L, Bortolotto ZA, Bannister N, Collingridge GL, Lodge D, Volianskis A. A novel anti‐epileptic agent, perampanel, selectively inhibits AMPA receptor‐mediated synaptic transmission in the hippocampus. Neurochem Int. 2012. [DOI] [PubMed] [Google Scholar]
- 136. Wu T, Ido K, Osada Y, Kotani S, Tamaoka A, Hanada T. The neuroprotective effect of perampanel in lithium‐pilocarpine rat seizure model. Epilepsy Res. 2017;137:152–8. [DOI] [PubMed] [Google Scholar]
- 137. Twele F, Bankstahl M, Klein S, Römermann K, Löscher W. The AMPA receptor antagonist NBQX exerts anti‐seizure but not antiepileptogenic effects in the intrahippocampal kainate mouse model of mesial temporal lobe epilepsy. Neuropharmacology. 2015;95:234–42. [DOI] [PubMed] [Google Scholar]
- 138. Apland J, Aroniadou Anderjaska V, Figueiredo T, Green C, Swezey R, Yang C, et al. Efficacy of the GluK1/AMPA receptor antagonist LY293558 against seizures and neuropathology in a soman‐exposure model without pretreatment and its pharmacokinetics after intramuscular administration. J Pharmacol Exp Therap. 2013;344:133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zheng P, Shultz S, Hovens C, O'Brien TJ. In Pitkanen A, editor. Animal Models of Acquired Epilepsy and Tauopathies. 2 ed. Academic Press; 2017. [Google Scholar]
- 140. Lee G, Leugers CJ. Tau and tauopathies. Prog Mol Biol Transl Sci. 2012;107:263–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Bi M, Gladbach A, van Eersel J, Ittner A, Przybyla M, van Hummel A, et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat Communicat. 2017;8:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Engel T, Goni‐Oliver P, Lucas JJ, Avila J, Hernandez F. Chronic lithium administration to FTDP‐17 tau and GSK‐3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre‐formed neurofibrillary tangles do not revert. J Neurochem. 2006;99:1445–55. [DOI] [PubMed] [Google Scholar]
- 144. Puvenna V, Engeler M, Banjara M, Brennan C, Schreiber P, Dadas A, et al. Is phosphorylated tau unique to chronic traumatic encephalopathy? Phosphorylated tau in epileptic brain and chronic traumatic encephalopathy. Brain Res. 2016;1630:225–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Tai XY, Koepp M, Duncan JS, Fox N, Thompson P, Baxendale S, et al. Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: a study of temporal lobe resections. Brain. 2016;139:2441–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Thom M, Liu JY, Thompson P, Phadke R, Narkiewicz M, Martinian L, et al. Neurofibrillary tangle pathology and Braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: a post‐mortem study. Brain. 2011;134:2969–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Sanchez MP, Garcia‐Cabrero AM, Sanchez‐Elexpuru G, Burgos DF, Serratosa JM. Tau‐induced pathology in epilepsy and dementia: notions from patients and animal models. Int J Mol Sci. 2018;19:E1092 10.3390/ijms19041092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Sen A, Thom M, Martinian L, Harding B, Cross JH, Nikolic M, et al. Pathological tau tangles localize to focal cortical dysplasia in older patients. Epilepsia. 2007;48:1447–54. [DOI] [PubMed] [Google Scholar]
- 149. Hawkins BE, Krishnamurthy S, Castillo‐Carranza DL, Sengupta U, Prough DS, Jackson GR, et al. Rapid accumulation of endogenous tau oligomers in a rat model of traumatic brain injury. J Biol Chem. 2013;288:17042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Monti G, Tondelli M, Giovannini G, Bedin R, Nichelli PF, Trenti T, et al. Cerebrospinal fluid tau proteins in status epilepticus. Epilepsy Behav. 2015;49:150–4. [DOI] [PubMed] [Google Scholar]
- 151. Palmio J, Suhonen J, Keranen T, Hulkkonen J, Peltola J, Pirttila T. Cerebrospinal fluid tau as a marker of neuronal damage after epileptic seizure. Seizure. 2009;18:474–7. [DOI] [PubMed] [Google Scholar]
- 152. Shahim P, Rejdak R, Ksiazek P, Blennow K, Zetterberg H, Mattsson N, et al. Cerebrospinal fluid biomarkers of beta‐amyloid metabolism and neuronal damage in epileptic seizures. Eur J Neurol. 2014;21:486–91. [DOI] [PubMed] [Google Scholar]
