

Abstract
Previously, we proposed a new perspective of triptolide (TP)-associated hepatotoxicity: liver hypersensitivity upon lipopolysaccharide (LPS) stimulation. However, the mechanisms for TP/LPS-induced hepatotoxicity remained elusive. The present study aimed to clarify the role of LPS in TP/LPS-induced hepatotoxicity and the mechanism by which TP induces liver hypersensitivity upon LPS stimulation. TNF-α inhibitor, etanercept, was injected intraperitoneally into mice to investigate whether induction of TNF-α by LPS participated in the liver injury induced by TP/LPS co-treatment. Mice and hepatocytes pretreated with TP were stimulated with recombinant TNF-α to assess the function of TNF-α in TP/LPS co-treatment. Additionally, time-dependent NF-κB activation and NF-κB-mediated pro-survival signals were measured in vivo and in vitro. Finally, overexpression of cellular FLICE-inhibitory protein (FLIP), the most potent NF-κB-mediated pro-survival protein, was measured in vivo and in vitro to assess its function in TP/LPS-induced hepatotoxicity. Etanercept counteracted the toxic reactions induced by TP/LPS. TP-treatment sensitized mice and hepatocytes to TNF-α, revealing the role of TNF-α in TP/LPS-induced hepatotoxicity. Mechanistic studies revealed that TP inhibited NF-κB dependent pro-survival signals, especially FLIP, induced by LPS/TNF-α. Moreover, overexpression of FLIP alleviated TP/LPS-induced hepatotoxicity in vivo and TP/TNF-α-induced apoptosis in vitro. Mice and hepatocytes treated with TP were sensitive to TNF-α, which was released from LPS-stimulated immune cells. These and other results show that the TP-induced inhibition of NF-κB-dependent transcriptional activity and FLIP production are responsible for liver hypersensitivity.
Key words: Triptolide, LPS, TNF-α, NF-κB, FLIP
Abbreviations: CIAPs, cellular inhibitor of apoptosis proteins; Etan, etanercept; FADD, FAS-associated protein with death domain; FLIP, cellular FLICE-inhibitory protein; IκB-α, NF-κB inhibitor alpha; LDH, lactate dehydrogenase; LPS, lipopolysaccharide; MLKL, mixed lineage kinase domain like pseudokinase; MPO, myeloperoxidase; PAS, periodic acid-schiff; RIPK1/3, receptor-interacting protein kinase 1/3; TNFAIP3, TNF-α-induced protein 3; TNF-R1, tumor necrosis factor receptor type 1; TP, triptolide; TRADD, TNF receptor-associated death domain; TRAF2, TNF receptor-associated factor 2; XIAP, X-linked inhibitor of apoptosis protein
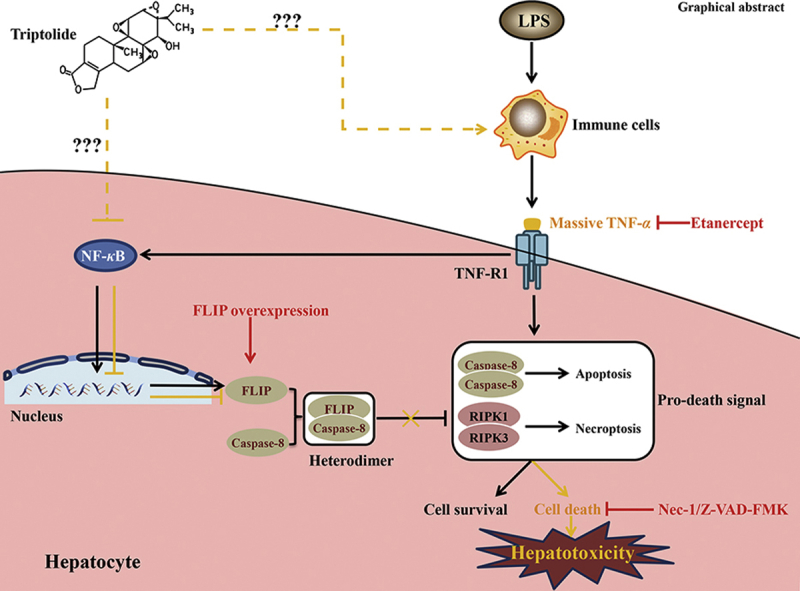
Graphical abstract
Triptolide (TP) treatment increased the sensitivity of liver upon lipopolysaccharide (LPS), which participated in TP/LPS-induced hepatotoxicity through promoting the TNF-α release. Inactivation of NF-κB and NF-κB-mediated cellular FLICE-inhibitory protein expression by TP were responsible for this sensitization. It is necessary to pay attention to the hepatotoxicity of drugs that block TNF-α-TNF-R1 signaling.
