

Abstract
Integrating single-cell RNA sequencing (scRNA-seq) data with genotypes obtained from DNA sequencing studies facilitates the detection of functional genetic variants underlying cell type-specific gene expression variation. Unfortunately, most existing scRNA-seq studies do not come with DNA sequencing data; thus, being able to call single nucleotide variants (SNVs) from scRNA-seq data alone can provide crucial and complementary information, detection of functional SNVs, maximizing the potential of existing scRNA-seq studies. Here, we perform extensive analyses to evaluate the utility of two SNV calling pipelines (GATK and Monovar), originally designed for SNV calling in either bulk or single-cell DNA sequencing data. In both pipelines, we examined various parameter settings to determine the accuracy of the final SNV call set and provide practical recommendations for applied analysts. We found that combining all reads from the single cells and following GATK Best Practices resulted in the highest number of SNVs identified with a high concordance. In individual single cells, Monovar resulted in better quality SNVs even though none of the pipelines analyzed is capable of calling a reasonable number of SNVs with high accuracy. In addition, we found that SNV calling quality varies across different functional genomic regions. Our results open doors for novel ways to leverage the use of scRNA-seq for the future investigation of SNV function.
Introduction
Accurate measurement of genetic variants is key for investigating the relationship between genotypes and molecular level phenotypes such as gene expressions. Genotype arrays and recent developments of whole exon or whole genome sequencing techniques (1–3) have allowed us to accurately measure genotypes, often in terms of SNV, at the genome-wide scale (4). High throughput genomic sequencing studies have also allowed us to provide accurate measurements of different omic phenotypes such as transcriptomics. Pairing these two parallel technical developments have enabled the routine performance of large-scale molecular quantitative trait loci (QTL) mapping studies such as expression QTL (eQTL) studies, providing unprecedented insights into the molecular function of genetic variants (5–8). While most existing eQTL studies are performed at the tissue or organism level, with the development of single-cell RNA-seq, we are now able to characterize the function of genetic variants at the single-cell resolution or at sub-cell-type level (9, 10). For example, a few recent studies have collected a large number of individuals to perform eQTL mapping studies in scRNA-seq, identifying many functional variants that influence gene expression levels in a cell type-specific fashion (11–13).
Performing single-cell eQTL studies requires us to collect genotype information from either WGS or genotype array in conjunction with scRNA-seq (14). Unfortunately, due to limited starting material, sequencing cost, or the biological problem of focus, studies that collect both scRNA-seq data and genotype data are still a minority. Most existing scRNA-seq studies do not collect genotype data in accompany with RNA-seq data, which limits our ability to investigate the function of SNVs in the majority of existing scRNA-seq data. However, the sequencing reads collected in scRNA-seq contain valuable SNV information that could potentially allow us to call SNVs from scRNA-seq. Indeed, many previous studies have demonstrated that calling SNVs from bulk-RNA-seq data or other genomic sequencing data (e.g. ChIP-SEQ) is feasible and can maximize the use of data (11, 12, 15). Calling SNVs in genomic sequencing studies allow us to make full use of the same data to obtain both gene expression measurement and SNVs, facilitating the investigation of their relationship. For example, by determining the SNVs present in each ChIP-seq read, researchers are able to assign each read to an allele and study the methylation marks inherited from each parent to the offspring (15). As another example, calling SNVs in bulk RNA-seq facilitates powerful eQTL mapping and allelic-specific expression (ASE) analysis in natural primate populations, where samples are challenging to obtain, arrays are unavailable and DNA sequencing remains expensive (16). The only relevant methods in single-cell settings were developed to call SNVs in single-cell DNA-seq data (scDNA-seq) (12, 17). However, calling SNVs in scRNA-seq is likely more challenging than calling SNVs in scDNA-seq, as scRNA-seq often suffers from extremely low capture efficiency and low sequencing depth with reads covering only a fraction of the entire genome. Up to now, there is limited investigation and comparison of the accuracy of genotype calls in scRNA-seq data using different approaches.
Therefore, we performed a comprehensive analysis to compare the accuracy of different existing approaches for calling SNVs in scRNA-seq data and to characterize the property of SNVs called from scRNA-seq. In particular, we examined two approaches that were originally designed to call SNVs using DNA sequencing data: GATK that was developed using bulk tissue analysis, and Monovar that was developed for single-cell exome-seq data. We analyzed bulk and single-cell RNA sequencing data with accompanying DNA sequencing data to determine the optimal criteria to reliably identify SNVs using both approaches (Supplementary Material, Fig. S1A) (18). In the present study, we primarily focus on calling SNVs from each individual by combining scRNA-seq across cells within the individual, which serves as the first important step towards cell type-specific eQTL mapping using scRNA-seq data alone. However, we also explore the more challenging approach of calling SNVs at the single-cell level, which, while not directly relevant to eQTL mapping, could be important in other analysis settings such as cancer studies. Our results can aid researchers in determining the best SNV calling strategy for scRNA-seq studies.
Results
Performance of variant callers on bulk samples
We first examined the SNV calling accuracy of two variant callers in the True Bulk and Pseudo Bulk RNA-seq samples for each of the three Yoruba individuals. In this dataset, single cells from each individual replicate sequencing using the Fluidigm C1 system (18). For the bulk sample replicate, an aliquot of ~ 10 000 cells were collected in parallel with the single-cell replicate and processed using the same protocols. Sequencing libraries were prepared using tagmentation and isolation of 5′ fragments for each sample. This allows all samples to be sequenced in the same method to decrease any downstream analyses bias for library preparation methods. The bulk sample replicate is analyzed here as the True Bulk sample, whereas the reads from the individual cells are combined into a Pseudo Bulk sample. We analyzed each individual’s replicate separately and combined the results to provide a range of results for each individual for all downstream analyses. The two examined variant callers are Monovar, which is specifically designed for SNV calling in scDNA-seq, and GATK, which is a general SNV calling tool kit that has been widely applied for SNV calling in bulk RNA-seq (17, 19). With both callers, more SNVs were identified in the True Bulk than the Pseudo Bulk despite more reads in the Pseudo Bulk (Fig. 1A and B; Supplementary Material, Fig. S1B, Table S1). The overlap of SNVs between True Bulk and Pseudo Bulk for each individual is similar across both callers (Fig. 1C; Supplementary Material, Table S2). Comparing between the two callers, GATK called over 10-fold more SNVs than Monovar in each sample (Fig. 1A and B; Supplementary Material, Table S1). However, 98% of the overlapping SNVs between the True Bulk and Pseudo Bulk called by Monovar are also genotype concordant, compared to those called by GATK at 90% (Fig. 1D). We additionally investigated the fidelity of SNV calls across the three technical replicates for each individual. On average, 40.8% of the True Bulk calls and 35.6% of the Pseudo Bulk calls are shared in all three chips with Monovar, whereas GATK averaged 35.8% and 26.4%, respectively (Supplementary Material, Fig. S2). These data suggest both GATK and Monovar call a similar list of SNVs across the replicates. Additionally, both methods had a similar overlap between the two samples across all three individuals even though GATK identified a larger number of SNVs compared to Monovar. For the rest of the analyses, we included all the replicates for all individuals in all results presented.
Figure 1.