- 153. Gheyara AL, Ponnusamy R, Djukic B, Craft RJ, Ho K, Guo W, et al. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann Neurology. 2014;76:443–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, et al. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J Neurosci. 2013;33:1651–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Yan XX, Cai Y, Shelton J, Deng SH, Luo XG, Oddo S, et al. Chronic temporal lobe epilepsy is associated with enhanced Alzheimer‐like neuropathology in 36 x Tg‐AD mice. PloS One. 2012;7: e48782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Liu SJ, Shen Y, Shultz SR, Nguyen A, Hovens C, Adlard PA, et al. Accelerated kindling epileptogenesis in Tg4510 tau transgenic mice, but not in tau knockout mice. Epilepsia. 2017;58:E136–E141. [DOI] [PubMed] [Google Scholar]
- 157. Jones NC, Nguyen T, Corcoran NM, Velakoulis D, Chen T, Grundy R, et al. Targeting hyperphosphorylated tau with sodium selenate suppresses seizures in rodent models. Neurobiol Dis. 2012;45:897–901. [DOI] [PubMed] [Google Scholar]
- 158. Bhowmik M, Khanam R, Saini N, Vohora D. Activation of AKT/GSK3beta pathway by TDZD‐8 attenuates kainic acid induced neurodegeneration but not seizures in mice. Neurotoxicology. 2015;46:44–52. [DOI] [PubMed] [Google Scholar]
- 159. Roberson ED, Scearce‐Levie K, Palop JJ, Yan FR, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid beta‐induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–4. [DOI] [PubMed] [Google Scholar]
- 160. Matsumoto G, Sakai H. Microtubules inside the plasma membrane of squid giant axons and their possible physiological function. J Membr Biol. 1979;50:1–14. [DOI] [PubMed] [Google Scholar]
- 161. DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 2013;33:12887–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Bi M, Gladbach A, van Eersel J, Ittner A, Przybyla M, van Hummel A, et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat Commun. 2017;8:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Hatch RJ, Wei Y, Xia D, Gotz J. Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol. 2017;133:717–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Mondragon Rodriguez S, Salas Gallardo A, Gonzalez Pereyra P, Macias M, Ordaz B, Pena Ortega F, et al. Phosphorylation of tau protein correlates with changes in hippocampal theta oscillations and reduces hippocampal excitability in Alzheimer's model. J Biol Chem. 2018; 293:8462–8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Liang Z, Liu F, Iqbal K, Grundke‐Iqbal I, Gong CX. Dysregulation of tau phosphorylation in mouse brain during excitotoxic damage. J Alzheimer's Dis. 2009;17:531–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. van Eersel J, Ke Y, Liu X, Delerue F, Kril J, Götz J, et al. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer's disease models. Proc Natl Acad Sci USA. 2010;107:13888–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Corcoran N, Martin D, Hutter Paier B, Windisch M, Nguyen T, Nheu L, et al. Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer's disease model. J Clin Neurosci. 2010;17:1025–33. [DOI] [PubMed] [Google Scholar]
- 168. Corcoran NM, Hovens CM, Michael M, Rosenthal MA, Costello AJ. Open‐label, phase I dose‐escalation study of sodium selenate, a novel activator of PP2A, in patients with castration‐resistant prostate cancer. Br J Cancer. 2010;103:462–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Malpas C, Vivash L, Genc S, Saling M, Desmond P, Steward C, et al. A phase IIa randomized control trial of VEL015 (sodium selenate) in mild‐moderate Alzheimer's disease. J Alzheimers Dis. 2016;54:223–32. [DOI] [PubMed] [Google Scholar]
- 170. Masters C, Bateman R, Blennow K, Rowe C, Sperling R, Cummings J. Alzheimer's disease. Nat Rev Dis Primers. 2015;1:15056. [DOI] [PubMed] [Google Scholar]
- 171. Masters C, Selkoe D. Biochemistry of amyloid β‐protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Selkoe D, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, et al. Amyloid beta‐protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–52. [DOI] [PubMed] [Google Scholar]
- 174. Walsh D, Selkoe D. A beta oligomers – a decade of discovery. J Neurochem. 2007;101:1172–84. [DOI] [PubMed] [Google Scholar]
- 175. Hauser WA, Morris ML, Heston LL, Anderson VE. Seizures and myoclonus in patients with Alzheimer's disease. Neurology. 1986;36:1226–30. [DOI] [PubMed] [Google Scholar]
- 176. Risse SC, Lampe TH, Bird TD, Nochlin D, Sumi SM, Keenan T, et al. seizures, and paratonia in Alzheimer disease. Alzheimer Dis Assoc Disord. 1990;4:217–25. [DOI] [PubMed] [Google Scholar]
