

Abstract
Background/Aim: The clinical response rate of prostate cancer to tyrosine kinase inhibitor (TKI) monotherapy is low. The mechanisms of resistance to TKI are unclear. This study aimed to examine if the tumor microenvironment (TME) is involved in the resistance. Materials and Methods: The anti-vascular effect of Sutent was examined by immunofluorescent staining in TRAMP-C1 tumor. The percentage of CD11b+ population were analyzed by flow cytometry. The level of cytokines and chemokines were measured by multiplex immunoassay. Results: The Sutent monotherapy caused 1.5 days of tumor growth delay, chronic hypoxia, and more mature vasculature. Sutent monotherapy increased the percentage of polymorphonuclear myeloid-derived suppressor cells (MDSCs) in peripheral blood. The evolved TME triggered the re-distribution of myeloid cells in chronically hypoxic areas. The multiplex immunoassay indicated higher levels of several cytokines and chemokines both in tumors and the blood. Conclusion: Sunitinib treatment induced a distinct tumor microenvironment that impaired the efficient reduction of MDSCs by TKI.
Keywords: Tyrosine kinase inhibitor, sunitinib, angiogenesis, tumor-associated macrophage, myeloid-derived suppressor cells, hypoxia, peripheral blood
The family of receptor tyrosine kinases (RTK) regulates physiological signaling in normal cells as well as oncological progression in cancer cells (1). In the past decades, many drugs, such as antibodies and small molecules, have been developed to target RTK for cancer therapy (2,3). Sunitinib (SU11248, is known as Sutent has been developed by Pfizer) is a multi-target tyrosine kinase inhibitor (TKI) that has been shown to target vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), stem cell growth factor (SCF, c-kit), etc. (4,5), and has been approved for renal cell carcinoma (RCC), the neuroendocrine tumor of the pancreas, and as second-line treatment for gastrointestinal stromal tumors (GIST). Many studies have demonstrated that the combination of TKI with other treatment modalities, such as radiotherapy and immunotherapy, could improve tumor control (6-10). However, the response rate and the efficacy of TKI monotherapy for prostate cancer (7,11) and other tumor types (5,12) has been low in preclinical and clinical trials. The dominating mechanisms for the resistance of these cancer types to TKI treatment are still unclear.
The tumor microenvironment plays an essential role in tumor response to treatments, and TKI treatment has been shown to remodel it through its effect on the vascular network or the immune system. Many previous studies have shown that sunitinib and other TKI could disrupt angiogenesis by targeting vascular-associated pathways, including VEGF and PDGF. Furthermore, the concept of “vessel normalization” has been introduced to explain why the tumor response could be improved by a combination of an anti-vascular drug with other intervention modalities (13-16). However, the continuous administration of TKI or anti-VEGF therapy could induce a more hypoxic microenvironment that disturbs the efficiency of drug delivery and provides a niche for tumor cells (17-19). Consequently, the chronological observation of the vascular response within TKI- or anti-VEGF-treated tumor will indicate the window of investigating extrinsic drug-resistance.
In addition to the vascular response, the CD11b+ myeloid lineages, which comprise tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), and myeloid-derived suppressor cells (MDSCs), also play important roles in tumor progression and response to treatment and progression (20-22). TAMs are abundant in many types of tumors and might assist tumor cells in developing resistance to TKI treatment. A previous study has suggested that the activation of CCAAT/enhancer-binding protein-beta (C/EBPβ) factors could polarize TAMs toward the M2 type, which supports GIST progression despite TKI treatment (23). Recruitment of bv8-dependent CD11b+Gr-1+ cells into tumors has also been shown to be the predominant factor for the resistance to anti-VEGF treatment (24,25). Recently, sunitinib therapy was introduced as an immunomodulator to reduce the quantity of MDSCs in animal models (26) and patients (27,28). This reduction of MDSCs by sunitinib could consequently improve immunotherapy and be beneficial in combination with other tumor intervention protocols. A previous study has suggested that tumor resistance to sunitinib might be attributed to the inefficiency of reducing MDSCs (29). Therefore, studying the responses of myeloid lineage in TKI-treated tumors could be crucial for understanding the mechanism of resistance to TKI treatment.
In this study, we verified the changes of the tumor microenvironment following treatment of the murine prostate tumor model TRAMP-C1 with Sutent. Surprisingly, we found that the abundance of MDSCs in TRAMP-C1 tumors was unchanged after Sutent treatment, but was dramatically increased in peripheral blood. The underlying mechanism for maintenance of MDSCs’ frequency was attributed to the increased levels of systemic cytokines and chemokines and the mature vasculature of Sutent-treated tumor.
Materials and Methods
Cell line and tumor model. The murine prostate cancer cell line, TRAMP-C1 (ATCC® CRL-2730™), was maintained in DMEM medium containing 10% fetal bovine serum, 50 U/ml penicillin, 50 μg/ml streptomycin (Thermo Fisher Scientific, Gibco™, Waltham, MA, USA), 5 μg/ml bovine insulin (Sigma-Aldrich, St. Louis, MO, USA) and 10–8 M dehydroisoandrosterone. The TRAMP-C1 tumors were generated by intramuscular injection into the legs of male C57BL/6J mice (3×106 per mice).
Sutent treatment. Sutent was prepared in PBS containing 0.4% Tween-80 and 0.5% carboxyl methylcellulose (Sigma-Aldrich). Tumor bearing mice were intraperitoneally injected with Sutent (20mg/kg per day; Pfizer, New York, NY, USA) when tumor diameter reached 4mm in average. All the animal experiments were approval by the Institutional Animal Care and Use Committee in Chang Gung University, Taiwan (IACUC approval No. CGU107-044).
