

Abstract
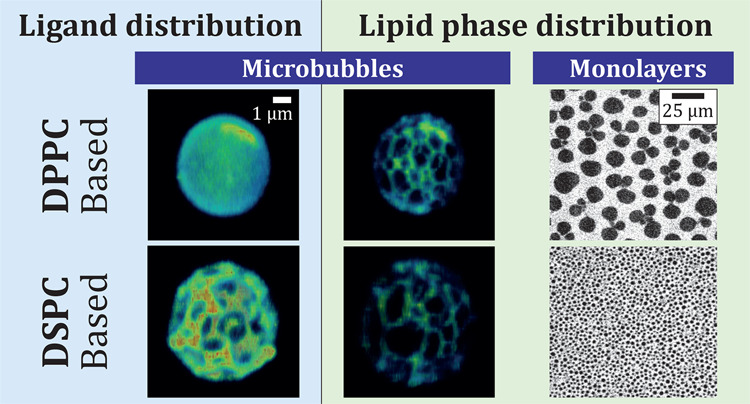
Phospholipid-coated targeted microbubbles are ultrasound contrast agents that can be used for molecular imaging and enhanced drug delivery. However, a better understanding is needed of their targeting capabilities and how they relate to microstructures in the microbubble coating. Here, we investigated the ligand distribution, lipid phase behavior, and their correlation in targeted microbubbles of clinically relevant sizes, coated with a ternary mixture of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) or 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), with PEG40-stearate and DSPE-PEG2000. To investigate the effect of lipid handling prior to microbubble production in DSPC-based microbubbles, the components were either dispersed in aqueous medium (direct method) or first dissolved and mixed in an organic solvent (indirect method). To determine the lipid-phase behavior of all components, experiments were conducted on monolayers at the air/water interface. In comparison to pure DSPC and DPPC, the ternary mixtures had an additional transition plateau around 10–12 mN/m. As confirmed by infrared reflection absorption spectroscopy (IRRAS), this plateau was due to a transition in the conformation of the PEGylated components (mushroom to brush). While the condensed phase domains had a different morphology in the ternary DPPC and DSPC monolayers on the Langmuir trough, the domain morphology was similar in the coating of both ternary DPPC and DSPC microbubbles (1.5–8 μm diameter). The ternary DPPC microbubbles had a homogenous ligand distribution and significantly less liquid condensed (LC) phase area in their coating than the DSPC-based microbubbles. For ternary DSPC microbubbles, the ligand distribution and LC phase area in the coating depended on the lipid handling. The direct method resulted in a heterogeneous ligand distribution, less LC phase area than the indirect method, and the ligand colocalizing with the liquid expanded (LE) phase area. The indirect method resulted in a homogenous ligand distribution with the largest LC phase area. In conclusion, lipid handling prior to microbubble production is of importance for a ternary mixture of DSPC, PEG40-stearate, and DSPE-PEG2000.
Introduction
Microbubbles with a diameter of 1 to 10 μm have been used as ultrasound contrast agents for noninvasive diagnostic imaging of perfusion since they became available for clinical use in the 1990 s.1 When administered intravenously, these microbubbles are too large to extravasate and therefore function as blood pool agents.2 The gas core of a microbubble compresses and expands in response to ultrasound. This feature not only provides contrast for ultrasound imaging but can also induce bioeffects in nearby cells, resulting in locally enhanced drug delivery.3,4 The gas core of the microbubble is usually stabilized by a phospholipid, protein, or polymer coating, which prolongs its lifetime by reducing surface tension and gas diffusion. The coating can be functionalized by incorporating a ligand such that these microbubbles can be targeted to specific biomarkers expressed by cells. Novel targeted microbubbles are being developed for ultrasound molecular imaging of cancer and cardiovascular disease and for therapeutic applications.3,5−7 However, before there can be widespread use of targeted microbubbles in the clinic, a better understanding and control is needed of the acoustic response and targeting, especially the ligand distribution on the microbubble coating.
A common type of coating for clinically approved microbubbles consists of a monolayer of phospholipids and emulsifiers, such as in Definity (Luminity in Europe; coating composition: 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dihexadecanoyl-sn-glycero-3-phosphate (DPPA), and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (polyethylene glycol) (DPPE-PEG5000))8 and Lumason (SonoVue in Europe; coating composition: 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), polyethylene glycol (PEG4000), and 1,2-dihexadecanoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DPPG)).9 Many experimental microbubbles are in-house produced analogues of these clinically approved microbubbles, consisting of a main phospholipid component such as DPPC (C16 tail) or DSPC (C18 tail) and an emulsifier such as polyoxyethylene(40) stearate (PEG40-stearate) and/or 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-carboxy(polyethylene glycol) (DSPE-PEG2000).10−13 For the production of targeted microbubbles, a ligand is typically coupled to DSPE-PEG2000 by biotin–avidin bridging or an alternative method of chemical coupling.5
Although it is generally assumed that the ligand, that is, DSPE-PEG2000 lipid, covers the microbubble surface uniformly, heterogeneous ligand distributions have been reported for DSPC-based microbubbles coated with binary and ternary mixtures.12,15 One study illustrated that the ligand distribution could be altered from heterogeneous to homogenous in microbubbles coated with DSPC and DSPE-PEG2000 (9:1) by different heating–cooling protocols.12
We have previously shown that the main phospholipid component influences the ligand distribution because for DPPC-based microbubbles the ligand distribution in ternary coating mixtures was homogenous in contrast to a heterogeneous distribution in DSPC-based microbubbles.15 Increased probability of successful binding of a targeted microbubble is expected when the ligand is distributed homogeneously over the microbubble coating. This is especially important in large vessels where blood flow is high and targeting is more challenging.16 It therefore remains to be explored if there are other ways to tune the ligand distribution in DSPC-based ternary coated microbubbles.
Next to a homogeneous ligand distribution for optimal targeting, the acoustic response of the microbubble is important for safe and effective use in therapeutic applications.4 Microbubbles coated with a DPPC-based ternary mixture proved to be less acoustically stable than those coated with a DSPC-based ternary mixture.17 Both these types of microbubbles show a large variation in their response to ultrasound,17,18 even when their size distribution is monodisperse.19 Kim et al.10 proposed that the acoustical properties of the microbubbles are influenced by microstructures in the coating. Microstructures are formed as a result of the phase behavior and miscibility of the different components20 and can be influenced by the conformation of the polymer chain for PEGylated components, which can be either in a brush or mushroom state.21 The degree of phase separation in the microbubble coating was also found to influence the subharmonic response.22
The phospholipid DPPC can transition from the liquid expanded (LE) to the liquid condensed (LC) phase during monolayer compression,23 whereas the phospholipid DSPC is always in the LC phase at room temperature.24 In addition, the emulsifier PEG40-stearate is known to be in the LE phase only,11 whereas DSPE-PEG2000 in a binary mixture with DSPC could transition from the LE to LC phase depending on the concentration and surface pressure.25 Studies using binary lipid mixtures (phospholipid with C18 to C24 tail and PEG40-stearate) demonstrated that microbubble coatings had microstructures with larger domain sizes when they were cooled at a slower rate after production by probe sonication.10 Another study confirmed LE and LC phase coexistence in binary microbubble coatings (phospholipids with C12 to C24 tail and PEG40-stearate), and domain morphologies varied depending on the cooling rate as well.11
Next to the lipid phase behavior, microstructures in the microbubble coating are also influenced by the miscibility and conformation (mushroom or brush) of the PEGylated components. The most widely used emulsifiers PEG40-stearate and DSPE-PEG2000 have one and two acyl chains, respectively.13 When microbubbles coated with a binary mixture of DSPC and PEG40-stearate (9:1) were studied with confocal microscopy, no domains were observed in microbubbles smaller than 5 μm,26 suggesting that there was no phase separation. Lozano and Longo concluded from their phase diagrams of monolayers at an air/water interface that DSPC and DSPE-PEG2000 are immiscible at all relevant pressures because DSPC is always in LC phase, whereas DPPC and DSPE-PEG2000 were miscible in both LE and LC phases.25 Another study focused on the distribution of DSPE-PEG2000, with a fluorescent ligand attached, in microbubbles (diameter > 10 μm) coated with DSPC and DSPE-PEG2000 (9:1). DSPE-PEG2000, that is, the ligand, was heterogeneously distributed and colocalized with the LE phase as reported for a single example, yet no quantification was performed.12 Up to now, the ligand distribution and lipid phase coexistence in microbubble coatings have not been quantified simultaneously in individual microbubbles.
A major difference between studies that evaluated microbubbles coated with a binary mixture and those coated with a ternary mixture is the handling of phospholipid components during microbubble production. For binary mixtures, the components were generally dissolved and premixed in organic solvent first, then dried to form a lipid film, and dispersed in aqueous medium before microbubbles were produced11,12,26 (i.e., indirect method). For ternary mixtures, the components were generally dispersed directly in aqueous medium before microbubble production15,27−29 (i.e., direct method). Based on the effect of cooling rate on microstructures in large microbubbles (>10 μm) after microbubble production,11 we hypothesize that the method of handling the lipids prior to microbubble production may also influence the ligand distribution and/or lipid phase in the coating of microbubbles. The ligand distribution, that is, the location of the ligand on the microbubble surface, is important for the binding probability while the lipid phases are expected to affect the elasticity and viscosity of the coating and thereby influence the acoustical performance.
The main objective of this study was to determine the DSPE-PEG2000 (i.e., ligand) distribution and lipid-phase behavior in microbubbles of clinically relevant sizes (diameter 2–8 μm) coated with a ternary mixture of DPPC or DSPC as main component and both PEG40-stearate and DSPE-PEG2000 as emulsifiers. Microbubbles were made by probe sonication after which the ligand distribution and lipid phase behavior in the microbubble coatings were visualized with high axial resolution 4Pi confocal microscopy. In addition, the relationship between the ligand distribution and the lipid phase behavior was investigated by quantifying the co-localization of ligand and LE phase. Previous studies have shown that DSPC-based microbubbles were acoustically more stable than DPPC-based microbubbles,17 but they had a heterogeneous ligand distribution.15 We therefore also investigated the effect of lipid handling on the ligand distribution in DSPC-based microbubbles. To gain insights into the physicochemical properties of the ternary mixtures, we first focused on characterizing the lipid phase behavior and PEG conformation in monolayers at an air/water interface. Because the phospholipid molecules (1 nm2)14 in the microbubble coating are so much smaller than the total surface area (0.3–0.8 × 108 nm2 for microbubbles of 3–5 μm diameter), the coating can be regarded as a flat monolayer, despite the spherical shape of the microbubble. Compression isotherms were obtained and were used together with fluorescence microscopy to visualize the lipid phase behavior of the ternary mixtures. Infrared reflection absorption spectroscopy (IRRAS) was performed to determine the phase and conformation of the individual components during monolayer compression.
