

Abstract
Background and Purpose
Larotrectinib is a FDA‐approved oral small‐molecule inhibitor for treatment of neurotrophic tropomyosin receptor kinase fusion‐positive cancer. We here investigated the functions of the multidrug efflux transporters ABCB1 and ABCG2, the SLCO1A/1B (OATP1A/1B) uptake transporters, and the multispecific drug‐metabolizing enzyme CYP3A in larotrectinib pharmacokinetic behaviour.
Experimental Approach
In vitro, transepithelial drug transport and uptake assays were performed.
In vivo, larotrectinib (10 mg·kg−1) was administered orally to relevant genetically modified mouse models. Cell medium, plasma samples, and organ homogenates were measured by a sensitive and specific LC–MS/MS larotrectinib assay.
Key Results
In vitro, larotrectinib was avidly transported by human (h) ABCB1 and mouse (m) Abcg2 efficiently by hABCG2 and modestly by hOATP1A2. In vivo, both mAbcb1a/1b and mAbcg2 markedly limited larotrectinib oral availability and brain and testis accumulation (by 2.1‐fold, 10.4‐fold, and 2.7‐fold, respectively), with mAbcb1a/1b playing a more prominent role. mOatp1a/1b also restricted larotrectinib oral availability (by 3.8‐fold) and overall tissue exposure, apparently by mediating substantial uptake into the liver, thus likely facilitating hepatobiliary excretion. Additionally, larotrectinib is an excellent substrate of CYP3A, which restricts the oral availability of larotrectinib and hence its tissue exposure.
Conclusions and Implications
ABCG2 and especially ABCB1 limit the oral availability and brain and testis penetration of larotrectinib, while OATP1A/1B transporters restrict its systemic exposure by mediating hepatic uptake, thus allowing hepatobiliary excretion. CYP3A‐mediated metabolism can strongly limit larotrectinib oral availability and hence its tissue concentrations. These insights may be useful in the further clinical development of larotrectinib.
Abbreviations
- ABC
ATP‐binding cassette
- ABCB1
ATP‐binding cassette sub‐family B member 1
- ABCG2
ATP‐binding cassette sub‐family G member 2
- BCRP
breast cancer resistance protein
- CYP
cytochrome P450
- Cyp3aXAV
Cyp3a knockout mice with specific expression of human CYP3A4 in the liver and intestine
- m (as prefix)
mouse
- NTRK
neurotrophic tropomyosin receptor kinase
- OATP
organic‐anion‐transporting polypeptide
- P‐gp
P‐glycoprotein
- SLCO
solute carrier organic anion family
- TKI
TK inhibitor
What is already known
Oncogenic fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) genes occur in diverse malignancies.
Larotrectinib is the first FDA‐approved oral inhibitor highly selective for treatment of NTRK fusion‐positive cancers.
What this study adds
ABCB1 and ABCG2 restrict the brain accumulation and oral availability of larotrectinib.
OATP1A/1B and CYP3A contribute to the elimination of larotrectinib.
What is the clinical significance
The insights obtained may be used to further enhance the therapeutic application of larotrectinib.
This could be especially relevant for treatment of NSCLC brain metastases and glioblastoma.
1. INTRODUCTION
The neurotrophic tropomyosin receptor kinase (NTRK) family of three nerve growth factor receptor genes (NTRK1, NTRK2, and NTRK3) encodes the single‐transmembrane receptor tyrosine kinases, TrkA, TrkB, and TrkC respectively (Nakagawara, 2001). These receptors are composed of an extracellular domain for ligand binding, a transmembrane region, and an intracellular kinase domain. Nerve growth factor, brain‐derived growth factor, and neurotrophin 3 specifically bind to TrkA, TrkB, and TrkC respectively (Huang & Reichardt, 2003). This ligand binding triggers the oligomerization of the receptors and phosphorylation of tyrosine residues in the intracytoplasmic kinase domain, thus activating signal transduction pathways leading to growth, differentiation, and survival of neurons (Nakagawara, 2001). Trk receptors, which are mainly expressed in human neuronal tissues, therefore play critical roles in the development and homeostasis of the nervous system.
Oncogenic fusions involving one or more of the NTRK genes occasionally occur in diverse adult malignancies and paediatric cancers, such as glioblastoma, non‐small‐cell lung cancer (NSCLC), and colorectal cancer (Creancier et al., 2015; Stransky, Cerami, Schalm, Kim, & Lengauer, 2014; Wu et al., 2014). In certain rare paediatric tumours, including infantile fibrosarcoma, cellular congenital mesoblastic nephroma, and papillary thyroid cancer, the frequencies of Trk fusions are considerably higher. Thus, the presence of Trk fusions defines a unique molecular subgroup of advanced solid tumours in adult malignancies and paediatric cancers.
Several multikinase inhibitors inhibiting the function of TrkA, TrkB, and TrkC fusion proteins are under evaluation in clinical trials, for example, entrectinib (RXDX‐101), altiratinib (DCC‐2701), sitravatinib (MGCD516), TSR‐011, PLX7486, DS‐6051b, F17752, and cabozantinib (Amatu, Sartore‐Bianchi, & Siena, 2016). Unlike those multikinase inhibitors, larotrectinib is the first selective pan‐TrK inhibitor (Vitrakvi, LOXO‐101, Figure S1) and has been approved by the Food and Drug Administration (FDA) in November 2018 (FDA, 2018). Larotrectinib is an orally administered ATP‐competitive inhibitor of TrkA, TrkB, and TrkC and has been reported to induce marked tumour shrinkage in patients with NTRK‐rearranged cancers. In Phase 1/2 clinical trials, larotrectinib showed marked and durable antitumor activity in patients with TrK fusion‐positive cancers and an overall response rate of 75% regardless of tumour type, age, or gender (Drilon et al., 2018).
Transmembrane transporters can affect drug absorption, distribution, metabolism, and excretion. They can thus influence the pharmacokinetics and hence the safety and efficacy profiles of specific drugs. There are two major super‐families of transmembrane transporters affecting drug disposition—the ATP‐binding cassette (ABC) efflux transporter family and the solute carrier (SLC) uptake transporter family (Giacomini et al., 2010; Nigam, 2014).
The ABC drug efflux transporters P‐glycoprotein (P‐gp; ABCB1) and breast cancer resistance protein (BCRP; ABCG2) are highly expressed in the luminal membrane of the small intestine, blood–brain barrier (BBB), blood–testis barrier (BTB), and blood–placenta barrier and in the apical membranes of excretory cells such as renal proximal tubule epithelial cells and hepatocytes (Schinkel & Jonker, 2003). ABCB1 and ABCG2 can therefore influence the intestinal absorption, biliary and urinary excretion, and also the penetration in the CNS of specific drugs. Many anti‐tumour drugs, including numerous tyrosine kinase inhibitors (TKIs), are transported substrates of ABCB1 and/or ABCG2. This interaction often results in reduced oral availability or poor brain penetration (Li et al., 2018a, b; Tang et al., 2013, 2014). As larotrectinib targets different tumour types with NTRK fusions, which may develop brain metastases or even occur primarily in the brain (glioblastoma), it is important to know whether larotrectinib interacts with ABCB1 and/or ABCG2 in vivo, potentially affecting its oral availability and brain accumulation.
Organic‐anion‐transporting polypeptides (OATPs) are sodium‐independent transmembrane uptake transporters encoded by SLCO genes. They are expressed mainly in organs such as the liver, kidney, and small intestine, where they mediate the tissue uptake of many endogenous compounds, including bile acids and steroids, as well as exogenous compounds. The OATP1A/1B transporters have broad substrate specificities and are highly expressed in the liver (Hagenbuch & Meier, 2004). They can therefore have key roles in hepatic uptake and hence plasma clearance of several drug substrates including statins, cardiac glycosides, antibiotics, and many chemotherapeutic agents with highly diverse structures (Kalliokoski & Niemi, 2009; Shitara et al., 2013; van de Steeg et al., 2008, 2010, 2012; van de Steeg, van Esch, Wagenaar, Kenworthy, & Schinkel, 2013). While there are no direct one‐to‐one orthologues between the mouse and human OATP1A/1B proteins, mouse Oatp1a1, Oatp1a4, Oatp1a5, and Oatp1a6 are most related to the single human OATP1A2 protein, whereas mouse Oatp1b2 is most related to human OATP1B1 and OATP1B3. Thus, we wanted to know whether larotrectinib is a substrate of OATP1A/1B and whether this can influence the oral availability and organ distribution of larotrectinib.
The multidrug‐metabolizing cytochrome P450 3A (CYP3A) enzyme complex is responsible for most phase I drug metabolism. CYP3A4 is the most abundant CYP enzyme in human liver, the main detoxification organ. Many (pro‐)drugs are substrates of CYP3A4, resulting in drug inactivation or sometimes also activation (Guengerich, 1999; van Herwaarden et al., 2007). Thus, the oral availability and plasma exposure of many drugs can be strongly affected by CYP3A4, which may dramatically influence their therapeutic efficacy.
The primary aim of this study was to clarify the effects, in vivo, of ABCB1 and ABCG2, SLCO1A/1B (OATP1A/1B), and CYP3A enzymes on larotrectinib pharmacokinetic behaviour, including oral availability and organ distribution by using appropriate genetically modified mouse models.
1. METHODS
1.1. Cell lines and transport assays
Polarized MDCK‐II cells stably transduced with human (h) ABCB1, hABCG2, or mouse (m) Abcg2 cDNA were used and cultured as described (Bakos et al., 2000; Evers et al., 1998). Transepithelial transport assays were performed on microporous polycarbonate membrane filters (3.0‐μm pore size, 12‐mm diameter, Transwell 3414). Parental and variant subclones were seeded at a density of 2.5 × 105 cells per well and cultured for 3 days to form an intact monolayer. Membrane tightness was assessed by measurement of transepithelial electrical resistance before and after the transport phase.
For inhibition experiments, 5‐μM zosuquidar (ABCB1 inhibitor) and/or 5‐μM Ko143 (ABCG2/Abcg2 inhibitor) were used during the transport experiments. Cells were pre‐incubated with one or combination of the inhibitors for 1 hr in both apical and basolateral compartments. The transport phase was started (t = 0) by replacing the medium in either the apical or the basolateral compartment with fresh DMEM including 10% (v/v) FBS and larotrectinib at 5 μM, as well as the appropriate inhibitor(s). Plates then were kept at 37°C in 5% (v/v) CO2 during the experiment, and 50‐μl aliquots were taken from the acceptor compartment at 1, 2, 4, and 8 hr and stored at −30°C until LC–MS/MS measurement of the larotrectinib concentrations. Experiments were performed in independent triplicates, and the mean transport is shown in the figure. Active transport was expressed using the transport ratio r, that is, the amount of apically directed drug transport divided by basolaterally directed drug translocation after 8 hr. Our extensive previous studies with these cell lines have demonstrated that the transport data are highly reproducible. Transport ratios (r) above 3 therefore normally can be reliably assessed without the need for statistical analysis. For this reason, n = 3 was considered sufficient for the larotrectinib transport results.