Immunofluorescence staining. To detect hypoxic regions within tumor, mice were intraperitoneally injected with the hypoxia indicator pimonidazole (PIMO, 80 mg/kg; Hypoxyprobe™, Burlington, MA, USA), 1 hour prior to sacrifice. Tumors were frozen in optimal cutting temperature compound (Tissue-Tek, SAKURA, Alphen aan den Rijn, the Netherlands) before cryosectioning. Tumor sections were stained with specific primary antibodies against rat anti-mouse CD31, hamster anti-mouse I-CAM1 (BD Bioscience, San Jose, CA, USA), rabbit anti-mouse CD31 (abcam, Cambridge, UK), mouse Mab-1 (Hypoxyprobe™, Burlington, MA, USA), mouse anti-mouse α-smooth muscle actin conjugated with FITC (Sigma-Aldrich), rat anti-mouse Ly6G (BioLegend, San Diego, CA, USA), rabbit anti-mouse carbonic anhydrase IX (CA9, Novus biologicals, Centennial, CO, USA), rat anti-mouse CD68 and rabbit anti-mouse V-CAM1 (BIO-RAD, Hercules, CA, USA). Primary antibodies were detected by secondary antibodies conjugated with fluorescent dyes. The images were acquired by ImageXpress® micro confocal high-content system and analyzed by Image-Pro Plus 6.0 software (MediaCybernetics, Rockville, MD, USA).
Flow cytometry analysis. The tumors were minced by a scalpel, digested in PBS buffer containing dispase (Invitrogen, Waltham, MA, USA) and DNase1 (Sigma-Aldrich) and subsequently filtered through a 100 μm- and a 40 μm-pore cell strainer (BD Bioscience) to obtain single cells. The blood samples were obtained by cardiac puncture with EDTA-coated syringe. The cell suspensions from digested tumors and blood samples were re-suspended in red blood cell (RBC) lysis buffer (BioLegend). The cells were firstly blocked with anti-mouse CD16/CD32 antibody (BD Biosciences) and then labeled with specific antibodies: anti-mouse CD11b-percp cy5.5, Ly6G-PE, Ly6C-FITC (BD Biosciences), CD68-alexa fluor700 (BIO-RAD) and CD206-APC (BioLegend). The stained cells were analyzed by FACSCanto or FACSFortessa flow cytometer. Data was acquired by BD FACSDiva™ software and analyzed by FACSDiva™ and FlowJo.
ELISA and multiplex immunoassay. Whole blood samples obtained from cardiac puncture were centrifuged to collect the plasma. Tumors were excised, minced and suspended in tissue lysis buffer (ebioscience, affymetrix, Santa Clara, CA, USA). After homogenization, the tumor lysate was centrifuged to collect the supernatant. Target proteins participating in myeloid cell recruitment and inflammatory response were selected for the panel of multiplex immunoassay: M-CSF, GM-CSF, G-CSF, CCL2, CCL3, CCL5, CXCL5, IFN-γ, IL1α, IL1β, IL-6, IL17a, TNF-α, and VEGF-a. To analyze their levels, all samples were labeled with magnetic microsphere by Procartaplex immunoassay kit and acquired data by the Luminex 200 system instrument (ebioscience, affymetrix). The protein levels of CXCL12 were quantified by the mouse quantikine ELISA kit (R&D systems, Minneapolis, MN, USA).
Statistical analysis. The T-test or One-way ANOVA were used to determine the statistical significance of differences and performed using GraphPad Prism6 software. All the results are expressed as the mean±SD with significance set at p<0.05.
Results
Sutent monotherapy decreased micro-vascular density and subsequently delayed TRAMP-C1 tumor growth. We first examined the efficacy of Sutent monotherapy in the TRAMP-C1 tumor model. The mice were classified into the control group injected with vehicle solution (abbreviated to VE) and the experimental group treated with Sutent (abbreviated to SU). The tumor growth curve showed that the SU-treated mice had slower tumor growth than the VE mice and the difference in volume was 30% decrease at day 7 (p=0.0253, Figure 1A); tumor growth delay was only 1.5 days. This data demonstrated that Sutent monotherapy could significantly delay TRAMP-C1 tumor growth, but not to a large extent.
Figure 1. Effect of Sutent monotherapy on tumor growth, MVD and the tumor microenvironment. (A) SU tumors had 1.5 days growth delay compared to size-matched VE tumors, at day-7. (N=5 in vehicle group; N=8 in Sutent-treated group). (B) Reconstituted fluorescent images of whole tumors in pre-treated, vehicle-treated group (VE) and Sutent-treated group (SU). (red:CD31. Green: pimonidazole; bar: 2 mm) (C) Representative fluorescent images of CD31 and PIMO staining in VE and SU-treated tumors. Avascular chronically hypoxic region is observed in both SU2D and SU9D tumors. (red: CD31, green: PIMO, star: central necrotic region; bar=100 μm) (D) Quantitative analysis from reconstituted whole tumor sections to reflect changes in MVD, necrosis, acute hypoxic and chronically hypoxic regions in size-matched tumors (N=5 for all groups).

