

Abstract
Background
Deletions removing 100s–1000s kb of DNA, and variable numbers of poorly characterised genes, are often found in patients with a wide range of developmental abnormalities. In such cases, understanding the contribution of the deletion to an individual’s clinical phenotype is challenging.
Methods
Here, as an example of this common phenomenon, we analysed 41 patients with simple deletions of ~177 to ~2000 kb affecting one allele of the well-characterised, gene dense, distal region of chromosome 16 (16p13.3), referred to as ATR-16 syndrome. We characterised deletion extents and screened for genetic background effects, telomere position effect and compensatory upregulation of hemizygous genes.
Results
We find the risk of developmental and neurological abnormalities arises from much smaller distal chromosome 16 deletions (~400 kb) than previously reported. Beyond this, the severity of ATR-16 syndrome increases with deletion size, but there is no evidence that critical regions determine the developmental abnormalities associated with this disorder. Surprisingly, we find no evidence of telomere position effect or compensatory upregulation of hemizygous genes; however, genetic background effects substantially modify phenotypic abnormalities.
Conclusions
Using ATR-16 as a general model of disorders caused by CNVs, we show the degree to which individuals with contiguous gene syndromes are affected is not simply related to the number of genes deleted but depends on their genetic background. We also show there is no critical region defining the degree of phenotypic abnormalities in ATR-16 syndrome and this has important implications for genetic counselling.
Keywords: ATR16, thalassemia, CNV, developmental delay
Introduction
Cytogenetic, molecular genetic and more recently, next-generation sequencing (NGS) approaches have revealed CNVs in the human genome ranging from 1 to 1000s of kilobases (kb).1 2 CNVs are common in normal individuals and have been identified in ~35% of the human genome.1 When present as hemizygous events, in phenotypically ‘normal’ individuals, these imbalances are considered benign; however, CNVs are also among the most common causes of human genetic disease and they have been associated with a wide range of developmental disabilities present in up to 14% of the population.3
CNVs have been shown to play an important role in neurodevelopmental disorders including autism spectrum disorder, bipolar disorder and schizophrenia as well as influencing broader manifestations such as learning disabilities, abnormal physical characteristics and seizures.4 Some CNVs occur recurrently in association with one particular phenotype: for example, deletions within 16p11.2 and/or chromosome 22q are frequently associated with autism, and deletions within 15q13.3 and 1q21.1 are found in schizophrenia. However, the impact of most CNVs on phenotype is much less clear.4 Difficulty in interpreting CNVs particularly occurs when they result from complex rearrangements such as those associated with unbalanced translocations, inversions and imprinting effects.
To understand the principles and mechanisms by which CNVs lead to developmental abnormalities we have simplified the issue by studying the relationship between uncomplicated deletions within the region ~0.3 to ~2 Mb in the subtelomeric region of chromosome 16 and the resulting phenotypes. The 41 individuals studied here (comprising 12 new and 29 previously reported cases) represent a cohort of patients with the α-thalassaemia mental retardation (MR) contiguous gene syndrome, involving the chromosomal region 16p13.3, termed ATR-16 syndrome (MIM 141750).5
Individuals studied here have monosomy for various extents of the gene-rich distal region at 16p13.3 and all individuals with ATR-16 syndrome have α-thalassaemia because two of the four paralogous α-globin genes are deleted (--/αα) and this manifests as mild hypochromic microcytic anaemia. In combination with a common small deletion involving one α-gene on the non-paralogous allele (--/-α), patients may have a more severe form of α-thlassaemia referred to as HbH disease.6 Some patients also have MR, developmental abnormalities and/or speech delay and facial dysmorphism. The most severe cases also manifest abnormalities of the axial skeleton. By precisely defining the 16p13.3 deletions in 11 cases (with a further 8 characterised by microarray) we address whether the associated neurological and developmental defects are simply related to the size of the deletions and the number of genes removed and whether there are critical haploinsufficient genes within this region. Our findings suggest that while the loss of an increased number of genes tends to underlie more severe phenotypic abnormalities, the genetic background in which these deletions occur contributes to the occurrence of MR and developmental abnormalities.
Finally, this subgroup of ATR-16 patients also allowed us to address two long-standing questions associated with large subcytogenetic deletions: those of compensatory gene expression and telomere proximity effect (TPE) in cases of telomere repaired chromosomal breakages.
Methods
Patients
Here, we focus on a cohort of patients with pure monosomy within 16p13.3 to clarify the effect of the deletion. In this work, we identify or refine the breakpoints of 14 deletions in a total of 19 individuals (including 9 cases from 4 families designated TN, TY, CS and SH and 10 singleton cases (OY, LA, YA, BA, NL, CJ, MY, BAR, IM and LIN)). In addition, we review 11 cases from 2 families (designated BF and F) and 11 singleton cases (JT, CV, AB, GZ, GIB, DO, SCH, PV, FT, BO, HN). Together this amounts to 20 familial cases from 5 pedigrees and 21 singletons amounting to a total of 41 patients.
Fluorescent in situ hybridisation
FISH studies were carried out on fixed chromosome preparations as previously described7 from each patient using a series of cosmids covering the terminal 2 Mb of chromosome 16p (online supplementary table 1). FISH studies were also performed using probes specific for the subtelomeric regions of each chromosome in order to exclude any cryptic chromosomal rearrangements. Subtelomeric rearrangements were detected as previously described.8 9
jmedgenet-2019-106528supp001.pdf (852.8KB, pdf)
Southern blotting
Single copy probes labelled with α32P-dCTP were synthesised and used to hybridise Southern blots of DNA isolated from transformed lymphoblastoid cells.
PCR detection of chromosomal deletions
DNA was extracted from mouse/human hybrid cell lines or transformed lymphoblastoid lines. Based on FISH results with chromosome 16 cosmids, primer pairs were designed located at regular intervals across the breakpoint clone. To refine the 16p breakpoint, PCR amplification was performed using normal and abnormal patient hybrid DNA obtained from mouse erythroleukaemia (MEL cells) fused to patient cells and selected to contain a single copy of human chromosome 16 generated as previously described10 as template. A positive PCR indicated the sequence was present; a negative PCR indicated it was deleted.
Telomere-anchored PCR amplification
Telomere-anchored PCR was undertaken using a primer containing canonical telomeric repeats in conjunction with a reverse primer specific for the normal 16p sequence (primer sequences provided in online supplementary table 2). Telomere repeat primers hybridise at any location in telomere repeats so heterogeneous amplification products are produced. Amplification products were purified and digested with restriction endonucleases BamHI or EcoRI and products ligated into appropriately prepared pBluescript. Resulting colonies were screened for inserts and DNA Sanger sequenced.