1.2. Cellular uptake assays
HEK293 cells (RRID:CVCL_0045) transduced with vector control and human SLCO1A2, SLCO1B1, or SLCO1B3 cDNAs were a kind gift from Prof. Werner Siegmund and Dr Markus Keiser (University of Greifswald, Greifswald, Germany). HEK293 cells transduced with vector (mock) or human SLCO2B1 cDNA were a kind gift from Prof. Per Artursson and Dr Maria Karlgren (Uppsala University, Uppsala, Sweden). All the HEK293 cell lines were grown in DMEM supplemented with 10% (v/v) FBS and 1% penicillin–streptomycin mix at 37°C in 5% (v/v) CO2. Moreover, for HEK293‐control, HEK293‐SLCO1A2, HEK293‐SLCO1B1, and HEK293‐SLCO1B3 cell lines, 500 μg·ml−1 of G418 was added, while for HEK293‐mock and HEK293‐SLCO2B1 cell lines, 75 μg·ml−1 of hygromycin was added. For the uptake experiments, cells were first seeded in 12‐well plates (coated with 50 mg·L−1 of poly(l‐lysine) and 50 mg·L−1 of poly(l‐ornithine)) at a density of 1.0 × 105 cells per well. For the transport study, the cell culture medium was replaced with culture medium supplemented with 5‐mM sodium butyrate 24 hr before the uptake assay in order to induce the expression of OATP transporters.
The uptake transport study was carried out as described previously (Durmus et al., 2014). After cells had been washed twice and pre‐incubated with Krebs–Henseleit solution at 37°C for 15 min, uptake was initiated by adding Krebs–Henseleit buffer containing 5‐μM larotrectinib or 0.2‐μM rosuvastatin as positive control. The Krebs–Henseleit solution was prepared from Krebs–Henseleit buffer modified powder and supplemented with 25‐mM NaHCO3 and 2.5‐mM CaCl2 adjusted to pH 6.4 with 1‐M HCL. At 2.5 min, the incubation buffer was removed, and uptake was terminated by adding 1 ml of ice‐cold Krebs–Henseleit buffer, followed by two times washing with 1 ml of ice‐cold Krebs–Henseleit buffer. Afterwards, cells were lysed with 150 μl of 0.2‐N NaOH 15 min at room temperature, and cell lysates were transferred into 1.5‐ml Eppendorf tubes and stored at −20°C until the next day. The cellular protein amount was determined by the Bradford method using 10 μl of the cell lysates with BSA as a standard. LC–MS/MS measurements of the larotrectinib and rosuvastatin concentrations were performed for cell lysates. Experiments were performed in independent triplicates, and the mean transport is shown in the figure. As for the transepithelial transport assay, n = 3 was considered sufficient for the larotrectinib and rosuvastatin uptake results.
1.3. Animals
All animal care followed institutional guidelines complying with Dutch and EU legislation and experimental protocols were evaluated and approved by the institutional animal care and use committee. Experimental procedures were optimized to follow the principles of 3Rs (replacement, refinement, or reduction), as also required by Dutch law. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology.
Wild‐type, Abcb1a/1b −/− (RRID:IMSR_TAC:1487), Abcg2 −/− (RRID:IMSR_TAC:2767), Abcb1a/1b;Abcg2 −/− (RRID:IMSR_TAC:3998), Slco1a/1b −/− (RRID:MGI:4830142), Cyp3a −/− (RRID:IMSR_TAC:9011), and Cyp3aXAV (RRID:IMSR_TAC:9049) male mice, all of a >99% FVB genetic background, were used between 9 and 16 weeks of age. All the mouse strains were well established and characterized before. The mouse strains are viable, fertile, and stable and have previously been successfully used in extensive pharmacokinetic studies. The body weight was 30 ± 5 g. Animals were kept in specific pathogen‐free animal facility, in a temperature‐controlled environment with 12‐hr light and 12‐hr dark cycle, and they received a standard diet (TransBreed, SDS Diets, Technilab—BMI) and acidified water ad libitum. Male and female mice were housed separately, and 2–5 mice were housed per cage.
In this study, only male mice were randomly allocated for experiments. Because the main readout of these experiments was objective (larotrectinib plasma and tissue concentrations as measured by LC–MS/MS), we applied no blinding method. The mouse number may differ slightly among different groups in different experiments (usually six based on the power calculation and our extensive experience, but one more mouse was occasionally added when multiple mouse groups were tested and needed to be compared at the same time).
1.4. Drug solutions
For oral administration, larotrectinib was dissolved in DMSO at a concentration of 50 mg·ml−1 and further diluted with 5% glucose in water to yield a concentration of 1.0 mg·ml−1. Final concentrations for DMSO and glucose in the dosing solution were 2% (v/v) and 4.75% (w/v), respectively. All dosing solutions were prepared freshly on the day of experiment.
1.5. Plasma and organ pharmacokinetics of larotrectinib in mice
In order to minimize variation in absorption because of oral administration, mice were first fasted for 3 hr before larotrectinib (10 mg·kg−1) was administered orally, using a blunt‐ended needle. For the transporter experiments over 8‐ or 1‐hr, approximately 50‐μl tail vein blood samples were collected at 0.25‐, 0.5‐, 1‐, 2‐, 4‐, and 8‐hr time points or 0.125‐, 0.25‐, 0.5‐, 0.75‐, and 1‐hr time points after oral administration, respectively, using microvettes containing dipotassium EDTA. For the CYP3A experiments (8‐ or 4‐hr), tail vein blood sampling was performed at 0.25, 0.5, 1, 2, and 4 hr or 0.125, 0.25, 0.5, 1, and 2 hr, respectively. At the last time point in each experiment (1, 4, or 8 hr), mice were anaesthetized with 5% isoflurane and blood was collected by cardiac puncture. Blood samples were collected in Eppendorf tubes containing heparin as an anticoagulant. The mice were then killed by cervical dislocation, and the brain, liver, kidney, lung, small intestine, and testis were rapidly removed. Plasma was isolated from the blood by centrifugation at 9,000 g for 6 min at 4°C, and the plasma fraction was collected and stored at −30°C until analysis. Organs were homogenized with 4% (w/v) BSA and stored at −30°C until analysis. Relative tissue‐to‐plasma ratio after oral administration was calculated by determining the larotrectinib tissue concentration relative to larotrectinib plasma concentration at the last time point.
1.6. LC–MS/MS analysis
Larotrectinib concentrations in DMEM/FBS (9/1, v/v; Invitrogen) cell culture medium, plasma samples, and organ homogenates were determined using a validated LC–MS/MS assay (Sparidans, Wang, Schinkel, Schellens, & Beijnen, 2018).
1.7. Materials
Larotrectinib was purchased from Carbosynth (Oxford, UK). Zosuquidar and elacridar HCl were obtained from Sequoia Research Products (Pangbourne, UK). Ko143 was from Tocris Bioscience (Bristol, UK). BSA Fraction V was obtained from Roche Diagnostics GmbH (Mannheim, Germany). Glucose water 5% w/v was from B. Braun Medical Supplies, Inc. (Melsungen, Germany). Isoflurane was purchased from Pharmachemie (Haarlem, The Netherlands), and heparin (5,000 IU·ml−1) was from Leo Pharma (Breda, The Netherlands). All other chemicals used in the larotrectinib detection assay were described earlier (Sparidans et al., 2018). All other chemicals and reagents were obtained from Sigma‐Aldrich (Steinheim, Germany).
1.8. Data and statistical analysis
Pharmacokinetic parameters were calculated by non‐compartmental methods using the PK solver software (Zhang, Huo, Zhou, & Xie, 2010). The area under the plasma concentration–time curve (AUC) was calculated using the trapezoidal rule, without extrapolating to infinity. The peak plasma concentration (C max) and the time of maximum plasma concentration (T max) were estimated from the original (individual mouse) data. For mouse experiments, one‐way ANOVA was used when multiple groups were compared and the Bonferroni post hoc correction was used to accommodate multiple testing (with F achieved P < .01 and no significant variance inhomogeneity). The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. The two‐sided unpaired Student's t test was used when treatments or differences between two specific groups were compared using the software GraphPad Prism7 (GraphPad Software Inc., La Jolla, CA, USA, RRID:SCR_002798). All the data were log transformed before statistical tests were applied. Statistical analysis was undertaken only for studies where each group size was at least n = 5. For in vitro transport and uptake experiments, the number of each group is 3, and no statistical analysis was performed for these data. Differences were considered statistically significant when P < .01. All data are presented as geometric mean ± SD.
1.9. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Fabbro et al., 2019a, b; Alexander, Kelly et al., 2019).
2. RESULTS
2.1. In vitro transport of larotrectinib
Transepithelial transport of larotrectinib was tested using polarized monolayers of MDCK‐II parental cells and its subclones overexpressing human (h) ABCB1, hABCG2, and mouse (m) Abcg2. Larotrectinib (5 μM) was modestly transported in the apical direction in the parental MDCK‐II cell line (r = 3.8, Figure 1a), and this was virtually abolished by addition of the ABCB1 inhibitor zosuquidar (r = 1.1, Figure 1b). These data are compatible with modest transport of larotrectinib by the low‐level endogenous canine Abcb1 that is known to be present in the parental MDCK‐II cells (Simoff et al., 2016). In cells overexpressing hABCB1, there was strong apically directed transport of larotrectinib (r = 33, Figure 1c), which was markedly inhibited by zosuquidar (r = 1.0, Figure 1d).
FIGURE 1.
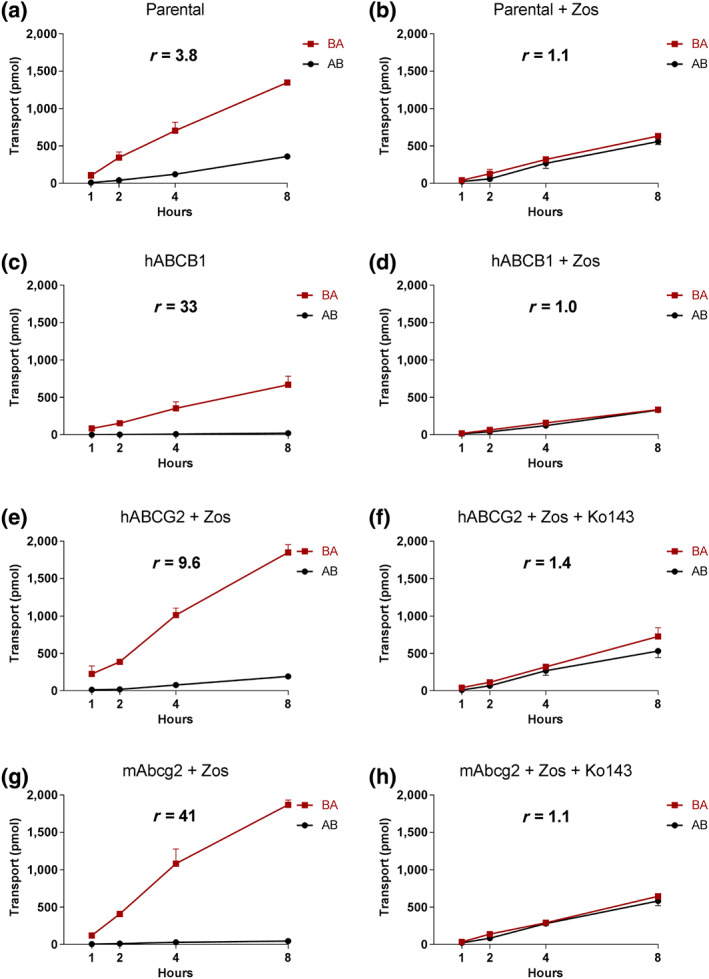
Transepithelial transport of larotrectinib (5 μM) assessed in MDCK‐II cells either non‐transduced (a, b) or transduced with hABCB1 (c, d), hABCG2 (e, f) or mAbcg2 (g, h) cDNA. At t = 0 hr, drug was applied in the donor compartment and the concentrations in the acceptor compartment at t = 1, 2, 4, and 8 hr were measured and plotted as larotrectinib transport (pmol) in the graph (n = 3). (b, d–h) Zosuquidar (Zos, 5 μM) was applied to inhibit human and/or endogenous canine ABCB1. (f, h) The ABCG2 inhibitor Ko143 (5 μM) was applied to inhibit ABCG2/Abcg2‐mediated transport. AB, translocation from the apical to the basolateral compartment; BA, translocation from the basolateral to the apical compartment. Data shown are means ± SD; r, relative transport ratio
Zosuquidar was added to inhibit any possible contribution of endogenous canine Abcb1 in subsequent experiments with MDCK‐II cells overexpressing hABCG2 and mAbcg2. The ABCG2 inhibitor Ko143 was used to inhibit the transport activity of hABCG2 and mAbcg2. In hABCG2‐overexpressing MDCK‐II cells, there was clear apically directed transport of larotrectinib (r = 9.6, Figure 1e), and transport was inhibited by Ko143 (r = 1.4, Figure 1f). We also observed strong apically directed transport of larotrectinib in cells overexpressing mouse Abcg2 (r = 41), and this transport activity could be completely inhibited by Ko143 (r = 1.1, Figure 1g,h).