Since TKI could target VEGF signaling and are regarded as anti-vascular therapy, the decrease of micro-vascular density (MVD) and the increase of hypoxia within tumors are considered as indices to evaluate the efficacy of TKI treatment. The tumor sections obtained from the VE and SU groups were co-stained with CD31 and pimonidazole (PIMO). The reconstituted images of the whole tumor are shown in Figure 1B. In pre-treated tumors, only few hypoxic regions were found in small size tumors (tumor diameter=4 mm), whereas many hypoxia regions associated with vasculatures were found both in the post-2days vehicle-treated (abbreviated to VE2D) and post-7days vehicle-treated (VE7D) groups; this hypoxia could be acute/transient and the result of “vascular inefficiency” (Figure 1C). In contrast, tumors in the post-2days Sutent-treated (SU2D, size-matched to VE2D group) and post-9days Sutent-treated (SU9D, size-matched to VE7D group) groups had much lower MVD and there was no vasculature in the hypoxic regions; this hypoxia should be chronic and is caused by “vascular deficiency”. The fluorescent image in Figure 1C also shows the formation of small central necrosis (marked by star) within both acute hypoxia in VE tumors and chronic hypoxia in size-matched SU tumors. MVD, percentage of necrosis and hypoxia were quantified from the reconstituted whole tumor images (Figure 1D). Compared to size-matched VE tumors, the administration of Sutent resulted in 45% decrease of the MVD in both SU2D and SU9D tumors (p=0.0031 and 0.0165, respectively). Moreover, the percentage of chronic hypoxia was concurrently increased in SU tumors (about 44-fold increase in both the SU2D and the SU9D group, p<0.001 and 0.0128, respectively). However, the percentage of acute hypoxia and perceivable necrosis in VE and SU tumors didn’t reach statistically significant difference. This emerging data indicated that Sutent treatment could rapidly impair tumor angiogenesis and induce higher percentage of avascular chronic hypoxia in TRAMP-C1 tumors.
Sutent treatment has minor impact on recruitment of myeloid cells into tumors but dramatically increased PMN-MDSCs in peripheral blood. Circulating white blood cells could be used to assess the immune response to treatment. Some studies have indicated that the percentage of circulating MDSCs could be a prognostic index in clinical patients (30,31). Moreover, many studies have revealed that Sutent treatment not only decreases the recruitment of myeloid cells into tumors but also the percentage of MDSCs in peripheral blood (26,29,32). We next investigated whether the percentage and systemic mobilization of myeloid cells was changed after TKI therapy in the TRAMP-C1 model. The subpopulations of the myeloid lineage were classified by flow cytometry analysis. The gating strategy of CD11b+ myeloid cells in the tumor are shown in Figure 2A. CD11b+ myeloid cells in the tumor were classified into three subpopulations, including CD11b+Ly6G–Ly6C– TAMs (symbol a), CD11b+Ly6G+Ly6C+ PMN-MDSCs (symbol b) and CD11b+Ly6G–Ly6C+ TAMs/M-MDSCs (symbol c). We further examined the expression of conventional macrophage markers, such as CD68 and CD206 (mannose receptor, also known as pro-tumoral M2 marker), for these CD11b+ subpopulations. Most of CD11b+Ly6G–Ly6C– cells were double positive for CD68 and CD206 staining, which indicated that they belong to a TAM subpopulation. A fraction of CD11b+Ly6G–Ly6C+ cells expressed CD68 and/or CD206. In contrast, the CD11b+Ly6G+Ly6C+ PMN-MDSCs lacked macrophage markers. Moreover, we further utilized t-Distributed Stochastic Neighbor Embedding (t-SNE) plot and showed the heterogeneity of total CD11b+ myeloid cells in VE7D tumor (Figure 2B). Moreover, the classification of the circulated CD11b+ population in the blood could be also separated to three groups (Figure 2C): CD11b+Ly6G+Ly6C+ PMN-MDSCs (symbol d), CD11b+Ly6G–Ly6Chi M-MDSCs (symbol e) and CD11b+Ly6G–Ly6C– resident monocytes (symbol f).
Figure 2. The influence of Sutent monotherapy on the CD11b+ populations in tumors and peripheral blood. (A) The gating scheme showed that tumor-associated CD11b+ myeloid cells could be classified into three subpopulations including CD11b+Ly6G+Ly6C+ PMN-MDSCs (symbol a), CD11b+Ly6G–Ly6C+ M-MDSCs/TAMs (symbol b) and CD11b+Ly6G–Ly6C– TAMs (symbol c). These subpopulations were further examined by macrophage markers, such as CD68 and CD206 (mannose receptor, also known as pro-tumoral M2 marker). (B) The t-distributed stochastic neighbor embedding (tSNE) illustration showed the heterogeneity of total CD11b+ myeloid cells in VE7D tumors. (C) The gating scheme revealed that the circulating CD11b+ myeloid cells in peripheral blood could be briefly classified into three subpopulations, such as CD11b+Ly6G+Ly6C+ PMN-MDSCs (symbol d), CD11b+Ly6G–Ly6Chi M-MDSCs (symbol e) and CD11b+Ly6G–Ly6C– monocytes (symbol f). The percentages of (D) total CD11b+ cells, (E) CD11b+Ly6G–Ly6C– TAMs, (F) CD11b+Ly6G+Ly6C+ PMN-MDSCs and (G) CD11b+Ly6G–Ly6C+ TAMs/M-MDSC in tumors were quantified by flow cytometry (N=5 in each group). The populations of (H) total CD11b+ cells, (I) CD11b+Ly6G–Ly6C– resident monocyte, (J) CD11b+Ly6G+Ly6C+ PMN-MDSCs and (K) CD11b+Ly6G–Ly6C+ M-MDSCs, in blood samples were determined (N≥4 in each group).