Quantification of gene expression
Total RNA was isolated from Epstein-Barr virus transformed lymphoblastoid cell lines for 11 patients (OY, TY, BA, MY, BO, CJ, YA, TN (Pa), SH (Pa), LIN and IM) and 20 control individuals using TRI reagent. In the case of OY, the genes MRPL28, TMEM8, NME4, DECR2 and RAB11FIP3 were excluded from the analysis as they are proximal to the deleted region and are not hemizygous in this patient. In the case of CJ, POLR3K, C16ORF33, MPG and C16ORF35 are excluded as they are distal to the interstitial deletion in this patient. cDNA synthesis was performed with the AffinityScript kit (Stratagene). Where gene expression was measured by quantitative real-time PCR, TaqMan Gene Expression Assays Applied Biosystems (ABI, www.appliedbiosystems.com) were used. Genes and assay numbers are given in online supplementary information.
Microarray analysis
Details of the microarray platforms used for each patient are given in online supplementary information.
Whole genome sequencing
Whole genome sequencing (WGS) was carried out, using DNA from the three affected members of the TN family and YA, at Edinburgh Genomics, The University of Edinburgh. The pathogenicity of each variant was given a custom deleterious score based on a six-point scale,11 calculated using output from ANNOVAR.12 This was used to prioritise variants present in the hemizygous region of chr16p13.3 in each patient and also genome-wide.
Results
Clinical features of ATR-16 syndrome
In addition to the α-thalassaemia, manifesting as a microcytic anaemia (identified by full blood count) that is always present, common features of ATR-16 syndrome include speech delay, developmental delay and a variable degree of facial dysmorphism and, in severe cases, abnormalities of the axial skeleton. Individual case reports are provided in online supplementary information; newly cloned breakpoint sequences are shown in figure 1; deletions are shown in figure 2 and phenotypic abnormalities are summarised in table 1. Deletions larger than 2000 kb including the PKD1 and TSC2 genes lead to severe MR with polycystic kidney disease and tuberous sclerosis, respectively.13
Figure 1.
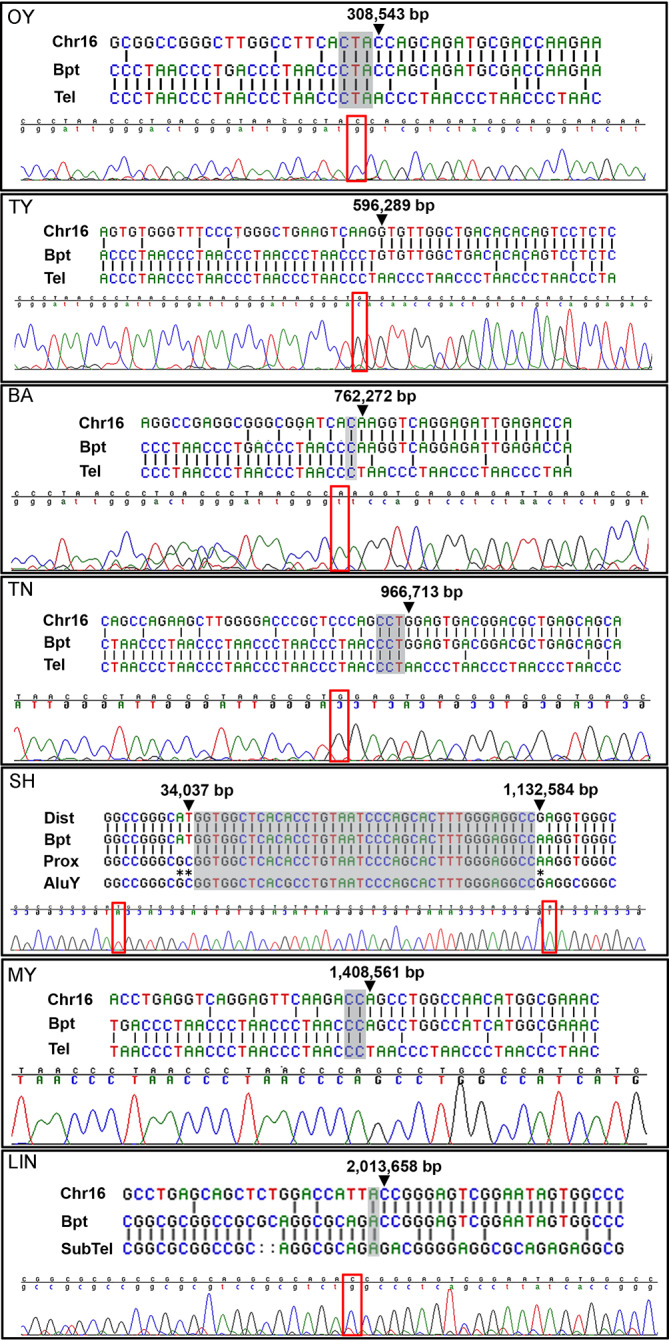
Chromosome 16 breakpoint sequences. DNA sequences at ATR-16 breakpoints. Patient codes are given in the upper left of each panel. For each case, alignment of the two normal sequences is shown with sequence from the derivative chromosome (upper) with chromatogram traces traversing each breakpoint (lower). Areas of ambiguity are highlighted with grey boxes and the location of the last unambiguous base pair(s) are denoted by arrowheads and red boxes. Chr16, normal chromosome 16 sequence; Bpt, breakpoint sequence; Tel, telomere repeat sequence; SubTel, subtelomere repeat sequence; Prox, proximal chromosome 16 sequence; Dist, distal chromosome 16 sequence; AluY, AluY repetitive element. Asterisks indicate informative polymorphisms allowing sequence origins to be identified. For patients MY and OY, a telomere primer with a mismatched G nucleotide was used.
Figure 2.

Summary of ATR-16 deletions. Upper: HiC interaction map showing interactions across the terminal 2 Mb of chromosome 16 at a 5 kb resolution in K562 cells (data from Rao et al 40). This shows how the ATR-16 deletions detailed in the lower section may impact the genome organisation. Middle: the positions of the α-globin cluster and other genes within this region are indicated. The α-globin genes and genes that, when mutated, are associated with tuberous sclerosis and adult polycystic kidney disease are shown in shaded boxes. Lower: the extent of each deletion is shown with the patient code (left). Deletions shown in green cause no other abnormalities apart from α-thalassaemia and those in red cause at least one other abnormality present in ATR-16. Solid bars indicate regions known to be deleted and fine lines show regions of uncertainty. Asterisks indicate individuals whose deletion breakpoints have been cloned or refined in this work.
