

Abstract
Background and Purpose
Th17 cells play critical roles in chronic inflammation, including fibrosis. Histone acetyltransferase p300, a bromodomain‐containing protein, acetylates RORγt and promotes Th17 cell development. The bromodomain inhibitor JQ1 was shown to alleviate Th17‐mediated pathologies, but the underlying mechanism remains unclear. We hypothesized that JQ1 suppresses the response of Th17 cells by impairing p300‐mediated acetylation of RORγt.
Experimental Approach
The effect of JQ1 on p300‐mediated acetylation of RORγt was investigated in HEK293T (overexpressing Flag‐p300 and Myc‐RORγt) and human Th17 cells through immunoprecipitation and western blotting. To determine the regions of p300 responsible for JQ1‐mediated suppression of HAT activity, we performed HAT assays on recombinant p300 fragments with/without the bromodomain, after exposure to JQ1. Additionally, the effect of JQ1 on p300‐mediated acetylation of RORγt and Th17 cell function was verified in vivo, using murine Schistosoma‐induced fibrosis models. Liver injury was assessed by histopathological examination and measurement of serum enzyme levels. Expression of Th17 effectors was detected by qRT‐PCR, whereas IL‐17‐ and RORγt‐positive granuloma cells were detected by FACS.
Key Results
JQ1 impaired p300‐mediated RORγt acetylation in human Th17 and HEK293T cells. JQ1 failed to suppress the acetyltransferase activity of p300 fragments lacking the bromodomain. JQ1 treatment attenuated Schistosoma‐induced fibrosis in mice, by inhibiting RORγt acetylation and IL‐17 expression.
Conclusions and Implications
JQ1 impairs p300‐mediated RORγt acetylation, thus reducing the expression of RORγt target genes, including Th17‐specific cytokines. JQ1‐mediated inhibition of p300 acetylase activity requires the p300 bromodomain. Strategies targeting p300 may provide new therapeutic approaches for controlling Th17‐related diseases.
Abbreviations
- BRD
bromodomain
- BET
bromodomain and extra‐terminal
- CBP
CREB binding protein
- HATs
histone acetyltransferases
- KAT
lysine (K) acetyl transferases
What is already known
JQ1 inhibits Th17‐mediated pathology.
p300 acetylates RORγt and promotes Th17 differentiation and development.
What this study adds
JQ1 inhibits p300‐mediated RORγt acetylation via bromodomain, reducing RORγt's ability to drive Th17 cytokines expression.
What is the clinical significance
Strategies targeting p300 may provide a new therapeutic approach for controlling Th17‐related diseases.
1. INTRODUCTION
Activation of naive CD4+ T cells, mediated by specific cytokines, develops into distinct effector TH subsets. Cells belonging to IL‐17‐producing CD4+ T subset are referred to as “Th17” (T‐help 17) cells. Development of naive T cells into Th17 cells is different in mice and humans. In mice, Th17 cell differentiation requires combined exposure to TGF‐β and IL‐6 or TGF‐β and IL‐21. In humans, Th17 cells differentiation requires combinations of TGF‐β and IL‐1β or TGF‐β and IL‐6 (Korn, Bettelli, Oukka, & Kuchroo, 2009). During Th17 development, IL‐21 functions in an autocrine loop and mediates its own amplification (Yang et al., 2008), whereas IL‐23 is required to expand and stabilize the differentiated effector phenotype both in mice and humans (Volpe et al., 2008).
Aberrant generation and activation of Th17 cells are involved in multiple chronic inflammatory pathologies, including autoimmune diseases (Korn et al., 2009; Patel & Kuchroo, 2015) and liver fibrosis, as well as the murine models of these diseases (Park et al., 2018; Zhang et al., 2012; Zhang et al., 2015). Retinoic acid receptor‐related orphan receptor gamma t (RORγt) is signature transcription factor that directly activates the transcription of Th17‐specific genes, including IL‐17A (commonly referred to as IL‐17), IL‐17F, IL‐21, IL‐22,and GM‐CSF (Ciofani et al., 2012; Ivanov et al., 2006; Yang et al., 2008) in both mouse and human Th17 cells. These cytokines are critical for the pro‐inflammatory function of Th17 cells (Yasuda, Takeuchi, & Hirota, 2019). Since Th17 cells are strongly associated with various pro‐inflammatory responses, the expression and activity of RORγt are tightly regulated in vivo. Multiple post‐translational modifications, such as acetylation, phosphorylation, methylation, and ubiquitination, modulate the functions of RORγt and interaction with various proteins (Huh & Littman, 2012). Recently, as a nuclear receptor, RORγt has become a prime target for repressing the function of Th17 cells and their downstream cytokines. Until now, several potent RORγt inverse agonists have been developed (Fauber & Magnuson, 2014; Isono, Fujita‐Sato, & Ito, 2014; Rutz, Eidenschenk, Kiefer, & Ouyang, 2016).
Co‐transcription factor p300, bromodomain (BRD)‐containing protein, belongs to the type 3 family of lysine (K) acetyl transferases (KAT3, also known as KAT3B). This protein is known to regulate chromatin accessibility by acetylating lysine residues from histones (Delvecchio, Gaucher, Aguilar‐Gurrieri, Ortega, & Panne, 2013; Ebrahimi et al., 2019). In addition, p300 can also up‐regulate transcription by acetylating various transcription factors, such as p53, Foxp3 and RelA (Gu & Roeder, 1997; van Loosdregt et al., 2010; Mukherjee et al., 2013). We previously showed that p300 acetylates K81 of RORγt and up‐regulates RORγt‐mediated transcriptional activation of IL‐17, thereby promoting Th17 differentiation and development (Wu et al., 2015).
JQ1, an inhibitor of bromodomain and extra‐terminal (BET) family proteins (including bromodomain2, bromodomain3, bromodomain4 and bromodomain testis‐specific protein), was produced by James Bradner (Bid & Kerk, 2016). Through competitive binding to bromodomain, JQ1 prevents the interaction of bromodomain4 and acetylated lysine residues, therefore altering gene transcription. Previous reports have revealed a variety of alterations in cytokine production in Th17 cell following JQ1 treatment (Bandukwala et al., 2012; Mele et al., 2013). However, the mechanism underlying these alterations remains unclear. In this study, we demonstrated that JQ1 interacts with bromodomain to inhibit p300‐mediated acetylation of RORγt, thereby down‐regulating multiple Th17 specific genes, including IL17, IL21 and GM‐CSF, in vitro. The inhibitory effect of JQ1 on Th17 function in vivo was further verified using the Schistosoma japonicum‐infected mouse model.
2. METHODS
2.1. HEK293T cell transfection and JQ1 treatment
The Human Embryonic Kidney 293 T (HEK293T, RRID: CVCL_0063) cell line is immortalized with the Simian Virus 40 T‐antigen and represents a highly transfectable cell system, which allows overexpression of interest proteins for detailed biochemical analyses (Naz et al., 2014). In our study, we co‐transfected HEK293T cells with p300 and RORγt expression plasmids to study the interaction between p300 and RORγt and the effects of JQ1 treatment on p300‐mediated acetylation of RORγt. Briefly, HEK293T cells were co‐transfected with the p300‐expression (pIPFlag2 vector) and RORγt‐expression plasmids (pIPMyc2 vector) using polyethylenimine (#23966, Polysciences), according to the manufacturer's instructions. Transfected cells were cultured at 37°C, in a humidified atmosphere supplemented with 5% CO2, and were treated with DMSO or different concentrations of JQ1 for 9 hr prior to cell harvesting, 48 hr post‐transfection.
2.2. Co‐immunoprecipitation
HEK293T cells and liver tissues of S. japonicum ‐infected mice were used for co‐immunoprecipitation assays. HEK293T cells and liver fragments were lysed in a buffer containing 50‐mM Tris–HCl pH 7.5, 150‐mM NaCl, 1‐mM EDTA disodium salt (Na2‐EDTA), 1% Nonidet P‐40 (NP40), 0.5% sodium deoxycholate (NaDOC), and 10% glycerol, supplemented with 1% protease inhibitors cocktail (P8340, Sigma), 1‐mM Na3VO4, 10‐mM NaF, and 1‐mM phenylmethylsulfonyl fluoride. The resulting cell lysates were centrifuged at 10,000× g at 4°C, and the supernatants were submitted for immunoprecipitation. Samples were incubated rotating head over tail for 3 hr at 4°C with anti‐Flag (AT0022, CMC TAG) or anti‐RORγt (eBioscience, 14‐6988‐82) antibodies and then reacted overnight with protein A/G‐magnetic beads (LSKMAGA10, Millipore). These beads were then washed three times with lysis buffer, and immunoblotting was performed with anti‐Myc (#2276, Cell Signaling Technology), anti‐Flag (AT0022, CMC TAG), and anti‐Acetyl‐Lysine (#9441, Cell Signaling Technology) antibodies.