- 177. Friedman D, Honig L, Scarmeas N. Seizures and epilepsy in Alzheimer's disease. CNS Neurosci Therap. 2012;18:285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Scarmeas N, Honig L, Choi H, Cantero J, Brandt J, Blacker D, et al. Seizures in Alzheimer disease: who, when, and how common? Arch Neurol. 2009;66:992–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Amatniek J, Hauser WA, DelCastillo CC, Jacobs D, Marder K, Bell K, et al. Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia. 2006;47:867–72. [DOI] [PubMed] [Google Scholar]
- 180. Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron. 2012;76:886–99. [DOI] [PubMed] [Google Scholar]
- 181. Hayashi T, Ago K, Nakamae T, Higo E, Ogata M. Two different immunostaining patterns of beta‐amyloid precursor protein (APP) may distinguish traumatic from nontraumatic axonal injury. Int J Legal Med. 2015;129:1085–90. [DOI] [PubMed] [Google Scholar]
- 182. Hortobágyi T, Wise S, Hunt N, Cary N, Djurovic V, Fegan Earl A, et al. Traumatic axonal damage in the brain can be detected using beta‐APP immunohistochemistry within 35 min after head injury to human adults. Neuropathol Appl Neurobiol. 2007;33:226–37. [DOI] [PubMed] [Google Scholar]
- 183. Ikonomovic M, Uryu K, Abrahamson E, Ciallella J, Trojanowski J, Lee VMY, et al. Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190:192–203. [DOI] [PubMed] [Google Scholar]
- 184. Murakami N, Yamaki T, Iwamoto Y, Sakakibara T, Kobori N, Fushiki S, et al. Experimental brain injury induces expression of amyloid precursor protein, which may be related to neuronal loss in the hippocampus. J Neurotrauma. 1998;15:993–1003. [DOI] [PubMed] [Google Scholar]
- 185. Pierce JE, Trojanowski JQ, Graham DI, Smith DH, McIntosh TK. Immunohistochemical characterization of alterations in the distribution of amyloid precursor proteins and beta‐amyloid peptide after experimental brain injury in the rat. J Neurosci. 1996;16:1083–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Lewén A, Li GL, Nilsson P, Olsson Y, Hillered L. Traumatic brain injury in rat produces changes of beta‐amyloid precursor protein immunoreactivity. Neuroreport. 1995;6:357–60. [DOI] [PubMed] [Google Scholar]
- 187. Otsuka N, Tomonaga M, Ikeda K. Rapid appearance of beta‐amyloid precursor protein immunoreactivity in damaged axons and reactive glial cells in rat brain following needle stab injury. Brain Res. 1991;568:335–8. [DOI] [PubMed] [Google Scholar]
- 188. Kodam A, Ourdev D, Maulik M, Hariharakrishnan J, Banerjee M, Wang Y, et al. A role for astrocyte‐derived amyloid β peptides in the degeneration of neurons in an animal model of temporal lobe epilepsy. Brain Pathol. 2019;29:28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Kenney K, Iacono D, Edlow B, Katz D, Diaz Arrastia R, Dams O'Connor K, et al. Dementia after moderate‐severe traumatic brain injury: coexistence of multiple proteinopathies. J Neuropathol Experimental Neurol. 2018;77:50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Chan J, Jones N, Bush A, O'Brien T, Kwan P. A mouse model of Alzheimer's disease displays increased susceptibility to kindling and seizure‐associated death. Epilepsia. 2015;56:e73–e77. [DOI] [PubMed] [Google Scholar]
- 191. Sima X, Xu J, Li J, Zhong W, You C. Expression of β‐amyloid precursor protein in refractory epilepsy. Mol Med Report. 2014;9:1242–8. [DOI] [PubMed] [Google Scholar]
- 192. Bakker A, Albert M, Krauss G, Speck C, Gallagher M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. NeuroImage Clin. 2015;7:688–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Vossel K, Tartaglia M, Nygaard H, Zeman A, Miller B. Epileptic activity in Alzheimer's disease: causes and clinical relevance. Lancet Neurol. 2017;16:311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Huijbers W, Mormino E, Schultz A, Wigman S, Ward A, Larvie M, et al. Amyloid‐β deposition in mild cognitive impairment is associated with increased hippocampal activity, atrophy and clinical progression. Brain. 2015;138:1023–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Vossel K, Beagle A, Rabinovici G, Shu H, Lee S, Naasan G, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Villemagne V, Burnham S, Bourgeat P, Brown B, Ellis K, Salvado O, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12:357–67. [DOI] [PubMed] [Google Scholar]