Table 1.
ATR-16 syndrome phenotypic severity
| Case* | Sex | Deletion coordinates (hg18) | Methods | Origin | Mechanism | MR | AT | SD | DD | FD | SA | Reference(s) |
| JT | F | 34,113 bp to 301,556 bp† | F, SB, S | Mat | De novo | – | + | – | – | – | – | Horsley, 20019 |
| OY‡ | F | 0 bp to 308,540 bp | F, SB, S | Pat | De novo | – | + | – | – | – | – | This study |
| CS‡(+1)§ | F | ~36,766 bp to 328,247 bp | A | Pat | Inherited | – | + | – | – | – | – | This study |
| BF (+5)§ | M | 166,680 bp to 342,681 bp | WGS | na | De novo | – | + | – | – | – | – | Heireman etal 24 |
| CV | F | ~1 22 000 bp to 2 99 000–3 75 000 bp | M | na | na | – | + | – | – | – | – | Coelho etal 41 |
| AB | na | 0–45,799 bp to 3 50 916–4 00 279 bp | M | na | na | – | + | – | – | – | – | Harteveld etal 42 |
| LA‡ | M | ~94,214bp to 502,227 bp | A | Pat | Inherited | – | + | + | +/- | – | – | This study |
| TY(MI)‡ | M | 0 bp to 596,289 bp | F, SB, S | na | na | – | + | – | – | – | – | This study |
| TY(Mi)‡ | F | 0 bp to 596,289 bp | F, SB, S | Pat | Inherited | – | + | – | – | – | – | This study |
| YA‡ | F | 0 bp to 747,840 bp | F, A | na | na | na | + | + | + | + | – | This study |
| BA‡ | F | 0 bp to 762,370 bp | F, SB, S | Pat | De novo | – | + | – | + | – | – | Daniels etal 43 |
| GZ | M | 0–45,799 bp to 8 69 698–9 00 907 bp | M | Mat | Inherited | + | – | – | – | – | Harteveld etal 18 | |
| TN(Pa)‡ | F | 0 bp to 966,710 bp | F, SB, S | Mat | De novo | +/- | + | + | + | + | – | Daniels etal 43 |
| TN(Pe)‡ | M | 0 bp to 966,710 bp | F, SB, S | Mat | Inherited | + | + | + | + | + | – | Daniels etal 43 |
| TN(Al)‡ | M | 0 bp to 966,710 bp | F, SB, S | Mat | Inherited | + | + | + | + | + | – | Daniels etal 43 |
| FI.2 | F | 0–45,799 bp to~9 76 591 bp | M | na | na | – | + | – | – | – | – | Bezerra etal 20 |
| FII.1 | M | 0–45,799 bp to~9 76 591 bp | M | Mat | Inherited | – | + | – | – | – | – | Bezerra etal 20 |
| FII.2 | M | 0–45,799 bp to~9 76 591 bp | M | Mat | Inherited | – | + | – | – | – | – | Bezerra etal 20 |
| FII.4 | F | 0–45,799 bp to~9 76 591 bp | M | Mat | Inherited | – | + | – | – | – | – | Bezerra etal 20 |
| FIII.1 | M | 0–45,799 bp to~9 76 591 bp | M | Mat | Inherited | – | + | – | – | – | – | Bezerra etal 20 |
| GIB | F | ~1 00 000 bp to~1,000,000 bp | F, A | na | De novo | + | + | + | + | + | – | Gibson, 200844 |
| SH(Pa)ঠ| M | 34,037 bp to 1,132,584 bp | F, SB, S | Mat | Inherited | + | + | na | + | + | + | This study |
| SH(Ju)ঠ| F | 34,037 bp to 1,132,584 bp | F, SB, S | na | na | Р| + | na | Р| Р| Р| This study |
| NL‡ | M | 0–23 949 bp to~1,246,849 bp | A, M | na | De novo | – | + | – | – | – | – | Phylipsen etal 45; This study |
| DO | F | 0 bp to 1,175,000–1,805,487 bp | SB | Mat | Unknown | + | + | + | + | + | – | Wilkie etal 5 |
| CJ‡ | M | 120,000 bp to 1,357,000 bp | F, A | Mat | De novo | + | + | + | + | + | + | This study |
| MY‡ | F | 0 bp to 1,408,950 bp | F, SB, S | Mat | De novo | + | + | + | + | + | – | This study |
| BAR‡ | M | 0–23,949 bp to~1,440,000 bp | A, M | na | De novo | – | + | – | – | – | – | This study |
| SCH | M | ~281,65 bp to 1,447,989 bp | F, A, M | na | De novo | + | + | + | + | + | + | Scheps etal 28 |
| PV | M | 0–45,799 bp to 1,615,979–1,730,426 bp | M | na | De novo | + | + | + | + | + | + | Harteveld etal 18 |
| FT | F | 0–45,799 bp to 1,880,277–1,913,866 bp | M | na | De novo | + | + | + | + | + | + | Harteveld etal 18 |
| BO | M | 0 bp to 1,886,763 bp | C, F, SB, S | Pat | De novo | + | + | na | + | + | + | Wilkie etal 5; Lamb etal 46; Daniels etal 43 |
| HN | M | 0–45,799 bp to 1,913,923–1,928,982 bp | M | na | De novo | + | + | + | + | + | + | Harteveld etal, 200718 |
| IM‡ | F | 0 bp to 2,011,646 bp | F, SB, A | na | na | + | + | – | + | +/- | + | Felice 47; Fei etal 48; Daniels etal 43 |
| LIN‡ | F | 0 bp to 2,013,657 bp | F, SB, S | Pat | De novo | + | + | + | + | + | – | Lindor etal 49; Daniels etal 43 |
+ indicates presence of an abnormality; – indicates absence and +/− indicates borderline assessment.
Methods column summarises the methods used to refine or identify the breakpoint:C, cytogenetics; F, FISH; WGS, Whole Genome Sequencing; M, MLPA; SB, Southern blot; A, microarray, S, breakpoint has been DNA sequenced.
*ATR-16 individuals are identified by unique codes, references are shown in figure 2. Pale green rows indicate ATR-16 individuals with only alpha-thalassemia, yellow rows indicate ATR-16 individuals also have at least one other abnormality but no defects of the axial skeleton and orange rows indicate the individual also has skeletal defects.