Materials and Methods
Materials
DPPC, DSPE-PEG2000, and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-biotinyl(polyethylene glycol) (DSPE-PEG2000-biotin) as well as the chain deuterated lipids DPPC-d62 and DSPC-d70 were purchased from Avanti Polar Lipids (Alabaster, Alabama, USA). DSPC and PEG40-stearate were obtained from Sigma-Aldrich (Zwijndrecht, the Netherlands). Perfluorobutane (C4F10) was purchased from F2 Chemicals (Preston, UK) and argon gas was purchased from Linde Gas Benelux (Schiedam, the Netherlands). Streptavidin Oregon Green 488 was purchased from BioSynthesis (Louisville, Texas, USA), and Lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (rhodamine-DHPE) was purchased from Thermo Fisher (Waltham, Massachusetts, USA).
Monolayer Compression Isotherms
Monolayer compression isotherms were obtained at 20 °C with a Langmuir trough (sample trough 6.8 × 80 cm2) purchased from Riegler and Kirstein GmbH (Berlin, Germany) equipped with movable barriers and a Wilhelmy pressure sensor with a filter paper functioning as pressure probe. The pressure sensor was calibrated prior to each experiment to a surface pressure of 0 mN/m in water and 72 mN/m in air. The temperature was maintained at 20 °C by a circulation water bath. The complete setup was placed inside a hood to reduce dust deposition and water evaporation. Monolayers of pure DPPC or DSPC, a binary mixture (composition in mol %: DPPC or DSPC 92.4; DSPE-PEG2000 7.6), or ternary mixture (composition in mol %: DPPC or DSPC 84.8; PEG40-stearate 8.2; DSPE-PEG2000 5.9; DSPE-PEG2000-biotin 1.1) was spread on a surface of phosphate-buffered saline (PBS) as the subphase buffer solution. The binary mixture with 7.6 mol % DSPE-PEG2000 was chosen based on previously published work on microbubbles coated with a binary mixture of DPPC or DSPC (92.4 mol %) and DSPE-PEG2000 (7.6 mol %),30 having the same molar ratio of 12:1 for the main lipid to DSPE-PEG2000 as the ternary mixtures studied. Chloroform/methanol (9:1 v/v) was used as the spreading solvent31 and allowed to evaporate for at least 15 min32 before starting compression with a speed of 2 Å2 molecule–1 min–1. The surface pressure was recorded during compression with a time resolution of 2 s with RUK trough control software (Riegler and Kirstein GmbH).
Monolayer Fluorescence Microscopy
To study the lipid organization with fluorescence microscopy, rhodamine-DHPE (0.01 mol %) was added to the DPPC- and DSPC-based ternary mixtures before spreading the monolayer. Because this dye does not diffuse into the LC phase,33 all dark areas are lipids in the LC phase and all areas with a fluorescent signal are in a more fluid phase, that is, LE phase. The monolayers were spread on a Langmuir trough (sample trough 9.9 × 26 cm2; Riegler and Kirstein GmbH) and imaged during compression with an Axio Scope A1 Vario epifluorescence microscope (Carl Zeiss MicroImaging, Jena, Germany) equipped with a mercury arc lamp (HXP 120 C) for excitation, a long working distance objective (NEOFLUAR 50×), and a filter/beam splitter set (Zeiss Filter Set 09), which allows excitation between 450 and 490 nm and detection of emitted light above 515 nm. Images were recorded with an EMCCD Camera (ImageEM, C9100-13, Hamamatsu, Herrsching, Germany) and the surface pressure was recorded as described previously.
IRRAS Experiments
On the basis of the isotherms and the fluorescence microscopy images, it is not possible to distinguish the phase state of the individual components in the ternary mixtures. To investigate the phase behavior of the individual components, IRRAS experiments were performed. The use of chain-deuterated phospholipids (DPPC-d62 or DSPC-d70) in the ternary mixtures allowed us to distinguish the signal from the PEGylated (CH2 vibrations) and non-PEGylated (CD2 vibrations) components. The IRRAS measurements were performed using a Bruker Vector 70 FT–IR spectrometer equipped with a nitrogen-cooled MCT detector and an A511 reflection unit (Bruker Optics, Ettlingen, Germany), placed over a Langmuir trough setup (Riegler and Kierstein GmbH). The sample trough (6 × 30 cm2) was set up according to the protocol described above. A circular reference trough (3 cm radius) placed next to the sample trough could be brought into the focus of the IR beam by means of a shuttle. Both troughs were filled with PBS as the subphase buffer solution and lipid mixtures were spread in the sample trough as described above. The filling levels of both troughs were kept equal and constant by means of an automated, laser reflection-controlled, pumping system connected to PBS reservoirs. The IR beam was coupled out from the spectrometer and focused by mirrors onto the buffer or film surface at an incidence angle of φ = 60°. A KRS-5 polarizer was used to generate perpendicular polarized light. The compression of the monolayer was performed at 2 Å2 molecule–1 min–1. The compression was stopped at several predefined areas per lipid chain, as indicated in Figure 3B,D, to record at least three IRRA spectra at constant molecular area before the compression was continued. Spectra were recorded with a spectral resolution of 4 cm–1 and 160 kHz scanner velocity. One thousand single interferograms were zero-padded with a factor of two and averaged, followed by fast Fourier transformation, resulting in a nominal spectral resolution of 2 cm–1. IRRAS spectra were calculated from the reflectivity on the monolayer covered surface (R) and the bare buffer surface (R0) according to reflection absorption RA = −log10(R/R0). All IRRA spectra were corrected for atmospheric water vapor absorption using OPUS software (Bruker Optics GmbH, Ettlingen, Germany) and set to a common baseline in a spectral range where no absorptions occurred (4500–4600 cm–1). The maxima of the CH2 (2800–2950 cm–1) and CD2 (2020–2270 cm–1) stretching vibrational bands were determined using the standard method of the OPUS software. The peak positions were averaged for spectra recorded at the same lipid molecular area and surface pressure and are presented together with their standard deviation. The presented spectra are averages of all spectra recorded at the same monolayer state. To identify the contribution of the phospholipid headgroups to the transitions, principal component analysis (PCA)34,35 was done with the princomp function of MATLAB. PCA was chosen to analyze the variation in the data by computing principal components (PC), which can be used to determine the variable responsible for the largest variance in the dataset. Corresponding scores were used to determine the contribution of each spectrum to this main variance. For the analysis, the spectral regions between 1050 and 1300 cm–1 for headgroup vibrations and 2020–2270 cm–1 for CD2 stretching vibrations were selected from the IRRA spectra recorded at various surface pressures. From both spectral ranges, a linear baseline was subtracted before they were normalized to a vector norm of unity. Subsequently, both subspectra were combined to a single input vector for the PCA, where the wavenumbers were the variables and the surface pressures the conditions. The first principal components and the respective scores are presented in the Results and Discussion section. Scores of the higher principal components did not change systematically with film compression.
Figure 3.

Spectra obtained by IRRAS of a monolayer composed of the ternary mixture (A) DPPC-d62/DSPE-PEG2000/PEG40-stearate (84.8:7.0:8.2 mol %) and (C) DSPC-d70/DSPE-PEG2000/PEG40-stearate (84.8:7.0:8.2 mol %), reflection absorption (RA = −log10(R/R0)) as a function of wavenumber (ν̃) for different surface pressures (π). CH2 and CD2 stretching bands are zoomed in; other bands are labeled: (a) OH stretching, (b) C=O stretching, (c) HOH bending, (d) PO2 antisymmetric stretching, (e) PO2 symmetric stretching and C–O stretching. (B,D) surface pressure (π) as a function of area per lipid chain (AM) (black curve, left y-axis); wavenumbers of symmetric (orange line and symbols, right y-axis) and antisymmetric (blue line and symbols, right y-axis) CD2 stretching vibration of the (B) DPPC-d62-based or (D) DSPC-d70-based ternary mixtures. Note the wider area range in (D) as compared to (B). Wavenumbers of the CH2 stretching vibrational bands are given in Supplementary Figure 3.
Microbubble Production
Biotinylated lipid-coated microbubbles (composition in mol %: DSPC or DPPC 84.8; PEG40-stearate 8.2; DSPE-PEG2000 5.9; DSPE-PEG2000-biotin 1.1) with a C4F10 gas core were made by probe sonication at 20 kHz with a Sonicator Ultrasonic Processor XL2020 at power setting 10 (HeatSystems, Farmingdale, NY, USA) for 10 s as described previously.27 The coating components were prepared in two different ways. (1) For the direct method, all components were dissolved in PBS with a final concentration of 2.5 mg/mL DSPC or DPPC, 0.625 mg/mL PEG40-stearate, 0.625 mg/mL DSPE-PEG2000, and 0.125 mg/mL DSPE-PEG2000-biotin. Fluorescent dye rhodamine-DHPE (0.01 mol %) was added to study the lipid phase organization in the microbubble coating. (2) For the indirect method, DSPC, PEG-40 stearate, DSPE-PEG2000, and DSPE-PEG2000-biotin were dissolved in chloroform/methanol (9:1 vol/vol). The organic solvent was then evaporated with argon gas and the obtained lipid film was dried under vacuum overnight. Finally, the lipid film was dispersed in PBS with a final concentration of 2.5 mg/mL DSPC or DPPC, 0.625 mg/mL PEG40-stearate, 0.625 mg/mL DSPE-PEG2000, and 0.125 mg/mL DSPE-PEG2000-biotin, fluorescent dye rhodamine-DHPE (0.01 mol %) was added, the solution was placed in a sonicator bath for 10 min, and a probe sonicator was used at power setting 3 for 5 min. The three types of microbubbles produced are referred to as “direct DPPC”, “direct DSPC”, or “indirect DSPC” microbubbles.