- 197. Kam K, Duffy ÁM, Moretto J, LaFrancois JJ, Scharfman H. Interictal spikes during sleep are an early defect in the Tg2576 mouse model of β‐amyloid neuropathology. Sci Rep. 2016;6:20119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Ziyatdinova S, Rönnbäck A, Gurevicius K, Miszczuk D, Graff C, Winblad B, et al. Increased epileptiform EEG activity and decreased seizure threshold in arctic APP transgenic mouse model of Alzheimer's disease. Curr Alzheimer Res. 2016;13:817–30. [DOI] [PubMed] [Google Scholar]
- 199. Minkeviciene R, Rheims S, Dobszay M, Zilberter M, Hartikainen J, Fülöp L, et al. Amyloid beta‐induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Busche M, Chen X, Henning H, Reichwald J, Staufenbiel M, Sakmann B, et al. Critical role of soluble amyloid‐β for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2012;109:8740–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Busche M, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold K‐H, Haass C, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008;321:1686–9. [DOI] [PubMed] [Google Scholar]
- 202. Brown JT, Chin J, Leiser SC, Pangalos MN, Randall AD. Altered intrinsic neuronal excitability and reduced Na+ currents in a mouse model of Alzheimer's disease. Neurobiol Aging. 2011;32:e2101–2114. [DOI] [PubMed] [Google Scholar]
- 203. Sawada M, Ichinose M. Amyloid beta proteins reduce the GABA‐induced Cl‐ current in identified Aplysia neurons. Neurosci Lett. 1996;213:213–5. [DOI] [PubMed] [Google Scholar]
- 204. Gurevicius K, Lipponen A, Tanila H. Increased cortical and thalamic excitability in freely moving APPswe/PS1dE9 mice modeling epileptic activity associated with Alzheimer's disease. Cereb Cortex. 2013;23:1148–58. [DOI] [PubMed] [Google Scholar]
- 205. Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fulop L, et al. Amyloid beta‐induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Ziyatdinova S, Gurevicius K, Kutchiashvili N, Bolkvadze T, Nissinen J, Tanila H, et al. Spontaneous epileptiform discharges in a mouse model of Alzheimer's disease are suppressed by antiepileptic drugs that block sodium channels. Epilepsy Res. 2011;94:75–85. [DOI] [PubMed] [Google Scholar]
- 207. Ziyatdinova S, Viswanathan J, Hiltunen M, Tanila H, Pitkanen A. Reduction of epileptiform activity by valproic acid in a mouse model of Alzheimer's disease is not long‐lasting after treatment discontinuation. Epilepsy Res. 2015;112:43–55. [DOI] [PubMed] [Google Scholar]
- 208. Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta‐amyloid peptide in vivo: evidence for augmentation of a 42‐specific gamma secretase. Hum Mol Genet. 2004;13:159–70. [DOI] [PubMed] [Google Scholar]
- 209. Costa C, Parnetti L, D'Amelio M, Tozzi A, Tantucci M, Romigi A, et al. Epilepsy, amyloid‐β, and D1 dopamine receptors: a possible pathogenetic link? Neurobiol Aging. 2016;48:161–71. [DOI] [PubMed] [Google Scholar]
- 210. Ren S‐Q, Yao W, Yan J‐Z, Jin C, Yin J‐J, Yuan J, et al. Amyloid β causes excitation/inhibition imbalance through dopamine receptor 1‐dependent disruption of fast‐spiking GABAergic input in anterior cingulate cortex. Sci Rep. 2018;8:302–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Wu J, Khan G, Nichols R. Dopamine release in prefrontal cortex in response to beta‐amyloid activation of alpha7 * nicotinic receptors. Brain Res. 2007;1182:82–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Born H, Kim J‐Y, Savjani R, Das P, Dabaghian Y, Guo Q, et al. Genetic suppression of transgenic APP rescues Hypersynchronous network activity in a mouse model of Alzheimer's disease. J Neurosci. 2014;34:3826–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Cirrito J, Yamada K, Finn M, Sloviter R, Bales K, May P, et al. Synaptic activity regulates interstitial fluid amyloid‐beta levels in vivo. Neuron. 2005;48:913–22. [DOI] [PubMed] [Google Scholar]
- 214. Yun SH, Gamkrelidze G, Stine WB, Sullivan PM, Pasternak JF, Ladu MJ, et al. Amyloid‐beta1‐42 reduces neuronal excitability in mouse dentate gyrus. Neurosci Lett. 2006;403:162–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Thornton E, Vink R, Blumbergs PC, Van Den Heuvel C. Soluble amyloid precursor protein alpha reduces neuronal injury and improves functional outcome following diffuse traumatic brain injury in rats. Brain Res. 2006;1094:38–46. [DOI] [PubMed] [Google Scholar]
- 216. Van den Heuvel C, Blumbergs PC, Finnie JW, Manavis J, Jones NR, Reilly PL, et al. Upregulation of amyloid precursor protein messenger RNA in response to traumatic brain injury: an ovine head impact model. Exp Neurol. 1999;159:441–50. [DOI] [PubMed] [Google Scholar]