Larotrectinib thus appears to be avidly transported by hABCB1 and mAbcg2 and efficiently by hACBG2. It could also be modestly transported by canine ABCB1.
2.2. ABCB1 and ABCG2 limit larotrectinib plasma, brain, and testis exposure
To investigate the possible effects of ABCB1A/1B and ABCG2 on oral bioavailability and tissue disposition of larotrectinib, we performed an 8‐hr pharmacokinetic pilot study in male wild‐type and Abcb1a/1b;Abcg2 −/− mice, using oral administration of 10 mg·kg−1 of larotrectinib. As shown in Figure S2 and Table S1, absorption was very rapid and the plasma exposure of larotrectinib over 8 hr (AUC0–8 hr) was significantly higher (2.1‐fold) in Abcb1a/1b;Abcg2 −/− mice than in wild‐type mice, especially before 4 hr.
Brain, liver, kidney, lung, small intestine, and testis concentrations of larotrectinib 8 hr after oral administration were analysed. The brain concentration and brain‐to‐plasma ratio in Abcb1a/1b;Abcg2 −/− mice were increased by 15.8‐fold and 11.5‐fold, respectively, compared with those in wild‐type mice (Figure S3 and Table S1). The larotrectinib brain‐to‐plasma ratio (0.09) in wild‐type mice was quite low, suggesting poor brain penetration of larotrectinib at 8 hr (Table S1). We further observed a slightly lower liver‐to‐plasma ratio and a markedly lower small intestine‐to‐plasma ratio in Abcb1a/1b;Abcg2 −/− mice compared with wild‐type mice (Figure S4).
We next studied the separate and combined functions of Abcb1a/1b and Abcg2 in oral bioavailability and tissue distribution of larotrectinib. Larotrectinib (10 mg·kg−1) was administered orally to wild‐type, Abcb1a/1b −/−, Abcg2 −/−, and Abcb1a/1b/Abcg2 −/− mice, and the experiment was terminated at 1 hr, when larotrectinib plasma levels were still comparatively high. As shown in Figure 2 and Table 1, single deficiency of either mAbcb1 or mAbcg2 resulted in higher larotrectinib plasma exposure, with the plasma AUC0–1 hr increased in both Abcb1a/1b −/− (2.3‐fold, P < .01) and Abcg2 −/− (1.7‐fold, P < .01) mice (Table 1). In combination Abcb1a/1b/Abcg2 −/− mice, the larotrectinib plasma AUC0–1 hr was even further increased up to 3.4‐fold compared with wild‐type mice (P < .01). The data suggest that Abcb1a/1b, but to some extent also Abcg2, can limit the oral availability of larotrectinib.
FIGURE 2.
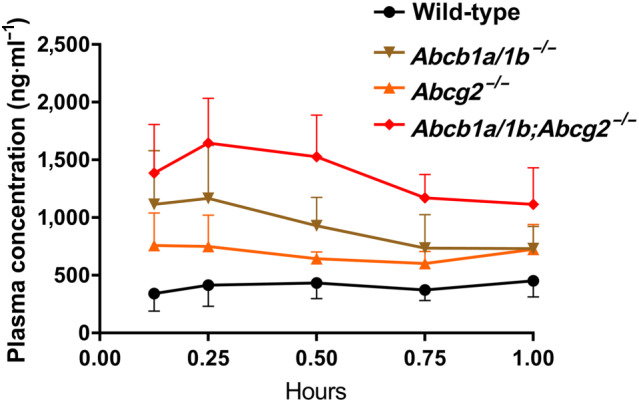
Plasma concentration–time curves of larotrectinib in male wild‐type, Abcb1a/1b −/−, Abcg2 −/−, and Abcb1a/1b;Abcg2 −/− mice over 1 hr after oral administration of 10 mg·kg−1 of larotrectinib. Data are given as mean ± SD (wild‐type and Abcb1a/1b −/−, n = 7; Abcg2 −/− and Abcb1a/1b;Abcg2 −/−, n = 6)
TABLE 1.
Plasma, brain, and testis pharmacokinetic parameters of larotrectinib over 1 hr after oral administration of 10 mg·kg−1 of larotrectinib to male wild‐type, Abcb1a/1b −/−, Abcg2 −/−, and Abcb1a/1b;Abcg2 −/− mice
| Parameter | Genotype | |||
|---|---|---|---|---|
| Wild‐type | Abcb1a/1b −/− | Abcg2 −/− | Abcb1a/1b;Abcg2 −/− | |
| AUC0–1 hr (ng·ml−1·hr−1) | 379 ± 120 | 867 ± 282 * | 639 ± 129 * , # | 1,297 ± 266 * |
| Fold‐change AUC0–1 hr | 1.0 | 2.3 | 1.7 | 3.4 |
| C max (ng·ml−1) | 494 ± 156 | 1,140 ± 431 * | 856 ± 276 # | 1,684 ± 349 * |
| T max (hr) | 0.7 ± 0.3 | 0.2 ± 0.1 | 0.5 ± 0.4 | 0.3 ± 0.1 |
| C brain (ng·g−1) | 16.7 ± 3.9 | 69.7 ± 16.2 * , # | 20.9 ± 2.7 # , ^ | 393.3 ± 58.6 * |
| Fold‐change C brain | 1.0 | 4.4 | 1.3 | 25 |
| Brain‐to‐plasma ratio | 0.036 ± 0.01 | 0.10 ± 0.01 * , # | 0.03 ± 0.01 # , ^ | 0.38 ± 0.12 * |
| Fold‐change ratio | 1.0 | 2.6 | 0.9 | 10.4 |
| C testis (ng·g−1) | 81.7 ± 15.8 | 301.2 ± 187.1 * , # | 99.3 ± 17.3 # , ^ | 564.6 ± 133.8 * |
| Fold‐change C testis | 1.0 | 3.7 | 1.2 | 6.9 |
| Testis‐to‐plasma ratio | 0.19 ± 0.06 | 0.41 ± 0.22 * | 0.14 ± 0.03 # , ^ | 0.52 ± 0.09 * |
| Fold‐change ratio | 1.0 | 2.1 | 0.7 | 2.7 |
Note. Data are given as mean ± SD (wild‐type and Abcb1a/1b −/−, n = 7; Abcg2 −/− and Abcb1a/1b;Abcg2 −/−, n = 6). Statistical analysis was applied after log transformation of linear data.
Abbreviations: AUC0–1 hr, area under the plasma concentration–time curve; C brain, brain concentration; C max, maximum concentration in plasma; C testis, testis concentration; T max, time point (hr) of maximum plasma concentration.
P < .01 compared with wild‐type mice.
P < .01 compared with Abcb1a/1b;Abcg2 −/− mice.
P < .01 compared between Abcb1a/1b −/− and Abcg2 −/− mice.
Interestingly, larotrectinib brain concentrations in Abcb1a/1b −/− and Abcb1a/1b/Abcg2 −/− mice were clearly increased, by 4.4‐fold and 25‐fold, respectively, compared with wild‐type mice, but not in single Abcg2 −/− mice. The brain‐to‐plasma ratio of larotrectinib was again very low (0.036) in wild‐type mice but could be increased to 0.10 (2.6‐fold) due to single mAbcb1 deficiency and further up to 0.38 (10.4‐fold) by combined mAbcb1 and mAbcg2 deficiency (Figure 3a,b and Table 1). The further increase in Abcb1a/1b/Abcg2 −/− brain demonstrates that, in the absence of Abcb1a/1b activity, Abcg2 still markedly limits larotrectinib brain penetration. The apparent lack of Abcg2 effect in the single Abcg2 knockout strain can be readily explained if its absolute transport contribution is considerably lower than that of Abcb1a/1b in the mouse BBB. However, once this Abcb1a/1b transport background is removed, the more modest transport contribution of Abcg2 becomes apparent (compare Abcb1a/1b and Abcb1a/1b;Abcg2 knockout data). A straightforward quantitative pharmacokinetic model to explain this behaviour of shared Abcb1a/1b and Abcg2 substrate drugs has been developed (Kodaira, Kusuhara, Ushiki, Fuse, & Sugiyama, 2010). Qualitatively similar results were obtained for larotrectinib testis penetration, although the wild‐type testis‐to‐plasma ratio was substantially higher (0.19), and the relative increases in ratios in Abcb1a/1b −/− (2.1‐fold) and Abcb1a/1b/Abcg2 −/− (2.7‐fold) mice were more modest than for the brain (Figure 3c,d and Table 1). The data indicate that Abcb1a/1b and, to a lesser extent, Abcg2 can strongly reduce brain accumulation of larotrectinib, while testis accumulation was more modestly affected.
FIGURE 3.