Microbubble Fluorescence Imaging
The fluorescent ligand streptavidin Oregon Green 488 was conjugated to the biotinylated microbubbles as described previously by Kooiman et al.,15 allowing us to determine the distribution of DSPE-PEG2000–biotin over the lipid phases in the microbubble coating. Briefly, 0.9 mL of microbubble suspension was placed in a 3 mL syringe and topped with 2.1 mL of PBS saturated with C4F10 for washing by flotation. The subnatant was drained after 45 min, and the microbubbles were resuspended in 0.3 mL PBS saturated with C4F10 and collected. Next, 22.5 μL of streptavidin (2 mg/mL) was added to 0.7–1.0 × 108 microbubbles. After incubation on ice for 30 min, the excess streptavidin was washed by flotation, as described above, and the microbubbles were resuspended in 0.2 mL of PBS saturated with C4F10.
A Coulter Counter Multisizer 3 (Beckman Coulter, Mijdrecht, the Netherlands) was used to measure the microbubble size distribution and concentration. A 50 μm aperture tube was used for quantification of particles between 1 and 30 μm with a linear spacing between the 256 channels. The size distribution of the samples was evaluated by the span value, which illustrates the width of the distribution, defined as (d90–d10%)/d50%, where d90, d10, and d50% are the microbubble diameters below which 90, 10, and 50% of the cumulative number of microbubbles was found.
After conjugation with streptavidin Oregon Green 488, microbubbles were visualized as described by Kooiman et al.15 To reduce Brownian motion, microbubbles were placed in 87% glycerol (v/v in PBS) and visualized using a Leica TCS 4Pi confocal laser-scanning microscope.36 The 87% was chosen because this has the same refractive index as the quartz glass and glycerol objective of the 4Pi microscope. This high-resolution imaging system has a matched pair of aligned opposing 100× glycerol HCX PL APO objective lenses (Numerical aperture 1.35), increasing the axial resolution up to 90 nm. A 488 nm laser was used for excitation of Oregon Green 488 and a 561 nm laser was used for excitation of rhodamine-DHPE. Image stacks were recorded as y-stacked xz-scans in a green (500–550 nm) and red (580–640 nm) spectral channel. The software AMIRA (Version 2019.1, FEI, Mérignac Cedex, France) was used to volume-render the image stacks with the “voltex” function.
Microbubble Data Analysis
Custom-developed image analysis software in MATLAB (Mathworks, Natick, MA, USA) was used for quantitative analysis of the 4Pi microscopy data. The ligand distribution was analyzed based on the method described by Kooiman et al.15 First, a circle was fitted through the fluorescence intensity maxima of the green channel (Oregon Green 488, 500–550 nm) and per xz-slice a region of interest (ROI) was defined in a band of 7 pixels around the fitted circle. Only slices with an ROI radius larger than 75% of the radius in the equatorial plane ROI were included in the analysis. Each of the ROIs was divided into 32 angular parts and the mean fluorescence pixel intensity (Ipart) was calculated for each of those parts. The Ipart values were plotted per microbubble as a function of the axial plane and the microbubble circumference in 2D color-coded heatmaps (Supporting Information Figure 1A). On average, 30 xz-slices were included per microbubble, resulting in an average of 960 angular parts per microbubble. The median intensity of all of the angular parts (Imedian) was calculated for each microbubble. The image analysis software classified an individual angular part as inhomogeneous when the absolute difference between Ipart and Imedian was more than two-third times the value of Imedian (i.e., |Ipart – Imedian| > 2/3 × Imedian). The percentage of parts classified as inhomogeneous was calculated per microbubble as a measure for the inhomogeneity of the ligand distribution. After this analysis, an adapted version of the software was used to analyze the lipid phase behavior in the red channel (rhodamine-DHPE, 580–640 nm). The same xz-slices and ROIs were used as those obtained during the ligand distribution analysis. Again, the ROIs were divided in 32 angular parts and the mean fluorescence pixel intensity (Ipart-rhod) in each part was calculated. From these, the median part intensity (Imedian-rhod) was calculated per microbubble and plotted as 2D color-coded heatmaps (Supporting Information Figure 1B). The software classified an individual angular part as LC phase when the value of Ipart-rhod was less than one-third of Imedian-rhod (i.e., Ipart-rhod < 1/3 × Imedian-rhod) (Supporting Information Figure 1C). The LC phase surface area was determined per microbubble in μm2 and presented as percentage of the total analyzed surface area per microbubble. To study if the ligand colocalized with the parts classified as LC areas, the median fluorescence intensity of the green channel (ligand) was calculated for all parts in LC phase and for those not in LC phase (Supporting Information Figure 1D,E). The ratio between these two values was defined as the colocalization ratio.
IBM SPSS Statistics 25 was used to perform statistical analysis. The distribution of the data was assessed using a Shapiro–Wilk test. The data on ligand inhomogeneity was not normally distributed (p < 0.001) for all microbubble types. The data on the LC phase area was only normally distributed for direct DPPC (p = 0.228); not for direct DSPC (p = 0.002) and indirect DSPC (p < 0.001) microbubbles. The colocalization ratio was normally distributed (DPPC: p = 0.168, DSPC direct method: p = 0.203, DSPC indirect method: p = 0.334). Therefore, the Mann–Whitney U test was used to test if the microbubble types had a significant difference in inhomogeneity of the ligand distribution and LC phase area. For the colocalization ratio, a regular t-test was used to analyze the differences between the microbubble types. Differences were regarded as significant at p-value < 0.01.
Results and Discussion
Monolayer Compression Isotherms
The results of the Langmuir trough experiments are presented in Figure 1. All curves are representative of the results from three or more experiments. In accordance with the literature,23,25 there was a clear difference between the isotherms of pure DPPC (Figure 1A, black line) and DSPC (Figure 1C, black line) as DPPC had a transition from the LE to LC phase at a surface pressure (π) of ∼5 mN/m, whereas DSPC did not form an LE phase and therefore underwent a direct gaseous to LC phase transition. The binary mixture with 7.6 mol % DSPE-PEG2000 was chosen based on previously published work on microbubbles coated with a binary mixture of DPPC or DSPC (92.4 mol %) and DSPE-PEG2000 (7.6 mol %).30 For DPPC in mixtures with PEGylated compounds, we observed two transitions (Figure 1A,B). The transition from the LE to LC phase of DPPC occurred almost at the same surface pressure in the binary mixture (DPPC/DSPE-PEG2000), whereas it was slightly shifted to a lower pressure in the ternary mixture containing PEG40-stearate. This shift to a lower surface pressure is due to the long stearoyl chain of the PEG40-stearate increasing the stability of an LC phase. In the binary mixture, a second transition at ∼10 mN/m was observed (Figure 1B, orange line). In the ternary mixture with a higher content of PEGylated components, the second transition moved to ∼12 mN/m (Figure 1B, blue line). For DSPC in mixtures with PEGylated lipid components, we observed phase transition plateaus only at ∼10 mN/m (Figure 1B, blue line, binary mixture) and ∼12 mN/m (Figure 1D, blue line, ternary mixture), similar to the second transition in DPPC-based mixtures (Figure 1C,D, orange and blue lines). Again, the transition pressure increased with the increasing content of PEGylated components. As the transition ≥10 mN/m is independent of the type of phospholipid, we assume that it is due to the so-called mushroom to brush transition of the PEG chains attached to the lipid headgroups.37−39 Theoretical calculations of the mushroom to brush transition40 for the binary mixture in this study is 45 Å2 (lipid chain)−1 (see Supporting Information Figure 2) which is in agreement with the experimental findings of 30–60 Å2 (lipid chain)−1. For the ternary mixture, the calculated mushroom to brush transition is 87 Å2 (lipid chain)−1 (see Supporting Information Figure 2) which is only slightly higher than the experimentally observed 60–80 AM/Å2 (lipid chain)−1. The difference could be explained by the polydispersity of the PEG40-stearate41 because a decrease in chain length lowers the Flory radius.
Figure 1.

Langmuir isotherms of pure, binary (92.4:7.6 mol %), and ternary (84.8:7.0:8.2 mol %) mixtures with (A) DPPC or (C) DSPC as main lipid components and DSPE-PEG2000 and/or PEG40-stearate (PEG40-S) as additional components. (A,C) Surface pressure (π) as a function of the area per molecule (AM). (B,D) Derived compressibility (κ) as a function of the surface pressure (π) where the peaks indicate transition plateaus. Representative curves are shown of at least three repeated experiments.
The isotherms of the binary mixtures presented here are in agreement with literature for the same binary mixtures.25,42 By contrast, another study on binary mixtures of DSPC with DSPE-PEG2000 or PEG40-stearate (9:1) found that the mixture with DSPE-PEG2000 had an isotherm similar to that of pure DSPC, while the binary mixture with PEG40-stearate had an extra transition plateau around 35 mN/m.26 This was attributed to expulsion of material from the monolayer, sometimes referred to as squeeze-out.11 However, in the present study, we observed no squeeze-out plateau in the ternary mixtures that contained PEG40-stearate. This may be explained by the differences in concentration of PEG40-stearate (10 vs 8 mol %) and the addition of DSPE-PEG2000 as the third component.
Monolayer Fluorescence Microscopy
Fluorescent micrographs of monolayers containing ternary mixtures with DSPC or DPPC at different surface pressures during compression are shown in Figure 2. The DPPC containing mixture was homogenous with some bright spots at the starting surface pressure (<1 mN/m). These bright spots could be due to the coexistence of gaseous and LE phases, with the fluorescent lipid dye enriched in the LE phase spots. With the increasing surface pressure, the bright fluorescent spots disappeared and at 5 mN/m the fluorescent dye was homogenously distributed, indicating that all components were in the same phase, namely, the LE phase. Above 5 mN/m, dark domains of LC-phase lipids appeared and grew larger as the compression of the monolayer advanced. Initially, these dark domains were clustered like flower petals connected to a central point (Figure 2, DPPC 10 mN/m). As the surface pressure increased, the LC domains separated and the interdomain region became brighter because a fixed amount of fluorescent dye was distributed over a smaller surface area. These micrographs show the same morphology of dark domains as in previously published micrographs on a binary mixture of DPPC and PEG40-stearate (9:1).11 Interestingly, the dark LC domains containing mainly DPPC did not form the characteristic bean- or propeller-like shapes with defined chirality as observed for pure DPPC.43,44 This seems to be an indication that the LC phase is not pure DPPC but contains some achiral PEG40-stearate or DSPE-PEG2000, thus preventing the formation of chiral domains.
