

Abstract
Objective:
Microcephalic primordial dwarfism (MPD) is a group of clinically and genetically heterogeneous disorders which result in severe prenatal and postnatal growth failure. X-ray repair cross-complementing protein 4 (XRCC4) is a causative gene for an autosomal recessive form of MPD. The objective of this report is to describe novel XRCC4 mutations in a female infant with MPD, dilated cardiomyopathy, and subclinical hypothyroidism.
Methods:
Genetic testing was performed using a comprehensive next generation sequencing panel for MPD, followed by targeted XRCC4 gene sequencing.
Results:
We report the case of a 970-gram, 35-cm, female infant (weight z score −5.05, length z score −4.71) born at 36 weeks and 3 days gestation. Physical examination revealed triangular facies, micrognathism, clinodactyly, and second and third toe syndactyly. Initial echocardiogram at birth was normal. Follow-up echocardiogram at 60 days of life revealed dilated cardiomyopathy with moderate left ventricular systolic dysfunction (ejection fraction was 40 to 45%), and anticongestive therapy was initiated. Thyroid testing revealed subclinical hypothyroidism with elevated thyroid-stimulating hormone of 13.0 μIU/mL (reference range is 0.3 to 5.0 μIU/mL) and normal free thyroxine by dialysis of 1.6 ng/dL (reference range is 0.8 to 2.0 ng/dL). Levothyroxine was initiated. Postnatal growth remained poor (weight z score at 3 months −4.93, length z score at 3 months −6.48), including progressive microcephaly (head circumference z score at 3 months −10.94). Genetic testing revealed novel compound heterozygous XRCC4 variants in trans: c.628A>T and c.638+3A>G. The child ultimately had cardiopulmonary arrest and died at 6 months of life.
Conclusion:
Molecular diagnosis in MPD is key to defining the natural history, management, and prognosis for patients with these rare disorders.
INTRODUCTION
Microcephalic primordial dwarfism (MPD) is a group of clinically and genetically heterogeneous disorders which result in severe prenatal and postnatal growth failure. Genetic mutations found to cause MPD result in decreased cellular proliferation due to decreased efficiency of cell cycle progression or increased programmed cell death, ultimately resulting in globally reduced cell numbers and smaller organism size (1).
The gene X-ray repair cross-complementing protein 4 (XRCC4) has recently been identified as a causative gene for a form of MPD (2). The XRCC4 protein is instrumental for nonhomologous end-joining, a major pathway for DNA repair after double strand breakage from genotoxic exposure. XRCC4 also plays a role in sealing breakage during V(D)J recombination in lymphocyte development (3). It functions by stabilizing DNA ligase IV, which works to ligate DNA ends, and binding XRCC4-like factor to form a complex that helps bridge broken ends and promote ligation by DNA ligase IV. Interestingly, despite the importance of the XRCC4 protein in V(D)J recombination, individuals with MPD due to XRCC4 gene mutation do not demonstrate overt immunodeficiency (3–5). Previous reports have noted a possibly increased malignancy risk (6).
In this case report, we describe an infant female with MPD, subclinical hypothyroidism, and dilated cardiomyopathy found to have novel compound heterozygous XRCC4 variants, c.628A>T and c.638+3A>G. Ultimately, she died from a cardiopulmonary arrest at 6 months of life. Of the 15 described cases of XRCC4 mutations in the literature, 1 was reported to have hypothyroidism. To our knowledge this is the first report of childhood-onset cardiomyopathy and early death in an individual with XRCC4 mutations, helping to expand the known phenotype of this rare disorder.
CASE REPORT
A 970-gram (z score −5.05) infant female was born at 36 weeks and 3 days gestation to a 29-year-old, G1P0→1 (first-time) mother via induced vaginal delivery for impaired fetal growth and non-reassuring fetal heart tracing. She was conceived on clomiphene. Prenatal ultra-sound noted the fetus was small for gestational age at 18 weeks of gestation and oligohydramnios was noted soon thereafter. The parents were non-consanguineous. There was no family history of growth failure, thyroid disease, or cardiac disease, and both parents were healthy and of average height.
The mother received 2 doses of betamethasone prior to delivery. Maternal placental pathology showed uneven villous maturity, multiple foci of parenchymal infarcts, and intraparenchymal perivillous and intravillous fibrin deposition comprising 3% of the total placental volume. Birth length was 35 cm (z score −4.71) and head circumference was 25.5 cm (z score −4.83). Physical examination revealed triangular facies, micrognathism, right clinodactyly, left fifth digit brachyclinodactyly, and left second and third toe syndactyly. She had normal first and second heart sounds with no audible murmurs or clicks. She had no hepatosplenomegaly and had normal pulses with no jugular venous distention.