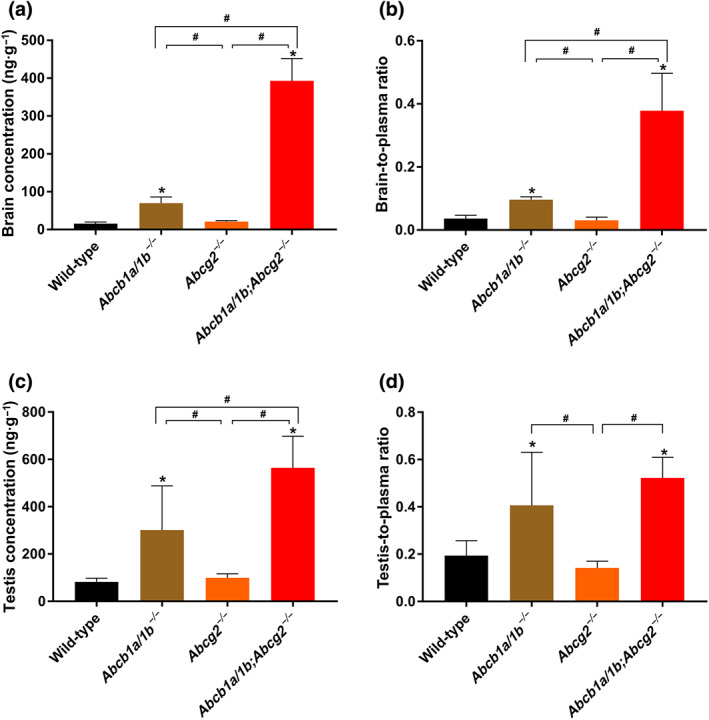
Organ concentration (a, c) and organ‐to‐plasma ratio (b, d) of larotrectinib in male wild‐type, Abcb1a/1b −/−, Abcg2 −/−, and Abcb1a/1b;Abcg2 −/− mice 1 hr after oral administration of 10 mg·kg−1 of larotrectinib (wild‐type and Abcb1a/1b −/−, n = 7; Abcg2 −/− and Abcb1a/1b;Abcg2 −/−, n = 6). * P < .01, significantly different from wild‐type mice. # P < .01, significantly different from knockout mouse strains. Statistical analysis was applied after log transformation of linear data
Whereas the liver‐to‐plasma, kidney‐to‐plasma, and lung‐to‐plasma ratios were not significantly altered between the four strains, we observed markedly lower small intestine‐to‐plasma ratios in all three knockout strains compared with wild‐type mice, especially in the Abcb1a/1b −/− and Abcb1a/1b/Abcg2 −/− mice (Figure S5B,D,F,H). The latter result was also observed in the 8‐hr experiment. The small intestinal tissue concentration often reflects the amount of drug still present in the intestinal lumen. These results may therefore point to more rapid absorption of intestinal larotrectinib in the absence of Abcg2 and especially Abcb1a/1b, or to reduced hepatobiliary excretion of the absorbed larotrectinib, or to a combination of both processes.
It is worth noting that in wild‐type mice, the tissue‐to‐plasma ratios for the liver, kidney, lung, and small intestine (all >1) were far higher than observed for the brain (0.036) and even the testis (0.19), illustrating the marked effects of the BBB and BTB on tissue accumulation of larotrectinib. In spite of the clearly increased plasma and overall tissue exposure of larotrectinib in Abcb1a/1b;Abcg2 −/− mice, and the dramatically increased brain levels, we did not observe any sign of acute spontaneous toxicity. This contrasts with the TKI drug brigatinib, for which we recently observed severe and even lethal acute toxicity in Abcb1a/1b;Abcg2 −/− mice (Li et al., 2018a).
2.3. In vitro uptake of larotrectinib
As the OATP1A/1B and OATP2B1 transporters, which are highly expressed in the liver, can have profound effects on tissue distribution and elimination of their substrates, we evaluated whether larotrectinib is a substrate of human OATP1A2, OATP1B1, OATP1B3, or OATP2B1 in vitro using HEK293 cells overexpressing these transporter proteins. Interestingly, OATP1A2 increased the in vitro uptake of larotrectinib (5 μM) by 2.4‐fold compared with the vector control cells (Figure S6A). However, we observed no significant increase in the uptake of larotrectinib in OATP1B1‐ and OATP1B3‐expressing cells compared with their vector control cells or OATP2B1‐expressing cells compared with their mock control cells. The positive control substrate rosuvastatin was avidly taken up by all the OATP‐overexpressing cell lines, confirming functional OATP expression in each cell line (Figure S6B). These results suggest that in vitro larotrectinib is a transport substrate of human OATP1A2, but not of OATP1B1, OATP1B3, or OATP2B1 as measured in HEK293 cells.
2.4. Effects of SLCO1A/1B on larotrectinib plasma pharmacokinetics and tissue disposition
Following up on the in vitro OATP uptake results, we investigated whether Slco1a/1b deficiency would influence larotrectinib pharmacokinetics in vivo. Aiming for a relatively high larotrectinib plasma level when assessing tissue distribution, we performed a 1‐hr pharmacokinetic experiment in wild‐type and Slco1a/1b −/− mice, receiving oral larotrectinib at 10 mg·kg−1. Interestingly, Slco1a/1b −/− mice showed a markedly higher larotrectinib plasma AUC0–1 hr than wild‐type mice (3.8‐fold, Figure 4 and Table 2). Larotrectinib tissue concentrations were similarly increased in Slco1a/1b −/− mice compared with wild‐type mice. In the brain, testis, kidney, and lung of Slco1a/1b −/− mice, the larotrectinib tissue concentrations were 4.2‐fold, 3.7‐fold, 4.4‐fold, and 3.2‐fold increased, respectively. No substantial changes were found in tissue‐to‐plasma ratios in these organs, suggesting that tissue concentrations mainly reflected the increased plasma levels of larotrectinib in Slco1a/1b −/− mice (Figures 5 and S7 and Table 2).
FIGURE 4.
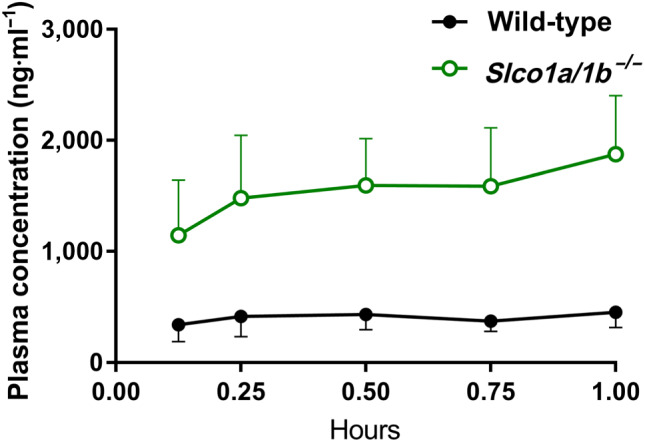
Plasma concentration–time curves of larotrectinib in male wild‐type and Slco1a/1b −/− mice over 1 hr after oral administration of 10 mg·kg−1 of larotrectinib. Data shown means ± SD (n = 7)
TABLE 2.
Plasma and organ pharmacokinetic parameters of larotrectinib in male wild‐type and Slco1a/1b −/− mice over 1 hr after oral administration of 10 mg·kg−1 of larotrectinib
| Parameter | Genotype | |
|---|---|---|
| Wild‐type | Slco1a/1b −/− | |
| AUC0–1 hr (ng·ml−1·hr−1) | 379 ± 120 | 1,452 ± 443 * |
| Fold‐change AUC0–1 hr | 1.0 | 3.8 |
| C max (ng·ml−1) | 494 ± 156 | 1,791 ± 494 * |
| T max (hr) | 0.7 ± 0.3 | 0.9 ± 0.2 |
| C brain (ng·g−1) | 16.7 ± 3.9 | 66.5 ± 17.4 * |
| Fold‐increase C brain | 1.0 | 4.2 |
| Brain‐to‐plasma ratio | 0.036 ± 0.01 | 0.046 ± 0.01 |
| Fold‐increase ratio | 1.0 | 1.0 |
| C Liver (ng·g−1) | 4,614 ± 862 | 7,393 ± 2,215 * |
| Fold‐increase C liver | 1.0 | 1.6 |
| Liver‐to‐plasma ratio | 11.2 ± 4.8 | 3.9 ± 0.2 * |
| Fold‐change ratio | 1.0 | 0.35 |
| C SI (ng·g−1) | 7,140 ± 2,645 | 9,879 ± 1,487 |
| Fold‐increase C SI | 1.0 | 1.4 |
| SI‐to‐plasma ratio | 16.0 ± 4.2 | 5.6 ± 1.7 * |
| Fold‐change ratio | 1.0 | 0.35 |
| C testis (ng·g−1) | 81.7 ± 15.8 | 301.5 ± 98.0 * |
| Fold‐increase C testis | 1.0 | 3.7 |
| Testis‐to‐plasma ratio | 0.19 ± 0.06 | 0.16 ± 0.04 |
| Fold‐change ratio | 1.0 | 0.85 |
Note. Data are given as mean ± SD (n = 7). Statistical analysis was applied after log transformation of linear data.
Abbreviations: AUC0–8 hr, area under the plasma concentration–time curve; C brain, brain concentration; C liver, liver concentration; C max, maximum concentration in plasma; C SI, small intestine tissue concentration; C testis, testis concentration; SI, small intestine (tissue); T max, time point (hr) of maximum plasma concentration.
P < .01 compared with wild‐type mice.
FIGURE 5.
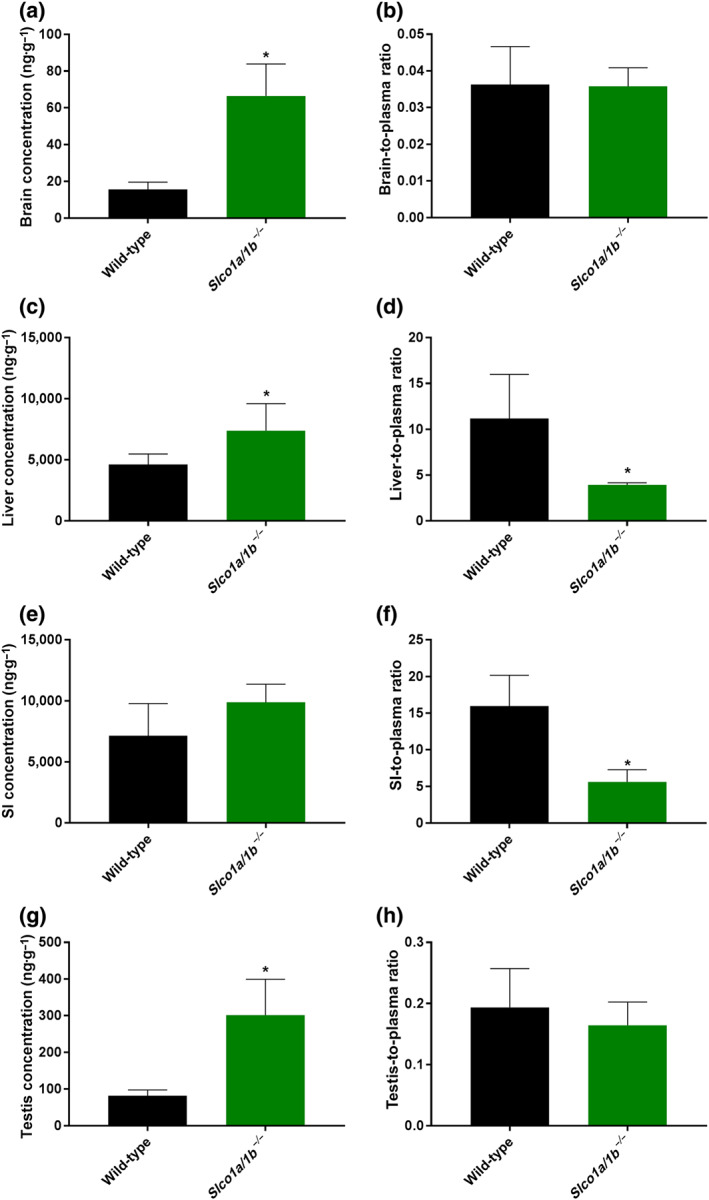
Tissue concentrations (a, c, e, g) and tissue‐to‐plasma ratios (b, d, f, h) of larotrectinib in male wild‐type and Slco1a/1b −/− mice 1 hr after oral administration of 10 mg·kg−1 of larotrectinib (n = 7). SI, small intestinal tissue. * P < .01, significantly different from wild‐type mice. Statistical analysis was applied after log transformation of linear data
However, in the liver, even though the absolute larotrectinib liver concentration was increased by 1.6‐fold (P < .01) in Slco1a/1b −/− mice compared with wild‐type mice, the liver‐to‐plasma ratio was 2.9‐fold decreased (P < .01), suggesting relatively reduced liver uptake of larotrectinib in Slco1a/1b −/− mice. Perhaps related to this, in small intestinal tissue, the absolute larotrectinib tissue concentration was also increased by 1.4‐fold, but the tissue‐to‐plasma ratio was significantly decreased by 2.9‐fold (P < .01) in Slco1a/1b −/− mice compared with wild‐type mice (Figure 5 and Table 2). It thus appears that mouse Oatp1a/1b proteins have substantial effects on oral larotrectinib pharmacokinetics, most likely by mediating efficient (first‐pass) liver uptake of absorbed larotrectinib. The in vitro uptake results suggested that larotrectinib is a substrate of OATP1A2. Considering that mouse Oatp1a1, Oatp1a4, Oatp1a5, and Oatp1a6 are most related to the single human OATP1A2 protein and that Oatp1a1 and Oatp1a4 are both highly expressed in the sinusoidal (basolateral) membrane of hepatocytes (Cheng, Maher, Chen, & Klaassen, 2005; Iusuf, van de Steeg, & Schinkel, 2012), it is very likely that larotrectinib is a hepatic uptake transport substrate of Oatp1a1 and/or Oatp1a4. The deficiency of mouse Oatp1a/1b thus results in reduced uptake of larotrectinib into the liver, which may further reduce the hepatobiliary excretion towards the small intestinal lumen. However, as we found that there was still a large amount of most likely unabsorbed larotrectinib (10–15% of the administered dose) present in small intestinal tissue plus contents at 1 hr after administration in both wild‐type and Slco1a/1b −/− mice (data not shown), we could not directly test the latter idea.