Figure 2.

Fluorescent micrographs of monolayers of ternary mixtures containing DSPE-PEG2000 (7.0 mol %), PEG40-stearate (8.2 mol %), and either DPPC (84.8 mol %, top row) or DSPC (84.8 mol %, bottom row) at various surface pressures, taken during monolayer compression. Scale bars (black) represent 25 μm and apply to all images.
A major difference between DPPC- and DSPC-based mixtures is the presence of LC domains in the DSPC-containing monolayer at low surface pressures (<1 mN/m). This is consistent with the isotherms of pure DSPC where a direct transition from the gaseous phase to LC phase is observed. Interestingly, in the DSPC mixture there appeared to be three phases at low surface pressures (<1–5 mN/m). Previous work on bilayers has indeed shown that three-phase co-existence can occur in a ternary mixture of DSPC with 1,2-dioleoyl-sn-glycero-3-phosphocholine and cholesterol.45 With the increasing surface pressure, the dark LC domains grew larger. However, the size of the LC domains at different surface pressures is much smaller than in the DPPC containing monolayers. The size of LC domains is dependent on the line tension between the LC domains and the surrounding phase and the excess dipole density in the LC domains.46−48 The latter effect leads to a repulsion between the domains and prevents the domain growth driven by the line tension. The DSPC-containing LC domains probably have a larger excess dipole density with respect to the surrounding gaseous phase, than DPPC-containing LC domains with respect to the surrounding LE phase, which prevents further LC domain growth of DSPC. High excess dipole density leading to small LC domains has been reported for DMPC/DSPC monolayers containing 60 mol % DSPC46 and for DSPC monolayers containing 1–9 mol % DSPE-PEG2000.49 The transition around 10 mN/m that was identified in the isotherms was less apparent in the fluorescence micrographs. With increasing surface pressure, the most noticeable change was an increase in the relative surface area of the LC domains, indicating that the surrounding LE phase is being compressed without molecules transitioning into the LC phase. This suggests that a transition in the headgroup region occurs instead, namely a mushroom-brush transition of the PEG chains in the aqueous phase.
IRRAS Experiments
Figure 3 shows the results of the IRRAS experiments we performed to attribute the different transition plateaus of the isotherms to specific phase transitions. The position of the methylene stretching vibrational bands in the IRRA spectra is dependent on the phase state of the respective lipid, with a downshift in their wavenumbers being indicative for an LE to LC transition.50−52 The stretching vibrations of the CD2 groups of the deuterated lipids are well separated from the CH2 stretching vibrations of the PEGylated components and any other vibrational bands, allowing separate analysis of the phase state of the main phospholipid component and the PEGylated components (Figure 3A,C). Isotherms of pure DPPC-d62 and DSPC-d70 were measured to confirm that deuteration had only little effect on the lipid phase behavior (Supporting Information Figure 3, compared to Figure 1A,C). However, the LE to LC phase transition of pure DPPC-d62 shifted to slightly higher surface pressure, that is, toward the second transition detected for DPPC-based ternary mixtures, resulting in a slight overlap of both transitions. Nevertheless, IRRA spectra of the monolayers of ternary mixtures containing DPPC-d62 showed a transition from LE to LC phase concomitant with the plateau (at ∼12 mN/m) in the isotherm (Figure 3B). In contrast, no DSPC-d70 molecules were found in the LE phase, indicating a direct transition from the gaseous to the LC phase (Figure 3D, blue and orange line). The position of the CD2 stretching vibrational bands at low wavenumbers throughout the examined compression range indicates that DSPC-d70 is already in the LC phase at high molecular areas. This unambiguously shows that the transition plateau we found around ∼10–12 mN/m is not due to a phase transition of DSPC itself and must thus be caused by a reorganization of the PEGylated components, probably the mushroom to brush transition of the PEG chains.25 The CH2 stretching vibrations of the PEGylated components were analyzed as well (Supporting Information Figure 4B,D). However, because of low signal, we only have data from surface pressures above 12.5 mN/m; thus, we cannot distinguish the transition that occurs below this surface pressure. The CH2 vibrational bands arise from the CH2 groups in the chains and in the headgroups of the PEGylated components, mainly PEG40-stearate, with the majority of the CH2 groups being located in the PEG groups of PEG40-stearate and DSPE-PEG2000. However, because the conformation of the CH2 groups in the flexible PEG chains is not well defined, their contribution to the CH2 stretching vibrational band is broad and the band position is still dominated by the vibrations of the higher ordered lipid acyl chains. The CH2 vibrational bands of pure DPPC before and after transition can be used as a reference for characteristic LE and LC phase wavenumbers (Supporting Information Figure 5). The CH2 bands in the ternary mixtures were observed at a wavenumber characteristic for neither an LC nor an LE phase, but in between; namely, at 2852 cm–1 (symmetric CH2-vibration) and at 2922 cm–1 (antisymmetric CH2-vibration) (Supporting Information Figure 4B, 4D). This suggests that part of the PEGylated molecules was in the LE phase and part was in the LC phase. When comparing the CH2 vibrational bands of the PEGylated molecules to the CH2 vibrational band of pure DPPC during transition, the observed wavenumbers (ν̃) suggest that the majority (about 60%) of the PEGylated lipids are still in LE phase (Supporting Information Figure 5). In case of the DSPC-based ternary mixtures, the LE phase is consequently formed only by the PEGylated molecules, whereas in the DPPC-based mixtures the LE phase contains DPPC and/or PEGylated molecules.
To identify the contributions of the lipid headgroups to the transitions, we performed a PCA on the IRRA spectra in the region of the PO2 and C–O stretching vibrations (symmetric and antisymmetric; Figure 3A,C, label d and e), originating from the headgroup attached PEG chains (1050–1300 cm–1), and the CD2 stretching vibrations, originating from the acyl chains (2020–2270 cm–1) (Figure 4). This type of analysis identifies the main variances in the spectra during compression of the mixed monolayers and attributes them to different spectral regions, influential in the reorganization of different molecular moieties. The extent of these variations is expressed in first principal component (PC1) scores (Figure 4A,C). In the here presented analysis, these scores change systematically with the surface pressure, with a pronounced step at about 10 mN/m. This corroborates our finding that both mixed monolayers show a transition when compressed above this surface pressure. Interestingly, the reason for this transition is different in DPPC-d62 containing and DSPC-d70 containing monolayers, as can be deduced from the PC1 depicted in Figure 4B,D. The DPPC-d62 containing monolayer spectra show simultaneous changes in the CD2 stretching vibrational region and in the region of the headgroup vibrations. This suggests that both the acyl chains and the PEGylated headgroups contributed to the transition. In contrast, for the DSPC-d70 containing monolayer, spectral changes corresponding to the transition were only identified in the head group region because the PC1 (red line in Figure 4D) shows variations in the spectral range of the PO2 and C–O stretching vibrations but is essentially zero in the range of the CD2 stretching vibrations. With this finding, the transition in the ternary mixtures containing DSPC can clearly be attributed to reorganizations in the PEGylated headgroups, presumably a PEG mushroom to brush transition.
Figure 4.
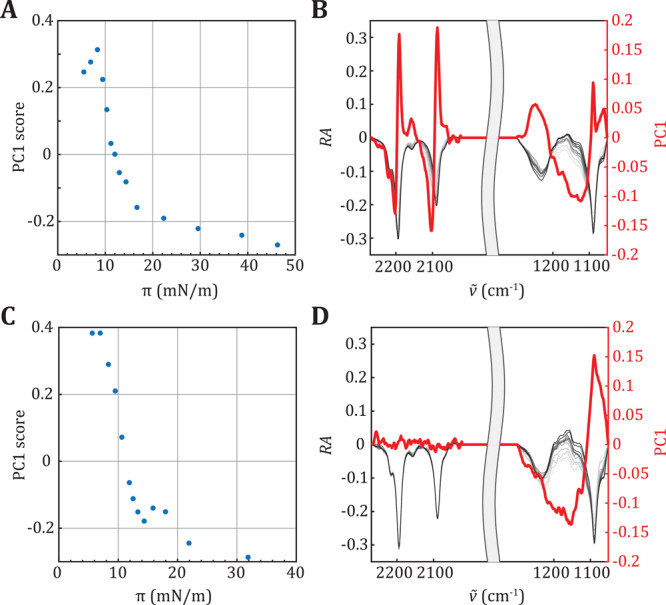
PCA of IRRA spectra recorded during the compression of a PL/DSPE-PEG2000/PEG40-stearate (84.8:7.0:8.2 mol %) mixed monolayer, where PL = DPPC-d62 (A and B) and DSPC-d70 (C and D). IRRA spectra were simultaneously analyzed in the range of the headgroup vibrations (1050–1300 cm–1) and the CD2 stretching vibrations (2020–2270 cm–1) after separate vector normalization in the two respective ranges (gray lines in (B and D); low surface pressures (light gray) to high surface pressures (dark gray)). Panels (A and C) show the scores of the first principal components (PC1) as function of the surface pressure (π). Panels (B and D) show the reflection absorption (RA, gray lines, left y-axis) and PC1 score (red lines, right y-axis) as a function of the wavenumbers (ν̃).
To verify that the changes in the headgroup region are not only due to the phospholipid phosphate groups but contain contributions of PEG chain reorganization, we repeated the IRRAS compression experiment and PCA with a pure DPPC-d62 monolayer (see Supporting Information Figure 6). Comparison of the first principal components shows a lower PC1 in the range of the headgroup vibrations for pure DPPC-d62, indicating that only phosphate reorganization would not be sufficient to explain the variations in the above presented spectra. Thus, we conclude that PEG chain conformational changes must be involved in the transitions of the monolayers containing ternary mixtures of DSPE-PEG2000 and PEG40-stearate with DPPC or DSPC.