2.3. Human Th17 cell differentiation, JQ1 treatment, flow cytometry analysis and immunoblotting analysis
For human Th17 differentiation, naive CD4+ T cells were isolated from cord blood of healthy donors using EasySep™ Human Naive CD4+ T Cell Isolation Kit (#19555, Stem Cell). Subsequently, naive CD4+ T cells were differentiated into Th17 cells in X‐VIVO15 medium (04‐418Q, Lonza), containing 10% FBS, 1% non‐essential amino acids, 1% sodium pyruvate, 1% l‐glutamine, and 1% penicillin/streptomycin by stimulation with anti‐CD3/CD28 dynabeads (11161D, Gibco) at a cell to bead ratio of 1:1 in presence of the following cocktail: 50 ng·ml−1 of rhIL‐6 (206‐IL‐010, R&D), 100 ng·ml−1 of rhIL‐23 (1290‐IL‐010, R&D), 1 ng·ml−1 of rhTGF‐β (240‐B‐002, R&D), 10 ng·ml−1 of rhIL‐1β (201‐LB‐005, R&D), neutralizing antibodies anti‐IL‐4 (10 μg·ml−1, BD Biosciences Cat# 554481, RRID:AB_395421) and anti‐IFN‐γ (10 μg·ml−1, 554698, BD Biosciences). Cells were cultured in an incubator at 37°C with 5% CO2 for 6 days, before treatment with JQ1 (50, 100, and 250 nM) or DMSO for 9 hr.
For flow cytometry analysis, human Th17 cells were stimulated for 4 hr with phorbol 12‐myristate 13‐acetate (PMA; 25 ng·ml−1, Sigma), ionomycin (1 μg·ml−1, Sigma), in the presence of GolgiPlug (1 μg·ml−1, BD Pharmingen). Cells were then blocked with 10% rat serum and stained for surface markers CCR6 PE (R6H1, eBioscience), followed by fixation, permeabilization using the Cytofix/Cytoperm kit (554714; BD) and staining for intracellular IL‐17A APC (BL168, Biolegend) and RORγt BV‐421(Q21‐559, BD). Cells were submitted for flow cytometry on an FACS Calibur system (BD Biosciences). Data were analysed using the Flowjo V10 software.
The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018). For immunoblotting analysis, Th17 cells were harvested and lysed on ice for 30 min with protein lysis buffer (50‐mM Tris–HCl pH 7.5, 150‐mM NaCl, 1‐mM Na2‐EDTA, 1% NP40, 0.5% NaDOC, and 10% glycerol), containing protease inhibitors (1% cocktail, 1‐mM Na3VO4, 10‐mM NaF, and 1‐mM phenylmethylsulfonyl fluoride). Cell lysates were centrifuged at 10,000× g for 15 min. Supernatants were then separated through SDS‐PAGE, transferred to nitrocellulose membranes, and then immunoblotted with the following antibodies: anti‐p300 (Santa Cruz Biotechnology Cat# sc‐48343, RRID:AB_628075), anti‐RORγt (# 14‐6988‐82, eBioscience), anti‐Acetyl‐Lysine (ICP0380, ImmuneChem, Canada) and anti‐β‐actin (BM0627, BOSTER Biological Technology).
2.4. RNA isolation and quantitative PCR
Total RNA was extracted from cells or tissues using TRIzol (Sigma) and was reverse transcribed to cDNA using TB Green™ Premix Ex Taq™ II (RR820A, Takara). Quantitative real‐time PCR to assess expression of p300, IL‐17A, IL‐17F, IL‐2, and GM‐CSF was performed using the PrimeScript™ RT Reagent Kit (RR036A, Takara) on a StepOnePlus™ Real Time PCR System (Applied Biosystems). Relative mRNA expression was normalized to the expression of β‐actin. The sequences of primers are listed in Table 1.
TABLE 1.
Primers sequences of qRT‐PCR used in this study
| Gene | Forward | Reverse |
|---|---|---|
| Il17a | ACCGCAATGAAGACCCTGAT | CAGGATCTCTTGCTGGATGAGA |
| Il17f | TGCTACTGTTGATGTTGGGAC | CAGAAATGCCCTGGTTTTGGT |
| Il1β | CTGAACTCAACTGTGAAATGC | TGATGTGCTGCTGCGAGA |
| Il6 | ACACATGTTCTCTGGGAAATCGT | AAGTGCATCATCGTTGTTCATACA |
| Rorc | TGCAAGACTCATCGACAAGG | AGGGGATTCAACATCAGTGC |
| Ep300 | CAGCAGCGACTTCTTCAACAACAG | GGAGAGCGCACTTGATTGGAGAG |
| Actb | AGAGGGAAATCGTGCGTGAC | CAATAGTGATGACCTGGCCGT |
| IL17A | CCATCCCCAGTTGATTGGAA | CTCAGCAGCAGTAGCAGTGACA |
| IL17F | CCTCCCCCTGGAATTACACT | ACCAGCACCTTCTCCAACTG |
| RORC | AGTTCTGCTGACGGGTGC | CTGCTGAGAAGGACAGGGAG |
| IL21 | CACCTTCCACAAATGCAGGG | ACCGTGAGTAACTAAGAAGCAAA |
| IL22 | CGCACCTTCATGCTGGCTAA | ACCACCTCCTGCATATAAGGC |
| IL6 | ATGCAATAACCACCCCTGAC | CATGCTACATTTGCCGAAGA |
| CSF2 | TTCCTGTGCAACCCAGATTA | CTTGGTCCCTCCAAGATGAC |
| IL1B | GCTGAAGGAGATGCCTGAGAT | CACACCCAGTAGTCTTGCTTTGT |
| IL23R | CATGACTTGCACCTGGAATG | GCTTGGACCCAAACCAAGTA |
| ACTB | TGACGTGGACATCCGCAAAG | CTGGAAGGTGGACAGCGAGG |
2.5. Cell Counting Kit‐8 (CCK8) test for cell viability
Cell viability was evaluated using the CCK8 Detection Kit (Dojindo, Kumamoto, Japan), according to the manufacturer's instructions. Briefly, HEK293T cells were seeded in 96‐well plates at a density of 5,000 cells per well and cultured with high‐glucose DMEM containing 10% FBS. After 24 hr, cells were treated for 3 hr with JQ1 at different concentrations (50, 100, 150, 200, and 250 nM). Then, 10 μl of CCK‐8 solution was added to each well, and plates were incubated for 1 hr at 37°C. The absorbance was measured on a micro‐plate reader (Power Wave XS2; BioTek Instruments, Inc., Winooski, VT, USA) at 450 nm.
2.6. ELISA
The concentrations of IL‐17 from cell supernatants were assessed using the corresponding human ELISA kits (DY317 R&D SYSTEM), according to the manufacturer's protocol.
2.7. Acetyltransferase activity assays
Recombinant p300 fragments without the histone acetyltransferase (HAT) domain (p300 HAT Del), without the bromodomain (p300 HAT) or containing bromodomain (p300 BRD HAT) were independently expressed in ExpiCHO cells using the pcDNA™ 3.1 expression system and purified as described previously (Henry, Kuo, & Andrews, 2013). Protein purity and quantification were assessed by SDS‐PAGE followed by Coomassie Blue staining alongside a BSA standard. The acetyltransferase activity of recombinant p300 proteins (5 mg·ml−1) was measured in the presence or absence of JQ1 (250 nM) or (−)‐JQ1, using a commercially available kit (ab65352, Abcam), according to the manufacturer's instructions.
2.8. Infection of mice and JQ1 treatment
Animal care and experimental protocols complied with the guidelines of Institutional Animal Care and Use Committee (IACUC), Anhui Medical University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology. The group sizes were decided according to the experiment protocol (Curtis et al., 2018). Mice were killed by cervical dislocation or CO2 inhalation.