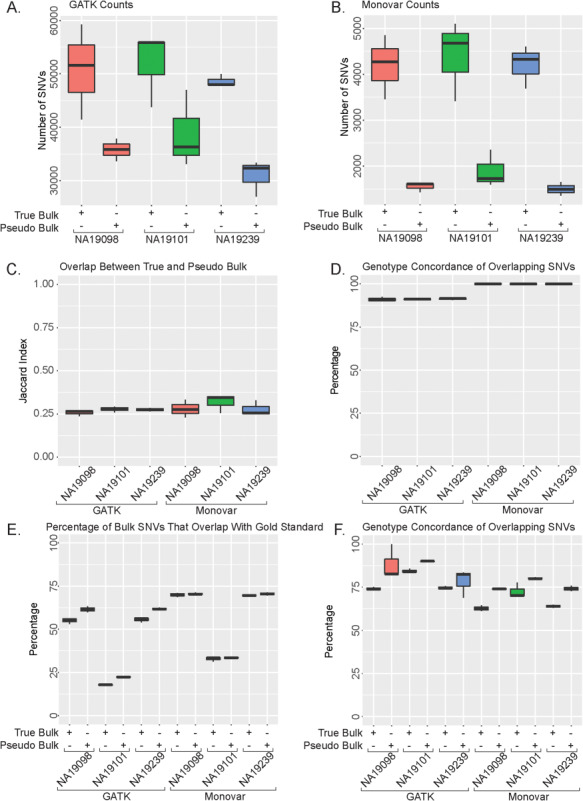
Analysis of True Bulk and Pseudo Bulk SNV calls. (A) Number of SNVs called using GATK. (B) Number of SNVs called Using Monovar. (C) Jaccard Index of overlap between True Bulk and Pseudo Bulk samples. (D) Percentage of overlapping SNVs in (C) with concordance. (E) Percentage of Bulk SNVs calls that identified in gold standard. (F) Percentage of overlapping concordant SNVs with the gold standard.
To determine how accurately both variant callers identified known SNVs, we compared the called SNVs to multiple existing SNP databases. First, we compared the SNV calls to the list of known SNPs in dbSNP 146 and 1000 Genomes Project (phase 1 SNP calls) (20). The overlap percentage of Monovar called SNVs from either bulk sample compared with dbSNP 146 ranged from 70%–71%, whereas GATK ranged from 57%–64% (Supplementary Material, Fig. S3A). Additionally, the overlap percentage of Monovar called SNVs ranged from 60%–64% in either bulk sample and 48%–57% in GATK called SNVs when compared to the variants identified in 1000 Genomes Project (phase 1) (21) (Supplementary Material, Fig. S3B). However, in either case, despite a lower percentage, a higher number of SNVs called by GATK were identified in both dbSNP 146 and 1000 Genomes Project (phase 1) than in Monovar (Supplementary Material, Table S1). Next, because the three individuals are also part of the 1000 Genomes Project (phase 3), we compared our list of SNVs called by the two variant callers directly to the gold standard data set compiled from the 1000 Genomes Project (phase 3) for these individuals. Over 50% of two of the individuals’ SNV calls (NA19098 and NA19239) were identified in the gold standard while NA19101 displays a significantly lower percentage (Fig. 1E). This is due to the methods used to identify the gold standard SNPs during phase 3: in both NA19098 and NA19239, sequencing with an average coverage of 50X was used resulting in 77 629 796 SNVs for each individual while in NA19101 an array-based Omni chip was used resulting in only 2 162 597 SNPs being identified. Among the SNVs overlap with the gold standard data, we also directly measured genotype accuracy by calculating the concordance rate. We found that all individuals had a high percentage of concordance with the gold standard variants (Fig. 1F). The results suggest both variant callers identified known SNVs with high accuracy and precision as demonstrated by the overlap with both the gold standard and other SNP databases. Additionally, using RNA-sequencing reads, both callers likely identified SNVs not documented in either dbSNP 146 or the 1000 Genomes Project.
We explored the choice of two important parameters, which include 1) coverage and 2) quality score, in determining the quality of the final call set of SNVs. Since scRNA-seq can have a variation in sequencing depth and coverage across the genome, investigating these two parameters can uncover whether the level of coverage and depth found in scRNA-seq is compatible with calling high-quality SNVs. First, we varied the number of reads coverage required for calling an SNV in both GATK and Monovar. In both methods, increasing the coverage requirement results in a dramatic decrease in the number of SNVs in the final call set with only a moderate increase in overlap with dbSNP 146 and the gold standard at the strictest cut-offs tested (Supplementary Material, Fig. S4A and B). In both callers, Pseudo Bulk performed equal to or poorer than the True Bulk sample in all comparisons (Supplementary Material, Fig. S4C–F). We observe that increasing the coverage minimum improves the overlap percentage with both the dbSNP 146 and the gold standard; however, the co-current decrease in the number of identified SNVs results in high variation in the overlap. A coverage minimum of 10 reads for GATK or 30 reads for Monovar results in increase in overlap with dbSNP146 and the gold standard. However, the number of SNVs identified is dramatically decreased. Next, we varied the quality score cut off for both methods. The quality score produced in GATK (range of 10–1778) had a wider range than that of Monovar (range of 1–31) (Supplementary Material, Fig. S5, Table S4). Increasing the quality score requirement resulted in decreased number of SNVs in the final call set in both callers (Supplementary Material, Fig. S5A and B). At the strictest quality score, the overlap with dbSNP 146 was 100% in the samples using both callers (Supplementary Material, Fig. S5C and D). However, the percentage of overlap with the gold standard decreased using GATK (from 55% to 40%), whereas it increased using Monovar (from 60% to 75%) (Supplementary Material, Fig. S5E and F). Using the quality score minimum of 10 for GATK and 1 for Monovar produced the highest number of SNVs with a minimum of 50% overlap with dbSNP 146 and gold standard. Together, the results demonstrate that increasing the coverage and quality score parameters in both calling methods can improve the percentage of called SNVs also be identified previously in dbSNP146 or the gold standard.
Variation in variant caller performance across different genomic regions
SNVs are located in different regions of the genome (e.g. promoter region or exons). While all samples were sequenced using the same protocol and from the 5′ ends, it is unknown whether either SNV calling methods will preferably identify SNVs in different genomic regions. To investigate the quality of SNV calls from RNA-seq in different genomic regions, we performed stratified analysis of called SNVs by dividing them into 15 genomic regions and examining SNV calling quality in each region. Both variant callers identified the highest number of SNVs in the exonic, intergenic, intronic and UTR regions (Fig. 2A and B; Supplementary Material, Fig. S6, Table S5). GATK identified the highest number of SNVs in the intronic and intergenic regions, whereas Monovar identified the highest number in the UTR3 regions (Fig. 2A and B; Supplementary Material, Table S5). When comparing the SNV calls in both True Bulk and Pseudo Bulk samples, the Jaccard Index, a measure of similarity, ranges from 0.12 to 0.50 with the UTR regions displaying the highest overlap across both callers (Fig. 2C; Supplementary Material, Fig. S7). In the exonic:splicing region, it is much higher in NA19098 compared to the other regions. In the other two individuals, this region has a Jaccard Index similar to that of the other remaining genomic regions (Supplementary Material, Fig. S7). These data suggest the RNA-sequencing data contains valuable information for calling SNVs located in specific genomic regions as both variant callers identified the majority of SNVs in five regions: exonic, intergenic, intronic, and the UTR regions.
Figure 2.