2.5. Effects of CYP3A on larotrectinib plasma pharmacokinetics and tissue disposition
CYP3A plays an important role in metabolism of many (pro‐)drugs and therefore in restricting oral availability of its substrates. To assess its effects in vivo on larotrectinib pharmacokinetics, we performed an 8‐hr pharmacokinetic pilot study in wild‐type and Cyp3a −/− mice. Larotrectinib (10 mg·kg−1) was administered orally after 2–3 hr of fasting, blood samples were taken at several time points, and at 8 hr, organs were collected. Following oral administration, plasma larotrectinib AUC0–8 hr in Cyp3a −/− mice was significantly higher (1.7‐fold, P < .01) than that in wild‐type mice (Figure S8 and Table S2). Interestingly, as seen with the ABC transporter‐deficient mice (Figure S2), the main differences in plasma exposure occurred before 4 hr. With respect to the tissue distribution at 8 hr, there were no significant/meaningful differences in tissue concentrations or tissue‐to‐plasma ratios between wild‐type and Cyp3a −/− mice for the brain, liver, kidney, small intestine, lung, and testis (Table S2 and Figure S9).
To study the in vivo effect of human CYP3A4 on larotrectinib metabolism, a 4‐hr experiment was performed in male wild‐type, Cyp3a −/−, and Cyp3aXAV mice (Cyp3a −/− mice with transgenic expression of human CYP3A4 in the liver and intestine). After oral administration of 10 mg·kg−1 of larotrectinib, blood and organs were collected and processed as described above. As shown in Figure 6 and Table 3, the oral larotrectinib plasma AUC0–4 hr in Cyp3a −/− mice was 1.7‐fold higher than that in wild‐type mice. Moreover, larotrectinib plasma exposure in Cyp3aXAV mice was decreased by twofold relative to that in wild‐type mice and by 3.5‐fold relative to Cyp3a −/− mice. Compared with the Cyp3a −/− and wild‐type mice, larotrectinib clearance from 30 min on was markedly higher in the Cyp3aXAV mice (Figure 6b). With respect to the tissue distribution of larotrectinib at 4 hr, the observed differences in absolute tissue concentrations between the strains in the brain, liver, kidney, small intestine, lung, and testis mostly reflected the different plasma concentrations (Figure 7), as the tissue‐to‐plasma ratios were not substantially altered between the strains (Figure S10). Note that the brain concentration at 4 hr in Cyp3aXAV mice was below the lower limit of detection due to low plasma exposure of larotrectinib, and the associated brain parameters were therefore not plotted.
FIGURE 6.
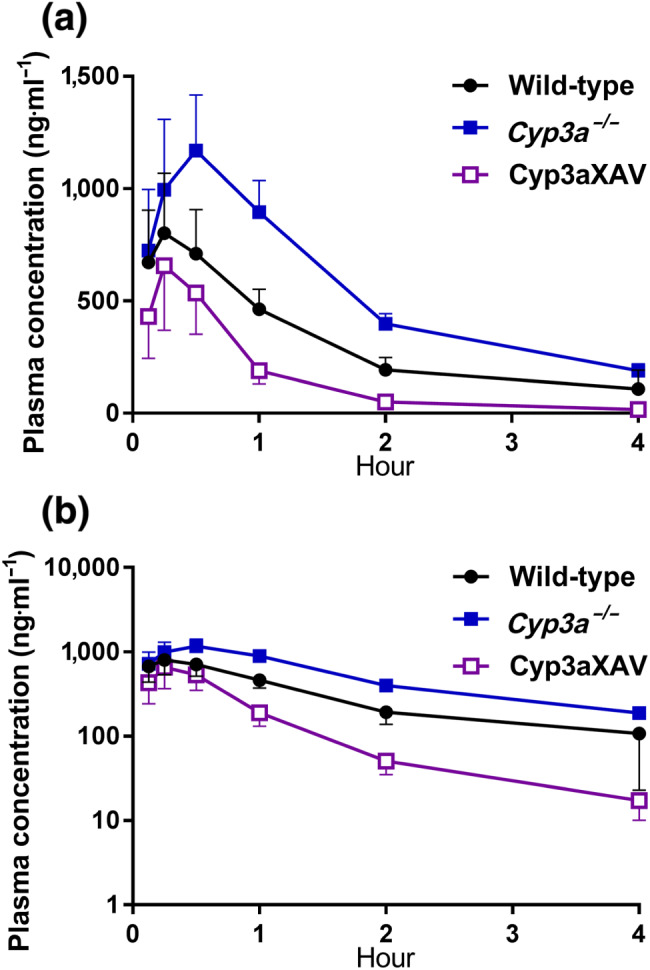
Plasma concentration–time curves (a) and semi‐log plot of plasma concentration–time curves (b) of larotrectinib in male wild‐type, Cyp3a −/−, and Cyp3aXAV mice 4 hr after oral administration of 10 mg·kg−1 of larotrectinib. Data shown are means ± SD (n = 6)
TABLE 3.
Pharmacokinetic parameters, brain concentrations, and brain‐to‐plasma ratios of larotrectinib in male wild‐type, Cyp3a −/−, and Cyp3aXAV mice over 4 hr after oral administration of 10 mg·kg−1 of larotrectinib
| Parameter | Genotype | ||
|---|---|---|---|
| Wild‐type | Cyp3a −/− | Cyp3aXAV | |
| AUC0–4 hr (ng·ml−1·hr−1) | 1,246 ± 228 | 2,174 ± 286 * | 614 ± 192 * , # |
| Fold‐change AUC0–4 hr | 1.0 | 1.7 | 0.5 |
| C max (ng·ml−1) | 816 ± 258 | 1,187 ± 236 | 666 ± 278 # |
| T max (hr) | 0.3 ± 0.1 | 0.5 ± 0.2 | 0.3 ± 0.1 |
| C brain (ng·g−1) | 4.6 ± 2.5 | 8.0 ± 1.6 | N.D. |
| Fold‐change C brain | 1.0 | 1.7 | N.A. |
| Brain‐to‐plasma ratio | 0.05 ± 0.02 | 0.04 ± 0.004 | N.A. |
| Fold‐change ratio | 1.0 | 0.8 | N.A. |
| C Liver, ng·g−1 | 930 ± 591 | 1,894 ± 450 * | 206 ± 78 # |
| Fold‐change C liver | 1.0 | 2.0 | 0.2 |
| C SI (ng·g−1) | 1,551 ± 925 | 4,131 ± 1,503 * | 418 ± 223 * , # |
| Fold‐change C SI | 1.0 | 2.7 | 0.3 |
| C testis (ng·g−1) | 50.4 ± 17.8 | 64.6 ± 14.0 | 21.6 ± 15.1 * , # |
| Fold‐change C testis | 1.0 | 1.3 | 0.3 |
| Testis‐to‐plasma ratio | 0.8 ± 0.5 | 0.3 ± 0.05 | 1.3 ± 0.6 |
| Fold‐change ratio | 1.0 | 0.4 | 1.6 |
Note. Data are given as mean ± SD (n = 6). Statistical analysis was applied after log transformation of linear data.
Abbreviations: AUC0–4 hr, area under plasma concentration–time curve; C brain, brain concentration. C liver, liver concentration; C max, maximum concentration in plasma; C SI, small intestine tissue concentration; C testis, testis concentration; N.A., not applicable; N.D., not detectable; SI, small intestine (tissue); T max, time point (hr) of maximum plasma concentration.
P < .01 compared with wild‐type mice.
P < .01 compared between Cyp3a −/− and Cyp3aXAV mice.
FIGURE 7.
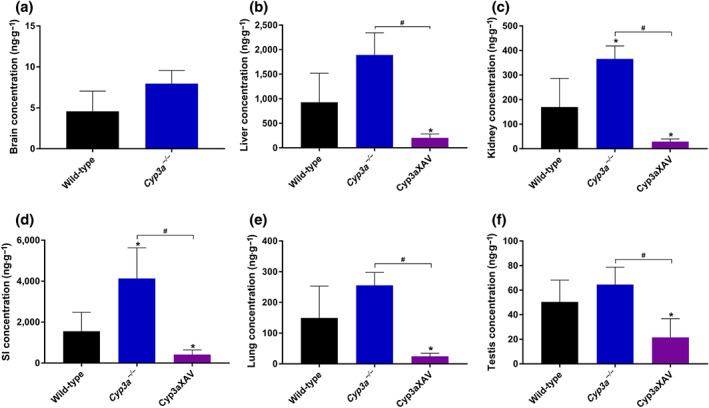
Tissue concentrations for the brain (a), liver (b), kidney (c), small intestine (d), lung (e), and testis (f) of larotrectinib in male wild‐type, Cyp3a −/−, and Cyp3aXAV mice 4 hr after oral administration of 10 mg·kg−1 of larotrectinib (n = 6). * P < .01, compared with wild‐type mice. # P < .01 significantly different as indicated. Statistical analysis was applied after log transformation of linear data
Collectively, these results indicate that larotrectinib is substantially metabolized by mouse CYP3A and human CYP3A4, which markedly affects the oral availability and, consequently, the tissue levels of larotrectinib.