†40 bp ambiguity, values taken from midpoint
‡Indicates individuals whose deletion breakpoints have been cloned or refined in this work.
§There numbers refer to other family members who carry this deletion and have no associated abnormalities apart from alpha-thalassemia
¶Individuals have discordant abnormalities, most likely due to a deletion in NRXN1.
A, microarray; AT, alpha-thalassaemia; C, cytogenetics; DD, developmental delay; F, FISH; FD, facial dysmorphism; M, Multiplex Ligation-dependent Probe Amplification (MLPA); MR, mental retardation; na, data not available; S, breakpoint has been DNA sequenced; SA, skeletal abnormalities; SB, Southern blot; SD, speech delay; WGS, whole genome sequencing.
Twelve individuals from 9 pedigrees are reported here for the first time (OY, CS, CS (father), LA, TY (MI), TY (Mi), YA, SH (P), SH (Ju), CJ, MY and BAR) and we refine or identify the breakpoints in five previously reported cases (BA, TN, IM, NL, LIN). We define breakpoints at the DNA sequence level in 7 of the 14 pedigrees studied (figure 1), 6 of which have been repaired by the addition of a telomere or subtelomere. In the remaining family (SH), the deletion is interstitial and mediated by repeats termed short interspersed nuclear elements.
Identification of co-inherited deleterious loci
Six individuals from four families (LA, BA, YA and TN) have 16p13.3 deletions smaller than 1 Mb and yet show relatively severe abnormalities. To test whether 16p13.3 deletions of <1 Mb may be unmasking deleterious mutations on the intact chromosome 16 allele in severely affected patients, we performed WGS where DNA was available (YA and the three affected members of the TN family) and considered only coding variants in the hemizygous region of chromosome 16. However, only common variants (allele frequency >5%) were present (online supplementary table 3) suggesting the cause(s) of the relatively severe phenotypes in these patients reside elsewhere in the genome. To identity rare variants genome-wide, we considered only those absent from the publicly available databases. This analysis yielded 14 variants shared between the three affected individuals of family TN (online supplementary table 4). Of these, only one (chr15:64 782 684 G>A) affects a gene possibly involved in the broader ATR-16 phenotypic abnormalities. This change leads to a R12X nonsense mutation in SMAD6, a negative regulator of the bone morphogenetic protein signalling pathway. Heterozygous mutations in SMAD6 have been reported to underlie craniosynostosis, speech delay, global developmental delay, fine motor impairment and aortic valve abnormalities with variable penetrance (see ‘Discussion’ section). We checked inheritance of the SMAD6 R12X variant in members of the TN family where samples were available and found it most likely came from the unaffected grandmother (individual I,2 in online supplementary figure 4). A phenotypically normal elder sister also inherited this variant. These findings suggest that coinheritance of this SMAD6 loss of function variant with the chromosome 16 deletion may lead to the increased severity of the ATR-16 syndrome.
Further evidence that the effect of ATR-16 deletions is modified by other loci comes from patients SH (Ju) and SH (Pa), who harbour the same chromosome 16 deletion. Patient SH (Pa) has developmental delay and skeletal abnormalities, however, his mother SH (Ju) does not have craniofacial nor skeletal abnormalities nor developmental delay, although she suffers from severe anxiety and depression (see figures 1 and 2 and online supplementary information). Genome-wide microarray analysis revealed that both SH (Ju) and SH (Pa) harbour a ~133 kb deletion on the short arm of chromosome 2 including exons 5–13 of NRXN1 (online supplementary figure 1).
Chromatin structure
Recent reports demonstrate chromosomal rearrangements, including deletions, can result in aberrant DNA domain topology and illegitimate enhancer-promoter contact causing gene misexpression.14 Chromatin contact frequency is shown for the terminal 2 Mb of chromosome 16 in figure 2 to illustrate the effect of the deletions reported here on the chromatin structure. The deletion in BA removes ~50% of the self-interacting domain in which CHTF18, RPUSD1, GNG13 and LOC388199 reside, thereby potentially removing cis-acting regulatory elements of these genes, although the genes themselves remain intact. In the case of CJ, the deletion brings the powerful α-globin enhancer cluster15 into proximity of CRAMP1L and may cause its aberrant expression in developing erythroblasts. Although topologically associating domains have been reported to be stable structures,14 many chromatin contacts are now known to vary in a tissue-specific fashion16 and therefore it is not possible to predict which genes may be aberrantly expressed in any given tissue as a result of the ATR-16 deletions.
Compensatory gene expression
One explanation for the relatively mild abnormalities in many cases of ATR-16 syndrome with deletions up to 900 kb may be compensatory transcriptional upregulation of the homologues of deleted genes on the undamaged chromosome 16. This has been described as part of the mechanism of genetic compensation, also termed genetic robustness.17 To assay for compensatory gene transcription, we used qPCR to measure expression of 12 genes within the terminal 500 kb of chromosome 16 in lymphoblastoid cells from 20 normal individuals and from 11 patients with monosomy for the short-arm of chromosome 16 and found no evidence of compensatory upregulation: transcripts of all deleted genes were present at ~50% of the normal levels in these cells (figure 3B). It is possible that other genes in downstream pathways affected by haploinsufficiency may be transcriptionally upregulated, however, the mechanisms underlying this are complex and beyond the scope of this study.
Figure 3.

Effect of breakpoints and deletions on gene expression. (A) Schematic view of breakpoint positions in three patients with nearby expressed polymorphic genes. Genes are represented by black bars and transcription direction is indicated by an arrow. Polymorphic bases are shown by red letters indicating variant alleles and the distance of the promoter of each measured gene from the breakpoint is shown. On the right of each panel chromatograms show the quantity of the allele present in genomic DNA and cDNA from patient lymphoblastoid cells. (B) Expression of 12 genes within 500 kb of the tip of the short arm of chromosome 16 in lymphoblastoid cells from 20 normal individuals, shown as reference (red column) and from 11 ATR-16 individuals hemizygous for each gene. Measurements in control cells are normalised to 1 (red column), relative expression in ATR-16 patient cells is shown in blue. Error bars show SD. Gene expression was measured in triplicate and data combined.
Telomere position effect
To determine the effect of telomere proximity on genes adjacent to telomere-healed breakpoints, we measured their expression relative to the allele present in a normal chromosomal context. To achieve this, we screened them for informative SNPs in EBV transformed lymphoblastoid cells generated from ATR-16 patients. The phase of polymorphisms was established using MEL cells fused to patient cells and selected to contain a single copy of human chromosome 16, generated as previously described.10 Expressed coding polymorphisms were present in genes whose promoters are <60 kb away from breakpoints in three patients: TY, MY and BA.