- 217. Miszczuk D, Debski KJ, Tanila H, Lukasiuk K, Pitkanen A. Traumatic brain injury increases the expression of Nos1, abeta clearance, and epileptogenesis in APP/PS1 mouse model of Alzheimer's disease. Mol Neurobiol. 2016;53:7010–27. [DOI] [PubMed] [Google Scholar]
- 218. Nakagawa Y, Nakamura M, McIntosh TK, Rodriguez A, Berlin JA, Smith DH, et al. Traumatic brain injury in young, amyloid‐beta peptide overexpressing transgenic mice induces marked ipsilateral hippocampal atrophy and diminished Abeta deposition during aging. J Comp Neurol. 1999;411:390–8. [PubMed] [Google Scholar]
- 219. Nakagawa Y, Reed L, Nakamura M, McIntosh TK, Smith DH, Saatman KE, et al. Brain trauma in aged transgenic mice induces regression of established abeta deposits. Exp Neurol. 2000;163:244–52. [DOI] [PubMed] [Google Scholar]
- 220. Corrigan F, Vink R, Blumbergs PC, Masters CL, Cappai R, van den Heuvel C. Characterisation of the effect of knockout of the amyloid precursor protein on outcome following mild traumatic brain injury. Brain Res. 2012;1451:87–99. [DOI] [PubMed] [Google Scholar]
- 221. Corrigan F, Vink R, Blumbergs PC, Masters CL, Cappai R, van,. den Heuvel C. sAPPalpha rescues deficits in amyloid precursor protein knockout mice following focal traumatic brain injury. J Neurochem. 2012;122:208–20. [DOI] [PubMed] [Google Scholar]
- 222. Mockett BG, Richter M, Abraham WC, Muller UC. Therapeutic potential of secreted amyloid precursor protein APPsalpha. Front Mol Neurosci. 2017;10:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Muller UC, Deller T, Korte M. Not just amyloid: physiological functions of the amyloid precursor protein family. Nat Rev Neurosci. 2017;18:281–98. [DOI] [PubMed] [Google Scholar]
- 224. Schenk D, Basi G, Pangalos M. Treatment strategies targeting amyloid β‐protein. Cold Spring Harb Perspect Med. 2012;2:a006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Loane D, Pocivavsek A, Moussa CEH, Thompson R, Matsuoka Y, Faden A, et al. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med. 2009;15:377–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Corrigan F, Vink R, Blumbergs P, Masters C, Cappai R, van,. den Heuvel C. sAPPα rescues deficits in amyloid precursor protein knockout mice following focal traumatic brain injury. J Neurochem. 2012;122:208–20. [DOI] [PubMed] [Google Scholar]
- 227. Corrigan F, Thornton E, Roisman L, Leonard A, Vink R, Blumbergs P, et al. The neuroprotective activity of the amyloid precursor protein against traumatic brain injury is mediated via the heparin binding site in residues 96–110. J Neurochem. 2014;128:196–204. [DOI] [PubMed] [Google Scholar]
- 228. Rehman S, Ahmad A, Yoon G‐H, Khan M, Abid M, Kim M. Inhibition of c‐Jun N‐terminal kinase protects against brain damage and improves learning and memory after traumatic brain injury in adult mice. Cereb Cortex. 2018;28:2854–72. [DOI] [PubMed] [Google Scholar]
- 229. Mehan S, Meena H, Sharma D, Sankhla R. JNK: a stress‐activated protein kinase therapeutic strategies and involvement in Alzheimer's and various neurodegenerative abnormalities. J Mol Neurosci. 2011;43:376–90. [DOI] [PubMed] [Google Scholar]
- 230. Shoji M, Iwakami N, Takeuchi S, Waragai M, Suzuki M, Kanazawa I, et al. JNK activation is associated with intracellular beta‐amyloid accumulation. Mol Brain Res. 2000;85:221–33. [DOI] [PubMed] [Google Scholar]
- 231. Williams RSB, Bate C. Valproic acid and its congener propylisopropylacetic acid reduced the amount of soluble amyloid‐β oligomers released from 7PA2 cells. Neuropharmacology. 2018;128:54–62. [DOI] [PubMed] [Google Scholar]
- 232. Ziyatdinova S, Viswanathan J, Hiltunen M, Tanila H, Pitkänen A. Reduction of epileptiform activity by valproic acid in a mouse model of Alzheimer's disease is not long‐lasting after treatment discontinuation. Epilepsy Res. 2015;112:43–55. [DOI] [PubMed] [Google Scholar]
- 233. Zhang M‐Y, Zheng C‐Y, Zou M‐M, Zhu J‐W, Zhang Y, Wang J, et al. Lamotrigine attenuates deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice. Neurobiol Aging. 2014;35:2713–25. [DOI] [PubMed] [Google Scholar]
- 234. Bomben V, Holth J, Reed J, Cramer P, Landreth G, Noebels J. Bexarotene reduces network excitability in models of Alzheimer's disease and epilepsy. Neurobiol Aging. 2014;35:2091–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Gersner R, Ekstein D, Dhamne SC, Schachter SC, Rotenberg A. Huperzine A prophylaxis against pentylenetetrazole‐induced seizures in rats is associated with increased cortical inhibition. Epilepsy Res. 2015;117:97–103. [DOI] [PubMed] [Google Scholar]