3. DISCUSSION AND CONCLUSIONS
We found that in vitro, larotrectinib is highly effectively transported by human ABCB1 and mouse Abcg2 and efficiently by human ACBG2. In vivo, upon oral administration of 10 mg·kg−1 of larotrectinib, plasma AUCs were increased by 2.3‐fold and 1.7‐fold in Abcb1a/1b −/− and Abcg2 −/− mice, respectively, and by 3.4‐fold in Abcb1a/1b;Abcg2 −/− mice compared with wild‐type mice. Thus, both mAbcb1a/1b and mAbcg2 can markedly limit the oral availability of larotrectinib, with a somewhat more pronounced role for Abcb1a/1b. Given our data, this effect of the ABC transporters may be due to their limiting the net intestinal absorption of larotrectinib, or mediating the hepatobiliary elimination, or a combination of both processes. The brain‐to‐plasma ratio of larotrectinib was low (0.036) in wild‐type mice, indicating poor brain penetration of larotrectinib. This could be increased to 0.10 (2.6‐fold) in Abcb1a/1b −/− mice and further up to 0.38 (10.4‐fold) in Abcb1a/1b;Abcg2 −/− mice (Figure 3a,b and Table 1), but not in single Abcg2 −/− mice. The data indicate that ABCB1 P‐gp in the BBB can strongly restrict the brain penetration of larotrectinib, while ABCG2 has a more modest effect. We obtained qualitatively similar results for larotrectinib testis penetration, indicating similar functions of ABCB1 and ABCG2 in the BTB. Despite the increased plasma and tissue exposure, no acute spontaneous toxicity was observed for larotrectinib in the Abcb1a/1b;Abcg2 −/− mice (nor in the Slco1a/1b −/− and Cyp3a −/− mice). We note that the plasma levels of larotrectinib we obtained in our mouse studies (C max ~500–800 ng·ml−1 in wild‐type mice, Tables 1, 2, 3) were close to the average C max obtained for larotrectinib during steady‐state treatment of patients (788 ng·ml−1).
We observed a markedly decreased concentration and tissue‐to‐plasma ratio of larotrectinib in the small intestine in the absence of Abcb1a/1b or Abcg2, and when both Abcb1a/1b and Abcg2 were deficient, these values were even lower. As explained above, as small intestinal tissue concentrations often reflect the amount of drug still present in the intestinal lumen, this suggests that Abcb1a/1b and Abcg2 can mediate either the direct efflux of larotrectinib across the intestinal wall (also reducing net intestinal uptake) or the hepatobiliary excretion of larotrectinib or a combination of both processes. No noticeable changes in tissue distribution due to the ABC transporter deficiencies were found in other tissues including the liver, kidney, and lung.
Although infrequent, NTRK fusions can occur in diverse types of cancer, including glioblastoma, NSCLC, and colorectal cancer, and frequencies are even higher in certain rare paediatric tumours, such as infantile fibrosarcoma, cellular congenital mesoblastic nephroma, and papillary thyroid cancer (Creancier et al., 2015; Stransky et al., 2014; Wu et al., 2014). As brain metastases can easily occur in NSCLC, and glioblastoma is a primary brain tumour, it is important to know whether larotrectinib can achieve high intrinsic BBB permeability and how it interacts with ABCB1 and ABCG2 in the BBB. While this project was ongoing, the FDA approved larotrectinib (FDA, 2018). According to its guidelines, larotrectinib is a good substrate of ABCB1 and ABCG2, in accordance with our data. The observed strong interactions with ABCB1 and ABCG2 could well result in poor brain penetration also in humans, potentially limiting therapeutic efficacy. So far, there is little human data on the brain penetration or accumulation of larotrectinib. Ziegler et al. (2018) reported that larotrectinib penetrates the BBB and may have potent activity in TRK‐driven high‐grade glioma in a 3‐year‐old child, but there is no evidence such activity could be translated to other cases and the drug concentration and activity in the brain are still unclear. Based on our data and previous in vivo inhibition studies, brain penetration of larotrectinib could perhaps be enhanced by up to 10‐fold by co‐administering an efficacious ABCB1 and ABCG2 inhibitor such as elacridar. As also tumours that express significant levels of ABCB1 and/or ABCG2 themselves might become relatively drug resistant, there could be an added benefit to such use of an inhibitor.
Unexpectedly, we found that larotrectinib is a likely substrate of human OATP1A2 in vitro, but not of human OATP1B1 or OATP1B3, the latter negative result in line with the FDA registration documentation. An in vivo pharmacokinetic study revealed a pronounced effect of mouse Slco1a/1b transporters on the oral availability and tissue distribution of larotrectinib. Slco1a/1b‐deficient mice showed a 3.8‐fold higher larotrectinib plasma AUC0–1 hr than wild‐type mice, with concomitantly increased tissue concentrations in the brain, testis, kidney, and lung but without changes in tissue‐to‐plasma ratios. However, in the liver and small intestine, although absolute tissue concentrations were slightly higher in Slco1a/1b −/− mice due to the higher larotrectinib plasma exposure, we observed a clear decrease (both 2.9‐fold) in tissue‐to‐plasma ratios in Slco1a/1b −/− compared with wild‐type mice.
These data indicate that larotrectinib is a good in vivo transport substrate of mOatp1a/1b. Taking the in vitro uptake results into consideration, we assume that this uptake effect is mainly due to mOatp1a1 and/or mOatp1a4 in the mouse liver. The deficiency of hepatic sinusoidal mOatp1a leads to relatively reduced larotrectinib uptake into the liver and consequently more larotrectinib retention in the blood. The decreased accumulation of larotrectinib in the liver may then also result in relatively reduced hepatobiliary excretion of larotrectinib, restricting the amount of larotrectinib returned to the intestinal lumen. However, we could not confirm the latter effect due to the high levels of remaining larotrectinib in the intestinal lumen 1 hr after administration. The unchanged tissue‐to‐plasma ratios in all other tested tissues further suggest that, despite the significant expression of some Slco1a/1b transporters in BBB and BTB, they do not have noticeable effects on larotrectinib brain and testis penetration. Also, the kidney and lung appeared to passively follow the larotrectinib plasma concentration. It is worth noting that in most cases, we used single‐time‐point data for tissue‐to‐plasma ratios, which may not necessarily reflect the behaviour over the whole exposure time. However, by choosing time points where plasma levels were still very substantial, and close to the C max for the transporter studies (see, Figures 2 and 4), we think that we obtained a reasonable estimate of qualitative changes in tissue exposure due to the transporter inactivation.
Larotrectinib is, to our knowledge, the first targeted TKI anticancer drug for which the disposition of the parent compound has been shown to be strongly affected by mouse Oatp1a/1b proteins. For some targeted drugs, such as sorafenib, the disposition of the negatively charged glucuronide metabolite can be strongly affected, but the effects on the parent compound is minimal (Vasilyeva et al., 2015; Zimmerman et al., 2013). It appears that pharmaceutical companies are relatively efficient in developing targeted anticancer drugs that are minimal substrates for OATP. This contrasts with efforts to develop drugs that are not significantly transported by ABCB1 and/or ABCG2, as we and others have found that the vast majority of new TKIs is transported by ABCB1 and/or ABCG2, to such an extent that their brain penetration is markedly restricted by these transporters (van Hoppe & Schinkel, 2017; van Hoppe, Sparidans, Wagenaar, Beijnen, & Schinkel, 2017). As this study was being finalized, larotrectinib was approved by the FDA, and the registration documentation states that larotrectinib was not significantly transported by human OATP1B1 or OATP1B3, in line with our own findings. It may therefore be that the effects of OATP1A/1B is more limited in humans, and perhaps absent. It should be noted, however, that OATP‐mediated uptake of certain substrates can be cell‐type dependent for as yet unknown reasons (de Graan et al., 2012), so a negative result does not necessarily mean that a substrate cannot be transported under any circumstances. We therefore would still recommend vigilance in recognizing possible OATP‐mediated drug–drug interactions during the clinical application of larotrectinib.
We further found that larotrectinib oral availability in mice was markedly restricted by mouse Cyp3a (1.7‐fold) and especially by human CYP3A4 (3.5‐fold), demonstrating that human CYP3A can play a substantial role in the metabolic clearance of larotrectinib. We did not observe any meaningful changes in tissue‐to‐plasma ratios, suggesting that the differences in tissue concentrations simply reflected the different plasma concentrations among the mouse strains. Our results are in line with the FDA data, indicating that larotrectinib is a good substrate of CYP3A4. The substantial intestinal and hepatic first‐pass metabolism and elimination functions of CYP3A for larotrectinib is likely to markedly influence its overall tissue exposure and thus efficacy. Various well‐known drug–drug interactions affecting CYP3A activity as well as some genetic polymorphisms may drastically affect the oral availability and subsequent tissue and tumour accumulation of larotrectinib, potentially compromising its therapeutic effect and safety. On the other hand, the clear in vivo interactions of larotrectinib with the ABCB1, ABCG2, and OATP1A/1B transporters, as well as with CYP3A4 that we observed, raise the possibility that larotrectinib may also partly inhibit these detoxifying systems in clearing other substrate anticancer drugs, and this might affect their efficacy and toxicity. Indeed, the FDA documentation cautions against co‐administering larotrectinib with CYP3A(4)‐sensitive substrates. However, at least in vitro, larotrectinib did not substantially inhibit ABCB1, ABCG2, OATP1B1, and OATP1B3, suggesting that these transporters may present less of a drug interaction risk with larotrectinib.
In summary, the transporters ABCG2 and especially ABCB1, can limit the oral availability and brain penetration of larotrectinib. To the best of our knowledge, this is the first study documenting that OATP1A/1B (most likely OATP1A proteins) can restrict the systemic exposure of larotrectinib by mediating its hepatic uptake and thus, presumably, facilitating hepatobiliary excretion. Additionally, CYP3A‐mediated metabolism can strongly reduce the oral availability of larotrectinib and thus its tissue concentrations. The insights and principles obtained from our data may potentially be used to further enhance the therapeutic application and efficacy of larotrectinib, especially for brain metastases in NSCLC and for glioblastoma patients.
AUTHOR CONTRIBUTIONS
Y.W. and A.H.S. designed the study, analysed the data, and wrote the manuscript. Y.W., R.W.S., and W.L. performed the experimental parts of the study. M.C.L. contributed the reagents, materials, and mice. J.H.B. and R.W.S. supervised the bioanalytical part of the studies and checked the content and language of manuscript. All authors commented on and approved the manuscript.