The infant's initial metabolic newborn screening was normal. Additional normal diagnostic testing included urine organic acids, acylcarnitine profile, 7-dehydrocholesterol to rule out Smith-Lemli-Opitz syndrome, and single nucleotide polymorphism DNA microarray. She underwent 3 days of phototherapy for jaundice. Screening brainstem auditory evoked responses were normal bilaterally. Head and abdominal ultrasounds were unrevealing. She had normal retinal examination. She had no reported apneas or hypopneas noted, and remained on room air throughout her neonatal intensive care unit stay. At 60 days of age, she was discharged in a car bed per neonatal intensive care unit protocol given her weight less than 4 pounds at the time of discharge. The plan was to transition to an infant car seat once her weight reached over 4 pounds.
Initial echocardiogram at birth was normal aside from a small patent ductus arteriosus, transitional pulmonary hypertension, and a patent foramen ovale. The inferior vena cava was not well visualized. As such, repeat echo-cardiogram at 60 days of life was obtained (Fig. 1), which demonstrated mild to moderately depressed left ventricular systolic function (estimated ejection fraction was 40 to 45%). The left atrium and left ventricle were moderately dilated (left ventricular end-diastolic diameter z score of +3.3, left ventricular end-systolic diameter z score of +4.7) with a normal mitral valve and no mitral regurgitation. Coronary arteries appeared normal.
Fig. 1.
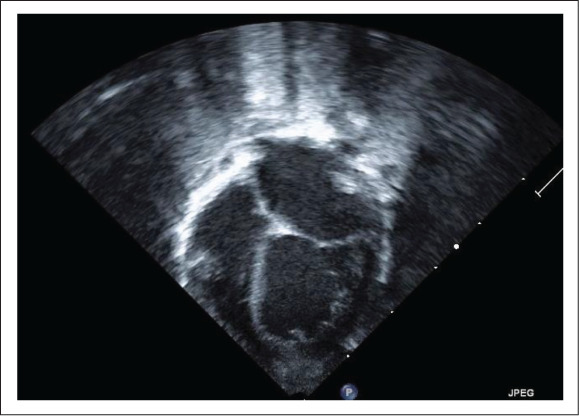
Left atrial and ventricular dilation on echocardiogram at 2 months of life.
At 3 months old, anti-congestive therapy was initiated including captopril twice daily at 0.3 mg/kg/day and metoprolol at 0.5 mg/kg/day. She had no clinical signs of heart failure. Thyroid testing was performed at 60 days of life and revealed subclinical hypothyroidism with thyroid-stimulating hormone of 13.0 μIU/mL (reference range is 0.3 to 5.0 μIU/mL) and free thyroxine by dialysis of 1.6 ng/dL (reference range is 0.8 to 2.0 ng/dL). Levothyroxine was initiated at 10 μg/kg/day and the dose was adjusted closely based on follow-up laboratory studies every 1 to 2 months.
Postnatal growth was poor (Table 1) with worsening of relative microcephaly (weight, length, and head circumference z scores were −4.93, −6.48, and −10.94 respectively at 3 months). Genetic testing in infancy included a comprehensive microcephalic primordial dwarfism next generation sequencing panel with no disease-causing mutations identified in multiple genes including ATR, ATRIP, CDC45, CDC6, CDT1, CENPJ, CEP63, CEP152, DNA2, GMNN, NIN, ORC1, ORC4, ORC6, PCNT, RBBP8, RNU4atac, and TRAIP. Subsequent testing revealed novel compound heterozygous XRCC4 variants in trans: c.628A>T and c.638+3A>G. The variant c.628A>T was predicted to result in premature protein termination, and c.638+3A>G was predicted to alter splicing.
Table 1.
Growth Parameters at Birth and 3 Months of Age
| Birth | 3 months | |
|---|---|---|
| Weight (g) | 970 | 1,890 |
| Weight z score* | −5.05 | −4.93 |
| Length (cm) | 35 | 43 |
| Length z score* | −4.71 | −6.48 |
| Head circumference (cm) | 25.5 | 28.7 |
| Head circumference z score* | −4.83 | −10.94 |
*Z scores are corrected for gestational age of 36 weeks, 3 days.
The child had cardiopulmonary arrest and died at 6 months of life. On the day of her arrest, she was more fussy than usual and was not tolerating her feeds. While en route to the hospital via car, she became apneic and unresponsive in her car seat. Cardiopulmonary resuscitation was initiated at the scene, but despite resuscitative efforts, no spontaneous circulation returned. Autopsy was not performed.
DISCUSSION
XRCC4 mutation as an etiology of MPD was first reported in 2014 (2), and there are 15 published reports to date describing patients with this rare condition (3). Clinical features include prenatal and postnatal growth restriction with disproportionate microcephaly, facial dysmorphisms such as micrognathia, hypotelorism, long face with prominent chin, long and beaked nose, high nasal bridge, prominent philtrum, fine sparse hair, broad nasal tip, and deep-set eyes. Developmental delay and neurological findings are commonly described, as well as primary gonadal failure. No overt cases of immunodeficiency have been described. We report novel XRCC4 mutations in a female infant with MPD, dilated cardiomyopathy, and subclinical hypothyroidism. This is the only known report of dilated cardiomyopathy in infancy associated with XRCC4 variants.