All experiments described above were performed at the air/buffer interface. However, during microbubble production, C4F10 gas is added to the air and the phospholipids are dispersed in PBS saturated with C4F10 gas. Recently published work demonstrates how the LE to LC transition of DPPC was shifted to higher surface pressures in the presence of C6F14 in the gas phase.53 Another study showed a shift of the LE to LC transition to higher surface pressures in a binary mixture of DPPC with 5 mol % DSPE-PEG2000, in the presence of C6F14 in the subphase and air in the gasphase.54 Taking this into consideration, the influence of a fluorinated hydrocarbon in the gas phase on the isotherms cannot be excluded, meaning that the isotherms would be slightly shifted to higher transition pressures.
Microbubbles
Figure 5 shows the number weighted size distribution of the streptavidin-conjugated microbubbles. The number-weighted mean diameter was 3.6 μm for direct DPPC microbubbles, 4.2 μm for direct DSPC microbubbles, and 5.17–5.22 μm (n = 2 batches) for indirect DSPC microbubbles. The volume-weighted mean diameter was 6.6 μm for direct DPPC microbubbles, 6.4 μm for direct DSPC microbubbles, and 7.9–8.4 μm (n = 2 batches) for indirect DSPC microbubbles. Direct and indirect DSPC microbubbles had a similar size distribution (span 1.0), whereas the direct DPPC microbubbles were more polydisperse (span 1.4). The size distributions of the direct microbubbles are in agreement with previously published work.15 The indirect method resulted in slightly larger DSPC microbubbles than direct DSPC microbubbles but did not affect the polydispersity.
Figure 5.
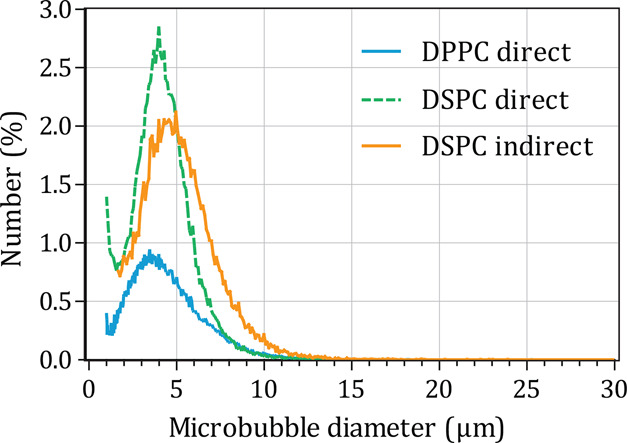
Number weighted size distribution of DPPC direct (blue line, n = 1 batch), DSPC direct (green dashed line, n = 1 batch), and DSPC indirect (orange line, representative for n = 2 batches) microbubbles with ternary coating composition containing DSPE-PEG2000 (7.0 mol %) and PEG40-stearate (8.2 mol %) as additional components.
Ligand Distribution in Microbubbles
The lipid phase and ligand distribution in the microbubble coating were imaged for direct DPPC (n = 50, 2 batches), direct DSPC (n = 47, 3 batches), and indirect DSPC microbubbles (n = 46, 2 batches) of 1.5–8 μm in diameter. Typical examples of the different types of microbubbles are shown in Figure 6. The ligand distribution, representative for the DSPE-PEG2000 distribution, is shown in the left column, the LE phase stained with rhodamine-DHPE in the middle column, and a composite of both signals is displayed in the right column. The calculated ligand distribution inhomogeneity is shown in Figure 7. In concurrence with our previous study,15 the direct DPPC microbubbles had a mostly homogenous ligand distribution (Figures 6A and 7), while there was a large variability in ligand distribution for the direct DSPC microbubbles ranging from heterogeneous with areas where the ligand was either lacking or enriched (Figure 6D), to a more homogenous ligand distribution (Figure 6G). Nevertheless, the indirect DSPC microbubbles all had a homogenous ligand distribution (Figures 6J and 7). No correlation was found between microbubble size and ligand inhomogeneity. A previous study, which focused on phase separation in phospholipid-coated microbubbles processed with different heating-cooling regimes, showed that the ligand was distributed heterogeneously in slowly cooled microbubbles and homogenously in rapidly cooled microbubbles.12 These microbubbles were coated with a binary mixture of DSPC and DSPE-PEG2000 (9:1) and made by mechanical shaking. In the present study, we investigated no heating-cooling regimes, yet we found that a different handling of phospholipids before microbubble production could also result in a more uniform ligand distribution.
Figure 6.
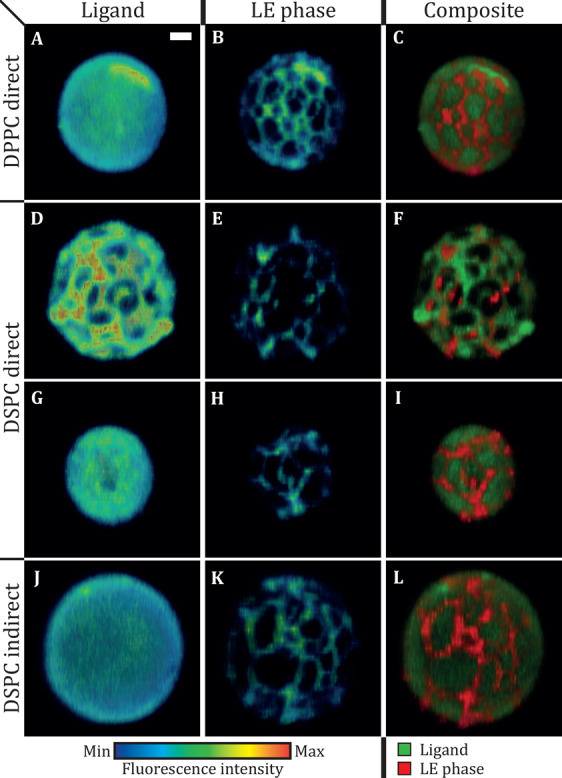
Selected views of 4Pi confocal microscopy y-stacks of direct DPPC (A–C, diameter (d) = 4.7 μm), direct DSPC (D–F, d = 4.9 μm; G–I, d = 3.4 μm), and indirect DSPC microbubbles (J–L, d = 5.3 μm) with ternary coating composition containing DSPE-PEG2000 (7.0 mol %) and PEG40-stearate (8.2 mol %) as additional components. The images show the ligand distribution (A, D, G, J; Oregon Green 488), LE phase (B, E, H, K; rhodamine-DHPE), and composite view (C, F, I, L). Scale bar is 1 μm and applies to all images. Full 3D reconstructions of these examples are provided as supplemental Videos 1–4.
Figure 7.
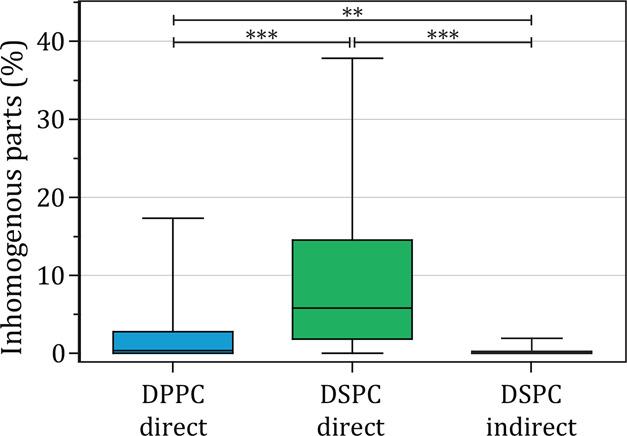
Parts classified as inhomogeneous (%) in the ligand distribution of direct DPPC (n = 50), direct DSPC (n = 47) and indirect DSPC (n = 46) microbubbles with ternary coating composition containing DSPE-PEG2000 (7.0 mol %) and PEG40-stearate (8.2 mol %) as additional components. Boxplots show the median, interquartile range and have whiskers from minimum to maximum. Statistical significance was indicated with **p < 0.01, ***p < 0.001.
Lipid Phase Distribution in Microbubbles
In all types of direct and indirect microbubbles, the lipids were phase-separated resulting in dark domains (i.e., LC phase) and bright interdomain regions (i.e., LE phase), when studying the fluorescence of rhodamine-DHPE (Figure 6B,E,H,K). Although the LC domains in the DPPC- and DSPC-based ternary mixture monolayers had different morphologies (Figure 2), the LC domains in the microbubble coatings were similar for all types of microbubbles. Fluorescent dyes have been used before to examine domain formation in microbubbles coated with binary mixtures of DPPC and DSPC with PEG40-stearate or DSPE-PEG2000, with a diameter larger than 10 μm.10−13,26 In these studies, the microstructures in the microbubble coating were tuned by varying the cooling rate after microbubble production or by varying the pure lipid to PEGylated molecule ratio. To the best of our knowledge, the present study is the first to include microbubbles coated with a ternary mixture and of clinically relevant sizes, namely, 1.5–8 μm in diameter.
The domain morphology of microbubbles coated with a ternary mixture presented here resembles that of microbubbles (diameter > 5 μm) coated with a binary mixture of DSPC and PEG40-stearate or DSPE-PEG2000 (9:1), despite the use of different fluorescent dyes and microbubble production methods.10,12 Others imaged phase separation with epifluorescence or confocal microscopy, in contrast to the high-resolution 4Pi confocal microscopy that was used for this study. Previous studies reported that no domain formation was observed in microbubbles with a binary mixture of DSPC and PEG40-stearate (9:1) smaller than 5 μm, even though domains smaller than 5 μm2 were observed in microbubbles larger than 5 μm.26 However, all microbubbles analyzed for the present study (1.5–8 μm diameter) had condensed domains in the coating. This is likely due to phase separation of the three components: the main lipid component DPPC (in LE/LC phase), or DSPC (in LC phase), PEG40-stearate in LE phase, and DSPE-PEG2000 in LC or LE phase. Microbubbles were mounted in 87% glycerol for 4Pi high-resolution imaging. Monolayer studies at the air/water interface showed that glycerol in the subphase had no effect on the phase behavior below the transition temperature.55 In our study, glycerol was added after microbubble production and the sample was kept at room temperature during imaging experiments. We therefore assume that the glycerol did not have an effect on the molecular structure of the lipid microbubble coating.