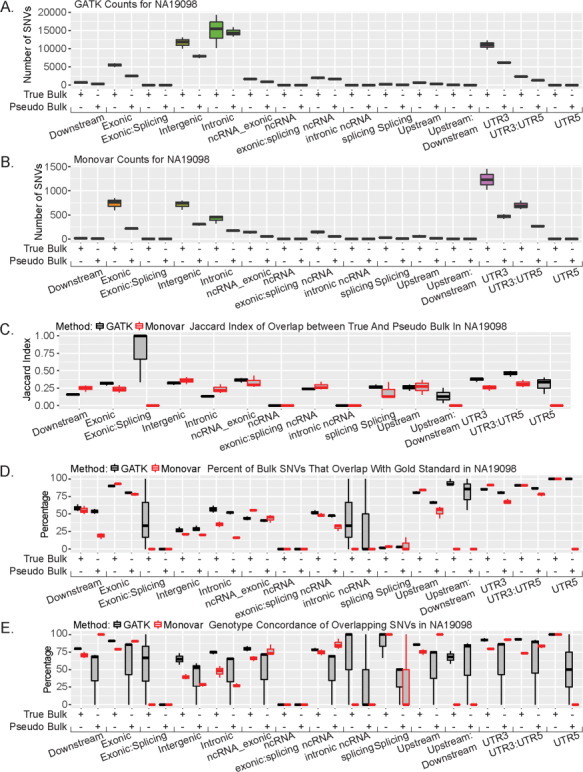
Genetic location affects SNV overlap. (A) Number of SNVs called using GATK for each genomic location in NA19098. (B) Number of SNVs called using Monovar for each genomic location in NA19098. (C) Jaccard Index of overlap between True and Pseudo Bulk at each genomic location in NA19098. (D) Percentage of either True Bulk or Pseudo Bulk SNVs that overlapped with gold standard in NA19098. (E) Percentage of overlapping concordant SNVs with gold standard in NA19098.
We further investigated each genomic region separately to determine the regions in which the SNVs have a better overlap with the known SNP databases. First, we compared the list of called SNVs in each genomic region to dbSNP 146 and 1000 Genomes Project (phase 1). Of the five regions with the highest number of SNVs (exonic, intronic, intergenic, UTR regions), only the intronic region had a better overlap using GATK instead of Monovar in both bulk samples while Monovar performed better in the remaining four regions when compared with both databases (Supplementary Material, Figs S8 and 9). Lastly, we compared to the gold standard set compiled from 1000 Genomes Project (phase 3). In both callers, there is high overlap in the UTR and exonic regions (Fig. 2D; Supplementary Material, Fig. S10A and B). In the other three regions with the highest number of SNVs, 25%–50% of these SNVs were also identified in the gold standard variant list (Fig. 2D; Supplementary Material, Fig. S10A and B). Furthermore, when we calculated the SNV concordance rate, both callers have similar rates of concordance across the five regions with the highest number of SNVs. To determine if a variation in sequencing depth could account for the variation in the number of SNVs identified across all the genomic regions, we calculated the sequencing depth of all SNVs. We observed similar read depth across all the genomic regions for both calling methods (Supplementary Material, Fig. S11). Together, these results suggest GATK-called SNVs are more likely to be previously identified SNVs and have a high level of concordance than Monovar-called SNVs.
Variant caller performance with identifying single-cell RNA sequencing variants
With the rise of single-cell sequencing, there has been an increased interest in calling SNVs in each single cell separately. However, due to the low sequencing coverage per cell, identifying these variants is challenging. Here, we test the performance of GATK and Monovar in calling SNVs in each single cell separately (Supplementary Material, Fig. S1A). GATK identified on average 1300 SNVs in an individual cell (Fig. 3A; Supplementary Material, Table S6), whereas Monovar identified on average 5642 SNVs (Fig. 3B; Supplementary Material, Table S6). There is little Jaccard Index overlap of SNVs with the True Bulk samples (Fig. 3C). However, over 75% of the GATK SNV calls in the individual cells but less than 25% of the Monovar SNV calls were identified in the True Bulk samples (Supplementary Material, Fig. S12A). Those overlapping individual cell and True Bulk SNVs over 80% were concordant in genotype (Fig. 3D). We observed similar results with compared with the Pseudo Bulk samples (Supplementary Material, Fig. S12B–D).
Figure 3.
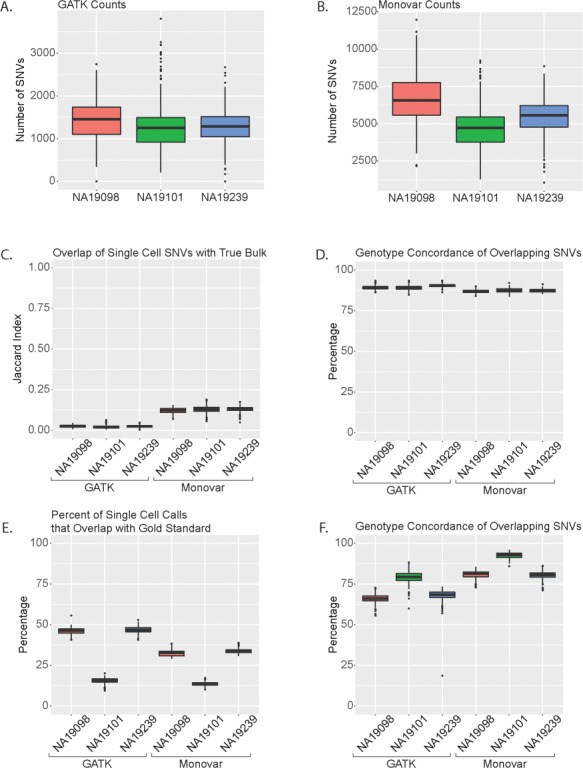
Analysis of single cell SNV calls. Number of SNVs in individual cells called using (A) GATK and (B) Monovar. (C) Jaccard Index of overlap between single cell SNVs and True Bulk. (D) Percentage of overlapping SNVs between single cell samples and True Bulk with genotype concordance. (E) Percentage of single cell SNVs that overlap with gold standard. (F) Percentage of overlapping genotype concordant SNVs between single cells and gold standard.
To determine how many of the individual cell SNVs calls are included in known SNP databases, we compared the SNV calls with dbSNP 146 and the 1000 Genome Project (phase 1 SNP list). Over 50% of the GATK SNV calls and over 30% of the Monovar SNV calls were identified in dbSNP 146 (Supplementary Material, Fig. S12E). However, over 30% of the GATK SNV calls and over 25% of the Monovar SNV calls were identified in 1000 Genome Project (phase 1) (Supplementary Material, Fig. S12F). To determine how well each calling method identified SNVs contained in the gold standard set, we compared each individual single cell’s SNV calls to the corresponding individual SNP list from 1000 Genome Project (phase 3). GATK identified a higher percentage of gold standard SNPs in NA19098 and NA19239 (>37% for GATK compared to > 26% for Monovar, Fig. 3E) while the two callers performed similarly for NA19101. When analyzing for genotype concordance between the gold standard set and the two SNV callers, Monovar had a higher percentage across all patients compared with GATK (Fig. 3F). Altogether, these data demonstrate it is possible to identify SNVs in individual cells with a lower level of accuracy as less than 50% of cells were previously identified in the different databases.