For TY, the nearest gene expressed in lymphoblastoid cells containing a coding polymorphism is WDR90, the promoter of which is ~43.1 kb from the abnormally appended telomere (figure 3A). For BA, CHTF18 is the closest expressed polymorphic gene with the promoter ~16.3 kb away from the breakpoint. For MY, CLCN7 is the closest gene expressed in lymphoblastoid cells to contain a polymorphism, the promoter of this gene is ~56.1 kb away from the telomere stabilised lesion. To determine whether either allele of each of these three genes is silenced we prepared genomic DNA and cDNA from each cell sample and Sanger sequenced amplified fragments containing informative polymorphisms. We compared peak heights of polymorphic bases in chromatograms derived from cDNA and genomic DNA. None of the alleles assayed in the three patients tested showed any evidence of a repressive effect (figure 3A).
Discussion
We characterised deletions leading to simple monosomy of the short arm of chromosome 16 that cause ATR-16 syndrome. Many ATR-16 patients suffer from neurodevelopmental abnormalities and one of the main questions in this disease, and in the study of CNVs in general, is how deletion size relates to phenotypic abnormalities. The monosomies analysed here show the likelihood and severity of neurological and developmental abnormalities increases with deletion size, however, there is no clear correlation.
The deletions in patients reported and reviewed here range from ~0.177 to ~2 Mb. Previous studies suggest the critical region leading to abnormalities in addition to α-thalassaemia is an 800 kb region between ~0.9 and~1.7 Mb from the telomere of chromosome 16 p18 and SOX8 has been proposed as the critical haploinsufficient gene.19 However, a report of a family with no developmental delay nor MR harbouring a 0.976 Mb deletion, suggests deletions of SOX8 may not lead to MR with complete penetrance and any ‘critical region’ for MR must start after this point20 (family ‘F’ in figure 2 and table 1). Supporting this we report patients NL and BAR, who have deletions of ~1.14 and ~1.44 Mb, respectively and show no abnormalities beyond α-thalassaemia.
By contrast, we find LA (deletion ~408 kb) has speech delay and YA (deletion ~748 kb) has speech and developmental delay and facial dysmorphism (figure 2, table 1). Family members of YA also have omphalocele, umbilical hernia and pyloric stenosis suggesting there are other loci rendering YA susceptible to developmental abnormalities. BA (deletion ~762 kb), who has a similarly sized deletion to YA, has developmental delay but no other abnormalities. Three other patients with deletions <1 Mb (TN (Pa), TN (Pe) and TN (Al)) have speech delay and facial dysmorphism. This suggests the risk of developmental and neurological abnormalities arises from much smaller terminal chromosome 16 deletions (~400 kb) than previously reported.
In family SH, we have identified a strong candidate for the discordant abnormalities: deletion of NRXN1. NRXN1 encodes a cell surface receptor involved in the formation of synaptic contacts and has been implicated in autism spectrum disorder, facial dysmorphism, anxiety and depression, developmental delay and speech delay.21 There is a higher incidence of autism in males than in females, with a ratio of 3.5 or 4.0 to 1.22 This phenomenon is also specifically found in individuals with autism resulting from rearrangements of NRXN1 23, where two affected siblings inherited a deletion of NRXN1 from their unaffected mother. It is therefore possible SH (Ju) is protected by her gender from the effects of NRXN1 disruption while the neurological and skeletal abnormalities in SH (Pa) arise from the complex interaction of NRXN1 perturbation with his gender and coinheritance of the 16p13.3 deletion. Abnormalities in siblings of YA and BF24 also suggest there may be other predisposing genes. Such loci compromise genetic robustness proposed to minimise the effect of deletions and loss of function mutations.17 Another example is the SMAD6 R12X nonsense mutation present in all three affected members of family TN. Some patients with loss of function mutations in SMAD6 have neurological abnormalities25 while others have not,26 suggesting variable penetrance. Our analysis shows there are no likely pathogenic variants on the hemizygous region of chromosome 16 in TN, suggesting modifying loci are present elsewhere in the genome. These may be rare variants (such as those identified in the TN and SH families) or common variation; a recent study shows that common genetic variants (allele frequency >5% in the general population) contribute 7.7% of the variance of risk to neurodevelopmental disorders,27 highlighting the complexity of this area.
Together these observations suggest that monosomy for 16p13.3 unmasks the effects of other variants genome-wide. This is supported by findings in SCH who has a very similar deletion to BAR and is more severely affected possibly owing to the presence of other CNVs.28 At the other end of the spectrum, large ATR-16 deletions may be associated with relatively mild abnormalities. In LIN (16p13.3 deletion ~2000 kb), there are no abnormalities of the axial skeleton and very mild facial dysmorphism. Similarly, in the case of IM (deletion size ~2000 kb), facial abnormalities are very mild and there is no evidence of language delay. Here, we propose chromosome 16p13.3 deletions larger than 400 kb predispose to MR and associated developmental abnormalities, however, we find no evidence for critical regions that incrementally worsen ATR-16 syndrome abnormalities.
In this work, we were able to provide evidence that CNVs and other variation genome-wide is likely to impact ATR-16 severity. However, we would not recommend this approach is yet widely applied as the impact of novel CNVs and sequence variants is challenging to interpret, especially when co-inherited with a terminal chromosome 16 deletion. We were unable to expand genome-wide analyses beyond the six patients (SH (Pa), SH (Ju), TN (Pa), TN (Pe), TN (Al) and YA) studied by microarray or WGS here and so cannot exclude the possibility variation genome-wide may influence the presentation of other ATR-16 patients reported and reviewed here. Previous work in human cells has shown that telomeres may affect chromatin interactions at distances of up to 10 Mb away from the chromosome ends29 reducing expression of the intervening genes. This phenomenon, termed TPE, is thought to be mediated by the spreading of telomeric heterochromatin to silence nearby genes. In budding yeast, this effect can extend a few kb towards the subtelomeres, although in some cases yeast telomeres can loop over longer distances30 and repress genes up to 20 kb away from the end of the chromosome. However, we could not detect compensatory upregulation of the homologues of deleted genes. Recently, a case of ATR-16 was reported with a~948 kb deletion who presented with a neuroblastoma in utero.31 These authors speculate that haploinsufficiency of the tumour suppressor AXIN1 may have contributed to the neuroblastoma. Our finding that the remaining AXIN1 allele shows no compensatory expression supports this hypothesis.