The flow cytometry analysis showed that upon Sutent treatment there was a trend to reduce the recruitment of CD11b+ cells in SU tumors, but the difference did not reach statistical significance compared with VE tumors (Figure 2D). This trend of decreased CD11b+ myeloid cells in SU tumors was mainly attributed to reduced percentage in TAM subpopulations (Figure 2E), not in PMN-MDSCs and M-MDSCs (Figure 2F and 2G). Regarding the blood samples, in addition to size-matched VE tumor-bearing mice, a non-tumor bearing group was treated with the same daily dose of Sutent as a reference group (abbreviated to SUw/oT group). Interestingly, the trend of change in the percentage of myeloid lineages in peripheral blood was not in agreement with that in tumors (Figure 2H-K). In the SU2D tumor-bearing group, the percentages of CD11b subpopulations didn’t change. However, at the late stage of the SU9D tumor-bearing group, the percentage of CD11b+ myeloid cells in peripheral blood was significantly increased (Figure 2H), which might result from the increase in PMN-MDSCs (Figure 2J). This data indicated that in the Sutent-treated mice the tumor microenvironment, which contained lower MVD and was chronically hypoxic, didn’t impair the recruitment of MDSCs, but dramatically facilitated the expansion of PMN-MDSCs in peripheral blood.
CD11b+ myeloid cells were re-distributed around necrotic regions in chronic hypoxia induced by Sutent treatment. The response of myeloid cells in TKI-treated tumors could be essential to the efficiency of subsequent therapeutic strategy. We next investigated the spatial distribution of myeloid cells such as CD68+ TAMs and Ly6G+ PMN-MDSCs within the SU-treated tumors. The endogenous marker, carbonic anhydrase 9 (CA9), was utilized to define the chronically hypoxic region. In the consecutive tumor sections, the Ly6G+ PMN-MDSCs and CD68+ TAMs were randomly distributed in acute/transient hypoxic region which was CA9-negative (Figure 3A). Surprisingly, those myeloid cells were re-distributed into different regions of the TME; CD68+ TAMS aggregated at chronically hypoxic regions and Ly6G+ PMN-MDSCs accumulated at necrotic regions, around CA9-positive chronically hypoxic regions within SU-tumors (Figure 3B). The quantitative data showed that the density of Ly6G+ PMN-MDSCs (Figure 3C) accumulated at central necrotic areas within Sutent-induced chronically hypoxic regions was significantly increased from 5.3±0.95% to 34.5±4.43%. The density of CD68+ TAMs in chronically hypoxic regions within SU tumors was higher than that in acute hypoxic regions within VE tumors (p=0.024). This data implied that Ly6G+ PMN-MDSCs and CD68+ TAMs, based on their inherent characteristics, migrated to different compartments of chronic hypoxic regions resulted from Sutent treatment, and might play roles in tumor regrowth.
Figure 3. The re-distribution of CD68+ TAMs and Ly6G+ PMN-MDSCs around PIMO+ chronically hypoxic areas. Representative section of TRAMPC1 tumors stained for hypoxia, CD31, CA9, CD68 and Ly6G in (A) VE7D and (B) SU9D tumors (bar: 100 μm). (C) Quantitative comparison of the Ly6G+ PMN-MDSCs population within the necrotic region in size-matched VE7D and SU9D tumors. (D) Quantitative comparison of CD68+ TAMs population within hypoxic regions in size-matched VE7D and SU9D tumors.

Sutent-treatment resulted in higher level of several chemokines and cytokines both in the tumor and peripheral blood. Since Sutent treatment didn’t impair recruitment of MDSCs in TRAMP-C1 tumors and reversely increased their percentage in peripheral blood, we hypothesized that this effect could be related to the levels of cytokines and chemokines released from the sunitinib-treated tumor. A multiplex immunoassay was utilized to assay for the levels of chemotaxis factors both in the tumor lysate and blood. Quantification of the data is shown in Table I. Even though the percentage of myeloid cells did not increase in SU tumors, the levels of cytokines and chemokines such as CCL2, CCL3, CCL5, CXCL5, M-CSF, GM-CSF, G-CSF, IL-17a and VEGF-a, in SU tumors were significantly increased. However, the levels of CXCL5, G-CSF, IL-1α and IL-6 were increased in the blood. Only the levels of CXCL5 and G-CSF were systemically increased both in SU tumors and blood. Therefore, the Sutent monotherapy could induce the expression of more cytokines and chemokines both in tumors and peripheral blood.
Table I. Cytokine and chemokine protein levels in tumor lysate and blood collected from SU9D tumor bearing mice. The levels of each protein were normalized to those in the control group. The relative ratio is shown as mean±SEM. (N≥3 in each group).

*Significant difference; -Non-determined, the levels of control group are non-detected; #If the level of control group is non-detected, the absolute value of protein level in the SU group would be shown at this column.
Administration of Sutent improved endothelial coverage with α-sma+ pericytes and increased the expression of adhesion molecules on endothelial cells. Excessive levels of VEGF in tumors could induce aberrant angiogenesis and cause acute hypoxia. Since the anti-VEGF strategy inhibit redundant vasculature formation, in addition to decreased MVD, vessel normalization is, therefore, another index in TKI therapy. To examine the vascular maturity, we examined the pericyte coverage of CD31+ endothelial cells in Sutent-treated tumors. The fluorescence images and quantification of this data showed that the coverage of endothelial cells with α-sma+ pericyte (the percentage of CD31+α-sma+ pixels/CD31+ pixels) was significantly increased in SU tumors (Figure 4A and B). Endothelial-derived adhesion molecules play a vital role in leukocyte transmigration from the blood stream into the tissues. Intercellular Adhesion Molecule 1 (ICAM-1, also known as CD54) is one of the adhesion molecules on endothelial cells and leukocytes. Interestingly, compared to VE tumors, the percentage of ICAM-1+ endothelial cells was increased in SU tumors (Figure 4C and 4D). Moreover, the percentage of VCAM-1 (vascular cell adhesion protein 1, another endothelial adhesion molecule also known as CD106) positive endothelial cells was significantly increased (about 15.6-fold, Figure 4E and 4F) in SU tumors. This fluorescence image data, therefore, suggested that Sutent treatment not only improved vasculature maturation but also increased the expression of adhesion molecules on CD31+ endothelial cells. This effect could improve the trafficking of MDSCs, which compensated for the reduction of MDSCs by Sutent treatment.