The rhodamine-DHPE fluorescence intensity (Ipart-rhod) and the surface area classified as LC phase were plotted as a function of the axial plane and the corresponding circumference (Figure 8A,B). The LC area fraction is presented in Figure 8C, as the percentage of the total surface area analyzed per microbubble. The mean percentage of LC area was significantly lower for the direct DPPC microbubbles than for both types of DSPC microbubbles. This was expected because DSPC is always in the LC phase, according to our monolayer results presented above and literature.24 The direct DSPC microbubbles had a significantly smaller LC phase area than the indirect DSPC microbubbles. Because DSPC is always in the LC phase and the other PEGylated components were the same, there must be a difference in the localization of these PEGylated components causing the differences in LC area between direct and indirect DSPC microbubbles. No correlation was found between microbubble size and LC phase area. These results indicate that the lipid handling affects the phase separation between different components. Previous studies that investigated domain characteristics focused mainly on the effect of cooling rates in microbubbles coated with binary mixtures of DSPC with PEG40-stearate,10,11 yet the microbubbles in those studies were much larger (>20 μm diameter) than the microbubbles investigated here.
Figure 8.
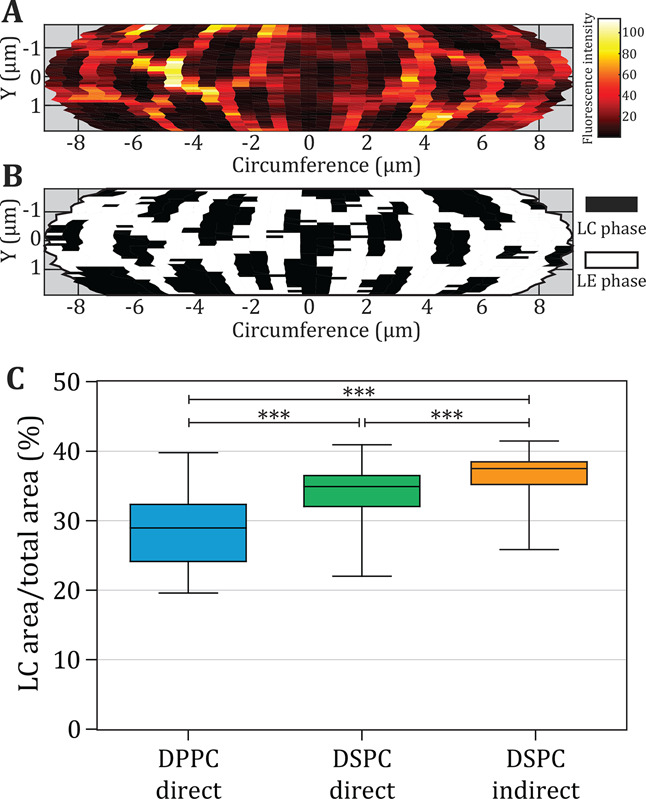
(A) Example of a heatmap of rhodamine-DHPE intensity over the analyzed surface area for an indirect DSPC microbubble (D = 5.74 μm). (B) Thresholded map of (A) showing the parts classified as LC area in black. (C) Size of the LC area (% of total surface area) of DPPC direct (n = 50), DSPC direct (n = 47), and DSPC indirect (n = 46) microbubbles with ternary coating composition containing DSPE-PEG2000 (7.0 mol %) and PEG40-stearate (8.2 mol %) as additional components. Boxplots show the median, interquartile range, and with whiskers from minimum to maximum. Statistical significance was indicated with *** for p < 0.001.
The right column of Figure 6 shows composites of the lipid phase and ligand distribution in the microbubble coating. For the direct DPPC and indirect DSPC examples, the green fluorescent ligand is distributed homogenously over the fluorescently stained LE phase and the LC phase (Figure 6C,L). For the direct DSPC microbubbles, two examples are shown to illustrate the variability within this group: heterogeneous distribution where the ligand is colocalized with the LE phase (Figure 6F) and homogenous ligand distribution similar to the other types of microbubbles (Figure 6I). Colocalization of the DSPE-PEG2000 with the LE phase has been reported before for a single example of a ∼20 μm diameter microbubble coated with a binary mixture of DSPC and DSPE-PEG2000 (9:1) without quantification.12 In our study, we quantified the colocalization between the LC phase (no rhodamine-DHPE fluorescence) and DSPE-PEG2000, the component where the fluorescent ligand Oregon Green 488 is attached to, which is presented in Figure 9A. For the direct DPPC and indirect DSPC microbubbles, the mean colocalization ratio was approximately 1, indicating that the amount of DSPE-PEG2000 in the LC phase domains was equal to the amount of DSPE-PEG2000 in the interdomain region. The colocalization ratio was significantly lower for the direct DSPC microbubbles, indicating that there was less DSPE-PEG2000 in the LC phase domains than in the interdomain regions. For these direct DSPC microbubbles, there was a negative correlation between the percentage of inhomogeneity in the ligand distribution and the colocalization ratio (Figure 9B). This suggests that in microbubbles with a heterogeneous ligand distribution, the ligand was depleted in the LC domains.
Figure 9.

(A) Colocalization ratio of direct DPPC (n = 50), direct DSPC (n = 47), and indirect DSPC (n = 46) microbubbles with ternary coating composition containing DSPE-PEG2000 (7.0 mol %) and PEG40-stearate (8.2 mol %) as additional components. Boxplots show the median, interquartile range, and with whiskers from minimum to maximum. Statistical significance was indicated with ***p < 0.001, **p < 0.01. (B) Colocalization ratio as a function of the parts classified as inhomogeneous (%) ligand distribution for direct DSPC microbubbles (n = 47).
Based on the differences that we found in LC area and ligand distribution, between the direct and indirect DSPC microbubbles, we expect that the DSPE-PEG2000 component is either excluded from preformed LC domains (for direct method, Figure 6D–F, supplemental Video 2) or equally distributed over the LE and LC phase (for indirect method), depending on the phospholipid handling prior to the microbubble production. This is in accordance with the IRRAS results, indicating that the PEGylated components were distributed over both the LE and LC phase, whereby we assume that the monolayer at the air/buffer interface was in thermodynamic equilibrium. With the indirect method for microbubble production, all components were dissolved and mixed in organic solvent. After evaporation of the solvent, the dried film of mixed lipids was dispersed in PBS buffer using a sonicator bath and a probe sonicator at low power. With the direct method, in contrast, the components were each dispersed in PBS buffer without use of sonication and then mixed together. Therefore, it is likely that the lipids in the precursors of the microbubbles, that is, in the liposomes and micelles,56 were more uniformly mixed with the indirect method than with the direct method. The lipids spontaneously self-assemble around the newly formed gas microbubbles during probe sonication,57,58 likely through membrane spreading.59 In other words, the indirect DSPC microbubbles are more in equilibrium than the direct DSPC microbubbles. This is in contrast to previous studies on monolayers at the air/water interface, which found that DSPC and DSPE-PEG2000 were immiscible at all surface pressures.25
For a fair comparison, the 4Pi confocal microscopy experiments were performed at room temperature, in accordance with all microscopy studies on lipid and ligand distribution on microbubble coatings. However, when developing microbubbles for in vivo applications, experiments at body temperature will be more translatable to human applications. Another important aspect for in vivo applications is the ligand distribution, because a more homogenous distribution could result in higher targeting efficiency. While the homogenous ligand distribution makes direct DPPC microbubbles a good candidate for in vivo applications, they are acoustically less stable than direct DSPC microbubbles.17 Our studies now show that homogenous ligand distributions are also possible for DSPC-based microbubbles. Future studies on the acoustical behavior of indirect DSPC microbubbles may give insight into the effect of LC area and ligand homogeneity on the acoustical stability, diversity in response to ultrasound, and efficacy to enhance molecular imaging and local drug delivery in a safe and effective way.
Conclusions
We investigated the ligand distribution and lipid phase state in microbubbles coated with a ternary phospholipid-based mixture of clinically relevant sizes. For better understanding of the lipid phases, we studied the lipid phase behavior in monolayers at the air/water interface of the same ternary mixtures that coated the microbubbles. Isotherms showed that DPPC had a transition from LE to LC phase during monolayer compression at ∼5 mN/m, which shifted to lower surface pressures in mixtures with DSPE-PEG2000 only or DSPE-PEG2000 and PEG40-stearate. In contrast, DSPC was always in the LC phase, also in the binary and ternary mixtures we studied. All binary and ternary mixtures had a transition plateau around 10–12 mN/m. As confirmed by IRRAS, this plateau was due to a conformational transition (mushroom to brush) in the PEGylated components. Based on 4Pi high-resolution imaging, direct DPPC microbubbles had a homogenous ligand distribution, with a significantly smaller LC phase area than the DSPC-based microbubbles. The lipid handling prior to microbubble production influenced both the ligand distribution and the LC phase area in the DSPC-based microbubbles. Microbubbles made by the direct method had a heterogeneous ligand distribution, while the ligand colocalized with the LE phase area. Microbubbles made by the indirect method had a significantly larger LC phase area and homogenous ligand distribution. By controlling the ligand distribution and microstructures in the microbubble coating, we can better understand the underlying mechanisms of targeting. This will lead to tailored microbubble formulations for specific clinical applications.
Acknowledgments
The authors are grateful to Prof. A. L. Klibanov from the University of Virginia, Cardiovascular Division, Charlottesville, Virginia, USA for discussions about the microbubble preparation. The authors thank the Erasmus Optical Imaging Centre of Erasmus MC for use of their facilities, and Dr. Gert van Cappellen and Alex Nigg for their help.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.langmuir.9b03912.