CONFLICT OF INTEREST
The research group of A.H.S. receives revenue from commercial distribution of some of the mouse strains used in this study. The remaining authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Figure S1. Molecular structure of larotrectinib (LOXO‐101)
Figure S2. Plasma concentration‐time curves (A), semi‐log plot of plasma concentration‐time curves (B) and plasma AUC0‐8h; (C) of larotrectinib in male wild‐type and Abcb1a/1b;Abcg2−/− mice over 8 h after oral administration of 10 mg/kg larotrectinib. Data are given as mean ± S.D. (wild‐type n = 6, Abcb1a/1b;Abcg2−/− n = 7). *, P < 0.01 compared to wild‐type mice. Statistical analysis was applied after log‐transformation of linear data
Figure S3. Brain concentration (A) and brain‐to‐plasma ratio (B) of larotrectinib in male wild‐type and Abcb1a/1b;Abcg2−/− mice 8 h after oral administration of 10 mg/kg larotrectinib (wild‐type n = 6, Abcb1a/1b;Abcg2−/− n = 7). *, P < 0.01 compared to wild‐type mice. Statistical analysis was applied after log‐transformation of linear data
Figure S4. Tissue concentrations (A, C, E, G, I) and tissue‐to‐plasma ratios (B, D, F, H, J) of larotrectinib in male wild‐type and Abcb1a/1b;Abcg2−/− mice 8 h after oral administration of 10 mg/kg larotrectinib (wild‐type n = 6, Abcb1a/1b;Abcg2−/− n = 7). SI, small intestinal tissue. *, P < 0.01 compared to wild‐type mice. Statistical analysis was applied after log‐transformation of linear data
Figure S5. Tissue concentrations (A, C, E, G) and tissue‐to‐plasma ratios (B, D, F, H) of larotrectinib in male wild‐type, Abcb1a/1b−/−, Abcg2−/− and Abcb1a/1b;Abcg2−/− mice 1 h after oral administration of 10 mg/kg larotrectinib (wild‐type and Abcb1a/1b−/− n = 7, Abcg2−/− and Abcb1a/1b;Abcg2−/− n = 6). SI, small intestinal tissue. *, P < 0.01 compared to wild‐type mice. #, P < 0.01 compared among knockout mouse strains. Statistical analysis was applied after log‐transformation of linear data
Figure S6. in vitro uptake of larotrectinib (A) and positive control rosuvastatin (B) by human OATP1A2, OATP1B1, OATP1B3 and OATP2B1. Uptake of 5 μM larotrectinib and 0.2 μM rosuvastatin were measured after 2.5 min incubation using vector‐transfected (control) or OATP1A2‐, OATP1B1‐ or OATP1B3‐ overexpressing HEK293 cells and vector‐transfected (mock) or OATP2B1‐overexpressing HEK293 cells. n = 3, data are given as mean ± S.D
Figure S7. Tissue concentrations (A, C) and tissue‐to‐plasma ratios (B, D) of larotrectinib in male wild‐type and Slco1a/1b−/− mice 1 h after oral administration of 10 mg/kg larotrectinib (n = 7). *, P < 0.01 compared to wild‐type mice. Statistical analysis was applied after log‐transformation of linear data
Figure S8. Plasma concentration‐time curves (A), semi‐log plot of plasma concentration‐time curves (B) and plasma AUC0‐8h; (C) of larotrectinib in male wild‐type and Cyp3a−/− mice over 8 h after oral administration of 10 mg/kg larotrectinib. Data are given as mean ± S.D. (wild‐type n = 6, Cyp3a−/− n = 7). *, P < 0.01 compared to wild‐type mice. Statistical analysis was applied after log‐transformation of linear data
Figure S9. Tissue concentrations for brain, liver, kidney, small intestine, lung and testis (A‐F) and tissue‐to‐plasma ratio (G‐L) of larotrectinib in male wild‐type and Cyp3a−/− mice 8 h after oral administration of 10 mg/kg larotrectinib (wild‐type n = 6, Cyp3a−/− n = 7). SI, small intestinal tissue. Statistical analysis was applied after log‐transformation of linear data
Figure S10. Tissue‐to‐plasma ratio for brain (A), liver (B), kidney (C), small intestine (D), lung (E) and testis (F) of larotrectinib in male wild‐type, Cyp3a−/− and Cyp3aXAV mice 4 h after oral administration of 10 mg/kg larotrectinib (n = 6). SI, small intestinal tissue. *, P < 0.01 compared to wild‐type mice. #, P < 0.01 compared between Cyp3a−/− and Cyp3aXAV mice. Statistical analysis was applied after log‐transformation of linear data
Table S1. Pharmacokinetic parameters, brain concentrations, and brain‐to‐plasma ratios of larotrectinib in male wild‐type and Abcb1a/1b;Abcg2−/− mice over 8 h after oral administration of 10 mg/kg larotrectinib
Table S2. Pharmacokinetic parameters, brain concentrations and brain‐to‐plasma ratios of larotrectinib in male wild‐type and Cyp3a−/− mice over 8 h after oral administration of 10 mg/kg larotrectinib
ACKNOWLEDGEMENT
This work was funded in part by the China Scholarship Council (CSC Scholarship No. 201506240107 to Y.W.).
Wang Y, Sparidans RW, Li W, Lebre MC, Beijnen JH, Schinkel AH. OATP1A/1B, CYP3A, ABCB1, and ABCG2 limit oral availability of the NTRK inhibitor larotrectinib, while ABCB1 and ABCG2 also restrict its brain accumulation. Br J Pharmacol. 2020;177:3060–3074. 10.1111/bph.15034
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … Davies, J. A. (2019a). The Concise Guide to PHARMACOLOGY 2019/20: Catalytic receptors. British Journal of Pharmacology, 176(Suppl 1), S247–s296. 10.1111/bph.14751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … Davies, J. A. (2019b). The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176(Suppl 1), S297–s396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , … Davies, J. A. (2019). The Concise Guide to PHARMACOLOGY 2019/20: Transporters. British Journal of Pharmacology, 176(Suppl 1), S397–s493. 10.1111/bph.14753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatu, A. , Sartore‐Bianchi, A. , & Siena, S. (2016). NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open, 1(2), e000023 10.1136/esmoopen-2015-000023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakos, E. , Evers, R. , Calenda, G. , Tusnady, G. E. , Szakacs, G. , Varadi, A. , & Sarkadi, B. (2000). Characterization of the amino‐terminal regions in the human multidrug resistance protein (MRP1). Journal of Cell Science, 113(Pt 24), 4451–4461. [DOI] [PubMed] [Google Scholar]
- Cheng, X. , Maher, J. , Chen, C. , & Klaassen, C. D. (2005). Tissue distribution and ontogeny of mouse organic anion transporting polypeptides (Oatps). Drug Metabolism and Disposition, 33(7), 1062–1073. 10.1124/dmd.105.003640 [DOI] [PubMed] [Google Scholar]
- Creancier, L. , Vandenberghe, I. , Gomes, B. , Dejean, C. , Blanchet, J. C. , Meilleroux, J. , … Kruczynski, A. (2015). Chromosomal rearrangements involving the NTRK1 gene in colorectal carcinoma. Cancer Letters, 365(1), 107–111. 10.1016/j.canlet.2015.05.013 [DOI] [PubMed] [Google Scholar]
- de Graan, A. J. , Lancaster, C. S. , Obaidat, A. , Hagenbuch, B. , Elens, L. , Friberg, L. E. , … Sparreboom, A. (2012). Influence of polymorphic OATP1B‐type carriers on the disposition of docetaxel. Clinical Cancer Research, 18(16), 4433–4440. 10.1158/1078-0432.Ccr-12-0761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon, A. , Laetsch, T. W. , Kummar, S. , DuBois, S. G. , Lassen, U. N. , Demetri, G. D. , … Hyman, D. M. (2018). Efficacy of larotrectinib in TRK fusion‐positive cancers in adults and children. The New England Journal of Medicine, 378(8), 731–739. 10.1056/NEJMoa1714448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durmus, S. , Naik, J. , Buil, L. , Wagenaar, E. , van Tellingen, O. , & Schinkel, A. H. (2014). In vivo disposition of doxorubicin is affected by mouse Oatp1a/1b and human OATP1A/1B transporters. International Journal of Cancer, 135(7), 1700–1710. 10.1002/ijc.28797 [DOI] [PubMed] [Google Scholar]
- Evers, R. , Kool, M. , van Deemter, L. , Janssen, H. , Calafat, J. , Oomen, L. C. , … Borst, P. (1998). Drug export activity of the human canalicular multispecific organic anion transporter in polarized kidney MDCK cells expressing cMOAT (MRP2) cDNA. The Journal of Clinical Investigation, 101(7), 1310–1319. 10.1172/jci119886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Food and Drug Administration . (2018). Center for Drug Evaluation and Research of the US Department of Health and Human Service, Food and Drug Administration multi‐discipline review. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/211710s000lbl.pdf.
- Giacomini, K. M. , Huang, S.‐M. , Tweedie, D. J. , Benet, L. Z. , Brouwer, K. L. R. , Chu, X. , … Zhang, L. (2010). Membrane transporters in drug development. Nature Reviews Drug Discovery, 9(3), 215–236. 10.1038/nrd3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich, F. P. (1999). Cytochrome P‐450 3A4: Regulation and role in drug metabolism. Annual Review of Pharmacology and Toxicology, 39, 1–17. 10.1146/annurev.pharmtox.39.1.1 [DOI] [PubMed] [Google Scholar]
- Hagenbuch, B. , & Meier, P. J. (2004). Organic anion transporting polypeptides of the OATP/SLC21 family: Phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflügers Archiv, 447(5), 653–665. 10.1007/s00424-003-1168-y [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res., 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, E. J. , & Reichardt, L. F. (2003). Trk receptors: Roles in neuronal signal transduction. Annual Review of Biochemistry, 72, 609–642. 10.1146/annurev.biochem.72.121801.161629 [DOI] [PubMed] [Google Scholar]
- Iusuf, D. , van de Steeg, E. , & Schinkel, A. H. (2012). Functions of OATP1A and 1B transporters in vivo: Insights from mouse models. Trends in Pharmacological Sciences, 33(2), 100–108. 10.1016/j.tips.2011.10.005 [DOI] [PubMed] [Google Scholar]
- Kalliokoski, A. , & Niemi, M. (2009). Impact of OATP transporters on pharmacokinetics. British Journal of Pharmacology, 158(3), 693–705. 10.1111/j.1476-5381.2009.00430.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodaira, H. , Kusuhara, H. , Ushiki, J. , Fuse, E. , & Sugiyama, Y. (2010). Kinetic analysis of the cooperation of P‐glycoprotein (P‐gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. The Journal of Pharmacology and Experimental Therapeutics, 333(3), 788–796. 10.1124/jpet.109.162321 [DOI] [PubMed] [Google Scholar]
- Li, W. , Sparidans, R. W. , Wang, Y. , Lebre, M. C. , Beijnen, J. H. , & Schinkel, A. H. (2018a). P‐glycoprotein and breast cancer resistance protein restrict brigatinib brain accumulation and toxicity, and, alongside CYP3A, limit its oral availability. Pharmacological Research, 137, 47–55. 10.1016/j.phrs.2018.09.020 [DOI] [PubMed] [Google Scholar]
- Li, W. , Sparidans, R. W. , Wang, Y. , Lebre, M. C. , Wagenaar, E. , Beijnen, J. H. , & Schinkel, A. H. (2018b). P‐glycoprotein (MDR1/ABCB1) restricts brain accumulation and cytochrome P450‐3A (CYP3A) limits oral availability of the novel ALK/ROS1 inhibitor lorlatinib. International Journal of Cancer, 143(8), 2029–2038. 10.1002/ijc.31582 [DOI] [PubMed] [Google Scholar]