Individual Cell Variant Calling Across All Genomic Locations
Similarly, to the analysis of the True Bulk and Pseudo Bulk samples, we investigated the distribution of SNVs called by either caller across all the genomic regions. Contrary to the analysis of the True Bulk and Pseudo Bulk samples, Monovar identified a higher number of SNVs per region than GATK (Fig. 4A and B; Supplementary Material, Table 7). However, the majority of SNVs were identified in the five main regions identified in the analysis of the True Bulk and Pseudo Bulk samples (exonic, intronic, intergenic and UTR regions). When comparing the individual cells with the SNVs called in the True Bulk samples, Monovar had a higher Jaccard Index than GATK (0.25 or lower versus 0.05 or lower; Fig. 4C; Supplementary Material, Fig. S13A and B). Of those SNVs that did overlap with True Bulk, at least 75% of them in both callers were genomic concordant (Fig. 4D; Supplementary Material, Fig. S13C and D). We observed similar results with comparing the individual cells with the Pseudo Bulk samples across all genomic locations (Supplementary Material, Fig. 14). To determine how many of the SNVs were previously documented SNPs, we compared all SNVs in each genomic location to SNP calls from dbSNP 146 and 1000 Genome Project (phase 1). In the top 5 genomic regions—exonic, intergenic, intronic, UTR3 and UTR5 (as determined by the number of SNVs), GATK had a higher percentage of overlapping SNV calls with dbSNP 146 (Supplementary Material, Fig. S15A–C). When compared with 1000 Genome Project (phase 1), GATK had a higher percentage of overlap in the exonic, UTR3 and UTR5 regions compared with Monovar (Supplementary Material, Fig. S15D–F). However, in the intergenic and intronic region, Monovar had a higher percentage (Supplementary Material, Fig. S15D–F). Lastly, we compared with the gold standard SNP sets for each patient. In the five top genomic regions, the highest percentages of overlap were identified in the exonic, UTR3 and UTR5 regions with GATK identified calls having a higher overlap than Monovar identified calls (Supplementary Material, Fig. S16A and B). Conversely, of those overlapping SNVs, a higher percentage of the Monovar calls were genotype concordant than GATK (90% or higher compared with 75% or higher, Supplementary Material, Fig. S16C and D). Altogether, these data suggest those SNVs that reside in the exonic and UTR regions are more likely to be known SNVs. This suggests the de novo SNVs in these regions are more likely to be true SNVs than those found in other genomic regions.
Figure 4.
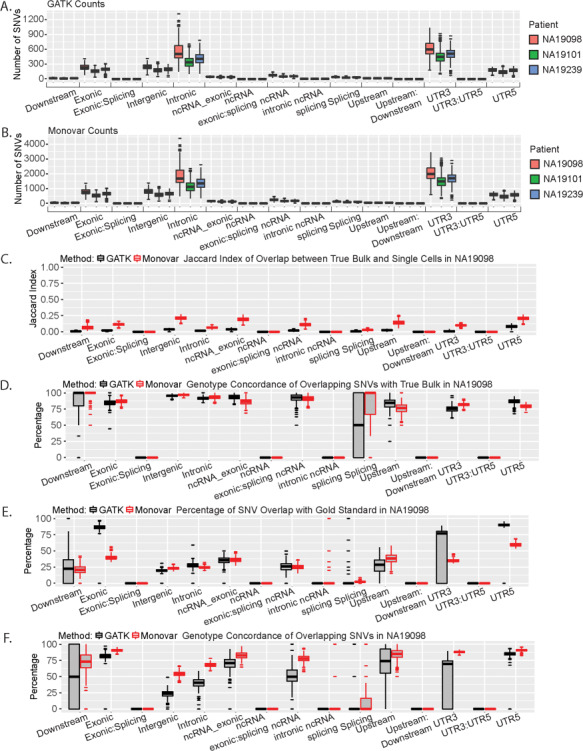
Genetic location distribution of SNVs called in single cells. All analysis results are for patient NA19098. (A) Number of SNVs in each genomic location called by GATK. (B) Number of SNVs in each genomic location called by Monovar. (C) Jaccard Index of overlap between single cell SNVs and True Bulk across all genomic locations. (D) Percentage of genotype concordance of overlapping SNVs between the single cell and True Bulk samples. (E) Percentage of single cells SNVs that overlap with gold standard. (F) Percentage of genotype concordance of overlapping SNVs between the single cell samples and the gold standard.
Performance Evaluation of Variant Callers With Combining SNV Calls Across Individual Cells
Many of the SNVs identified in the individual cells were not identified in either of the True Bulk or Pseudo Bulk or any of the 3 databases, due to the fact that some SNVs were identified with either 1) mixed genotypes across the cells or 2) identified in only a few cells. To test this, we examined two methods to combine the data to test the influence of these factors on the accuracy of the identified SNVs: 1) percentage of genotype agreement across cells and 2) number of cells each SNV is identified in. For the first method, we calculated the genotype agreement percentage for each SNV by identifying the most common genotype (either heterozygous or homozygous for reference or variant). The percentage is calculated by the number of cells with the most common genotype divided by the total number of cells in which the SNV is identified (Equation 3). For analysis, we varied the genotype agreement percentage cut-off value between 0.6 and 1.0. In our analysis, Monovar identified a higher number of SNVs across all cells at each ratio cut-off compared to GATK (Fig. 5A and B, Supplementary Material, Fig. S17, Table S8). For the first analysis, we compared SNVs across the individual cells to the SNVs called in the True Bulk. When measured with Jaccard Index, the overlap of SNVs called by Monovar increases with increasing cut-offs and reaches the highest Jaccard Index at 0.25 using the most stringent cut-off, 1.0, whereas the overlap of SNVs called by GATK remains the same at all cut-offs (Fig. 5C). Additionally, the SNV overlap called by both Monovar and GATK increases with increasing cut-offs. In particular, at the highest cut-off of 1.0, nearly 100% of the GATK variants called in the individual cells and 25%–35% of the Monovar variants called are also identified in True Bulk (Fig. 5D). At each cut-off, the called genotypes for over 75% of the SNVs are concordant with True Bulk, with nearly 100% concordance rate at the highest cut-off, for both variant callers (Fig. 5E). We investigated further the ratio cut-offs between 0.9 and 1.0 and observed an increase in overlap with True Bulk in the SNVs identified through Monovar until the cut-off reaches the value of 0.96, where it tapers off. In GATK, at the 0.96 cut-off, the overlap results increase dramatically until the 1.0 cut-off (Supplementary Material, Fig. S18). When compared with the Pseudo Bulk call set, we observed similar results to that from the comparison with True Bulk (Supplementary Material, Fig. 19).
Figure 5.
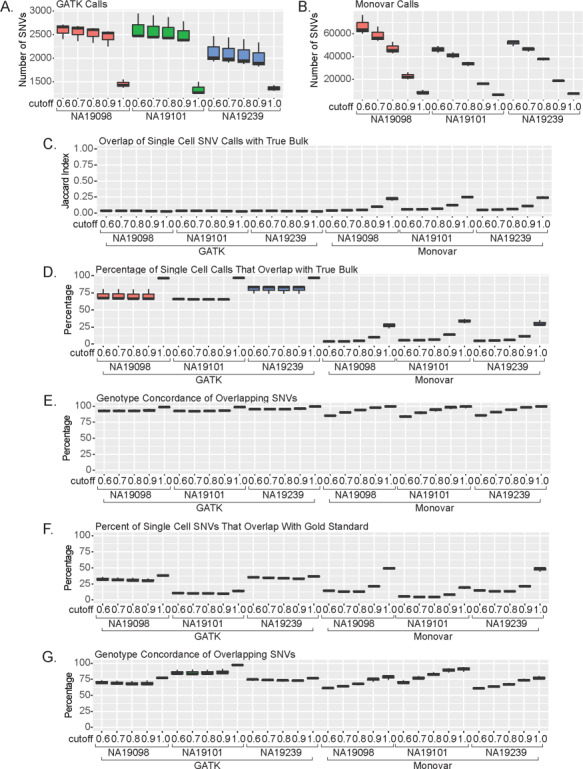
Single cell sequencing SNV calls vary based on percentage cut-off. Percent cut-off determined for each SNV based on what percentage of calls are concordant for each. Number of SNVs greater than percent cut-off called using (A) GATK and (B) Monovar. (C) Jaccard Index of overlap between single cell SNVs and True Bulk calls. (D) Percentage of single cell SNVs that overlap with True Bulk. (E) Percentage of overlapping concordant SNVs with True Bulk. (F) Percentage of single cell SNVs that overlap with gold standard. (G) Percentage of overlapping concordant SNVs with gold standard.