Female C57BL/6 mice, 6–8 weeks old, weighing 16–19 g (Experimental Animal Center, Chinese Science Academy, Shanghai, China) were used in all experiments. All mice were reared and housed under specific pathogen free conditions (20 ± 2°C; 12 hr light/12 hr dark) with free access to food (standard chow diet) and water. S. japonicum (Chinese mainland strain)‐infected Oncomelania hupensis snails were provided by Jiangxi Institute of Parasitic Diseases. Mice were anaesthetized by i.p. injection of ketamine (110 mg·kg−1) and subsequently infected percutaneously with 20 ± 2 freshly shed cercaria. To test the dynamic expression of p300, IL‐17A and RORγt in the liver of S. japonicum ‐infected mice, six mice were killed at 4, 6, and 9 weeks post infection (p.i.), respectively. To investigate the therapeutic effects of JQ1, some S. japonicum ‐infected mice were administered praziquantel (300 mg·kg−1) once by gavage on Day 42 p.i., to assure that the beneficial effect of JQ1 was not ascribed to its possible insecticidal effect. Thereafter, mice were randomly assigned to two groups (three mice each). Mice in one group were treated with JQ1 (50 mg·kg−1·day−1, dissolved in 2% DMSO) by i.p. injection for 15 days from 45 dpi. The control group mice received the same schedule of injections of 2% DMSO alone. At 60 dpi, mice were killed along with the age‐ and sex‐matched control mice. Blood samples and livers were collected for subsequent experiments. The JQ1 treatment experiments were repeated three times with three mice per group. There were nine mice in each group.
2.9. Liver mononuclear cells preparation and flow cytometry analysis
Liver granuloma cells were isolated from 60‐day infected mice using 0.2% type IV collagenase (Sigma) as previously described (Zhang et al., 2012). Control liver mononuclear cells were isolated from control mice as described by Blom et al. (2009) with some modifications. Briefly, mouse livers were perfused with PBS through the portal vein followed by cutting of the inferior vena cava. Livers were then cut into small pieces and mashed on 200‐gauge steel screens in cold PBS. Contaminating hepatocytes were removed by a brief centrifugation at 60× g for 1 min. The resulting supernatants were spun in 37.5% Percoll at 1,260× g for 30 min at 25°C to remove all nuclei and cell debris. New pellets containing liver mononuclear cells were treated with Ammonium‐Chloride‐Potassium (ACK) Lysis Buffer followed by two washes. Viable cells were counted by Trypan Blue exclusion assay.
For intracellular staining, liver granuloma cells from infected mice and liver mononuclear cells from control mice were stimulated with phorbol 12‐myristate 13‐acetate (PMA; 25 ng·ml−1, Sigma), ionomycin (1 μg·ml−1, Sigma), and GolgiPlug (1 μg·ml−1, BD Pharmingen) for 4 hr. Cells were stained for surface markers, such as CD3 PE‐Cy5.5 (17A2, BD) and CD4 FITC (GK1.5, BD, and then fixed, permeabilized and stained for intracellular IL‐17A PE (TC11‐18H10, BD) or RORγt PE. Cells were submitted for flow cytometry on a FACS Calibur system (BD Biosciences). Data were analysed using the Flowjo V10 software (FlowJo, RRID: SCR_008520).
2.10. Histology and morphological assessment
Liver samples from control or JQ1/DMSO treated S. japonicum ‐infected mice were fixed in 10% neutral buffered formalin and embedded in paraffin. Sections (4 μm) stained with haematoxylin and eosin or Sirius Red were examined under the microscope (Axioskop, Zeiss, Germany) for qualitative and quantitative alterations. The level of granulomatous inflammation was microscopically determined using granulomas area. The level of fibrosis was microscopically determined using collagen deposits area. Three discontinuous sections per mouse were analysed in nine mice per group. The percentage of positive staining was determined by two pathologists in a blind manner through digital morphometry (Image‐Pro Plus, RRID:SCR_007369).
2.11. Detection of liver transaminase activities
Serum samples from individual mice were obtained at Day 60 p.i. Liver injury was assessed by serum alanine aminotransferase and aspartate aminotransferase activities using a commercial kit (Jiancheng, Nanjing, China) according to the manufacturer's instructions.
2.12. Hepatic hydroxyproline content assay
Hepatic hydroxyproline content was quantified using a commercial hydroxyproline assay kit (Jiancheng, Nanjing, China) according to the manufacturer's instructions.
2.13. Data analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Human Th17 and HEK293T cells were assigned to the different groups, and the treatment was performed with randomization in a blind manner. We used normalized data to reduce the variations in the baseline between independent experiments. The expression of proteins and mRNA were normalized to fold over β‐actin and were presented as the mean ± SEM. Statistical analysis were performed using Student's t‐test or one‐way ANOVA. One‐way ANOVA with Bonferroni correction for multiple comparisons was used to detect significant differences, and there was no significant variance inhomogeneity. P values less than .05 were considered statistically significant. No outliers were excluded in the data analysis. Statistical analysis was performed using GraphPad Prism 7 (GraphPad Prism, RRID: SCR_002798). All experiments on cells were repeated at least five times. The number of independent animal experiments is indicated in the figure legends, with n referring to the number of mice in each group.
2.14. Materials
JQ1 (HY‐13030, MedChem Express, USA) and (−)‐JQ1 (HY‐13030A, MedChem Express, USA) was dissolved in 2% DMSO for use. The p300‐expression plasmid (pIPFlag2 vectors) and RORγt‐expression plasmid (pIPMyc2 vectors) were kind gifts from Prof. Bin Li (Shanghai Institute of Immunology, Shanghai JiaoTong University School of Medicine).
2.15. Nomenclature of targets and ligands
Key protein targets and ligands used in this article are hyperlinked to corresponding entries in http://www. guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018) and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. JQ1 suppresses acetylation of RORγt and down‐regulates Th17 cytokines
Previously, JQ1 was reported to strongly repress the production of effector cytokines in Th17 cells. Given the pivotal role of RORγt acetylation for the transcription of IL‐17A (Wu et al., 2015), we hypothesized that JQ1 might suppress Th17‐associated cytokines by inhibiting RORγt acetylation. To tackle this issue, human naive CD4+ T cells were differentiated into Th17 cells under standard polarizing conditions. After 6 days of culture, cells displayed classical human Th17 signature, as indicated by up‐regulation of CCR6, IL‐17A, IL‐17F, IL‐23R, IL‐22, IL‐21, and GM‐CSF (Figure S1). Consistent with previous reports (Chen et al., 2018; Wu et al., 2015), abundant expression of p300 protein was detected in Th17 cells (Figure 1a). Th17 cells were then treated with different concentrations of JQ1 for 9 hr. As expected, JQ1 treatment resulted in a reduction of RORγt acetylation, without interfering with RORγt and p300 expression (Figure 1a,b). Moreover, production of IL‐17 was blocked by JQ1 in a dose‐dependent manner, approaching significant suppression at a concentration of 200 nM (Figure 1b,c). Other critical Th17 genes, including IL‐17F, IL‐22, IL‐21, and GM‐CSF, were also suppressed after JQ1 treatment (Figure 1b). Thus, JQ1 might indeed decrease the production of Th17 lineage‐associated cytokines by reducing RORγt acetylation and, consequently, inhibiting the transcriptional activity of its target genes.
FIGURE 1.
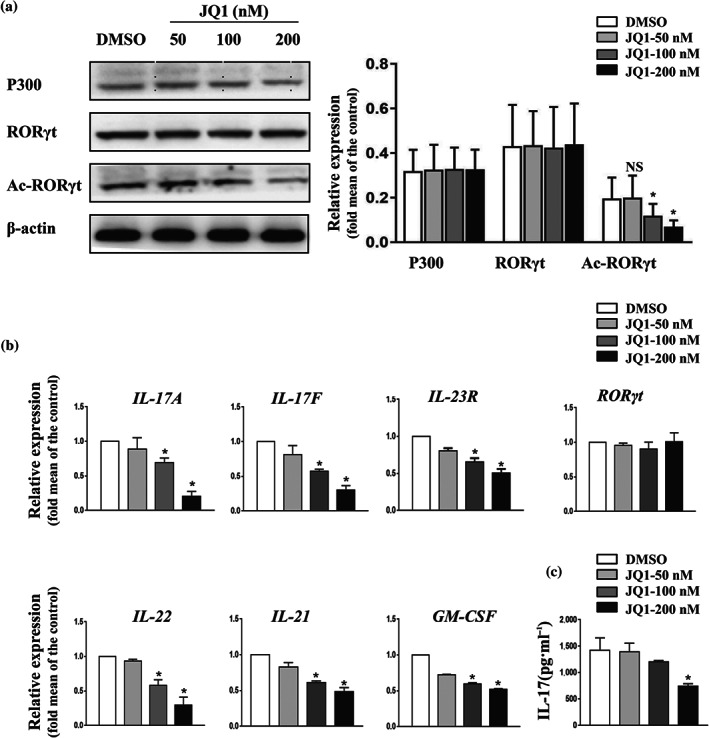
JQ1 reduces RORγt acetylation and down‐regulates the expression of human Th17 effector cytokines. Naive CD4+ T cells were cultured under Th17‐polarizing conditions for 6 days. Th17 cells were then treated with or without JQ1 at the indicated concentration for 9 hr. (a) Th17 cells were analysed by western blotting. Typical images are shown in the left panel; the average of protein relative level quantified by densitometry is shown in the right panel. (b) Expression of RORγt target genes was analysed by qRT‐PCR in Th17 cells. (c) IL‐17A level in the supernatant of cultured Th17 cells was detected by ELISA. Data were normalized to the expression level of β‐actin and presented as mean ± SEM from five independent donors, n = 5, * P < .05, compared to DMSO treatment (one‐way ANOVA)
3.2. JQ1 suppresses exogenous p300‐mediated acetylation of RORγt
We previously showed that p300 acetylates RORγt and that knockdown of p300 down‐regulates Th17‐related genes in HEK293T cells (Wu et al., 2015). To determine whether JQ1 suppresses p300‐mediated acetylation of RORγt, we transiently overexpressed Flag‐p300 and Myc‐RORγt, or Myc‐RORγt alone in HEK293T cells treated with JQ1 or DMSO for 9 hr. Then, we analysed cell lysates using anti‐Flag, anti‐Myc and anti‐Acetyl‐Lysine antibodies. We observed that JQ1 significantly decreased RORγt acetylation compared to the DMSO control in HEK293T cells co‐transfected with Flag‐p300 and Myc‐RORγt expression vectors (Figure 2a). In addition, acetylation of RORγt was not observed in HEK293T cells that overexpressed Myc‐RORγt alone (Figure 2b), altogether these data indicated that JQ1 suppresses p300‐mediated RORγt acetylation. Interestingly, p300 protein levels were not altered by JQ1, suggesting that altered p300 acetyltransferase activity was independent of expression changes. To rule out the possibility that JQ1‐induced reduction of RORγt acetylation could be associated with reduced cell survival, we compared the viability of HEK293T cells treated with JQ1 and DMSO. No significant difference in cell viability was observed between these groups, suggesting that JQ1 treatment (≤200 nM) had no effect on HEK293T cell survival (Figure 2c).