- Nakagawara, A. (2001). Trk receptor tyrosine kinases: A bridge between cancer and neural development. Cancer Letters, 169(2), 107–114. 10.1016/s0304-3835(01)00530-4 [DOI] [PubMed] [Google Scholar]
- Nigam, S. K. (2014). What do drug transporters really do? Nature Reviews Drug Discovery, 14(1), 29–44. 10.1038/nrd4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel, A. H. , & Jonker, J. W. (2003). Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Advanced Drug Delivery Reviews, 55(1), 3–29. 10.1016/s0169-409x(02)00169-2 [DOI] [PubMed] [Google Scholar]
- Shitara, Y. , Maeda, K. , Ikejiri, K. , Yoshida, K. , Horie, T. , & Sugiyama, Y. (2013). Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: Their roles in hepatic clearance and intestinal absorption. Biopharmaceutics & Drug Disposition, 34(1), 45–78. 10.1002/bdd.1823 [DOI] [PubMed] [Google Scholar]
- Simoff, I. , Karlgren, M. , Backlund, M. , Lindstrom, A. C. , Gaugaz, F. Z. , Matsson, P. , & Artursson, P. (2016). Complete knockout of endogenous Mdr1 (Abcb1) in MDCK cells by CRISPR‐Cas9. Journal of Pharmaceutical Sciences, 105(2), 1017–1021. 10.1016/s0022-3549(15)00171-9 [DOI] [PubMed] [Google Scholar]
- Sparidans, R. W. , Wang, Y. , Schinkel, A. H. , Schellens, J. H. M. , & Beijnen, J. H. (2018). Quantitative bioanalytical assay for the tropomyosin receptor kinase inhibitor larotrectinib in mouse plasma and tissue homogenates using liquid chromatography‐tandem mass spectrometry. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences, 1102‐1103, 167–172. 10.1016/j.jchromb.2018.10.023 [DOI] [PubMed] [Google Scholar]
- Stransky, N. , Cerami, E. , Schalm, S. , Kim, J. L. , & Lengauer, C. (2014). The landscape of kinase fusions in cancer. Nature Communications, 5, 4846 10.1038/ncomms5846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, S. C. , de Vries, N. , Sparidans, R. W. , Wagenaar, E. , Beijnen, J. H. , & Schinkel, A. H. (2013). Impact of P‐glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) gene dosage on plasma pharmacokinetics and brain accumulation of dasatinib, sorafenib, and sunitinib. The Journal of Pharmacology and Experimental Therapeutics, 346(3), 486–494. 10.1124/jpet.113.205583 [DOI] [PubMed] [Google Scholar]
- Tang, S. C. , Nguyen, L. N. , Sparidans, R. W. , Wagenaar, E. , Beijnen, J. H. , & Schinkel, A. H. (2014). Increased oral availability and brain accumulation of the ALK inhibitor crizotinib by coadministration of the P‐glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. International Journal of Cancer, 134(6), 1484–1494. 10.1002/ijc.28475 [DOI] [PubMed] [Google Scholar]
- van de Steeg, E. , Stranecky, V. , Hartmannova, H. , Noskova, L. , Hrebicek, M. , Wagenaar, E. , … Schinkel, A. H. (2012). Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. The Journal of Clinical Investigation, 122(2), 519–528. 10.1172/jci59526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Steeg, E. , van der Kruijssen, C. M. M. , Wagenaar, E. , Burggraaff, J. E. C. , Mesman, E. , Kenworthy, K. E. , & Schinkel, A. H. (2008). Methotrexate pharmacokinetics in transgenic mice with liver‐specific expression of human organic anion‐transporting polypeptide 1B1 (SLCO1B1). Drug Metabolism and Disposition, 37(2), 277–281. 10.1124/dmd.108.024315 [DOI] [PubMed] [Google Scholar]
- van de Steeg, E. , van Esch, A. , Wagenaar, E. , Kenworthy, K. E. , & Schinkel, A. H. (2013). Influence of human OATP1B1, OATP1B3, and OATP1A2 on the pharmacokinetics of methotrexate and paclitaxel in humanized transgenic mice. Clinical Cancer Research, 19(4), 821–832. 10.1158/1078-0432.ccr-12-2080 [DOI] [PubMed] [Google Scholar]
- van de Steeg, E. , Wagenaar, E. , van der Kruijssen, C. M. M. , Burggraaff, J. E. C. , de Waart, D. R. , Oude Elferink, R. P. , … Schinkel, A. H. (2010). Organic anion transporting polypeptide 1a/1b–knockout mice provide insights into hepatic handling of bilirubin, bile acids, and drugs. Journal of Clinical Investigation, 120(8), 2942–2952. 10.1172/jci42168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Herwaarden, A. E. , Wagenaar, E. , van der Kruijssen, C. M. , van Waterschoot, R. A. , Smit, J. W. , Song, J. Y. , … Schinkel, A. H. (2007). Knockout of cytochrome P450 3A yields new mouse models for understanding xenobiotic metabolism. The Journal of Clinical Investigation, 117(11), 3583–3592. 10.1172/jci33435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoppe, S. , & Schinkel, A. H. (2017). What next? Preferably development of drugs that are no longer transported by the ABCB1 and ABCG2 efflux transporters. Pharmacological Research, 123, 144 10.1016/j.phrs.2017.05.015 [DOI] [PubMed] [Google Scholar]
- van Hoppe, S. , Sparidans, R. W. , Wagenaar, E. , Beijnen, J. H. , & Schinkel, A. H. (2017). Breast cancer resistance protein (BCRP/ABCG2) and P‐glycoprotein (P‐gp/ABCB1) transport afatinib and restrict its oral availability and brain accumulation. Pharmacological Research, 120, 43–50. 10.1016/j.phrs.2017.01.035 [DOI] [PubMed] [Google Scholar]
- Vasilyeva, A. , Durmus, S. , Li, L. , Wagenaar, E. , Hu, S. , Gibson, A. A. , … Schinkel, A. H. (2015). Hepatocellular shuttling and recirculation of sorafenib–glucuronide is dependent on Abcc2, Abcc3, and Oatp1a/1b. Cancer Research, 75(13), 2729–2736. 10.1158/0008-5472.Can-15-0280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, G. , Diaz, A. K. , Paugh, B. S. , Rankin, S. L. , Ju, B. , Li, Y. , … Baker, S. J. (2014). The genomic landscape of diffuse intrinsic pontine glioma and pediatric non‐brainstem high‐grade glioma. Nature Genetics, 46(5), 444–450. 10.1038/ng.2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Huo, M. , Zhou, J. , & Xie, S. (2010). PKSolver: An add‐in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Computer Methods and Programs in Biomedicine, 99(3), 306–314. 10.1016/j.cmpb.2010.01.007 [DOI] [PubMed] [Google Scholar]
- Ziegler, D. S. , Wong, M. , Mayoh, C. , Kumar, A. , Tsoli, M. , Mould, E. , … Haber, M. (2018). Brief report: Potent clinical and radiological response to larotrectinib in TRK fusion‐driven high‐grade glioma. British Journal of Cancer, 119(6), 693–696. 10.1038/s41416-018-0251-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman, E. I. , Hu, S. , Roberts, J. L. , Gibson, A. A. , Orwick, S. J. , Li, L. , … Baker, S. D. (2013). Contribution of OATP1B1 and OATP1B3 to the disposition of sorafenib and sorafenib–glucuronide. Clinical Cancer Research, 19(6), 1458–1466. 10.1158/1078-0432.Ccr-12-3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Molecular structure of larotrectinib (LOXO‐101)
Figure S2. Plasma concentration‐time curves (A), semi‐log plot of plasma concentration‐time curves (B) and plasma AUC0‐8h; (C) of larotrectinib in male wild‐type and Abcb1a/1b;Abcg2−/− mice over 8 h after oral administration of 10 mg/kg larotrectinib. Data are given as mean ± S.D. (wild‐type n = 6, Abcb1a/1b;Abcg2−/− n = 7). *, P < 0.01 compared to wild‐type mice. Statistical analysis was applied after log‐transformation of linear data
Figure S3. Brain concentration (A) and brain‐to‐plasma ratio (B) of larotrectinib in male wild‐type and Abcb1a/1b;Abcg2−/− mice 8 h after oral administration of 10 mg/kg larotrectinib (wild‐type n = 6, Abcb1a/1b;Abcg2−/− n = 7). *, P < 0.01 compared to wild‐type mice. Statistical analysis was applied after log‐transformation of linear data
Figure S4. Tissue concentrations (A, C, E, G, I) and tissue‐to‐plasma ratios (B, D, F, H, J) of larotrectinib in male wild‐type and Abcb1a/1b;Abcg2−/− mice 8 h after oral administration of 10 mg/kg larotrectinib (wild‐type n = 6, Abcb1a/1b;Abcg2−/− n = 7). SI, small intestinal tissue. *, P < 0.01 compared to wild‐type mice. Statistical analysis was applied after log‐transformation of linear data
Figure S5. Tissue concentrations (A, C, E, G) and tissue‐to‐plasma ratios (B, D, F, H) of larotrectinib in male wild‐type, Abcb1a/1b−/−, Abcg2−/− and Abcb1a/1b;Abcg2−/− mice 1 h after oral administration of 10 mg/kg larotrectinib (wild‐type and Abcb1a/1b−/− n = 7, Abcg2−/− and Abcb1a/1b;Abcg2−/− n = 6). SI, small intestinal tissue. *, P < 0.01 compared to wild‐type mice. #, P < 0.01 compared among knockout mouse strains. Statistical analysis was applied after log‐transformation of linear data
Figure S6. in vitro uptake of larotrectinib (A) and positive control rosuvastatin (B) by human OATP1A2, OATP1B1, OATP1B3 and OATP2B1. Uptake of 5 μM larotrectinib and 0.2 μM rosuvastatin were measured after 2.5 min incubation using vector‐transfected (control) or OATP1A2‐, OATP1B1‐ or OATP1B3‐ overexpressing HEK293 cells and vector‐transfected (mock) or OATP2B1‐overexpressing HEK293 cells. n = 3, data are given as mean ± S.D
Figure S7. Tissue concentrations (A, C) and tissue‐to‐plasma ratios (B, D) of larotrectinib in male wild‐type and Slco1a/1b−/− mice 1 h after oral administration of 10 mg/kg larotrectinib (n = 7). *, P < 0.01 compared to wild‐type mice. Statistical analysis was applied after log‐transformation of linear data
Figure S8. Plasma concentration‐time curves (A), semi‐log plot of plasma concentration‐time curves (B) and plasma AUC0‐8h; (C) of larotrectinib in male wild‐type and Cyp3a−/− mice over 8 h after oral administration of 10 mg/kg larotrectinib. Data are given as mean ± S.D. (wild‐type n = 6, Cyp3a−/− n = 7). *, P < 0.01 compared to wild‐type mice. Statistical analysis was applied after log‐transformation of linear data
Figure S9. Tissue concentrations for brain, liver, kidney, small intestine, lung and testis (A‐F) and tissue‐to‐plasma ratio (G‐L) of larotrectinib in male wild‐type and Cyp3a−/− mice 8 h after oral administration of 10 mg/kg larotrectinib (wild‐type n = 6, Cyp3a−/− n = 7). SI, small intestinal tissue. Statistical analysis was applied after log‐transformation of linear data
Figure S10. Tissue‐to‐plasma ratio for brain (A), liver (B), kidney (C), small intestine (D), lung (E) and testis (F) of larotrectinib in male wild‐type, Cyp3a−/− and Cyp3aXAV mice 4 h after oral administration of 10 mg/kg larotrectinib (n = 6). SI, small intestinal tissue. *, P < 0.01 compared to wild‐type mice. #, P < 0.01 compared between Cyp3a−/− and Cyp3aXAV mice. Statistical analysis was applied after log‐transformation of linear data
Table S1. Pharmacokinetic parameters, brain concentrations, and brain‐to‐plasma ratios of larotrectinib in male wild‐type and Abcb1a/1b;Abcg2−/− mice over 8 h after oral administration of 10 mg/kg larotrectinib
Table S2. Pharmacokinetic parameters, brain concentrations and brain‐to‐plasma ratios of larotrectinib in male wild‐type and Cyp3a−/− mice over 8 h after oral administration of 10 mg/kg larotrectinib