Figure 4. Sutent monotherapy improved maturation of vasculature and expression of adhesion molecules associated with leukocyte transmigration. The representative images of VE7D and SU9D tumors stained with CD31 (red) and (A) α-sma+, (C) ICAM-1, (E) VCAM-1 (green) (bar: 100 μm). Quantitation of the data from fluorescent images showed increased expression of (B) α-sma+ (D) ICAM-1 (F) VCAM-1 on CD31+ endothelial cells within Sutent-treated tumors.

Discussion
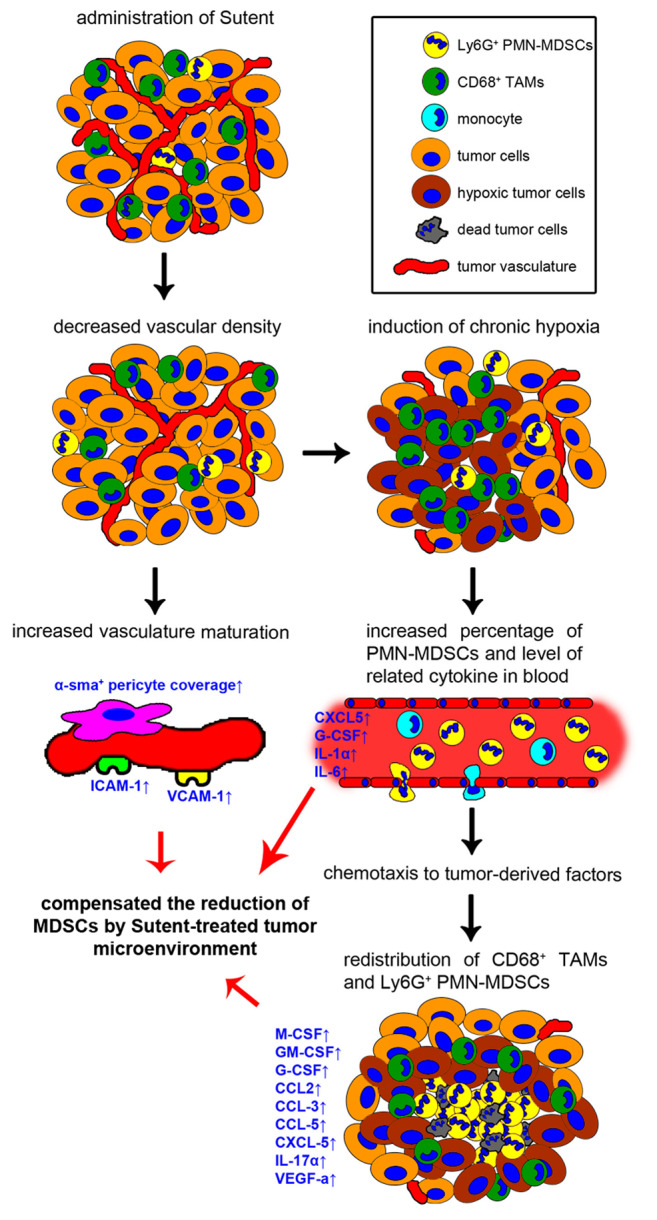
In this study, we examined changes in the tumor microenvironment after Sunitinib monotherapy in a murine TRAMP-C1 prostate tumor model. Herein, we graphically present the chronological response of a TRAMP-C1 tumor model to Sutent treatment (Figure 5). Sutent monotherapy could significantly decrease MVD and induce chronic hypoxia in 2 days. The CD68+ TAMs and Ly6G+ PMN-MDSCs re-distributed around chronically hypoxic regions as a result of decreased MVD. The percentage of Ly6G+ PMN-MDSCs in peripheral blood was increased, which is associated with the induction of cytokines, including G-CSF and IL-6. Upon treatment with SU tumors acquired a mature structure, and the coverage of endothelial cells with α-sma+ pericytes and expression of adhesion molecules, ICAM-1 and VCAM-1 were increased. Moreover, the Sutent-induced hypoxic tumor microenvironment possessed a higher level of several cytokines and chemokines, which were chemotactic for MDSCs.
Figure 5. A graphical flow chart recapitulating the alterations of tumor microenvironment after Sutent monotherapy. The Sutent treatment has anti-vascular effects, which not only result in formation of chronic hypoxic regions within the tumors, but also improves pericyte coverage and adhesion molecules expression on the vasculature. Increasing levels of CXCL5, G-CSF, IL-1α and IL-6 is correlated to PMN-MDSCs expansion in blood. In addition, increased expression of several cytokines and chemokines contributes to the maintenance of CD68+ TAMs and Ly6G+ PMN-MDSC within the tumor microenvironment and induces their redistribution around chronically hypoxic regions. These effects might compensate for the reduction of MDSCs by Sutent treatment.