FIGURE 2.

JQ1 suppresses p300‐mediated acetylation of RORγt. HEK293T cells were transfected with Flag‐p300 and Myc‐RORγt (a) or Myc‐RORγt plasmid alone (b). Thirty‐nine hours later, cells were treated with or without JQ1 as indicated for 9 hr. Cell lysates were immunoprecipitated with anti‐Flag antibodies and submitted for immunoblotting with the indicated antibodies. (a,b) Representative images from five independent experiments are shown in the left panels. In the right panels, the corresponding graphs show the average of protein relative level quantified by densitometry. (c) Dose‐dependent effect of JQ1 on the viability of HEK293T cells. HEK293T cells were treated with JQ1 at indicated concentration for 3 hr. Cell viability was determined using the CCK8 assay kit. Results are presented as mean ± SEM from five independent experiments: NS, no significant, n = 5, * P < .05, compared to DMSO treatment (one‐way ANOVA)
3.3. JQ1 suppresses p300 acetylase activity by interacting with bromodomain
The catalytic core of p300 consists of bromodomain, CH2 (including RING and PHD) region and HAT domain (Goodman & Smolik, 2000). In addition to the HAT domain, the bromodomain is required for effective substrate acetylation (Kraus, Manning, & Kadonaga, 1999). However, it is unclear whether JQ1 inhibits the acetylase activity of p300 by binding to bromodomain. To address this issue, we overexpressed various recombinant p300 constructions: a fragment lacking the HAT domain (p300 HAT Del, aa 950‐1285‐linker‐1644‐1673), a fragment lacking the bromodomain (p300 HAT, aa 1195‐1673) or a fragment containing both the bromodomain and HAT domain (p300 BRD HAT, aa 950‐1673; Figure 3a). Acetyltransferase activity of recombinant p300 was evaluated in the presence or absence of JQ1. As expected, deletion of HAT domain abolished the acetylase activity of p300 (Figure 3b). However, p300 displayed acetylase activity regardless of the presence/absence of a bromodomain. Reduction of acetylase activity after JQ1 treatment was observed only when bromodomain‐containing p300 fragment was overexpressed, as the acetyltransferase activity was not affected by JQ1 when the bromodomain‐lacking p300 fragment was overexpressed. In contrast, treatment with (−)‐JQ1, the inactive enantiomer of JQ1, had little impact on the acetylase activity of p300 fragments. Thus, we have shown that JQ1 impaired p300 acetyltransferase activity by interacting with the bromodomain.
FIGURE 3.
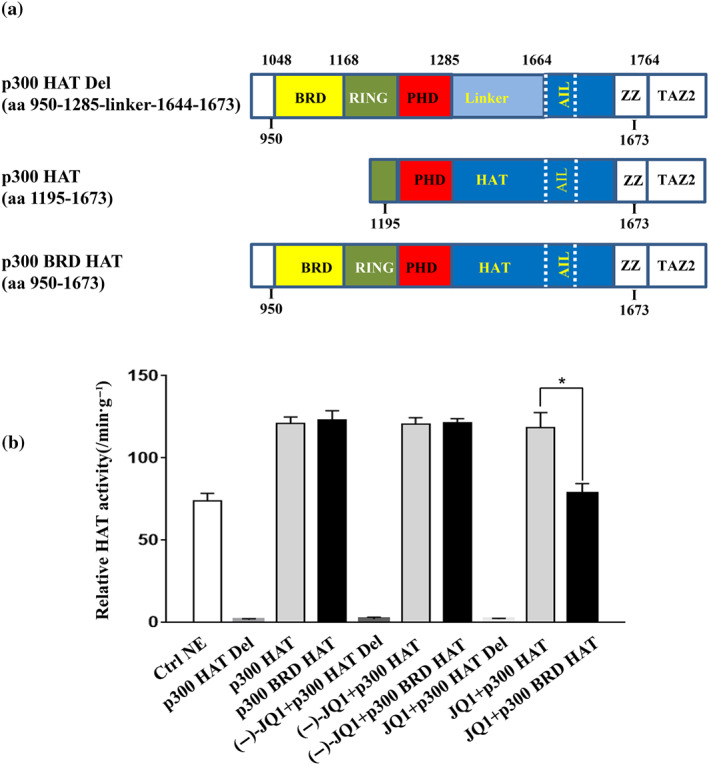
JQ1 reduces p300 acetylase activity, and the bromodomain (BRD) of p300 is required for this effect. (a) Schematic diagrams of recombinant p300 fragments lacking the HAT domain (p300 HAT Del), lacking the BRD (p300 HAT), and containing the BRD (p300 BRD HAT). These fragments were expressed in ExpiCHO cells using the pcDNA3.1 expression vector. The bromodomain, PHD (plant homeodomain), and HAT (histone acetyltransferase) domain of p300 are shown. Numbers indicate amino acid position in the sequence of p300 from the N‐terminus. (b) Relative HAT activity assay for recombinant p300 (5 μg·ml−1) with or without JQ1 (250 nM). Nuclear extract (NE) supplied in the kit was used as positive control, whereas the inactive enantiomer (−)‐JQ1 (250 nM) was used as control for JQ1. The HAT activity of p300 fragment was measured at 1 and 1.5 hr after the reaction start, and relative activities were calculated per minute and per gram of p300 protein. Results are shown as means ± SEM of triplicates from five independent experiments (n = 5). * P < .05 (one‐way ANOVA)
3.4. JQ1 suppresses acetylation of RORγt and pathogenic Th17 effect in vivo
In vitro results showed inhibitory effects of JQ1 on p300‐mediated RORγt acetylation, which in turn suppressed expression of Th17 effector cytokines. Next, we validated that JQ1 treatment inhibits the function of Th17 cells in vivo. Previous studies demonstrated the involvement of Th17 cells in the pathogenesis of S. japonicum egg‐induced liver fibrosis in mice (Shainheit et al., 2008; Zhang et al., 2012; Zhang et al., 2015). Consistent with previous reports, increased expression of IL‐17A and RORγt in the liver of S. japonicum ‐infected mice was observed. In addition, the expression of p300 was also increased after infection with S. japonicum (Figure 4). We hypothesized that JQ1 could have a protective effect against liver fibrosis in the mouse model of schistosomiasis. To test this hypothesis, we used the schistosomiasis liver fibrosis mouse model and therapeutically used JQ1 (dissolved in DMSO) or 2% DMSO alone. At 42 dpi, the infected mice were administrated praziquantel by gavage to eliminate adult worms. At 45 dpi, mice were randomly assigned to treatment with DMSO or JQ1. Due to JQ1 displaying excellent inhibitory effects without obvious toxicity when administered once daily with JQ1 50 mg·kg−1 (Filippakopoulos et al., 2010), in the present study, S. japonicum ‐infected mice were administered daily a dose of 50 mg·kg−1 of JQ1, for 15 days (Figure 5a). JQ1 treatment was partially protective against egg‐induced granulomatous inflammation and fibrosis, although the mice still have some areas of granuloma, collagen deposition and hydroxyproline (Figure 5b–d). JQ1 restored changes in liver function, as indicated by alanine aminotransferase levels (Figure 5e). Furthermore, in vivo administration of JQ1 decreased elevated liver and spleen index in S. japonicum‐infected mice (Figure 5f,g). In addition, no difference in the number of eggs between the two groups was observed (Figure S2).