Typical example of 2D heatmaps from an indirect DSPC microbubble with a diameter of 5.7 μm; theoretical calculation of the mushroom to brush transition for the binary and ternary mixture; compression isotherms of pure DPPC-d62 and DSPC-d70 at the air/water interface; wavenumbers of antisymmetric and symmetric CD2 and CH2 stretching vibration of the ternary mixtures DPPC-d62/DSPE-PEG2000/PEG40-stearate and DSPC-d70/DSPE-PEG2000/PEG40-stearate as function of the surface pressure; compression of a pure DPPC monolayers with simultaneous recording of the compression isotherm (black line, left y-axis) and IRRA spectra; and PCA of IRRA spectra recorded during the compression of a pure DPPC-d62 monolayer (PDF)
3D render of direct DPPC microbubble (diameter = 4.7 μm) coated with DPPC, PEG40-stearate, and DSPE-PEG2000 (84.8:8.2:7.0) (AVI)
3D render of direct DSPC microbubble (diameter = 4.9 μm) coated with DSPC, PEG40-stearate, and DSPE-PEG2000 (84.8:8.2:7.0) (AVI)
3D render of direct DSPC microbubble (diameter = 3.4 μm) coated with DSPC, PEG40-stearate, and DSPE-PEG2000 (84.8:8.2:7.0) (AVI)
3D render of indirect DSPC microbubble (diameter = 5.3 μm) coated with DSPC, PEG40-stearate, and DSPE-PEG2000 (84.8:8.2:7.0) (AVI)
Author Contributions
S.A.G.L. performed experiments, analyzed data, and wrote the manuscript. C.S. performed monolayer experiments, analyzed data, and contributed to the manuscript. I.B. contributed to data analysis. J.B. performed monolayer experiments and analyzed data. T.v.R., A.B., and K.K. conceived and initiated the project. K.K. supervised the project and all authors reviewed the manuscript.
This project was supported in part by the Phospholipid Research Center, Heidelberg, Germany; and the Thoraxcenter of Erasmus MC. C.S. and A.B. thank Deutsche Forschungsgemeinschaft (DFG) for financial support through the grant FOR1145.
The authors declare no competing financial interest.
Supplementary Material
References
- Feinstein S. B.; Cheirif J.; Ten Cate F. J.; Silverman P. R.; Heidenreich P. A.; Dick C.; Desir R. M.; Armstrong W. F.; Quinones M. A.; Shah P. M. Safety and efficacy of a new transpulmonary ultrasound contrast agent: initial multicenter clinical results. J. Am. Coll. Cardiol. 1990, 16, 316–324. 10.1016/0735-1097(90)90580-i. [DOI] [PubMed] [Google Scholar]
- Correas J.-M.; Bridal L.; Lesavre A.; Méjean A.; Claudon M.; Hélénon O. Ultrasound contrast agents: properties, principles of action, tolerance, and artifacts. Eur. Radiol. 2001, 11, 1316–1328. 10.1007/s003300100940. [DOI] [PubMed] [Google Scholar]
- Kooiman K.; Vos H. J.; Versluis M.; de Jong N. Acoustic behavior of microbubbles and implications for drug delivery. Adv. Drug Deliv. Rev. 2014, 72, 28–48. 10.1016/j.addr.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Roovers S.; Segers T.; Lajoinie G.; Deprez J.; Versluis M.; De Smedt S. C.; Lentacker I. The Role of Ultrasound-Driven Microbubble Dynamics in Drug Delivery: From Microbubble Fundamentals to Clinical Translation. Langmuir 2019, 35, 10173–10191. 10.1021/acs.langmuir.8b03779. [DOI] [PubMed] [Google Scholar]
- van Rooij T.; Daeichin V.; Skachkov I.; de Jong N.; Kooiman K. Targeted ultrasound contrast agents for ultrasound molecular imaging and therapy. Int. J. Hyperther. 2015, 31, 90–106. 10.3109/02656736.2014.997809. [DOI] [PubMed] [Google Scholar]
- Smeenge M.; Tranquart F.; Mannaerts C. K.; de Reijke T. M.; van de Vijver M. J.; Laguna M. P.; Pochon S.; de la Rosette J. J. M. C. H.; Wijkstra H. First-in-Human Ultrasound Molecular Imaging With a VEGFR2-Specific Ultrasound Molecular Contrast Agent (BR55) in Prostate Cancer. Invest. Radiol. 2017, 52, 419–427. 10.1097/rli.0000000000000362. [DOI] [PubMed] [Google Scholar]
- Willmann J. K.; Bonomo L.; Testa A. C.; Rinaldi P.; Rindi G.; Valluru K. S.; Petrone G.; Martini M.; Lutz A. M.; Gambhir S. S. Ultrasound Molecular Imaging With BR55 in Patients With Breast and Ovarian Lesions: First-in-Human Results. J. Clin. Oncol. 2017, 35, 2133–2140. 10.1200/jco.2016.70.8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA Definity label. https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021064s011lbl.pdf (accessed Nov 6, 2019).
- Schneider M.; Arditi M.; Barrau M.-B.; Brochot J.; Broillet A.; Ventrone R.; Yan F. BR1: a new ultrasonographic contrast agent based on sulfur hexafluoride-filled microbubbles. Invest. Radiol. 1995, 30, 451–457. 10.1097/00004424-199508000-00001. [DOI] [PubMed] [Google Scholar]
- Kim D. H.; Costello M. J.; Duncan P. B.; Needham D. Mechanical Properties and Microstructure of Polycrystalline Phospholipid Monolayer Shells: Novel Solid Microparticles. Langmuir 2003, 19, 8455–8466. 10.1021/la034779c. [DOI] [Google Scholar]
- Borden M. A.; Pu G.; Runner G. J.; Longo M. L. Surface phase behavior and microstructure of lipid/PEG-emulsifier monolayer-coated microbubbles. Colloids Surf., B 2004, 35, 209–223. 10.1016/j.colsurfb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Borden M. A.; Martinez G. V.; Ricker J.; Tsvetkova N.; Longo M.; Gillies R. J.; Dayton P. A.; Ferrara K. W. Lateral phase separation in lipid-coated microbubbles. Langmuir 2006, 22, 4291–4297. 10.1021/la052841v. [DOI] [PubMed] [Google Scholar]
- Lozano M. M.; Longo M. L. Microbubbles coated with disaturated lipids and DSPE-PEG2000: phase behavior, collapse transitions, and permeability. Langmuir 2009, 25, 3705–3712. 10.1021/la803774q. [DOI] [PubMed] [Google Scholar]
- Shaw A. S. Lipid rafts: now you see them, now you don’t. Nat. Immunol. 2006, 7, 1139–1142. 10.1038/ni1405. [DOI] [PubMed] [Google Scholar]
- Kooiman K.; Kokhuis T. J. A.; van Rooij T.; Skachkov I.; Nigg A.; Bosch J. G.; van der Steen A. F. W.; van Cappellen W. A.; de Jong N. DSPC or DPPC as main shell component influences ligand distribution and binding area of lipid-coated targeted microbubbles. Eur. J. Lipid Sci. Technol. 2014, 116, 1217–1227. 10.1002/ejlt.201300434. [DOI] [Google Scholar]
- Kornmann L. M.; Reesink K. D.; Reneman R. S.; Hoeks A. P. G. Critical Appraisal of Targeted Ultrasound Contrast Agents for Molecular Imaging in Large Arteries. Ultrasound Med. Biol. 2010, 36, 181–191. 10.1016/j.ultrasmedbio.2009.09.009. [DOI] [PubMed] [Google Scholar]
- van Rooij T.; Luan Y.; Renaud G.; van der Steen A. F. W.; Versluis M.; de Jong N.; Kooiman K. Non-linear response and viscoelastic properties of lipid-coated microbubbles: DSPC versus DPPC. Ultrasound Med. Biol. 2015, 41, 1432–1445. 10.1016/j.ultrasmedbio.2015.01.004. [DOI] [PubMed] [Google Scholar]
- van Rooij T.; Beekers I.; Lattwein K. R.; van der Steen A. F. W.; de Jong N.; Kooiman K. Vibrational Responses of Bound and Nonbound Targeted Lipid-Coated Single Microbubbles. IEEE Trans. Ultrason. Eng. 2017, 64, 785–797. 10.1109/tuffc.2017.2679160. [DOI] [PubMed] [Google Scholar]
- Emmer M.; Vos H.; Versluis M.; Jong N. Radial modulation of single microbubbles. IEEE Trans. Ultrason. Eng. 2009, 56, 2370–2379. 10.1109/tuffc.2009.1325. [DOI] [PubMed] [Google Scholar]
- Albrecht O.; Gruler H.; Sackmann E. Polymorphism of phospholipid monolayers. J. Phys. 1978, 39, 301–313. 10.1051/jphys:01978003903030100. [DOI] [Google Scholar]
- Marsh D.; Bartucci R.; Sportelli L. Lipid membranes with grafted polymers: physicochemical aspects. Biochim. Biophys. Acta, Biomembr. 2003, 1615, 33–59. 10.1016/s0005-2736(03)00197-4. [DOI] [PubMed] [Google Scholar]
- Helfield B. L.; Cherin E.; Foster F. S.; Goertz D. E. Investigating the subharmonic response of individual phospholipid encapsulated microbubbles at high frequencies: a comparative study of five agents. Ultrasound Med. Biol. 2012, 38, 846–863. 10.1016/j.ultrasmedbio.2012.01.011. [DOI] [PubMed] [Google Scholar]
- McConlogue C. W.; Vanderlick T. K. A Close Look at Domain Formation in DPPC Monolayers. Langmuir 1997, 13, 7158–7164. 10.1021/la970898e. [DOI] [Google Scholar]
- Kilic S.; Bolukcu E. S. Phase behavior of DSPC/PEG40St mixtures at higher emulsifier contents. Colloids Surf., B 2018, 171, 368–376. 10.1016/j.colsurfb.2018.07.046. [DOI] [PubMed] [Google Scholar]
- Lozano M. M.; Longo M. L. Complex formation and other phase transformations mapped in saturated phosphatidylcholine/DSPE-PEG2000 monolayers. Soft Matter 2009, 5, 1822. 10.1039/b820070j. [DOI] [Google Scholar]