- 236. Wang Y, Tang X, Zhang H. Huperzine A alleviates synaptic deficits and modulates amyloidogenic and nonamyloidogenic pathways in APPswe/PS1dE9 transgenic mice. J Neurosci Res. 2012;90:508–17. [DOI] [PubMed] [Google Scholar]
- 237. Peng Y, Lee DYW, Jiang L, Ma Z, Schachter SC, Lemere CA. Huperzine A regulates amyloid precursor protein processing via protein kinase C and mitogen‐activated protein kinase pathways in neuroblastoma SK‐N‐SH cells over‐expressing wild type human amyloid precursor protein 695. Neuroscience. 2007;150:386–95. [DOI] [PubMed] [Google Scholar]
- 238. Sanchez P, Zhu L, Verret L, Vossel K, Orr A, Cirrito J, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–E2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Perluigi M, Di Domenico F, Butterfield DA. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol Dis. 2015;84:39–49. [DOI] [PubMed] [Google Scholar]
- 240. Bove J, Martinez‐Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12:437–52. [DOI] [PubMed] [Google Scholar]
- 241. Meng XF, Yu JT, Song JH, Chi S, Tan L. Role of the mTOR signaling pathway in epilepsy. J Neurol Sci. 2013;332:4–15. [DOI] [PubMed] [Google Scholar]
- 242. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opinion Cell Biol. 2010;22:124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Heras‐Sandoval D, Perez‐Rojas JM, Hernandez‐Damian J, Pedraza‐Chaverri J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014;26:2694–701. [DOI] [PubMed] [Google Scholar]
- 244. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self‐digestion. Nature. 2008;451:1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Fulda S. Modulation of mitochondrial apoptosis by PI3K inhibitors. Mitochondrion. 2013;13:195–8. [DOI] [PubMed] [Google Scholar]
- 246. Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. [DOI] [PubMed] [Google Scholar]
- 247. Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha‐synucleinopathy. PloS One. 2010;5:e9313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 248. Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid‐beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. [DOI] [PubMed] [Google Scholar]
- 250. Santini E, Heiman M, Greengard P, Valjent E. Fisone G. Inhibition of mTOR signaling in Parkinson's disease prevents L‐DOPA‐induced dyskinesia. Sci Signal. 2009;2:ra36. [DOI] [PubMed] [Google Scholar]
- 251. Jiang X, Chen J, Song Q, Wang W, Zhang G, Li Y. Correlation between TSC1 gene polymorphism and epilepsy. Exp Therap Med. 2017;14:6238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Rosset C, Netto CBO, Ashton‐Prolla P. TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: a review. Genet Mol Biol. 2017;40:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Zeng LH, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Human Mol Genet. 2011;20:445–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, et al. Reversal of learning deficits in a Tsc2+/‐ mouse model of tuberous sclerosis. Nat Med. 2008;14:843–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Sunnen CN, Brewster AL, Lugo JN, Vanegas F, Turcios E, Mukhi S, et al. Inhibition of the mammalian target of rapamycin blocks epilepsy progression in NS‐Pten conditional knockout mice. Epilepsia. 2011;52:2065–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Tokuda S, Mahaffey CL, Monks B, Faulkner CR, Birnbaum MJ, Danzer SC, et al. A novel Akt3 mutation associated with enhanced kinase activity and seizure susceptibility in mice. Human Mol Genetics. 2011;20:988–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. D'Gama AM, Woodworth MB, Hossain AA, Bizzotto S, Hatem NE, LaCoursiere CM, et al. Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep. 2017;21:3754–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. D'Gama AM, Geng Y, Couto JA, Martin B, Boyle EA, LaCoursiere CM, et al. Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol. 2015;77:720–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Talos DM, Jacobs LM, Gourmaud S, Coto CA, Sun H, Lim KC, et al. Mechanistic target of rapamycin complex 1 and 2 in human temporal lobe epilepsy. Ann Neurol. 2018;83:311–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Chi X, Huang C, Li R, Wang W, Wu M, Li J, et al. Inhibition of mTOR pathway by rapamycin decreases P‐glycoprotein expression and spontaneous seizures in pharmacoresistant epilepsy. J Mol Neurosci. 2017;61:553–62. [DOI] [PubMed] [Google Scholar]