The anti-vascular effect is one of the essential characteristics of TKI adjuvant therapy. Despite the reduction of angiogenesis, the causal development of chronic hypoxia is another critical issue that results in further radio- or chemo-resistance in tumors. Regarding the molecular signaling, hypoxia-induced factor-1α (HIF-1α) plays a dominant role in tumor progression (33,34). We found that the levels of VEGF-a, a downstream factor of HIF-1α signaling, in SU9D tumors, was significantly increased about 6-fold compared to the size-matched VE tumors. This effect indicates a considerable activation of the HIF pathway, which could promote tumor growth. It should be noted that the percentage of total hypoxia (PIMO+ pixels) in the tumor section didn’t significantly increase after Sutent treatment. (p=0.4847, 19.76±5.48% in VE7D and 26.14±6.75% in SU9D tumor, respectively). The increase in chronic hypoxia caused by a vascular deficiency in SU tumors could be the reason for the increase in VEGF levels. A recent review indicated that the stabilization of HIF isoforms under different types of hypoxia could further activate an adaptive cellular mechanism (35), which might impact tumor control by targeted therapy.
Vasculature components, including endothelial cells, pericytes, and vessel associated-extracellular matrix, provide oxygen and nutrients to support tumor progression. Since vascular-related receptor tyrosine kinases (RTK), such as vascular endothelial growth factor receptor and platelet-derived growth factor receptor, are potent targets for TKI, the reduction of angiogenesis is a hallmark for TKI treatment. The essential concept of vessel normalization upon anti-vascular therapy could promote subsequent chemo- and radio-therapy (15). However, the use of anti-vascular drugs and the best time window to apply chemo- and radiotherapy is still a challenge for tumor intervention. In this study, we found that TKI treatment not only deceased MVD but significantly enhanced the coverage of endothelial cells with α-sma+ pericytes, which indicated a mature (or normalized) vasculature. In our previous work, TRAMP-C1 tumors inoculated at pre-irradiated tumor bed also showed lower MVD. The CD31+ endothelial cells covered with α-sma+ pericyte from the pre-irradiated tumor bed were highly resistant to subsequent radiotherapy or Sutent therapy (36). Interestingly, the formation of the mature vasculature was based on endothelial progenitor cells, not resident endothelial cells. Therefore, the formation of mature vasculature in Sutent-treated tumors may also be triggered by non-conventional angiogenesis, which needs further investigation. In addition, other pathways, such as angiopoietin receptors (also known as Tie receptors) receptor tyrosine kinase, could also participate in vessel formation. De Palma’s group has suggested that targeting Tie2+ macrophages that facilitate angiogenesis via the Ang2/Tie2 axis is effective for anti-VEGF resistant tumors (37). Moreover, a previous study has demonstrated that sunitinib or an anti-VEGF drug combined with a specific targeting of Ang2 could significantly delay tumor growth compared to individual monotherapy (38). Thus, the combination of TKI with agents targeting alternative pathways may overcome the resistance to TKI and be an effective strategy.
In the TRAMP-C1 tumor model, Sutent treatment seems less effective and the growth delay was only 1.5 days. The failure of reducing MDSCs could be the reason for resistance to Sutent treatment. MDSCs, known for their inherent immunosuppressive role, could be a prognostic factor in TKI treatment (39). We suggested that three presumable effects associated with MDSCs might diminish the efficiency of Sutent treatment: (I) The enhancement of endothelial-associated adhesion molecules, such as ICAM-1 and VCAM-1, that promotes leukocyte transmigration from the blood into tumor tissue. (II) The higher levels of pro-inflammatory factors and chemoattractants in the Sutent-treated tumor microenvironment, which facilitates the recruitment and expansion of MDSCs in tumors and in the blood, respectively. (III) The increased levels of chemokines, such as GM-CSF and IL-17a within SU-treated tumors, and polarization of MDSCs to support tumor progression. GM-CSF has been recognized as a potential target, especially for the expansion and polarization of MDSCs (29,39). A previous study has revealed that IL-17 signaling is a critical mediator of the accumulation and education of MDSCs in anti-VEGF therapy (40). Consequently, our data from the multiplex immunoassay is consistent with those previous findings, which indicated that these specific biomarkers could be an index for assessing tumor response to TKI therapy.
In addition to increased numbers, we found that CD11b+ myeloid cells re-distribute around chronically hypoxic regions induced by lower MVD within SU-treated tumors. The CD68+ TAMs aggregated at chronically hypoxic areas and the Ly6G+ PMN-MDSCs accumulated at central necrotic areas inside the chronically hypoxic regions, which indicates the dissemination of specific signals from dead (or hypoxic) tumor cells within SU-treated tumors. In our previous studies, similar re-distribution of CD68+ TAMs (41) and Ly6G+ PMN-MDSCs (manuscript submitted) around chronically hypoxic sites was also induced by a large single dose of radiation in the TRAMP-C1 model. Some studies have revealed that hypoxia could drive macrophages toward the M2 type, which plays a vital role in tumor growth (42). MDSCs are known to have an inherent immunosuppressive function. The accumulated MDSCs in necrotic regions within SU-treated tumors might disturb the immunity of T cells, which need tumor cell debris to stimulate the subsequent immune response. However, the distinct re-distribution of CD11b+ myeloid cells within the tumor is still unclear, and further investigations on myeloid cells and the physiological status of the local niche are needed.
We determined the chronological response of tumor microenvironment following Sutent treatment. The anti-vascular effect of TKI changed distinctly the tumor microenvironment; the vasculature was normalized, the myeloid cell lineage was re-distributed, the reduction of MDSCs both in tumor and blood was inefficient and the levels of cytokines and chemokines were increased. These effects might result in therapeutic resistance to TKI in prostate tumors.
Conflicts of Interest
The Authors have no conflicts of interest to declare regarding this study.