FIGURE 4.

Expression of p300, IL‐17A, and RORγt mRNA is significantly elevated in the livers from S. japonicum‐infected mice. The liver tissues were collected from mice post infection 0, 4, 6, 9 weeks. The mRNA levels were determined by qRT‐PCR. The data are normalized to the expression level of β‐actin and presented as the mean ± SEM pooled from two independent experiments (with three mice per group per time), n = 6 mice per group. * P < .05 by one‐way ANOVA
FIGURE 5.
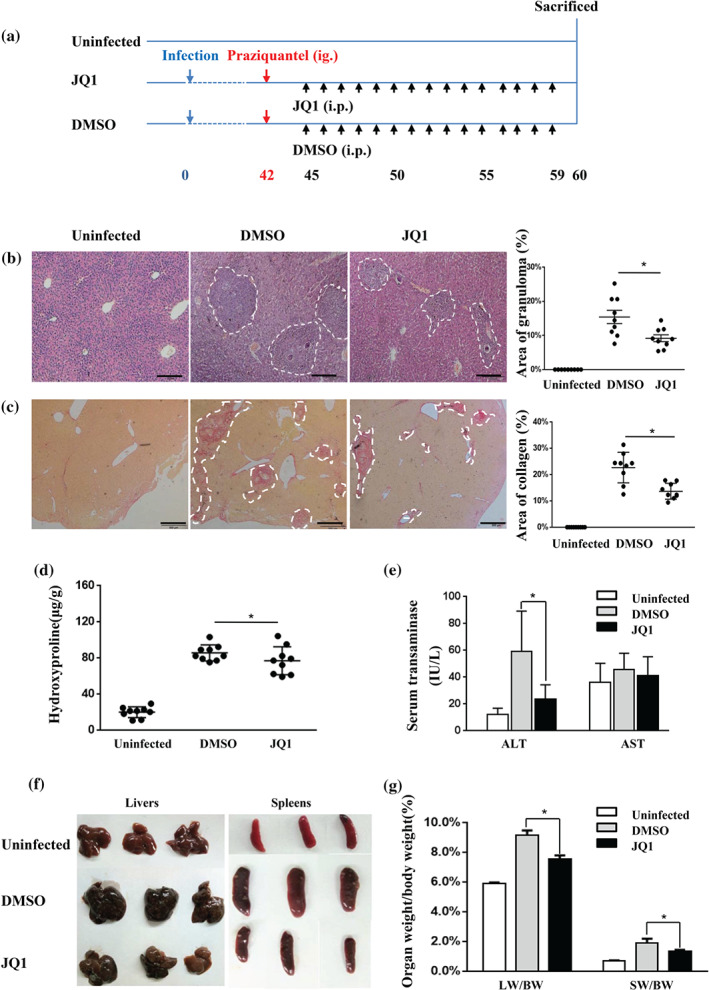
JQ1 treatment ameliorates hepatic immunopathology in S. japonicum‐infected mice. (a) Experimental design of JQ1 study on animals. Mice were infected percutaneously with S. japonicum cercaria at Day 0. Praziquantel (300 mg·kg−1) was administered once by gavage at 42 days. Daily i.p. injections of JQ1 or DMSO were conducted from Days 45 to 59. Animals were then killed for tissue collection at 60 dpi along with age‐ and sex‐matched control mice. Liver sections (4 μm) from infected and control mice were stained with either HE or Sirius Red. (b) Representative HE staining and (c) representative Sirius Red staining of liver sections of the indicated groups (three mice per group) are shown. Percentages of granulomatous and fibrosis areas were quantified and are shown on the right side in (b) and (c) respectively. Magnification was 40×. Bar = 200 μm. (d) Hydroxyproline was quantified in the livers of infected and control mice. (e) Serum ALT/AST levels are shown as the mean ± SEM. (f) Representative photograph of livers and spleens from S. japonicum‐infected mice treated with JQ1 or DMSO (three mice per group) are shown. (g) The liver and spleen weight to body weight ratio (LW/BW and SW/BW respectively) were calculated. All data were expressed as mean ± SEM pooled from three independent experiments (with three mice per group per time), n = 9 mice per group, * P < .05, compared to DMSO treatment (one‐way ANOVA)
To mechanistically address the impact of JQ1 in this mouse model, we isolated liver granulomatous cells and stimulated them with PMA/ionomycin. Compared to the control, only a decreased proportion of CD4+ T cells obtained from JQ1‐treated mice produced IL‐17A and RORγt (Figure 6a,b). Moreover, we observed a significant down‐regulation of IL‐17 and RORγt transcription (Figure 6c,d) in the livers of JQ1‐treated mice.
FIGURE 6.
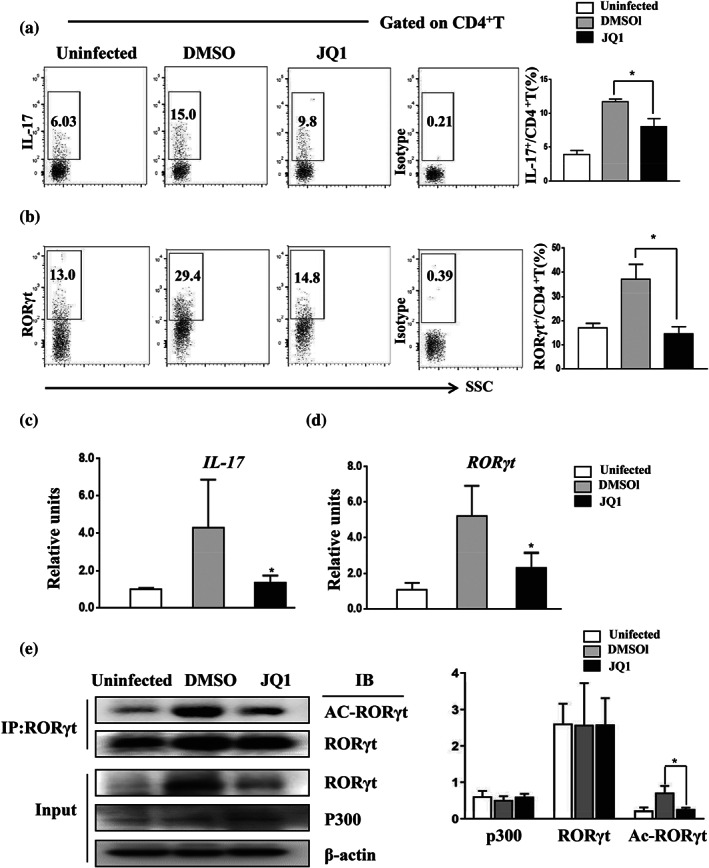
JQ1 treatment reduces the expression of IL‐17 and RORγt acetylation in the granulomatous livers of S. japonicum‐infected mice. Infected mice were treated with JQ1 or DMSO. (a,b) Liver granuloma cells were purified from 60‐day‐infected mice and stained with anti‐IL‐17 or RORγt antibodies. Data represent the percentage of IL‐17+ cells within the CD4+ lymphocyte gate in flow cytometry. (c,d) qRT‐PCR analysis of IL‐17 and RORγt mRNA from the liver of S. japonicum ‐infected mice. (e) Immunoprecipitation was performed using anti‐RORγt antibody from liver tissue lysates, followed by immunoblotting with the indicated antibodies. Representative images (left) and the average of protein relative level quantified by densitometry (right) are shown. All data are presented as means ± SEM, n = 9 mice per group pooled from three independent experiments, * P < .05, compared to DMSO treatment (one‐way ANOVA)
To investigate whether the anti‐fibrotic effect of JQ1 treatment was associated with reduced acetylation of RORγt, liver tissue from S. japonicum‐infected mice were used to examine RORγt acetylation levels. As shown in Figure 6e, JQ1 treatment resulted in a reduction of RORγt acetylation, consistent with in vitro data. These results suggested that JQ1 protected against S. japonicum egg‐induced liver fibrosis, at least in part, by decreasing the acetylation of RORγt, which in turn impaired generation and/or function of Th17 cells.