Terminal chromosome deletions are the most common subtelomeric abnormalities.32 The 16p deletions reported here are among the most common terminal deletions along with 1p36 deletion syndrome, 4p terminal deletion (causing Wolf-Hirschhorn syndrome), 5p terminal deletions (causing Cri-du-chat syndrome), 9q34 deletion syndrome and 22q terminal deletion syndrome. Despite their impact on human health, the mechanisms and timing underlying telomeric breakage remain unknown. Findings of terminal deletions of 16p reported and reviewed here and smaller deletions previously reported by our laboratory33 compared with more complex rearrangements at 1p, 22q and 9qter implies different chromosomes are predisposed to different breakage and rescue mechanisms. ATR-16 deletions are equally likely to have arisen on the maternal or paternal chromosome. There is no evidence that the parental origin affects the phenotypic severity of the ATR-16 syndrome, suggesting imprinting does not play a role in ATR-16 pathogenesis.
The presence of high and low copy number repeats at breakpoints may play a role in stimulating the formation of non-recurrent breakpoints.34 Low copy repeats (LCRs) are also mediators of non-allelic homologous recombination35 and could be involved in chromosome instability leading to terminal deletion. Following breakage, chromosomes can acquire a telomere by capture or de novo telomere addition, which is thought to be mediated by telomerase and this is stimulated by the presence of a telomeric repeat sequence to which the RNA subunit of telomerase can bind.36 We found 5 out of 6 telomere healed events share microhomology with appended telomeric sequence. This is the same ratio (5 out of 6 breakpoints with microhomology) described by Flint et al and supported by Lamb et al (1 out of 1) giving a total 11 out of 13 reported telomere-healed breaks characterised on 16p13.3 share microhomology with appended telomere sequences, strongly suggesting a role for internal telomerase binding sites.37 It may also be that telomerase binding to internal binding sites may inappropriately add telomeres and thereby contribute to the generation of the breakpoints.
The lack of evidence for TPE in silencing gene expression is surprising and at variance with previous findings,38 which show that TPE can influence gene expression at least 80 kb from the start of telomeric repeats. However, TPE is likely to be context and cell type dependent. Additionally, because of the lack of informative expressed polymorphisms in the patients studied here it was not possible for us to assay expression of genes immediately adjacent to telomeres and a more comprehensive screen may reveal TPE-mediated gene silencing closer to the telomere. Additionally, when the area of chromatin interaction (visualised by HiC) is considered (figure 2), contact domains for many genes adjacent to chromosomal breaks are severely disrupted. This is likely to include the loss of cis-acting regulatory elements and may bring the genes under the control of illegitimate regulatory elements.39 Therefore, it is likely that genes adjacent to breakpoints would be incorrectly spatiotemporally expressed.
This work substantially increases the number of fully characterised cases of ATR-16 syndrome reported and provides a uniquely well-characterised model for understanding how sporadic deletions giving rise to extended regions of monosomy may affect phenotype. The findings show larger deletions have a greater impact, but importantly our analysis suggesting there is no critical region defining the degree of phenotypic abnormalities has important implications for genetic counselling. Analysis of patients with uncomplicated deletions also revealed unexpected background genetic effects that alter phenotypic severity of CNVs.
Acknowledgments
The authors would like to thank Markissia Karagiorga-Lagana, MD, ex Director, Thalassaemia Unit, 'Aghia Sophia' Children's Hosital, Athens, Greece for referring case BAR.
Footnotes
Contributors: DRH and VJB conceived the project. CB, VJB and DRH wrote the paper. CB, JS, CLH, JB, EM, SWH, VJB, SK and AD-M performed experimental work. JT-S, PO, MK, AD-M and AOMW assessed patients and clinical data. All authors commented on the manuscript.
Funding: This work was supported by the Medical Research Council grant number: (MC_uu_12009).
Competing interests: None declared.
Patient consent for publication: Not required.
Ethics approval: This study was approved by the NRES Committee North West, Haydock, HRA NRES Centre, Manchester under the title ‘Establishing the molecular basis for atypical forms of thalassaemia’ (reference number MREC: 03/8/097) with written consent from patients and/or parents.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: All data relevant to the study are included in the article or uploaded as supplementary information.
References
- 1. Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet 2004;36:949–51. 10.1038/ng1416 [DOI] [PubMed] [Google Scholar]
- 2. MacDonald JR, Ziman R, Yuen RKC, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 2014;42:D986–92. 10.1093/nar/gkt958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG, Warren ST, Richard G, Compton JG, Fuller AE, Gliem TJ, Huang S, Collinson MN, Beal SJ, Ackley T, Pickering DL, Golden DM, Aston E, Whitby H, Shetty S, Rossi MR, Rudd MK, South ST, Brothman AR, Sanger WG, Iyer RK, Crolla JA, Thorland EC, Aradhya S, Ledbetter DH, Martin CL. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med 2011;13:777–84. 10.1097/GIM.0b013e31822c79f9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Merikangas AK, Corvin AP, Gallagher L. Copy-Number variants in neurodevelopmental disorders: promises and challenges. Trends Genet 2009;25:536–44. 10.1016/j.tig.2009.10.006 [DOI] [PubMed] [Google Scholar]