Next, we investigated the single-cell SNVs with the known SNP databases. Similar to our results with the True Bulk, increasing the cut-off value resulted in increasing percentage of single-cell SNVs also being identified in both dbSNP 146 and 1000 Genomes Project (phase 1) set (Supplementary Material, Fig. S20A and B). When compared to the gold standard, over 50% of the SNVs with the cut-off 1.0 are overlapping with nearly 100% concordance (Fig. 5F–G). Between the 0.9 and 1.0 cut-off, GATK results didn’t change across these cut-offs, whereas Monovar dramatically increased until the 0.96 cut-off (Supplementary Material, Fig. S20C and D). These results were the same when compared with the gold standard variant list between the 0.9 and 1.0 cut-off (Supplementary Material, Fig. S20E and F). Together, the results suggest the SNVs identified using single-cell sequencing are comparable to those identified in more common bulk sequencing. Furthermore, using the most stringent cut-off where all cells with the SNV also share the same genotype gives the highest confidence in the called SNVs.
Finally, we examined the importance of the second filtering criterion: the number of single cells for which an SNV is identified in. For this method, we calculated how many cells each SNVs was identified in and compared different thresholds in each of our analyses. Unsurprisingly, as we increase the cell number cut-off, both callers identified fewer SNVs (Supplementary Material, Fig. S21A and B, Table S9). When compared with True Bulk, there are no differences in Jaccard Index across all the different cut-off values (Supplementary Material, Fig. S21C). However, at the lowest cell number cut-off of 10 cells, we observe the highest percentage of overlapping and concordant SNVs (Supplementary Material, Fig. S21D). Similarly, when compared with Pseudo Bulk variants, there is a slight increase at the lowest cell number cut-off in the GATK calls but overall the cut-off value does not affect the overlap with Pseudo Bulk or concordance rate (Supplementary Material, Fig. S21E and F). Next, we compared the variant lists to the known SNP databases. The cell number cut-off did not improve the percentage of single-cell SNVs that identified in either dbSNP 146, 1000 Genomes Project (phase 1), or the gold standard set (Supplementary Material, Fig. S22A–C). Together, the results suggest that an SNV can be confidently called if it is identified in as few as 10 single cells.
Evaluation of Combination Methods for Single-Cell SNVs Across Each Genomic Region
To further investigate the SNVs called by GATK and Monovar in the individual cells, we performed stratified analysis to examine SNV calling accuracy in different genomic regions using the two methods to combined SNVs calls: genotype agreement cut-off or number of cell cut-off. We first investigated the distribution of SNVs included in the call set based on the genotype agreement cut-off method. Similar to our previous analyses, GATK mainly identified SNVs in the exonic, intergenic, intronic, non-coding RNA and UTR regions (Fig. 6A; Supplementary Material, Fig. S23A and B, Table S10). Monovar identified SNVs that located mainly in the exonic, intergenic, intronic and UTR regions (Fig. 6B; Supplementary Material, Fig. S23C and D, Table S10). When comparing with the True Bulk samples, all the regions had Jaccard Indices less than 0.12 except at the 1.0 cut-off (Fig. 6C; Supplementary Material, Fig. S24A and B). When there were overlapping SNVs, over 75% were concordant at all cut-off values. GATK did perform better in the concordance rate in the exonic and UTR regions (Fig. 6D; Supplementary Material, Fig. S24C and D). We observed similar results when compared with Pseudo Bulk (Supplementary Material, Fig. S25). However, Monovar performed better in the concordance rate of the SNVs in multiple regions including exonic, non-coding RNA intronic and UTR regions (Supplementary Material, Fig. S25). In the comparison with dbSNP 146, GATK performed better in the exonic, intergenic and UTR3 regions, whereas Monovar performed better in the UTR5 region (Supplementary Material, Fig. S26A–C). Second, when compared with 1000 Genomes Project (phase 1) variants, the highest overlapping regions were the upstream and UTR regions. In the 0.6 and 0.8 cut-off, GATK performed better. However, at the 1.0 cut-off, Monovar does as well if not out-performing GATK in every region (Supplementary Material, Fig. S26D–F). Lastly, in most of the regions, GATK and Monovar had similar performance in percentage overlap with the gold standard. In the UTR and exonic regions, GATK outperformed Monovar, whereas in the upstream region, Monovar performed better (Fig. 6; Supplementary Material, Fig. S27A and B). When investigating the concordance of the SNVs with the gold standard, Monovar performed better in the intergenic and intronic regions, whereas GATK performed better in the exonic, upstream and UTR regions (Fig. 6E; Supplementary Material, Fig. S27C and D). Together, these results suggest GATK and Monovar primarily identify SNVs in five regions with the best performance achieved using the most stringent cut-off of 1.0. Additionally, while GATK may outperform Monovar in overlapping percentage in multiple regions, Monovar identified a higher number of SNVs since Monovar identifies 10 times more SNVs in total compared to GATK.
Figure 6.
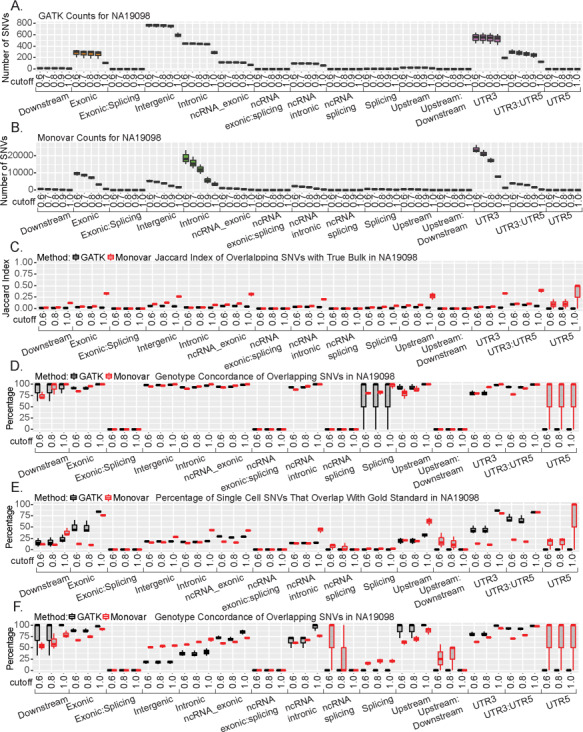
Genetic location effect on SNV calls from single cell data. These results are all for individual NA19098. Number of SNVs called across all genomic locations with varying SNV percent cut-offs by (A) GATK and (B) Monovar. (C) Jaccard Index of overlapping SNVs between single cell and True Bulk at each genomic location at varying percent cut-offs. (D) Percentage of overlapping concordant SNVs with varying percent cut-offs across all genomic locations with True Bulk. (E) Percentage of single cell SNVs identified in gold standard. (F) Percentage of overlapping concordant SNVs with gold standard across all genomic locations with varying percent cut-offs.
We next investigated how varying the cell number cut-off affects where SNVs are identified in the genome. Unsurprisingly, across all genomic regions as the cell number cut-off increases, the number of SNVs identified by both variant callers decreases. However, Monovar still identified more SNVs than GATK at each cut-off (Supplementary Material, Fig. S28, Table S11). When compared with True Bulk, all cell number cut-offs perform equally in terms of overlap with a slight increase in genotype concordance (Supplementary Material, Fig. S29, Table S11). We observed similar overlap results when compared with the Pseudo Bulk samples (Supplementary Material, Fig. S30). We next compared the overlap with known SNP databases across all genomic regions as we varied the cell number cut-off. However, increasing the cell number cut-off did not improve the performance of either variant caller significantly (Supplementary Material, Fig. S31A–C). We observe similar results when comparing to the 1000 Genomes Project (phase 1) set (Supplementary Material, Fig. S31D–F). We lastly compared each cut-off SNVs to the gold standard. GATK performed better in the exonic and UTR regions as compared to Monovar. Increasing the cell number cut-off did not improve performance in majority of regions except for the intergenic region where it reached 50% (Supplementary Material, Fig. S32A–C). However, increasing the cut-off value decreased the percentage of overlapping concordant SNVs in the exonic, intronic and UTR regions while not affecting any of the other genomic regions (Supplementary Material, Fig. S32D–F). Together, these results suggest that increasing the cell number cut-off doesn’t improve variant caller performance across all the genomic regions.