4. DISCUSSION
Deregulation of Th17 effector cells has been involved in the pathogenesis of multiple chronic inflammatory diseases (Littman & Rudensky, 2010), including Schistosoma egg‐induced liver granuloma and fibrosis (Wen et al., 2011; Zhang et al., 2012; Zhang et al., 2015). Previous studies demonstrated that JQ1 could alleviate Th17‐mediated immunopathologies. Here, we report that JQ1 suppressed p300‐mediated acetylation of RORγt, thereby down‐regulating RORγt‐target genes, such as IL‐17A, IL‐17F, GM‐CSF and IL‐21. In addition, by expressing recombinant p300 fragments containing or lacking the bromodomain, we found that the bromodomain of p300 is required for JQ1 to inhibit the acetyltransferase activity of p300.
Acetyltransferase p300 plays critical roles in transcriptional regulation, cell cycling, and inflammation (Conery et al., 2016; Fan et al., 2002; Iyer et al., 2004; Jin et al., 2011). In addition, previous studies demonstrated that p300 acetylated RORγt in human Th17 cells or when co‐expressed in HEK293T cells (Lim et al., 2015; Wu et al., 2015). p300‐mediated acetylation of RORγt at K81 increased its stabilization, which consequently enhanced RORγt‐mediated gene transcription. In the present study, we used a small‐molecule inhibitor of bromodomain, JQ1, and confirmed that p300 acetylates and functionally regulates RORγt. The acetylation level of RORγt may be an important regulator of its function, since its ability to mediate gene transcription was compromised when JQ1 decreased RORγt acetylation.
Based on western blotting results from human Th17 (Figure 1a) and HEK293T cells co‐transfected with RORγt and p300 expression vectors, we found that JQ1 did not significantly alter RORγt expression in vitro. However, the proportion of RORγt+ CD4+ T cells and RORγt protein level from JQ1‐treated S. japonicum‐infected mice was lower compared to control DMSO‐treated mice (Figure 6a,b). The reduction may result from the interaction of complex environmental factors, including IL‐6, which has been reported to promote STAT3‐mediated expression of RORγt (Bettelli et al., 2006). JQ1 was found to down‐regulate IL‐6 and pSTAT3 in human pancreatic ductal adenocarcinoma cells (Mazur et al., 2015) and IL‐6 is also up‐regulated in S. japonicum‐infected mice (Zhang et al., 2015). Thus, an intriguing possibility may be that JQ1 down‐regulated the expression of RORγt by inhibiting the IL‐6/STAT3 signal pathway.
Emerging studies showed that p300 plays important roles in the regulation of extracellular matrix homeostasis and myofibroblast transformation (Ghosh & Varga, 2007; Kaimori et al., 2010; Lim, Oh, Kim, Sohn, & Park, 2012). Ghosh et al. (2013) reported that TGF‐β stimulated fibroblasts by increasing p300 abundance and histone acetylation at the promoter of collagen gene. Accordingly, we found that the expression of p300 was markedly elevated in the liver of S. japonicum‐infected mice. JQ1 treatment protected against S. japonicum egg‐induced inflammation and fibrosis. The protective effects of JQ1 were accompanied by decreased levels of acetylated RORγt and down‐regulated expression of Th17 cell associated genes. Based on our in vitro and in vivo data, we proposed that p300‐mediated acetylation of RORγt might be a fundamental epigenetic mechanism in Schistosoma egg‐induced liver pathology. Targeting disruption of p300‐mediated acetylation might be a viable anti‐fibrosis strategy for schistosomiasis.
JQ1 is a potent pan‐bromodomain and extra‐terminal family bromodomain inhibitor. Recent evidence has shown that targeting bromodomains may be an important potential therapeutic approach for human inflammatory diseases (Muller, Filippakopoulos, & Knapp, 2011; Belkina, Nikolajczyk, & Denis, 2013; Filippakopoulos & Knapp, 2014). p300 contains a highly conserved bromodomain that was shown to bind acetylated lysine residues both from histones and transcription factors, such as p53 and Foxp3 (Gu et al., 1997; Ghosh & Varga, 2007; van Loosdregt et al., 2010). Hammitzsch et al. (2015) reported that CBP30, a selective p300 bromodomain blocker, inhibited secretion of IL‐17A in Th17 cells, which supports the view that p300 is a potential therapeutic target in Th17‐mediated diseases. In this study, we demonstrated that JQ1 inhibited IL‐17A secretion from human Th17 cells and expression of IL‐17A and other pro‐inflammatory cytokines in vitro. In addition, S. japonicum egg‐induced liver fibrosis, characterized by major Th17 contribution, was ameliorated after JQ1 treatment, in mice.
Recognition of acetylated lysine residues from histones by transcriptional co‐activator p300 plays a key role in the epigenetic regulation of gene transcription. It is well known that JQ1 prevents the interaction between bromodomain and acetylated lysine residues through competitive inhibition. The crystal structure of p300 including the bromodomain and HAT domain flanking a middle RING/PHD domain suggested the possibility of allosteric interactions regulating p300 HAT activity, however this has not been investigated in detail (Delvecchio et al., 2013; Rack et al., 2014). Here, we first demonstrated experimentally that JQ1 inhibited the acetyltransferase activity of p300 by interacting with the bromodomain, as JQ1 did not inhibit the acetyltransferase activity of p300 fragments lacking the bromodomain. Further studies are required to identify the precise mechanism underlying how JQ1 suppresses p300‐mediated acetylation of RORγt. However, the bromodomain occupancy per se is insufficient to account for the reduction in p300 enzymatic activity. It was reported that C‐terminal binding protein1 directly associated with p300 by binding to the PXDLS/K (where X indicates any residue) motif in the bromodomain of p300 and thus repressed p300‐mediated histone acetylation (Kim, Cho, Kim, & Youn, 2005). Similarly, JQ1 binds to PVDXXXL (where X indicates any residue) motif in bromodomain (Filippakopoulos et al., 2010) and thus may likely induce conformational changes or allosteric inactivation of p300.
JQ1 was previously shown to suppress the function of Th17 cells. In the present study, we found that its inhibitory effect is due to blocking p300‐mediated RORγt acetylation. Most importantly, we demonstrated the ability of JQ1 to suppress the acetyltransferase activity of p300 through interaction with the bromodomain, except for the well‐known mechanism that JQ1 competitively binds to acetyl‐lysine recognition motifs, reducing recruitment of p300 to chromatin and deregulating downstream transcription. Altogether, our findings increased pharmacological knowledge about JQ1 and unravelled its involvement in the regulation of p300 activity.
Our results showed that the bromodomain is required for JQ1‐mediated inhibition of p300 acetyltransferase activity and suppression of IL‐17 expression. It is likely that JQ1 induces conformational changes (such as bringing the HAT domain into an incorrect alignment) or allosteric inactivation of p300 through binding the bromodomain, which in turn, impair the generation and/or function of Th17 cells and lead to a reduction in liver fibrosis (see the model in Figure S3).
5. CONCLUSIONS
JQ1 alleviates Th17‐mediated immunopathologies by inhibiting p300‐catalysed acetylation of RORγt and consequently down‐regulating the transcription of Th17 cell specific genes. We believe that in the future, novel strategies targeting p300 may provide new therapeutic approaches for controlling Th17‐related diseases.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
J.S. and B.L. conceived and designed the experiments. Y.Y., X.W., D.R., Y.X., Y.D., W.H., and J.Z. performed most of the biological experiments. M.L. and C.R. analysed the data. Y.Z. and J.S. wrote and revised the main manuscript text. All the authors reviewed the manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1. Human Th17 differentiation and analysis. Human CD4+CD45RA+ naive T cells were purified from cord blood, cultured for 6 d under Th17 polarizing conditions, and analyzed by flow cytometry and qRT‐PCR. Data are representative of three independent experiments. (A) Representative flow cytometry plots from five donors show the percentage of CCR6‐, IL‐17‐, RORγt‐positive cells among differentiated Th17 cells. (B) Human Th17 signature genes were analyzed by qRT‐PCR, and normalized to the expression level of β‐actin. Data are presented as mean ± SEM compared to naive CD4+ T cells (harvested after isolation without culture), representative of three independent experiments.
Figure S2. No significant differences were observed in the number of eggs between JQ1‐treated and DMSO‐treated mice. Tissue removed from the left side of the liver of S. japonicum‐infected mice was digested with 5 ml of 5% KOH at 37°C for 5 h. The eggs recovered in 0.1 ml of digested tissue were quantified by smear examination under the microscope. Data are presented as mean ± SEM, n = 9 mice per group pooled from three independent experiments, NS = no significant, statistical analysis was performed by Student's t test.
Figure S3: Schematic model proposing JQ1 mode of action in Th17 cell and liver fibrosis.