- 5. Wilkie AO, Buckle VJ, Harris PC, Lamb J, Barton NJ, Reeders ST, Lindenbaum RH, Nicholls RD, Barrow M, Bethlenfalvay NC, Hutz MH, Tolmie JL, Weatherall DJ, Higgs DR. Clinical features and molecular analysis of the alpha thalassemia/mental retardation syndromes. I. cases due to deletions involving chromosome band 16p13.3. Am J Hum Genet 1990;46:1112–26. [PMC free article] [PubMed] [Google Scholar]
- 6. Harteveld CL, Higgs DR. Alpha-Thalassaemia. Orphanet J Rare Dis 2010;5:13 10.1186/1750-1172-5-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buckle VJ, Rack K. Fluorescent in situ hybridisation : Davies KE, Human genetic disease analysis: a practical approach. Oxford: Oxford University Press, 1993: 59–80. [Google Scholar]
- 8. Knight SJL, Horsley SW, Regan R, Lawrie NM, Maher EJ, Cardy DLN, Flint J, Kearney L. Development and clinical application of an innovative fluorescence in situ hybridization technique which detects submicroscopic rearrangements involving telomeres. Eur J Hum Genet 1997;5:1–8. 10.1159/000484724 [DOI] [PubMed] [Google Scholar]
- 9. Horsley SW, Daniels RJ, Anguita E, Raynham HA, Peden JF, Villegas A, Vickers MA, Green S, Waye JS, Chui DHK, Ayyub H, MacCarthy AB, Buckle VJ, Gibbons RJ, Kearney L, Higgs DR. Monosomy for the most telomeric, gene-rich region of the short arm of human chromosome 16 causes minimal phenotypic effects. Eur J Hum Genet 2001;9:217–25. 10.1038/sj.ejhg.5200610 [DOI] [PubMed] [Google Scholar]
- 10. Zeitlin HC, Weatherall DJ. Selective expression within the human alpha globin gene complex following chromosome-dependent transfer into diploid mouse erythroleukaemia cells. Mol Biol Med 1983;1:489–500. [PubMed] [Google Scholar]
- 11. Fu W, O'Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Rieder MJ, Altshuler D, Shendure J, Nickerson DA, Bamshad MJ, Akey JM, NHLBI Exome Sequencing Project . Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 2013;493:216–20. 10.1038/nature11690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. European Polycystic Kidney Disease Consortium The polycystic kidney disease 1 gene encodes a 14 KB transcript and lies within a duplicated region on chromosome 16. The European polycystic kidney disease Consortium. Cell 1994;77:881–94. 10.1016/0092-8674(94)90137-6 [DOI] [PubMed] [Google Scholar]
- 14. Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, Borschiwer M, Haas SA, Osterwalder M, Franke M, Timmermann B, Hecht J, Spielmann M, Visel A, Mundlos S. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015;161:1012–25. 10.1016/j.cell.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hay D, Hughes JR, Babbs C, Davies JOJ, Graham BJ, Hanssen LLP, Kassouf MT, Oudelaar AM, Sharpe JA, Suciu MC, Telenius J, Williams R, Rode C, Li P-S, Pennacchio LA, Sloane-Stanley JA, Ayyub H, Butler S, Sauka-Spengler T, Gibbons RJ, Smith AJH, Wood WG, Higgs DR. Genetic dissection of the α-globin super-enhancer in vivo. Nat Genet 2016;48:895–903. 10.1038/ng.3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szalaj P, Plewczynski D. Three-dimensional organization and dynamics of the genome. Cell Biol Toxicol 2018;34:381–404. 10.1007/s10565-018-9428-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. El-Brolosy MA, Stainier DYR. Genetic compensation: a phenomenon in search of mechanisms. PLoS Genet 2017;13:e1006780 10.1371/journal.pgen.1006780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harteveld CL, Kriek M, Bijlsma EK, Erjavec Z, Balak D, Phylipsen M, Voskamp A, Capua Edi, White SJ, Giordano PC. Refinement of the genetic cause of ATR-16. Hum Genet 2007;122:283–92. 10.1007/s00439-007-0399-y [DOI] [PubMed] [Google Scholar]
- 19. Pfeifer D, Poulat F, Holinski-Feder E, Kooy F, Scherer G. The Sox8 gene is located within 700 kb of the tip of chromosome 16p and is deleted in a patient with ATR-16 syndrome. Genomics 2000;63:108–16. 10.1006/geno.1999.6060 [DOI] [PubMed] [Google Scholar]
- 20. Bezerra MACB, Araujo AS, Phylipsen M, Balak D, Kimura EM, Oliveira DM, Costa FF, Sonati MF, Harteveld CL. The deletion of SOX8 is not associated with ATR-16 in an HbH family from Brazil. Br J Haematol 2008;142:324–6. 10.1111/j.1365-2141.2008.07187.x [DOI] [PubMed] [Google Scholar]
- 21. The Autism Genome Project Consortium Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet 2007;39:319–28. 10.1038/ng1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Volkmar FR, Lord C, Bailey A, Schultz RT, Klin A. Autism and pervasive developmental disorders. J Child Psychol & Psychiat 2004;45:135–70. 10.1046/j.0021-9630.2003.00317.x [DOI] [PubMed] [Google Scholar]
- 23. Kirov G, Gumus D, Chen W, Norton N, Georgieva L, Sari M, O'Donovan MC, Erdogan F, Owen MJ, Ropers H-H, Ullmann R. Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum Mol Genet 2008;17:458–65. 10.1093/hmg/ddm323 [DOI] [PubMed] [Google Scholar]
- 24. Heireman L, Luyckx A, Schynkel KD, Dheedene A, Delaunoy M, Adam A-S, Gulbis B, Dierick J. Detection of a large novel α-thalassemia deletion in an autochthonous Belgian family. Hemoglobin 2019;43:112–5. 10.1080/03630269.2019.1625786 [DOI] [PubMed] [Google Scholar]
- 25. Timberlake AT, Choi J, Zaidi S, Lu Q, Nelson-Williams C, Brooks ED, Bilguvar K, Tikhonova I, Mane S, Yang JF, Sawh-Martinez R, Persing S, Zellner EG, Loring E, Chuang C, Galm A, Hashim PW, Steinbacher DM, DiLuna ML, Duncan CC, Pelphrey KA, Zhao H, Persing JA, Lifton RP. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife 2016;5 10.7554/eLife.20125. [Epub ahead of print: 10.7554/eLife.20125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luyckx I, MacCarrick G, Kempers M, Meester J, Geryl C, Rombouts O, Peeters N, Claes C, Boeckx N, Sakalihasan N, Jacquinet A, Hoischen A, Vandeweyer G, Van Lent S, Saenen J, Van Craenenbroeck E, Timmermans J, Duijnhouwer A, Dietz H, Van Laer L, Loeys B, Verstraeten A. Confirmation of the role of pathogenic Smad6 variants in bicuspid aortic valve-related aortopathy. Eur J Hum Genet 2019;27:1044–53. 10.1038/s41431-019-0363-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Niemi MEK, Martin HC, Rice DL, Gallone G, Gordon S, Kelemen M, McAloney K, McRae J, Radford EJ, Yu S, Gecz J, Martin NG, Wright CF, Fitzpatrick DR, Firth HV, Hurles ME, Barrett JC. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 2018;562:268–71. 10.1038/s41586-018-0566-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scheps KG, Francipane L, Nevado J, Basack N, Attie M, Bergonzi MF, Cerrone GE, Lapunzina P, Varela V. Multiple copy number variants in a pediatric patient with Hb H disease and intellectual disability. Am J Med Genet A 2016;170:986–91. 10.1002/ajmg.a.37532 [DOI] [PubMed] [Google Scholar]