- 262. Park J, Zhang J, Qiu J, Zhu X, Degterev A, Lo EH, et al. Combination therapy targeting Akt and mammalian target of rapamycin improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metabol 2012;32:330–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Nikolaeva I, Crowell B, Valenziano J, Meaney D, D'Arcangelo G. Beneficial effects of early mTORC1 inhibition after traumatic brain injury. J Neurotrauma. 2016;33:183–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Zeng LH, Rensing NR, Wong M. Developing antiepileptogenic drugs for acquired epilepsy: targeting the Mammalian Target of Rapamycin (mTOR) pathway. Mol Cell Pharmacol. 2009;1:124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Baulac S. mTOR signaling pathway genes in focal epilepsies. Progress Brain Res. 2016;226:61–79. [DOI] [PubMed] [Google Scholar]
- 267. Muncy J, Butler I, Koenig M. Rapamycin reduces seizure frequency in tuberous sclerosis complex. J Child Neurol. 2009;24:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Guo D, Zeng L, Brody D, Wong M. Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PLoS One. 2013;8:e64078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Wang F, Chen F, Wang G, Wei S, Fang F, Kang D, et al. Rapamycin provides anti‐epileptogenic effect in a rat model of post‐traumatic epilepsy via deactivation of mTOR signaling pathway. Exp Ther Med. 2018;15:4763–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Talos D, Sun H, Zhou X, Fitzgerald E, Jackson M, Klein P, et al. The interaction between early life epilepsy and autistic‐like behavioral consequences: a role for the mammalian target of rapamycin (mTOR) pathway. PLoS One. 2012;7:e35885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Sliwa A, Plucinska G, Bednarczyk J, Lukasiuk K. Post‐treatment with rapamycin does not prevent epileptogenesis in the amygdala stimulation model of temporal lobe epilepsy. Neurosci Lett. 2012;509:105–9. [DOI] [PubMed] [Google Scholar]
- 272. Hester M, Hosford B, Santos V, Singh S, Rolle I, LaSarge C, et al. Impact of rapamycin on status epilepticus induced hippocampal pathology and weight gain. Exp Neurol. 2016;280:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Ali I, Chugh D, Ekdahl CT. Role of fractalkine‐CX3CR1 pathway in seizure‐induced microglial activation, neurodegeneration, and neuroblast production in the adult rat brain. Neurobiol Dis. 2015;74:194–203. [DOI] [PubMed] [Google Scholar]
- 274. Ali I, Silva JC, Liu S, Shultz SR, Kwan P, Jones NC, et al. Targeting neurodegeneration to prevent post‐traumatic epilepsy. Neurobiol Dis. 2019;123:100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Lotze MT, Tracey KJ. High‐mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–42. [DOI] [PubMed] [Google Scholar]
- 276. Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–43. [DOI] [PubMed] [Google Scholar]
- 277. Webster KM, Sun M, Crack P, O'Brien TJ, Shultz SR, Semple BD. Inflammation in epileptogenesis after traumatic brain injury. J Neuroinflammation. 2017;14:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Saletti PG, Ali I, Casillas‐Espinosa PM, Semple BD, Lisgaras C, Moshe SL, et al. In search of antiepileptogenic treatments for post‐traumatic epilepsy. Neurobiol Dis. 2019;123:86–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Semple BD, O'Brien TJ, Gimlin K, Wright DK, Kim SE, Casillas‐Espinosa PM, et al. Interleukin‐1 receptor in seizure susceptibility after traumatic injury to the pediatric brain. J Neurosci. 2017;37:7864–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Maroso M, Balosso S, Ravizza T, Iori V, Wright CI, French J, et al. Interleukin‐1beta biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics. 2011;8:304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Ravizza T, Noe F, Zardoni D, Vaghi V, Sifringer M, Vezzani A. Interleukin Converting Enzyme inhibition impairs kindling epileptogenesis in rats by blocking astrocytic IL‐1beta production. Neurobiol Dis. 2008;31:327–33. [DOI] [PubMed] [Google Scholar]
- 282. Noe FM, Polascheck N, Frigerio F, Bankstahl M, Ravizza T, Marchini S, et al. Pharmacological blockade of IL‐1beta/IL‐1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol Dis. 2013;59:183–93. [DOI] [PubMed] [Google Scholar]