ACKNOWLEDGEMENTS
The authors appreciate Mrs. Zhu Yalin and Cheng Wenhui, from Laboratory Animal Research Center, School of Basic Medical Sciences, for the excellent animal care. The authors thank Guanjun Chen and Dandan Zang of the centre for Scientific Research of Anhui Medical University for valuable help in our experiments. This study is supported by grants from National Natural Science Foundation of China (81471982), Natural Science Foundation of Anhui Province (1808085MH269), and Foundation of Education Department of Anhui province (KJ2018A0168).
Wang X, Yang Y, Ren D, et al. JQ1, a bromodomain inhibitor, suppresses Th17 effectors by blocking p300‐mediated acetylation of RORγt. Br J Pharmacol. 2020;177:2959–2973. 10.1111/bph.15023
Xiunan Wang, Yan Yang, and Dandan Ren contributed equally to this work.
Contributor Information
Jijia Shen, Email: shenjijia@ahmu.edu.cn.
Yuxia Zhang, Email: yuxiazh1999@163.com.
REFERENCES
- Alexander, S. P. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , … Southan, C. (2019). The Concise Guide to PHARMACOLGY 2019/20: Introduction and other protein targets. British Journal of Pharmacology, 176, S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175(3), 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandukwala, H. S. , Gagnon, J. , Togher, S. , Greenbaum, J. A. , Lamperti, E. D. , Parr, N. J. , … Rao, A. (2012). Selective inhibition of CD4+ T‐cell cytokine production and autoimmunity by BET protein and c‐Myc inhibitors. Proceedings of the National Academy of Sciences of the United States of America, 109(36), 14532–14537. 10.1073/pnas.1212264109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkina, A. C. , Nikolajczyk, B. S. , & Denis, G. V. (2013). BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. Journal of Immunology, 190(7), 3670–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli, E. , Carrier, Y. , Gao, W. , Korn, T. , Strom, T. B. , Oukka, M. , … Kuchroo, V. K. (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature, 441(7090), 235–238. 10.1038/nature04753 [DOI] [PubMed] [Google Scholar]
- Bid, H. K. , & Kerk, S. (2016). BET bromodomain inhibitor (JQ1) and tumor angiogenesis. Oncoscience, 3(11–12), 316–317. 10.18632/oncoscience.326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom, K. G. , Qazi, M. R. , Matos, J. B. , Nelson, B. D. , DePierre, J. W. , & Abedi‐Valugerdi, M. (2009). Isolation of murine intrahepatic immune cells employing a modified procedure for mechanical disruption and functional characterization of the B, T and natural killer T cells obtained. Clinical and Experimental Immunology, 155(2), 320–329. 10.1111/j.1365-2249.2008.03815.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Wang, D. , Zhao, Y. , Huang, B. , Cao, H. , & Qi, D. (2018). p300 promotes differentiation of Th17 cells via positive regulation of the nuclear transcription factor RORγt in acute respiratory distress syndrome. Immunology Letters, 202, 8–15. 10.1016/j.imlet.2018.07.004 [DOI] [PubMed] [Google Scholar]
- Ciofani, M. , Madar, A. , Galan, C. , Sellars, M. , Mace, K. , Pauli, F. , … Littman, D. R. (2012). A validated regulatory network for Th17 cell specification. Cell, 151(2), 289–303. 10.1016/j.cell.2012.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conery, A. R. , Centore, R. C. , Neiss, A. , Keller, P. J. , Joshi, S. , Spillane, K. L. , … Bommi‐Reddy, A. (2016). Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. eLife, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvecchio, M. , Gaucher, J. , Aguilar‐Gurrieri, C. , Ortega, E. , & Panne, D. (2013). Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nature Structural & Molecular Biology, 20(9), 1040–1046. 10.1038/nsmb.2642 [DOI] [PubMed] [Google Scholar]
- Ebrahimi, A. , Sevinc, K. , Gurhan Sevinc, G. , Cribbs, A. P. , Philpott, M. , Uyulur, F. , … Oppermann, U. (2019). Bromodomain inhibition of the coactivators CBP/EP300 facilitate cellular reprogramming. Nature Chemical Biology, 15(5), 519–528. 10.1038/s41589-019-0264-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, S. , Ma, Y. X. , Wang, C. , Yuan, R. Q. , Meng, Q. , Wang, J. A. , … Rosen, E. M. (2002). p300 modulates the BRCA1 inhibition of estrogen receptor activity. Cancer Research, 62(1), 141–151. [PubMed] [Google Scholar]
- Fauber, B. P. , & Magnuson, S. (2014). Modulators of the nuclear receptor retinoic acid receptor‐related orphan receptor‐γ (RORγ or RORc). Journal of Medicinal Chemistry, 57(14), 5871–5892. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos, P. , & Knapp, S. (2014). Targeting bromodomains: Epigenetic readers of lysine acetylation. Nature Reviews Drug Discovery, 13(5), 337–356. 10.1038/nrd4286 [DOI] [PubMed] [Google Scholar]
- Filippakopoulos, P. , Qi, J. , Picaud, S. , Shen, Y. , Smith, W. B. , Fedorov, O. , … Bradner, J. E. (2010). Selective inhibition of BET bromodomains. Nature, 468(7327), 1067–1073. 10.1038/nature09504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, A. K. , Bhattacharyya, S. , Lafyatis, R. , Farina, G. , Yu, J. , Thimmapaya, B. , … Varga, J. (2013). p300 is elevated in systemic sclerosis and its expression is positively regulated by TGF‐β: Epigenetic feed‐forward amplification of fibrosis. The Journal of Investigative Dermatology, 133(5), 1302–1310. 10.1038/jid.2012.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, A. K. , & Varga, J. (2007). The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. Journal of Cellular Physiology, 213(3), 663–671. [DOI] [PubMed] [Google Scholar]
- Goodman, R. H. , & Smolik, S. (2000). CBP/p300 in cell growth, transformation, and development. Genes & Development, 14(13), 1553–1577. [PubMed] [Google Scholar]
- Gu, W. , & Roeder, R. G. (1997). Activation of p53 sequence‐specific DNA binding by acetylation of the p53 C‐terminal domain. Cell, 90(4), 595–606. [DOI] [PubMed] [Google Scholar]
- Hammitzsch, A. , Tallant, C. , Fedorov, O. , O'Mahony, A. , Brennan, P. E. , Hay, D. A. , … Bowness, P. (2015). CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proceedings of the National Academy of Sciences of the United States of America, 112(34), 10768–10773. 10.1073/pnas.1501956112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46(D1), D1091–d1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry, R. A. , Kuo, Y. M. , & Andrews, A. J. (2013). Differences in specificity and selectivity between CBP and p300 acetylation of histone H3 and H3/H4. Biochemistry, 52(34), 5746–5759. 10.1021/bi400684q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh, J. R. , & Littman, D. R. (2012). Small molecule inhibitors of RORγt: targeting Th17 cells and other applications. European Journal of Immunology, 42(9), 2232–2237. 10.1002/eji.201242740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isono, F. , Fujita‐Sato, S. , & Ito, S. (2014). Inhibiting RORγt/Th17 axis for autoimmune disorders. Drug Discovery Today, 19(8), 1205–1211. 10.1016/j.drudis.2014.04.012 [DOI] [PubMed] [Google Scholar]
- Ivanov, I. I. , McKenzie, B. S. , Zhou, L. , Tadokoro, C. E. , Lepelley, A. , Lafaille, J. J. , … Littman, D. R. (2006). The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL‐17+ T helper cells. Cell, 126(6), 1121–1133. 10.1016/j.cell.2006.07.035 [DOI] [PubMed] [Google Scholar]
- Iyer, N. G. , Chin, S. F. , Ozdag, H. , Daigo, Y. , Hu, D. E. , Cariati, M. , … Caldas, C. (2004). p300 regulates p53‐dependent apoptosis after DNA damage in colorectal cancer cells by modulation of PUMA/p21 levels. Proceedings of the National Academy of Sciences of the United States of America, 101(19), 7386–7391. 10.1073/pnas.0401002101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Q. , Yu, L. R. , Wang, L. , Zhang, Z. , Kasper, L. H. , Lee, J. E. , … Ge, K. (2011). Distinct roles of GCN5/PCAF‐mediated H3K9ac and CBP/p300‐mediated H3K18/27ac in nuclear receptor transactivation. The EMBO Journal, 30(2), 249–262. 10.1038/emboj.2010.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaimori, A. , Potter, J. J. , Choti, M. , Ding, Z. , Mezey, E. , & Koteish, A. A. (2010). Histone deacetylase inhibition suppresses the transforming growth factor β1‐induced epithelial‐to‐mesenchymal transition in hepatocytes. Hepatology, 52(3), 1033–1045. [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , Altman, D. G. , & Group NCRRGW . (2010). Animal research: reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. H. , Cho, E. J. , Kim, S. T. , & Youn, H. D. (2005). CtBP represses p300‐mediated transcriptional activation by direct association with its bromodomain. Nature Structural & Molecular Biology, 12(5), 423–428. 10.1038/nsmb924 [DOI] [PubMed] [Google Scholar]