- 29. Robin JD, Ludlow AT, Batten K, Magdinier F, Stadler G, Wagner KR, Shay JW, Wright WE. Telomere position effect: regulation of gene expression with progressive telomere shortening over long distances. Genes Dev 2014;28:2464–76. 10.1101/gad.251041.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bystricky K, Laroche T, van Houwe G, Blaszczyk M, Gasser SM. Chromosome looping in yeast: telomere pairing and coordinated movement reflect anchoring efficiency and territorial organization. J Cell Biol 2005;168:375–87. 10.1083/jcb.200409091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quadrifoglio M, Faletra F, Bussani R, Pecile V, Zennaro F, Grasso A, Zandonà L, Alberico S, Stampalija T. A case of prenatal neurocytoma associated with ATR-16 syndrome. J Ultrasound Med 2016;35:1359–61. 10.7863/ultra.15.07045 [DOI] [PubMed] [Google Scholar]
- 32. Ballif BC, Sulpizio SG, Lloyd RM, Minier SL, Theisen A, Bejjani BA, Shaffer LG. The clinical utility of enhanced subtelomeric coverage in array CGH. Am J Med Genet A 2007;143A:1850–7. 10.1002/ajmg.a.31842 [DOI] [PubMed] [Google Scholar]
- 33. Flint J, Craddock CF, Villegas A, Bentley DP, Williams HJ, Galanello R, Cao A, Wood WG, Ayyub H, Higgs DR. Healing of broken human chromosomes by the addition of telomeric repeats. Am J Hum Genet 1994;55:505–12. [PMC free article] [PubMed] [Google Scholar]
- 34. Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics 2008;1:4 10.1186/1755-8417-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet 2002;18:74–82. 10.1016/S0168-9525(02)02592-1 [DOI] [PubMed] [Google Scholar]
- 36. Hannes F, Houdt JV, Quarrell OW, Poot M, Hochstenbach R, Fryns J-P, Vermeesch JR. Telomere healing following DNA polymerase arrest-induced breakages is likely the main mechanism generating chromosome 4p terminal deletions. Hum Mutat 2010;31:1343–51. 10.1002/humu.21368 [DOI] [PubMed] [Google Scholar]
- 37. Yang D, Xiong Y, Kim H, He Q, Li Y, Chen R, Songyang Z. Human telomeric proteins occupy selective interstitial sites. Cell Res 2011;21:1013–27. 10.1038/cr.2011.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stadler G, Rahimov F, King OD, Chen JCJ, Robin JD, Wagner KR, Shay JW, Emerson CP, Wright WE. Telomere position effect regulates DUX4 in human facioscapulohumeral muscular dystrophy. Nat Struct Mol Biol 2013;20:671–8. 10.1038/nsmb.2571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schöpflin R, Kraft K, Kempfer R, Jerković I, Chan W-L, Spielmann M, Timmermann B, Wittler L, Kurth I, Cambiaso P, Zuffardi O, Houge G, Lambie L, Brancati F, Pombo A, Vingron M, Spitz F, Mundlos S. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016;538:265–9. 10.1038/nature19800 [DOI] [PubMed] [Google Scholar]
- 40. Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014;159:1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Coelho A, Picanço I, Seuanes F, Seixas MT, Faustino P. Novel large deletions in the human alpha-globin gene cluster: Clarifying the HS-40 long-range regulatory role in the native chromosome environment. Blood Cells Mol Dis 2010;45:147–53. 10.1016/j.bcmd.2010.05.010 [DOI] [PubMed] [Google Scholar]
- 42. Harteveld CL, Voskamp A, Phylipsen M, Akkermans N, den Dunnen JT, White SJ, Giordano PC. Nine unknown rearrangements in 16p13.3 and 11p15.4 causing alpha- and beta-thalassaemia characterised by high resolution multiplex ligation-dependent probe amplification. J Med Genet 2005;42:922–31. 10.1136/jmg.2005.033597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Daniels RJ, Peden JF, Lloyd C, Horsley SW, Clark K, Tufarelli C, Kearney L, Buckle VJ, Doggett NA, Flint J, Sequence HDR. Sequence, structure and pathology of the fully annotated terminal 2 Mb of the short arm of human chromosome 16. Hum Mol Genet 2001;10:339–52. 10.1093/hmg/10.4.339 [DOI] [PubMed] [Google Scholar]
- 44. Gibson WT, Harvard C, Qiao Y, Somerville MJ, Lewis MES, Rajcan-Separovic E. Phenotype–genotype characterization of alpha-thalassemia mental retardation syndrome due to isolated monosomy of 16p13.3. Am J Med Genet A 2008;146A:225–32. 10.1002/ajmg.a.32056 [DOI] [PubMed] [Google Scholar]
- 45. Phylipsen M, Chaibunruang A, Vogelaar IP, Balak JRA, Schaap RAC, Ariyurek Y, Fucharoen S, den Dunnen JT, Giordano PC, Bakker E, Harteveld CL. Fine-tiling array CGH to improve diagnostics for α- and β-thalassemia rearrangements. Hum Mutat 2012;33:272–80. 10.1002/humu.21612 [DOI] [PubMed] [Google Scholar]
- 46. Lamb J, Harris PC, Wilkie AO, Wood WG, Dauwerse JG, Higgs DR. De novo truncation of chromosome 16p and healing with (TTAGGG)n in the alpha-thalassemia/mental retardation syndrome (ATR-16). Am J Hum Genet 1993;52:668–76. [PMC free article] [PubMed] [Google Scholar]
- 47. Felice AE, Cleek MP, McKie K, McKie V, Huisman TH. The rare alpha-thalassemia-1 of blacks is a zeta alpha-thalassemia-1 associated with deletion of all alpha- and zeta-globin genes. Blood 1984;63:1253–7. 10.1182/blood.V63.5.1253.1253 [DOI] [PubMed] [Google Scholar]
- 48. Fei YJ, Liu JC, Walker EL, Huisman TH. A new gene deletion involving the alpha 2-, alpha 1-, and theta 1-globin genes in a black family with Hb H disease. Am J Hematol 1992;39:299–303. 10.1002/ajh.2830390412 [DOI] [PubMed] [Google Scholar]
- 49. Lindor NM, Valdes MG, Wick M, Thibodeau SN, Jalal S. De novo 16p deletion: ATR-16 syndrome. Am J Med Genet 1997;72:451–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jmedgenet-2019-106528supp001.pdf (852.8KB, pdf)