Authors’ Contributions
Sheng-Yung Fu: manuscript – writing the first draft, experimental design, analysis and interpretation of the data, methodology; Chun-Chieh Wang: manuscript – organization, experimental design, concept of study, funding; Fang-Hsin Chen: manuscript - revision and editing, experimental design, methodology, concept of study, funding; Ching-Fang Yu: manuscript – revision and editing, experimental design, methodology; Ji-Hong Hong: manuscript – revision and editing, concept of study, advisor, funding; Chi-Shiun Chiang: manuscript – revision, editing and approval, concept of study, advisor, funding.
Acknowledgements
The Authors acknowledge the Radiation Biology Core Laboratory and Particle Physics and Irradiation Core Laboratory (Institute for Radiological Research, Chang Gung Memorial Hospital, Chang Gung University, Taoyuan, Taiwan) for scanning whole tumor sections. This study was supported by Chang Gung Memorial Hospital (CMRPG3H1691 and CMRPG3H1242) to Hong, J.H., Ministry of Science and Technology (MOST106-2627-M-007), National Health Research Institutes (NHRI-EX103-10132BI), and Frontier Research Center within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE 107QR001I5), Taiwan to Chiang, C.S, and Chang Gung Memorial Hospital (CMRPG3H1021 and CMRPG3H1022) to Wang, C.C. Chang Gung Memorial Hospital (CIRPD1D0063) and Ministry of Science and Technology (MOST107-2314-B-182-068-My2) to Chen F.H.
References
- 1.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353(2):172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 2.Morin MJ. From oncogene to drug: Development of small molecule tyrosine kinase inhibitors as anti-tumor and anti-angiogenic agents. Oncogene. 2000;19(56):6574–6583. doi: 10.1038/sj.onc.1204102. [DOI] [PubMed] [Google Scholar]
- 3.Ferguson FM, Gray NS. Kinase inhibitors: The road ahead. Nat Rev Drug Discov. 2018;17(5):353–377. doi: 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- 4.Roskoski R Jr. Sunitinib: A vegf and pdgf receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun. 2007;356(2):323–328. doi: 10.1016/j.bbrc.2007.02.156. [DOI] [PubMed] [Google Scholar]
- 5.Chow LQ, Eckhardt SG. Sunitinib: From rational design to clinical efficacy. J Clin Oncol. 2007;25(7):884–896. doi: 10.1200/JCO.2006.06.3602. [DOI] [PubMed] [Google Scholar]
- 6.Schueneman AJ, Himmelfarb E, Geng L, Tan J, Donnelly E, Mendel D, McMahon G, Hallahan DE. Su11248 maintenance therapy prevents tumor regrowth after fractionated irradiation of murine tumor models. Cancer Res. 2003;63(14):4009–4016. [PubMed] [Google Scholar]
- 7.Brooks C, Sheu T, Bridges K, Mason K, Kuban D, Mathew P, Meyn R. Preclinical evaluation of sunitinib, a multi-tyrosine kinase inhibitor, as a radiosensitizer for human prostate cancer. Radiat Oncol. 2012;7:154. doi: 10.1186/1748-717X-7-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY, Chen SH. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69(6):2514–2522. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, Wood L, Elson P, Garcia J, Dreicer R, Bukowski R. Sunitinib reverses type-1 immune suppression and decreases t-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14(20):6674–6682. doi: 10.1158/1078-0432.CCR-07-5212. [DOI] [PubMed] [Google Scholar]
- 10.Aparicio LMA, Fernandez IP, Cassinello J. Tyrosine kinase inhibitors reprogramming immunity in renal cell carcinoma: Rethinking cancer immunotherapy. Clin Transl Oncol. 2017;19(10):1175–1182. doi: 10.1007/s12094-017-1657-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michaelson MD, Oudard S, Ou YC, Sengelov L, Saad F, Houede N, Ostler P, Stenzl A, Daugaard G, Jones R, Laestadius F, Ullen A, Bahl A, Castellano D, Gschwend J, Maurina T, Chow Maneval E, Wang SL, Lechuga MJ, Paolini J, Chen I. Randomized, placebo-controlled, phase iii trial of sunitinib plus prednisone versus prednisone alone in progressive, metastatic, castration-resistant prostate cancer. J Clin Oncol. 2014;32(2):76–82. doi: 10.1200/JCO.2012.48.5268. [DOI] [PubMed] [Google Scholar]
- 12.Zhu AX, Sahani DV, Duda DG, di Tomaso E, Ancukiewicz M, Catalano OA, Sindhwani V, Blaszkowsky LS, Yoon SS, Lahdenranta J, Bhargava P, Meyerhardt J, Clark JW, Kwak EL, Hezel AF, Miksad R, Abrams TA, Enzinger PC, Fuchs CS, Ryan DP, Jain RK. Efficacy, safety, and potential biomarkers of sunitinib monotherapy in advanced hepatocellular carcinoma: A phase II study. J Clin Oncol. 2009;27(18):3027–3035. doi: 10.1200/JCO.2008.20.9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, Santosuosso M, Martin JD, Martin MR, Vianello F, Leblanc P, Munn LL, Huang P, Duda DG, Fukumura D, Jain RK, Poznansky MC. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012;109(43):17561–17566. doi: 10.1073/pnas.1215397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 16.Matsumoto S, Batra S, Saito K, Yasui H, Choudhuri R, Gadisetti C, Subramanian S, Devasahayam N, Munasinghe JP, Mitchell JB, Krishna MC. Antiangiogenic agent sunitinib transiently increases tumor oxygenation and suppresses cycling hypoxia. Cancer Res. 2011;71(20):6350–6359. doi: 10.1158/0008-5472.CAN-11-2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG, Wicha MS. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A. 2012;109(8):2784–2789. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bottsford-Miller JN, Coleman RL, Sood AK. Resistance and escape from antiangiogenesis therapy: Clinical implications and future strategies. J Clin Oncol. 2012;30(32):4026–4034. doi: 10.1200/JCO.2012.41.9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebos JM, Kerbel RS. Antiangiogenic therapy: Impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8(4):210–221. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gregory AD, Houghton AM. Tumor-associated neutrophils: New targets for cancer therapy. Cancer Res. 2011;71(7):2411–2416. doi: 10.1158/0008-5472.CAN-10-2583. [DOI] [PubMed] [Google Scholar]