- Owen J.; Kamila S.; Shrivastava S.; Carugo D.; Bernardino de la Serna J.; Mannaris C.; Pereno V.; Browning R.; Beguin E.; McHale A. P.; Callan J. F.; Stride E. The Role of PEG-40-stearate in the Production, Morphology, and Stability of Microbubbles. Langmuir 2019, 35, 10014. 10.1021/acs.langmuir.8b02516. [DOI] [PubMed] [Google Scholar]
- Klibanov A. L.; Rasche P. T.; Hughes M. S.; Wojdyla J. K.; Galen K. P.; Wible J. H.; Brandenburger G. H. Detection of Individual Microbubbles of Ultrasound Contrast Agents. Invest. Radiol. 2004, 39, 187–195. 10.1097/01.rli.0000115926.96796.75. [DOI] [PubMed] [Google Scholar]
- Chen G.; Yang L.; Zhong L.; Kutty S.; Wang Y.; Cui K.; Xiu J.; Cao S.; Huang Q.; Liao W.; Liao Y.; Wu J.; Zhang W.; Bin J. Delivery of Hydrogen Sulfide by Ultrasound Targeted Microbubble Destruction Attenuates Myocardial Ischemia-reperfusion Injury. Sci. Rep. 2016, 6, 30643. 10.1038/srep30643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinl D. C.; Xu L.; Ochoa-Espinosa A.; Punjabi M.; Kaufmann B. A. Non-invasive contrast enhanced ultrasound molecular imaging of inflammation in autoimmune myocarditis for prediction of left ventricular fibrosis and remodeling. PLoS One 2019, 14, e0224377 10.1371/journal.pone.0224377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daeichin V.; van Rooij T.; Skachkov I.; Ergin B.; Specht P. A. C.; Lima A.; Ince C.; Bosch J. G.; van der Steen A. F. W.; de Jong N.; Kooiman K. Microbubble Composition and Preparation for High-Frequency Contrast-Enhanced Ultrasound Imaging:In VitroandIn VivoEvaluation. IEEE Trans. Ultrason. Eng. 2017, 64, 555–567. 10.1109/tuffc.2016.2640342. [DOI] [PubMed] [Google Scholar]
- Blume A. Lipids at the air–water interface. ChemTexts 2018, 4, 3. 10.1007/s40828-018-0058-z. [DOI] [Google Scholar]
- Hosgor E.; Kucuk T.; Oksal I. N.; Kaymak D. B. Design and control of distillation processes for methanol-chloroform separation. Comput. Chem. Eng. 2014, 67, 166–177. 10.1016/j.compchemeng.2014.03.026. [DOI] [Google Scholar]
- Baumgart T.; Hunt G.; Farkas E. R.; Webb W. W.; Feigenson G. W. Fluorescence probe partitioning between Lo/Ld phases in lipid membranes. Biochim. Biophys. Acta 2007, 1768, 2182–2194. 10.1016/j.bbamem.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold S.; Esbensen K.; Geladi P. Principal component analysis. Chemom. Intell. Lab. Syst. 1987, 2, 37–52. 10.1016/0169-7439(87)80084-9. [DOI] [Google Scholar]
- Tatusov R. L.; Natale D. A.; Garkavtsev I. V.; Tatusova T. A.; Shankavaram U. T.; Rao B. S.; Kiryutin B.; Galperin M. Y.; Fedorova N. D.; Koonin E. V. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001, 29, 22–28. 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell S.; Stelzer E. H. K. Fundamental improvement of resolution with a 4Pi-confocal fluorescence microscope using two-photon excitation. Opt. Commun. 1992, 93, 277–282. 10.1016/0030-4018(92)90185-t. [DOI] [Google Scholar]
- Baekmark T. R.; Elender G.; Lasic D. D.; Sackmann E. Conformational Transitions of Mixed Monolayers of Phospholipids and Polyethylene Oxide Lipopolymers and Interaction Forces with Solid Surfaces. Langmuir 1995, 11, 3975–3987. 10.1021/la00010a058. [DOI] [Google Scholar]
- Hussain H.; Kerth A.; Blume A.; Kressler J. Amphiphilic Block Copolymers of Poly(ethylene oxide) and Poly(perfluorohexylethyl methacrylate) at the Water Surface and Their Penetration into the Lipid Monolayer. J. Phys. Chem. B 2004, 108, 9962–9969. 10.1021/jp0495702. [DOI] [Google Scholar]
- Baekmark T. R.; Wiesenthal T.; Kuhn P.; Albersdörfer A.; Nuyken O.; Merkel R. A Systematic Infrared Reflection–Absorption Spectroscopy and Film Balance Study of the Phase Behavior of Lipopolymer Monolayers at the Air–Water Interface. Langmuir 1999, 15, 3616–3626. 10.1021/la981360r. [DOI] [Google Scholar]
- Abou-Saleh R. H.; Swain M.; Evans S. D.; Thomson N. H. Poly(ethylene glycol) Lipid-Shelled Microbubbles: Abundance, Stability, and Mechanical Properties. Langmuir 2014, 30, 5557–5563. 10.1021/la404804u. [DOI] [PubMed] [Google Scholar]
- Theiss C.; Holzgrabe U. Characterization of polydisperse macrogols and macrogol-based excipients via HPLC and charged aerosol detection. J. Pharm. Biomed. Anal. 2018, 160, 212–221. 10.1016/j.jpba.2018.07.043. [DOI] [PubMed] [Google Scholar]
- Abou-Saleh R. H.; McLaughlan J. R.; Bushby R. J.; Johnson B. R.; Freear S.; Evans S. D.; Thomson N. H. Molecular Effects of Glycerol on Lipid Monolayers at the Gas-Liquid Interface: Impact on Microbubble Physical and Mechanical Properties. Langmuir 2019, 35, 10097–10105. 10.1021/acs.langmuir.8b04130. [DOI] [PubMed] [Google Scholar]
- McConnell H. M. Structures and transitions in lipid monolayers at the air-water interface. Annu. Rev. Phys. Chem. 1991, 42, 171–195. 10.1146/annurev.pc.42.100191.001131. [DOI] [Google Scholar]
- Scholtysek P.; Li Z.; Kressler J.; Blume A. Interactions of DPPC with Semitelechelic Poly(glycerol methacrylate)s with Perfluoroalkyl End Groups. Langmuir 2012, 28, 15651–15662. 10.1021/la3028226. [DOI] [PubMed] [Google Scholar]
- Feigenson G. W. Phase behavior of lipid mixtures. Nat. Chem. Biol. 2006, 2, 560–563. 10.1038/nchembio1106-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufeil-Fiori E.; Wilke N.; Banchio A. J. Dipolar interactions between domains in lipid monolayers at the air-water interface. Soft Matter 2016, 12, 4769–4777. 10.1039/c5sm02862k. [DOI] [PubMed] [Google Scholar]
- McConnell H. M. Structures and Transitions in Lipid Monolayers at the Air-Water Interface. Annu. Rev. Phys. Chem. 1991, 42, 171–195. 10.1146/annurev.pc.42.100191.001131. [DOI] [Google Scholar]
- Andelman D.; Broçhard F.; Joanny J. F. Phase transitions in Langmuir monolayers of polar molecules. J. Chem. Phys. 1987, 86, 3673–3681. 10.1063/1.451970. [DOI] [Google Scholar]
- Tanwir K.; Tsoukanova V. Lateral Distribution of a Poly(ethylene glycol)-Grafted Phospholipid in Phosphocholine Monolayers Studied by Epifluorescence Microscopy. Langmuir 2008, 24, 14078–14087. 10.1021/la802205y. [DOI] [PubMed] [Google Scholar]
- Mitchell M. L.; Dluhy R. A. In situ FT-IR investigation of phospholipid monolayer phase transitions at the air water interface. J. Am. Chem. Soc. 1988, 110, 712–718. 10.1021/ja00211a008. [DOI] [Google Scholar]
- Hunt R. D.; Mitchell M. L.; Dluhy R. A. The interfacial structure of phospholipid monolayer films: an infrared reflectance study. J. Mol. Struct. 1989, 214, 93–109. 10.1016/0022-2860(89)80007-9. [DOI] [Google Scholar]
- Blume A.; Kerth A. Peptide and protein binding to lipid monolayers studied by FT-IRRA spectroscopy. Biochim. Biophys. Acta 2013, 1828, 2294–2305. 10.1016/j.bbamem.2013.04.014. [DOI] [PubMed] [Google Scholar]
- Shi D.; Liu X.; Counil C.; Krafft M. P. Fluorocarbon Exposure Mode Markedly Affects Phospholipid Monolayer Behavior at the Gas/Liquid Interface: Impact on Size and Stability of Microbubbles. Langmuir 2019, 35, 10025–10033. 10.1021/acs.langmuir.8b03546. [DOI] [PubMed] [Google Scholar]
- Abou-Saleh R. H.; Peyman S. A.; Johnson B. R. G.; Marston G.; Ingram N.; Bushby R.; Coletta P. L.; Markham A. F.; Evans S. D. The influence of intercalating perfluorohexane into lipid shells on nano and microbubble stability. Soft Matter 2016, 12, 7223–7230. 10.1039/c6sm00956e. [DOI] [PubMed] [Google Scholar]
- Crowe J. H.; Whittam M. A.; Chapman D.; Crowe L. M. Interactions of phospholipid monolayers with carbohydrates. Biochim. Biophys. Acta, Biomembr. 1984, 769, 151–159. 10.1016/0005-2736(84)90018-x. [DOI] [PubMed] [Google Scholar]
- Ferrara K. W.; Borden M. A.; Zhang H. Lipid-shelled vehicles: engineering for ultrasound molecular imaging and drug delivery. Acc. Chem. Res. 2009, 42, 881–892. 10.1021/ar8002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirsi S. R.; Borden M. A. Microbubble Compositions, Properties and Biomedical Applications. Bubble Sci. Eng. Technol. 2009, 1, 3–17. 10.1179/175889709x446507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klibanov A. L.Ultrasound Contrast Agents: Development of the Field and Current Status. In Contrast Agents II: Optical, Ultrasound, X-Ray and Radiopharmaceutical Imaging, Krause W., Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2002; pp 73–106. [Google Scholar]
- Lee S.; Kim D. H.; Needham D. Equilibrium and Dynamic Interfacial Tension Measurements at Microscopic Interfaces Using a Micropipet Technique. 1. A New Method for Determination of Interfacial Tension. Langmuir 2001, 17, 5537–5543. 10.1021/la0103259. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.