Discussion
NGS technologies have provided researchers with access to unprecedented amounts of data. Being able to characterize genetic and transcriptomic features for the same individual with one sequencing sample opens up novel areas of study, such as eQTL mapping using scRNA-seq data alone. This study aimed to take one of the first steps to achieving this goal. We first evaluated the performance of two variant callers, GATK and Monovar, in identifying either True Bulk or a Pseudo Bulk comprised of reads from all single cells. GATK identified more SNVs than Monovar in the bulk samples. However, the performance of both callers in identifying known or de novo SNVs were similar (Fig. 1). Additionally, the genomic locations of the majority of the SNVs were located in regions that are expected to be enriched in the transcriptome, such as introns, exons and the UTR regions (Fig. 2). Due to the higher number of SNVs called and the similar overlap metrics as Monovar, GATK performed the best in either the true or pseudo bulk samples. However, when we analyzed the single-cell sequencing data alone, Monovar identified a higher number of SNVs than GATK (Fig. 3A) indicating that Monovar outperformed GATK. However, this was lower overlap with the known SNP databases in the individual cells compared with the bulks samples leading us to investigate two methods to increase the SNV accuracy from individual cells. The metrics for the two combination methods performed similarly to analyzing the cells individually (Fig. 5F–G; Supplementary Material, Fig. S22C–D). These results demonstrate the importance of choosing the correct variant caller for each sample type. Selecting the appropriate calling method for bulk versus single-cell RNA sequencing can optimize the identification of SNVs. In the case where all single cells come from the same individual and where there is no interest of identifying somatic mutations, we recommend combining all reads from the single cells into a Pseudo Bulk sample and following the GATK Best Practices (19) to identify the highest number and highest quality SNVs for a homozygous population of single cells. For a heterozygous population of single cells, we recommend using Monovar (17) to identify an average of 5642 SNVs per cell with an average of 33% of which are known SNVs (resulting in 1861 known SNVs per cell). Following GATK, an average of 1299 SNVs per cell were identified with 47% of those SNVs being previously known (resulting in 608 known SNVs per cell).
Single-cell sequencing is being employed more often in the research setting (22). Our results demonstrate three methods of calling SNVs from individual cells: 1) analyze each cell individually, 2) the SNV concordance rate across the single cells and 3) how many cells an SNV must be identified in. In our dataset, we are working with induced pluripotent stem cells (iPS). Normal embryonic stem cells exhibit a low level of mutations, resulting in a high homogenous cell population with effectively identical genotypes (23). iPS cells have a slightly evaluated mutation rate but still with very low level of SNP heterogeneity across cells in the population (e.g. over 99.44% matching SNPs between iPS clones) (24, 25). Therefore, an SNP would likely have identical genotypes across all single cells from the same individual. Such low level of heterogeneity is particularly suitable for validating SNV calling pipelines using RNA sequencing data, as is done in the present study. In such analysis, we found that increasing the ratio cut-off can lead to accurate SNVs in the final SNV call set. Furthermore, combining the sequencing reads from the single cells and using the GATK variant caller results in more SNVs (average 35 168 SNVs per sample with 53% being known SNVs) than analyzing the single cells using Monovar using the most stringent concordance ratio cut-off of 1.0% zygosity agreement (average 7750 SNVs per cell with 47% being known SNVs) (Figs 1A and 5A; Supplementary Material, Table S1 and S8). However, in other sample types such as cancer with a higher rate of mutation, this same assumption cannot be made. Genomic instability is one of the enabling hallmarks of cancer (26). In one event, tens to hundreds of genomic rearrangements occur in a one-time cellular crisis resulting in large amounts of genetic heterogeneity (27). Furthermore, a high level view of the genomic heterogeneity has been shown in breast tumors in which multiple tumor sub-clones are identified by genomic hybridization (28). With our analysis, we identified 5642 SNVs per cell using Monovar (or 1299 SNVs per cell using GATK) in the individual cells, which allows researchers to study the heterogeneity at those particular sites across all the single cells. Together, this provides evidence for the potential for individual cancer cells to have different SNVs. In this case, a stringent concordance ratio cut-off may remove true SNV calls from the final variant list and reduce the ability to detect somatic mutations. Due to the difficulty of calling SNVs in single cells as evidenced by the low percentages of known SNVs called in all analysis pipelines analyzed in this study, future methods are needed in this area.
This study opens up the potential to combine both the genetic and transcriptome information from a single cell using scRNA-seq alone. Researchers have begun to appreciate the power of combining different “omic” information (29, 30). For example, combining the genomic and transcriptome data allows researchers to identify regulatory elements such as transcription factors (TF) or SNVs (31). One computational method clusters genes based on their expression profiles (transcriptome data) and compares the upstream promoter sequence (genomic data) to identify statistically significant TF motifs (32). Computation methods are being developed to take advantage of multi-omic research (33). However, the acquisition for such studies can be difficult and expensive. Indeed, the association between an SNV and gene expression can only be fully investigated when both the genetic and transcriptome information are available for the same sample (34). By utilizing RNA-sequencing alone to extract genetic information while determining the transcriptome data allows research to more effectively utilize their data and answer new questions (16). Additionally, by combining this RNA-seq-based approach with insights only gained from single-cell data opens truly novel avenues of research. Single-cell sequencing allows researchers to study subpopulations of cells of heterogeneous tissues such as tumor. Different subpopulations of cells have been shown to play an important role in tumorigenesis. For example, cancer stem cells have been shown to be able to re-establish a tumor in vivo yet only comprise < 10% of the total tumor cell count (35). Furthermore, this same subpopulation was linked with increased therapeutic resistance (36, 37). With SNVs and other genomic features playing an important role in gene expression, understanding the role these features play in tumor subpopulations is an important avenue of research now open by the findings of this study.
In summary, our data demonstrate the ability to call SNVs from single-cell RNA-sequencing data. When using bulk samples or samples comprising all reads from single cells, GATK performed better in calling more numerous and reliable SNVs. However, when working with individual single cells, Monovar performed superior to GATK even at the most stringent of cut-off values. When working with single-cell data, the research must consider the importance of how stable an SNV’s concordance is with respect to changes of filtering parameters as such parameters may greatly affect the reliable and number of SNV calls. However, as we shown here, for SNVs called from scRNA-seq, the number of cells an SNV was identified in has a much lower effect on the resulting variant list. Further research including additional scRNA-seq data types in these comparisons as well as more SNV callers designed for single-cell analysis would be greatly beneficial in the future. This study’s results will provide important guidance to accurate and reliable variant identification in multiple types of RNA-sequencing data including single-cell samples, ultimately allowing researcher to gather more insights into the inner workings of the cell at the singular cell level. This methodology will ultimately allow researchers to gather both genomic and gene expression information from the same individual cells.