- 22.Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J Clin Invest. 2015;125(9):3356–3364. doi: 10.1172/JCI80005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cavnar MJ, Zeng S, Kim TS, Sorenson EC, Ocuin LM, Balachandran VP, Seifert AM, Greer JB, Popow R, Crawley MH, Cohen NA, Green BL, Rossi F, Besmer P, Antonescu CR, DeMatteo RP. Kit oncogene inhibition drives intratumoral macrophage m2 polarization. J Exp Med. 2013;210(13):2873–2886. doi: 10.1084/jem.20130875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N. Tumor refractoriness to anti-vegf treatment is mediated by cd11b+gr1+ myeloid cells. Nat Biotechnol. 2007;25(8):911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 25.Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, Blanchard D, Bais C, Peale FV, van Bruggen N, Ho C, Ross J, Tan M, Carano RA, Meng YG, Ferrara N. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450(7171):825–831. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- 26.Draghiciu O, Nijman HW, Hoogeboom BN, Meijerhof T, Daemen T. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology. 2015;4(3):e989764. doi: 10.4161/2162402X.2014.989764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, Dreicer R, Bukowski R, Finke JH. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–2157. doi: 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 28.Chen HM, Ma G, Gildener-Leapman N, Eisenstein S, Coakley BA, Ozao J, Mandeli J, Divino C, Schwartz M, Sung M, Ferris R, Kao J, Wang LH, Pan PY, Ko EC, Chen SH. Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clin Cancer Res. 2015;21(18):4073–4085. doi: 10.1158/1078-0432.CCR-14-2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finke J, Ko J, Rini B, Rayman P, Ireland J, Cohen P. Mdsc as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int Immunopharmacol. 2011;11(7):856–861. doi: 10.1016/j.intimp.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arihara F, Mizukoshi E, Kitahara M, Takata Y, Arai K, Yamashita T, Nakamoto Y, Kaneko S. Increase in cd14+hla-dr -/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother. 2013;62(8):1421–1430. doi: 10.1007/s00262-013-1447-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58(1):49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69(6):2506–2513. doi: 10.1158/0008-5472.CAN-08-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brahimi-Horn MC, Chiche J, Pouyssegur J. Hypoxia and cancer. J Mol Med (Berl) 2007;85(12):1301–1307. doi: 10.1007/s00109-007-0281-3. [DOI] [PubMed] [Google Scholar]
- 34.Rapisarda A, Melillo G. Role of the hypoxic tumor microenvironment in the resistance to anti-angiogenic therapies. Drug Resist Updat. 2009;12(3):74–80. doi: 10.1016/j.drup.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saxena K, Jolly MK. Acute vs. Chronic vs. Cyclic hypoxia: Their differential dynamics, molecular mechanisms, and effects on tumor progression. Biomolecules. 2019;9(8) doi: 10.3390/biom9080339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen FH, Chiang CS, Wang CC, Fu SY, Tsai CS, Jung SM, Wen CJ, Lee CC, Hong JH. Vasculatures in tumors growing from preirradiated tissues: Formed by vasculogenesis and resistant to radiation and antiangiogenic therapy. Int J Radiat Oncol Biol Phys. 2011;80(5):1512–1521. doi: 10.1016/j.ijrobp.2011.02.055. [DOI] [PubMed] [Google Scholar]
- 37.Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, Politi LS, Gentner B, Brown JL, Naldini L, De Palma M. Targeting the ang2/tie2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. 2011;19(4):512–526. doi: 10.1016/j.ccr.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 38.Huang H, Lai JY, Do J, Liu D, Li L, Del Rosario J, Doppalapudi VR, Pirie-Shepherd S, Levin N, Bradshaw C, Woodnutt G, Lappe R, Bhat A. Specifically targeting angiopoietin-2 inhibits angiogenesis, tie2-expressing monocyte infiltration, and tumor growth. Clin Cancer Res. 2011;17(5):1001–1011. doi: 10.1158/1078-0432.CCR-10-2317. [DOI] [PubMed] [Google Scholar]
- 39.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and t cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, Vernes JM, Jiang Z, Meng YG, Peale FV, Ouyang W, Ferrara N. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med. 2013;19(9):1114–1123. doi: 10.1038/nm.3291. [DOI] [PubMed] [Google Scholar]
- 41.Chen FH, Chiang CS, Wang CC, Tsai CS, Jung SM, Lee CC, McBride WH, Hong JH. Radiotherapy decreases vascular density and causes hypoxia with macrophage aggregation in tramp-c1 prostate tumors. Clin Cancer Res. 2009;15(5):1721–1729. doi: 10.1158/1078-0432.CCR-08-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiang CS, Fu SY, Wang SC, Yu CF, Chen FH, Lin CM, Hong JH. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front Oncol. 2012;2:89. doi: 10.3389/fonc.2012.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]