- 283. Iori V, Iyer AM, Ravizza T, Beltrame L, Paracchini L, Marchini S, et al. Blockade of the IL‐1R1/TLR4 pathway mediates disease‐modification therapeutic effects in a model of acquired epilepsy. Neurobiol Dis. 2017;99:12–23. [DOI] [PubMed] [Google Scholar]
- 284. Chugh D, Ali I, Bakochi A, Bahonjic E, Etholm L, Ekdahl CT. Alterations in brain inflammation, synaptic proteins, and adult hippocampal neurogenesis during epileptogenesis in mice lacking synapsin2. PloS One. 2015;10:e0132366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Akin D, Ravizza T, Maroso M, Carcak N, Eryigit T, Vanzulli I, et al. IL‐1beta is induced in reactive astrocytes in the somatosensory cortex of rats with genetic absence epilepsy at the onset of spike‐and‐wave discharges, and contributes to their occurrence. Neurobiol Dis. 2011;44:259–69. [DOI] [PubMed] [Google Scholar]
- 286. Amhaoul H, Ali I, Mola M, Van Eitveldt A, Szewczyk K, Missault S, et al. P2X7 receptor antagonism inhibits seizure severity in a chronic model of temporal lobe epilepsy. Neuropharmacology. 2016. [DOI] [PubMed] [Google Scholar]
- 287. Jimenez‐Pacheco A, Diaz‐Hernandez M, Arribas‐Blazquez M, Sanz‐Rodriguez A, Olivos‐Ore LA, Artalejo AR, et al. Transient P2X7 receptor antagonism produces lasting reductions in spontaneous seizures and gliosis in experimental temporal lobe epilepsy. J Neurosci. 2016; 36:5920–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Zhu M, Chen J, Guo H, Ding L, Zhang Y, Xu Y. High Mobility Group Protein B1 (HMGB1) and Interleukin‐1beta as Prognostic Biomarkers of Epilepsy in Children. J Child Neurol. 2018;33:909–17. [DOI] [PubMed] [Google Scholar]
- 289. Fu L, Liu K, Wake H, Teshigawara K, Yoshino T, Takahashi H, et al. Therapeutic effects of anti‐HMGB1 monoclonal antibody on pilocarpine‐induced status epilepticus in mice. Sci Rep. 2017;7:1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Ito M, Takahashi H, Yano H, Shimizu YI, Yano Y, Ishizaki Y, et al. High mobility group box 1 enhances hyperthermia‐induced seizures and secondary epilepsy associated with prolonged hyperthermia‐induced seizures in developing rats. Metab Brain Dis. 2017;32:2095–104. [DOI] [PubMed] [Google Scholar]
- 291. Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, et al. Toll‐like receptor 4 and high‐mobility group box‐1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–9. [DOI] [PubMed] [Google Scholar]
- 292. Walker LE, Frigerio F, Ravizza T, Ricci E, Tse K, Jenkins RE, et al. Molecular isoforms of high‐mobility group box 1 are mechanistic biomarkers for epilepsy. J Clin Invest. 2017;127:2118–32. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 293. Mazarati A, Maroso M, Iori V, Vezzani A, Carli M. High‐mobility group box‐1 impairs memory in mice through both toll‐like receptor 4 and receptor for advanced glycation end products. Exp Neurol. 2011;232:143–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Limatola C, Ransohoff RM. Modulating neurotoxicity through CX3CL1/CX3CR1 signaling. Front Cell Neurosci. 2014;8:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Louboutin JP, Strayer DS. Relationship between the chemokine receptor CCR5 and microglia in neurological disorders: consequences of targeting CCR5 on neuroinflammation, neuronal death and regeneration in a model of epilepsy. CNS Neurol Disord Drug Targets. 2013;12:815–29. [DOI] [PubMed] [Google Scholar]
- 296. Cerri C, Caleo M, Bozzi Y. Chemokines as new inflammatory players in the pathogenesis of epilepsy. Epilepsy Res. 2017;136:77–83. [DOI] [PubMed] [Google Scholar]
- 297. Benson MJ, Manzanero S, Borges K. Complex alterations in microglial M1/M2 markers during the development of epilepsy in two mouse models. Epilepsia. 2015;56:895–905. [DOI] [PubMed] [Google Scholar]
- 298. Okuneva O, Korber I, Li Z, Tian L, Joensuu T, Kopra O, et al. Abnormal microglial activation in the Cstb(‐/‐) mouse, a model for progressive myoclonus epilepsy, EPM1. Glia. 2015;63:400–11. [DOI] [PubMed] [Google Scholar]
- 299. Ali I, Aertgeerts S, Le Blon D, Bertoglio D, Hoornaert C, Ponsaerts P, et al. Intracerebral delivery of the M2 polarizing cytokine interleukin 13 using mesenchymal stem cell implants in a model of temporal lobe epilepsy in mice. Epilepsia. 2017;58:1063–72. [DOI] [PubMed] [Google Scholar]