Materials and Methods
Data sets
We obtained bulk and single-cell RNA-seq data for three individual human samples with three replicates each in the NCBI’s Gene Expression Omnibus (38) (accession number GSE77288) (18). In brief, single cells from each individual replicate were loaded onto a Fluidigm C1 instrument using manufacturer protocols (PIN 100–7168). Reverse transcription and cDNA library preparation was modified to allow for UMI labelling by including the UMI sequence in the template-switching RNA oligos in the reverse transcription mix (39). For the bulk sample replicate, an aliquot of ~10 000 cells were collected in parallel with the single-cell replicate and processed using the same protocols. Sequencing libraries were prepared using tagmentation and isolation of 5′ fragments for each sample. All sample libraries were sequenced on the Illumina HiSeq 2500 instrument and generated single end 100-bp reads along with 8-bp indexing reads corresponding to the cell-specific barcodes. For each individual in turn, we analyzed three sample conditions for each replicate: 1) the original bulk RNA-seq data (“True Bulk”), 2) all combined reads from the 96 single-cell RNA-seq (“Pseudo Bulk”) and 3) single-cell RNA-seq from each of the 96 single cells (“Single Cell”). For all samples, we mapped reads to the assembled human genome (hg38) (1) using the BWA aligner (40) (Supplementary Material, Fig. S1B and C), sorted and marked duplicates with Picard (http://broadinstitute.github.io/picard) (Table 1). For all analyses, all replicates were analyzed separately.
Table 1.
Mapping Quality of Samples
| Sample | In dividual | Number of Reads | Percentage of Mapped Reads | Number of Genes |
|---|---|---|---|---|
| True Bulk | NA19098 | 84 691 812 ± 7 088 563 | 86.61% ± 1.40% | 18 257 ± 295 |
| NA19101 | 81 869 062 ± 10 336 670 | 87.87% ± 0.19% | 18 459 ± 428 | |
| NA19239 | 85 139 927 ± 5 657 697 | 88.45% ± 0.65% | 18 190 ± 205 | |
| Pseudo Bulk | NA19098 | 625 455 881 ± 21 764 108 | 76.04% ± 8.48% | 17 032 ± 244 |
| NA19101 | 660 248 306 ± 19 378 657 | 82.20% ± 1.68% | 17 304 ± 409 | |
| NA19239 | 610 579 179 ± 22 727 346 | 81.72% ± 2.04% | 16 788 ± 309 | |
| Single Cell Samples | NA19098 | 6 212 597 ± 2 275 041 | 73.57% ± 11.62% | 17 032 ± 244 |
| NA19101 | 6 877 586 ± 2 336 461 | 80.17% ± 7.36% | 17 304 ± 409 | |
| NA19239 | 6 360 200 ± 2 111 833 | 79.46% ± 8.73% | 16 788 ± 309 |
The three individuals are from the Yoruba population and were part of the 1000 Genomes Project with imputed genotype data available from previous DNA sequencing. For comparisons with databases of known SNVs, we obtained allele and location information of 133 603 515 SNPs from dbSNP 146 (20) and 28 501 388 SNPs from the 1000 Genomes Project (phase 1) (21) on Broad Institute website (https://software.broadinstitute.org/gatk/download/bundle). SNPs included in dbSNP 146 were compiled from multiple sources including literature and genome project sequences. SNPs included in the 1000 Genomes Project (phase 1) were identified using whole genome sequencing of 180 samples at an average depth of 2–4×. In addition, to directly examine the genotype call accuracy from using RNAseq, we obtained genotype calls from previous DNA sequencing in the 1000 genomes project for the same three individuals to serve as the gold standard genotype database. These previous genotype calls were downloaded from phase 3 of the 1000 Genomes Project (21) (for NA19098 and NA19239: ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/supporting/GRCh38_positions/; for NA19101: ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/supporting/hd_genotype_chip). Individual NA19101 gold standard SNPs were identified using the Omni Platform (Illumina). Individuals NA19098 and NA19239 had whole genome sequencing conducted at an average depth of 50X. The gold standard for each individual contained the following number of SNVs: for NA19098 77 629 796 SNPs, for NA19101 2 162 597 SNPs and for NA19239 77 629 796 SNPs.
Examined SNV calling methods for scRNA-seq
GATK
We followed the recommended GATK Best Practices work flow (19) to call SNVs from RNA-seq data. Default setting was used during each step. In the filtering step, we excluded variants without a “PASS” filter tag, with more than two alleles, and along with insertions and deletions. For single-cell samples, each sample was individually analyzed using GATK, resulting in one filtered VCF file for each cell. A total of 96 single cells from the same chip were then consolidated into one VCF file using the GATK Joint Caller tool. Variants were further recalibrated and filtered based on joint call file (for single-cell samples) or individual file for the bulk samples. Variants were annotated using ANNOVAR (41).
Monovar
We used default settings for Monovar (17) algorithm. We used the same aligned reads as input as in GATK pipeline. For the True Bulk or Pseudo Bulk RNA-seq samples, each sample was analyzed individually. For single-cell samples, 96 cells from the same chip were analyzed together. In the filtering step, we again excluded variants without a “PASS” filter tag, with more than two alleles, and along with insertions and deletions. Variants were annotated using ANNOVAR (41).
Evaluation of SNV calling pipelines
To assess the performance of each variant calling pipeline, we first used the Jaccard Index to measure the commonality/overlap between SNVs called from scRNA-seq and SNVs in existing databases. Specifically, we refer to SNVs called from scRNA-seq that are also available in existing databases as known variants and refer to SNVs only in scRNA-seq as novel SNVs. The Jaccard Index is calculated as
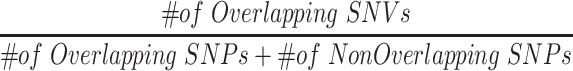 |
(1) |
Jaccard Index ranges from 0 to 1 with a score of one indicating that all SNVs are called in both lists.
We further used concordance rate to directly measure the genotype call accuracy of known SNVs called from scRNA-seq by comparing their genotypes to genotype calls from existing gold standard database obtained using DNA sequencing. SNVs were considered as concordant between scRNA-seq and the gold standard database if they meet the following criteria: 1) reside in the same genomic location, 2) have the same reference and variant allele types and 3) have the same genotype. The concordance rate was calculated as
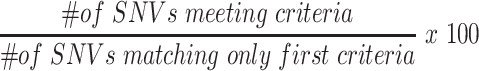 |
(2) |
We examined both Jaccard Index and concordance rate for SNVs called in 15 different genomic regions with functional annotations. For such analysis, SNVs were first annotated by ANNOVAR (41) for their genomic locations that include downstream, exonic, exonic:splicing, intergenic, intronic, ncRNA exonic, ncRNA exonic:splicing, ncRNA intronic, ncRNA splicing, splicing, upstream, upstream:downstream, UTR3, UTR5 and UTR3:UTR5 regions. Afterwards, we performed stratified analyses examining SNVs in each of the 15 regions one at a time.
SNV inclusion criteria for single-cell analysis
To determine the SNVs for analysis that were called in the single-cell sample, we varied two different cut-off criteria: 1) number of cells each SNV is identified in and 2) percentage of genotype agreement across cells. For the first cut-off criteria, individual SNVs must be called in at least 10 of the 96 cells. For the second cut-off criteria, we calculated the genotype agreement value for each SNV by:
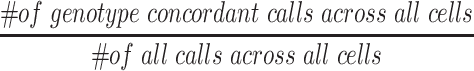 |
(3) |
The genotype of the concordant cells is used for each SNV in all downstream analyses. For the second criteria, each SNV must have a genotype agreement of 0.6 or higher to be included in the downstream analyses. The number of cells each SNV is called in (based on genomic location) is calculated.
Supplementary Material
Acknowledgments
The authors would like to thank the University of Michigan’s Advanced Research Computing Technology Services for providing the computing resources that supported this study. We would also like to thank Dr Lana Garmire for her comments on our manuscript. The code to reproduce all the analyses presented in the paper is available on GitHub (https://github.com/pschnepp/scRNA-seq-SNV). This work was supported by the National Institutes of Health P01 CA093900 to E.T.K.; Bioinformatic Institute and Rogel Cancer Center Single Cell Analysis Shared Resource P30 CA046592 to E.T.K. P.M.S. was supported by NCATS grant UL1TR002240. M.C. was supported by NIH grant R01GM126553. X.Z. was supported by NIH grant R01HG009124 and NSF grant DMS1712933.