- Korn, T. , Bettelli, E. , Oukka, M. , & Kuchroo, V. K. (2009). IL‐17 and Th17 cells. Annual Review of Immunology, 27, 485–517. 10.1146/annurev.immunol.021908.132710 [DOI] [PubMed] [Google Scholar]
- Kraus, W. L. , Manning, E. T. , & Kadonaga, J. T. (1999). Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Molecular and Cellular Biology, 19(12), 8123–8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, H. W. , Kang, S. G. , Ryu, J. K. , Schilling, B. , Fei, M. , Lee, I. S. , … Verdin, E. (2015). SIRT1 deacetylates RORγt and enhances Th17 cell generation. The Journal of Experimental Medicine, 212(5), 607–617. 10.1084/jem.20132378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, J. Y. , Oh, M. A. , Kim, W. H. , Sohn, H. Y. , & Park, S. I. (2012). AMP‐activated protein kinase inhibits TGF‐β‐induced fibrogenic responses of hepatic stellate cells by targeting transcriptional coactivator p300. Journal of Cellular Physiology, 227(3), 1081–1089. 10.1002/jcp.22824 [DOI] [PubMed] [Google Scholar]
- Littman, D. R. , & Rudensky, A. Y. (2010). Th17 and regulatory T cells in mediating and restraining inflammation. Cell, 140(6), 845–858. 10.1016/j.cell.2010.02.021 [DOI] [PubMed] [Google Scholar]
- Mazur, P. K. , Herner, A. , Mello, S. S. , Wirth, M. , Hausmann, S. , Sanchez‐Rivera, F. J. , … Trajkovic‐Arsic, M. (2015). Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics‐based therapy for pancreatic ductal adenocarcinoma. Nature Medicine, 21(10), 1163–1171. 10.1038/nm.3952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. British Journal of Pharmacology, 172(13), 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele, D. A. , Salmeron, A. , Ghosh, S. , Huang, H. R. , Bryant, B. M. , & Lora, J. M. (2013). BET bromodomain inhibition suppresses TH17‐mediated pathology. The Journal of Experimental Medicine, 210(11), 2181–2190. 10.1084/jem.20130376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee, S. P. , Behar, M. , Birnbaum, H. A. , Hoffmann, A. , Wright, P. E. , & Ghosh, G. (2013). Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF‐kappaB‐driven transcription. PLoS Biology, 11(9), e1001647 10.1371/journal.pbio.1001647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, S. , Filippakopoulos, P. , & Knapp, S. (2011). Bromodomains as therapeutic targets. Expert Reviews in Molecular Medicine, 13, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naz, S. , Bashir, M. , Ranganathan, P. , Bodapati, P. , Santosh, V. , & Kondaiah, P. (2014). Protumorigenic actions of S100A2 involve regulation of PI3/Akt signaling and functional interaction with Smad3. Carcinogenesis, 35(1), 14–23. [DOI] [PubMed] [Google Scholar]
- Park, M. J. , Moon, S. J. , Lee, E. J. , Jung, K. A. , Kim, E. K. , Kim, D. S. , … Cho, M. L. (2018). IL‐1‐IL‐17 signaling axis contributes to fibrosis and inflammation in two different murine models of systemic sclerosis. Frontiers in Immunology, 9, 1611 10.3389/fimmu.2018.01611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, D. D. , & Kuchroo, V. K. (2015). Th17 cell pathway in human immunity: Lessons from genetics and therapeutic interventions. Immunity, 43(6), 1040–1051. [DOI] [PubMed] [Google Scholar]
- Rack, J. G. M. , Lutter, T. , Kjaereng Bjerga, G. E. , Guder, C. , Ehrhardt, C. , Varv, S. , … Aasland, R. (2014). The PHD finger of p300 influences its ability to acetylate histone and non‐histone targets. Journal of Molecular Biology, 426(24), 3960–3972. 10.1016/j.jmb.2014.08.011 [DOI] [PubMed] [Google Scholar]
- Rutz, S. , Eidenschenk, C. , Kiefer, J. R. , & Ouyang, W. (2016). Post‐translational regulation of RORγt‐A therapeutic target for the modulation of interleukin‐17‐mediated responses in autoimmune diseases. Cytokine & Growth Factor Reviews, 30, 1–17. [DOI] [PubMed] [Google Scholar]
- Shainheit, M. G. , Smith, P. M. , Bazzone, L. E. , Wang, A. C. , Rutitzky, L. I. , & Stadecker, M. J. (2008). Dendritic cell IL‐23 and IL‐1 production in response to schistosome eggs induces Th17 cells in a mouse strain prone to severe immunopathology. Journal of Immunology, 181(12), 8559–8567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loosdregt, J. , Vercoulen, Y. , Guichelaar, T. , Gent, Y. Y. , Beekman, J. M. , van Beekum, O. , … Coffer, P. J. (2010). Regulation of Treg functionality by acetylation‐mediated Foxp3 protein stabilization. Blood, 115(5), 965–974. 10.1182/blood-2009-02-207118 [DOI] [PubMed] [Google Scholar]
- Volpe, E. , Servant, N. , Zollinger, R. , Bogiatzi, S. I. , Hupe, P. , Barillot, E. , & Soumelis, V. (2008). A critical function for transforming growth factor‐β, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)‐17 responses. Nature Immunology, 9(6), 650–657. 10.1038/ni.1613 [DOI] [PubMed] [Google Scholar]
- Wen, X. , He, L. , Chi, Y. , Zhou, S. , Hoellwarth, J. , Zhang, C. , … Su, C. (2011). Dynamics of Th17 cells and their role in Schistosoma japonicum infection in C57BL/6 mice. PLoS Neglected Tropical Diseases, 5(11), e1399 10.1371/journal.pntd.0001399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Q. , Nie, J. , Gao, Y. , Xu, P. , Sun, Q. , Yang, J. , … Li, B. (2015). Reciprocal regulation of RORγt acetylation and function by p300 and HDAC1. Scientific Reports, 5, 16355 10.1038/srep16355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L. , Anderson, D. E. , Baecher‐Allan, C. , Hastings, W. D. , Bettelli, E. , Oukka, M. , … Hafler, D. A. (2008). IL‐21 and TGF‐β are required for differentiation of human T(H)17 cells. Nature, 454(7202), 350–352. 10.1038/nature07021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. O. , Pappu, B. P. , Nurieva, R. , Akimzhanov, A. , Kang, H. S. , Chung, Y. , … Dong, C. (2008). T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR α and ROR γ. Immunity, 28(1), 29–39. 10.1016/j.immuni.2007.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda, K. , Takeuchi, Y. , & Hirota, K. (2019). The pathogenicity of Th17 cells in autoimmune diseases. Seminars in Immunopathology, 41(3), 283–297. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Chen, L. , Gao, W. , Hou, X. , Gu, Y. , Gui, L. , … Shen, J. (2012). IL‐17 neutralization significantly ameliorates hepatic granulomatous inflammation and liver damage in Schistosoma japonicum infected mice. European Journal of Immunology, 42(6), 1523–1535. 10.1002/eji.201141933 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Huang, D. , Gao, W. , Yan, J. , Zhou, W. , Hou, X. , … Shen, J. (2015). Lack of IL‐17 signaling decreases liver fibrosis in murine schistosomiasis japonica. International Immunology, 27(7), 317–325. 10.1093/intimm/dxv017 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Human Th17 differentiation and analysis. Human CD4+CD45RA+ naive T cells were purified from cord blood, cultured for 6 d under Th17 polarizing conditions, and analyzed by flow cytometry and qRT‐PCR. Data are representative of three independent experiments. (A) Representative flow cytometry plots from five donors show the percentage of CCR6‐, IL‐17‐, RORγt‐positive cells among differentiated Th17 cells. (B) Human Th17 signature genes were analyzed by qRT‐PCR, and normalized to the expression level of β‐actin. Data are presented as mean ± SEM compared to naive CD4+ T cells (harvested after isolation without culture), representative of three independent experiments.
Figure S2. No significant differences were observed in the number of eggs between JQ1‐treated and DMSO‐treated mice. Tissue removed from the left side of the liver of S. japonicum‐infected mice was digested with 5 ml of 5% KOH at 37°C for 5 h. The eggs recovered in 0.1 ml of digested tissue were quantified by smear examination under the microscope. Data are presented as mean ± SEM, n = 9 mice per group pooled from three independent experiments, NS = no significant, statistical analysis was performed by Student's t test.
Figure S3: Schematic model proposing JQ1 mode of action in Th17 cell and liver fibrosis.