Bee et al (7) reported monozygotic twin adult males with dilated cardiomyopathy and homozygous XRCC4 mutations (c.673C>T). Their parents were first-degree cousins. Case 1 had a significant history of alcoholism in young adulthood and was diagnosed with dilated cardiomyopathy at 27 years of age (his ejection fraction was 25%) and an implantable cardioverter defibrillator was placed at age 44. Case 2 was found on echocardiogram at 50 years of age to have a left ventricular diastolic defect (ejection fraction of 52%).
Thyroid disorders were reported in 2 of the 15 published cases of XRCC4 mutation in the literature; one was a 23-year-old female with XRCC4 compound heterozygous mutations (c. 673C>T, c.760delG) who was reported to have hypothyroidism (5), and multinodular thyroid hypertrophy was described in case 2 of the twin males noted above (7).
To our knowledge, sudden infant death has never been described in a patient with MPD due to XRCC4 mutations. However, individuals with Majewski osteodysplastic primordial dwarfism type II associated with mutations in the pericentrin gene (PCNT), are at risk of cerebral aneurysm and moyamoya angiopathy, which can lead to early death from stroke (8). Individuals with micrognathia can be at higher risk for obstructive apneas due to upper airway obstruction (9), placing them at increased morbidity and mortality risk. Our patient experienced cardiopulmonary arrest of unclear etiology despite no previous history of apneas or hypopneas. With only 15 published reports of XRCC4 mutations in the literature, specific genotypephenotype correlations are difficult, further emphasizing the importance of molecular diagnosis to help define the prognosis for patients with this rare disorder. This case report expands the knowledge of morbidity and mortality risks in this condition.
CONCLUSION
Molecular diagnosis in primordial dwarfism is key to defining the natural history, management, and prognosis for patients with this rare disorder. This unique case describes an infant with MPD with the novel compound heterozygous XRCC4 variants c.628A>T and c.638+3A>G found to have dilated cardiomyopathy and subclinical hypothyroidism, who experienced cardiopulmonary arrest of unknown etiology at 6 months of life. This case expands the described phenotype of this rare condition, with only 15 previously published reports. Further studies are required to better understand the morbidity and mortality risks in this rare condition, and help guide counseling for families.
Abbreviations
- MPD
microcephalic primordial dwarfism
- XRCC4
X-ray repair cross-complementing protein 4
Footnotes
DISCLOSURE
The authors have no multiplicity of interest to disclose.
REFERENCES
- 1.Klingseisen A, Jackson AP. Mechanisms and pathways of growth failure in primordial dwarfism. Genes Dev. 2011;25:2011–2024. doi: 10.1101/gad.169037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaheen R, Faqeih E, Ansari S et al. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res. 2014;24:291–299. doi: 10.1101/gr.160572.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saito S, Kurosawa A, Adachi N. Mutations in XRCC4 cause primordial dwarfism without causing immunodeficiency. J Hum Genet. 2016;61:679–685. doi: 10.1038/jhg.2016.46. [DOI] [PubMed] [Google Scholar]
- 4.de Villartay JP. When natural mutants do not fit our expectations: the intriguing case of patients with XRCC4 mutations revealed by whole-exome sequencing. EMBO Mol Med. 2015;7:862–864. doi: 10.15252/emmm.201505307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo C, Nakazawa Y, Woodbine L et al. XRCC4 deficiency in human subjects causes a marked neurological phenotype but no overt immunodeficiency. J Allergy Clin Immunol. 2015;136:1007–1017. doi: 10.1016/j.jaci.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 6.de Bruin C, Mericq V, Andrew SF et al. An XRCC4 splice mutation associated with severe short stature, gonadal failure, and early-onset metabolic syndrome. J Clin Endocrinol Metab. 2015;100:E789–E798. doi: 10.1210/jc.2015-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bee L, Nasca A, Zanolini A et al. A nonsense mutation of human XRCC4 is associated with adult-onset progressive encephalocardiomyopathy. EMBO Mol Med. 2015;7:918–929. doi: 10.15252/emmm.201404803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bober MB, Khan N, Kaplan J et al. Majewski osteodysplastic primordial dwarfism type II (MOPD II): expanding the vascular phenotype. Am J Med Genet A. 2010;152A:960–965. doi: 10.1002/ajmg.a.33252. [DOI] [PubMed] [Google Scholar]
- 9.Cielo CM, Montalva FM, Taylor JA. Craniofacial disorders associated with airway obstruction in the neonate. Semin Fetal Neonatal Med. 2016;21:254–262. doi: 10.1016/j.siny.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]