Conflict of Interest statement. None declared.
References
- 1. Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W. et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 2. Venter J.C., Adams M.D., Myers E.W., Li P.W., Mural R.J., Sutton G.G., Smith H.O., Yandell M., Evans C.A., Holt R.A. et al. (2001) The sequence of the human genome. Science, 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- 3. Melé M., Ferreira P.G., Reverter F., DeLuca D.S., Monlong J., Sammeth M., Young T.R., Goldmann J.M., Pervouchine D.D., Sullivan T.J. et al. (2015) The human transcriptome across tissues and individuals. Science, 348, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L. et al. (2014) The NHGRI GWAS catalog, a curated resource of SNP-trait associations. Nucleic Acids Res, 42, D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hwang S., Kim E., Lee I. and Marcotte E.M. (2015) Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci Rep, 5, 17875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sultan M., Schulz M.H., Richard H., Magen A., Klingenhoff A., Scherf M., Seifert M., Borodina T., Soldatov A., Parkhomchuk D. et al. (2008) A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science, 321, 956–960. [DOI] [PubMed] [Google Scholar]
- 7. Cui Y. and Paules R.S. (2010) Use of transcriptomics in understanding mechanisms of drug-induced toxicity. Pharmacogenomics, 11, 573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hoffmeyer S., Burk O., von Richter O., Arnold H.P., Brockmöller J., Johne A., Cascorbi I., Gerloff T., Roots I., Eichelbaum M. et al. (2000) Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. PNAS, 97, 3473–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pickrell J.K., Marioni J.C., Pai A.A., Degner J.F., Engelhardt B.E., Nkadori E., Veyrieras J.-B., Stephens M., Gilad Y. and Pritchard J.K. (2010) Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature, 464, 768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buettner F., Natarajan K.N., Casale F.P., Proserpio V., Scialdone A., Theis F.J., Teichmann S.A., Marioni J.C. and Stegle O. (2015) Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotech, 33, 155–160. [DOI] [PubMed] [Google Scholar]
- 11. Sun W. (2012) A statistical framework for eQTL mapping using RNA-seq data. Biometrics, 68, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wills Q.F., Livak K.J., Tipping A.J., Enver T., Goldson A.J., Sexton D.W. and Holmes C. (2013) Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat Biotechnol, 31, 748–752. [DOI] [PubMed] [Google Scholar]
- 13. Kang H.M., Subramaniam M., Targ S., Nguyen M., Maliskova L., McCarthy E., Wan E., Wong S., Byrnes L., Lanata C.M. et al. (2018) Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol, 36, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Borel C., Ferreira P.G., Santoni F., Delaneau O., Fort A., Popadin K.Y., Garieri M., Falconnet E., Ribaux P., Guipponi M. et al. (2015) Biased allelic expression in human primary fibroblast single cells. Am J Hum Genet, 96, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mikkelsen T.S., Ku M., Jaffe D.B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.-K., Koche R.P. et al. (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature, 448, 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tung J., Zhou X., Alberts S.C., Stephens M. and Gilad Y. (2015) The genetic architecture of gene expression levels in wild baboons. ELife, 4, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zafar H., Wang Y., Nakhleh L., Navin N. and Chen K. (2016) Monovar: single-nucleotide variant detection in single cells. Nat Meth, 13, 505–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tung P.Y., Blischak J.D., Hsiao C.J., Knowles D.A., Burnett J.E., Pritchard J.K. and Gilad Y. (2017) Batch effects and the effective design of single-cell gene expression studies. Sci Rep, 7, 39921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Van der G.A., Carneiro M.O., Hartl C., Poplin R., del G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J. et al. (2013) From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics, 43, 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M. and Sirotkin K. (2001) Db SNP: the NCBI database of genetic variation. Nucleic Acids Res, 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. 1000 Genomes Project Consortium, Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. and Abecasis G.R. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gawad C., Koh W. and Quake S.R. (2016) Single-cell genome sequencing: current state of the science. Nat Rev Genet, 17, 175–188. [DOI] [PubMed] [Google Scholar]
- 23. Cervantes R.B., Stringer J.R., Shao C., Tischfield J.A. and Stambrook P.J. (2002) Embryonic stem cells and somatic cells differ in mutation frequency and type. PNAS, 99, 3586–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gore A., Li Z., Fung H.L., Young J.E., Agarwal S., Antosiewicz-Bourget J., Canto I., Giorgetti A., Israel M.A., Kiskinis E. et al. (2011) Somatic coding mutations in human induced pluripotent stem cells. Nature, 471, 63–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Masaki H., Ishikawa T., Takahashi S., Okumura M., Sakai N., Haga M., Kominami K., Migita H., McDonald F., Shimada F. et al. (2008) Heterogeneity of pluripotent marker gene expression in colonies generated in human iPS cell induction culture. Stem Cell Research, 1, 105–115. [DOI] [PubMed] [Google Scholar]
- 26. Hanahan D. and Weinberg R.A. (2011) Hallmarks of cancer: the next generation. Cell, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 27. Stephens P.J., Greenman C.D., Fu B., Yang F., Bignell G.R., Mudie L.J., Pleasance E.D., Lau K.W., Beare D., Stebbings L.A. et al. (2011) Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell, 144, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Navin N., Krasnitz A., Rodgers L., Cook K., Meth J., Kendall J., Riggs M., Eberling Y., Troge J., Grubor V. et al. (2009) Inferring tumor progression from genomic heterogeneity. Genome Res. 20, 68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joyce A.R. and Palsson B.Ø. (2006) The model organism as a system: integrating ‘omics’ data sets. Nat Rev Mol Cell Biol, 7, 198–210. [DOI] [PubMed] [Google Scholar]
- 30. Poirion O., Zhu X., Ching T. and Garmire L.X. (2018) Using single nucleotide variations in single-cell RNA-seq to identify subpopulations and genotype-phenotype linkage. Nat Commun, 9, 4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li H. and Wang W. (2003) Dissecting the transcription networks of a cell using computational genomics. Curr Opin Genet Dev, 13, 611–616. [DOI] [PubMed] [Google Scholar]
- 32. Bussemaker H.J., Li H. and Siggia E.D. (2001) Regulatory element detection using correlation with expression. Nat Genet, 27, 167–171. [DOI] [PubMed] [Google Scholar]
- 33. Berger B., Peng J. and Singh M. (2013) Computational solutions for omics data. Nat Rev Genet, 14, 333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stranger B.E., Forrest M.S., Dunning M., Ingle C.E., Beazley C., Thorne N., Redon R., Bird C.P., de Grassi A., Lee C. et al. (2007) Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science, 315, 848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Prince M.E., Sivanandan R., Kaczorowski A., Wolf G.T., Kaplan M.J., Dalerba P., Weissman I.L., Clarke M.F. and Ailles L.E. (2007) Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. PNAS, 104, 973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bao S., Wu Q., McLendon R.E., Hao Y., Shi Q., Hjelmeland A.B., Dewhirst M.W., Bigner D.D. and Rich J.N. (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature, 444, 756–760. [DOI] [PubMed] [Google Scholar]
- 37. Schatton T., Frank N.Y. and Frank M.H. (2009) Identification and targeting of cancer stem cells. Bio Essays, 31, 1038–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Edgar R., Domrachev M. and Lash A.E. (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res, 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Islam S., Zeisel A., Joost S., La G., Zajac P., Kasper M., Lönnerberg P. and Linnarsson S. (2014) Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods, 11, 163–166. [DOI] [PubMed] [Google Scholar]
- 40. Li H. and Durbin R. (2009) Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang H. and Wang K. (2015) Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protocols, 10, 1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.