1. Introduction
Triptolide (TP), one of the fat-soluble components extracted from the Chinese medicinal herb Tripterygium wilfordii Hook F. (TWHF), has been proposed to be beneficial for a variety of diseases such as cancer, inflammatory disorders, immunosuppression, neurodegeneration and cardiovascular disorders1,2. Drugs containing TP, such as Tripterygium Glycosides and Leigongteng Tables, have been clinically used for the treatment of rheumatoid arthritis, nephrotic syndrome, and other autoimmune diseases. However, the wide application of TP and the drugs derived from TWHF has been restricted because of their severe side effects, especially hepatotoxicity3,4. Administration of the drugs containing TP (including Tripterygium Glycosides and Leigongteng Tables) caused severe hepatotoxicity with significant elevation of serum transaminases according to clinical reports, but published articles from other groups and the results from our laboratory revealed that mice or rats treated with high dose of Tripterygium Glycosides or TP expressed a slight increase in serum transaminases with insignificant liver damage. Thus, direct liver damage by these drugs may not be the only reason for their hepatotoxicity3,5, 6, 7. Our previous studies proposed a new perspective of TP-induced hepatotoxicity: liver hypersensitivity upon lipopolysaccharide (LPS) stimulation. We proposed that TP (500 μg/kg) treatment disrupted liver immune homeostasis, which limited hepatic function to detoxify the harmful response induced by the non-toxic dose of LPS (0.1 mg/kg), ultimately leading to liver hypersensitivity upon LPS stimulation8. However, the actual role of LPS in TP/LPS co-treatment and TP-induced liver hypersensitivity had not yet been established.
A vital immune organ, the liver, is constantly exposed to portal venous blood from gastrointestinal tract which is rich in the digestive products along with bacterial products, especially lipopolysaccharide endotoxin (also referred as LPS)9. Under physiological conditions, only small amounts of LPS succeed in reaching the liver and these are quickly removed by phagocytic hepatic cells without initiating the harmful response. Thus, the immune system can normally neutralize harmful responses induced by small amounts of LPS10,11. Interestingly, treatment of mice with d-galactosamine (d-GalN) was shown to inhibit NF-κB-dependent target genes, also when combined with a non-toxic dose of LPS-induced acute hepatotoxicity with massive destruction of hepatocytes. Inhibition of the production of TNF-α by Kupffer cells alleviated such d-GalN/LPS-induced hepatotoxicity12, 13, 14, 15. Furthermore, other research showed that the application of a transcription inhibitor or protein synthesis inhibitor, such as actinomycin D and cycloheximide, promoted cell death in the presence of TNF-α due to the deficiency of pro-survival proteins16, 17, 18, 19.
A rapidly inducible transcription factor, NF-κB, has been widely studied among researchers because of its central role in immunological regulation and also control of a wide range of gene expression20. Cascade activation by pro-inflammatory molecules such as LPS or TNF-α leads to immediate phosphorylation and subsequent degradation of IκB-α, followed by translocation of NF-κB subunits from cytoplasm to nucleus21. Published work documents that the cytoprotective effect of NF-κB is closely related to NF-κB-dependent anti-apoptosis (also referred as pro-survival) gene transcription. Products of this activated pathway include cellular FLICE-inhibitory protein (FLIP), X-linked inhibitor of apoptosis protein (XIAP), cellular inhibitor of apoptosis protein 1 (CIAP1), and cellular inhibitor of apoptosis protein (CIAP2)22,23. Recombinant mice lacking NF-κB P65 and the upstream signals of NF-κB activation, such as IKKβ or IKKγ, resulted in embryonic lethality due to the death of hepatocytes, suggesting an indispensable function of NF-κB in protecting hepatocytes from TNF-α-induced cell death24,25. Furthermore, deletion of P65 in hepatocytes using Cre/Lox system decreased the expression of FLIP that is associated with an increase in the sensitivity of liver upon LPS stimulation in vivo as well as the stimulation of TNF-α both in vivo and in vitro26. It was also reported that inactivation of NF-κB promoted cell death in the presence of TNF-α and overexpression of FLIP dampened the cytotoxicity of TNF-α, demonstrating that the cytoprotective effect of NF-κB depends on FLIP22. The protective effect of FLIP against TNF-α or FasL mediated hepatic cell death was also confirmed by the application of Fas agonistic Jo2 or GalN/LPS27. As a catalytically inactive homolog of caspase-8, FLIP was up-regulated by TNF-α22. The formation of FLIP–caspase-8 heterodimer inhibited the assembly of caspase-8 homologous complex and subsequent caspase-8-dependent apoptosis, which revealed the anti-apoptosis function of FLIP28. Recently, the combination of FLIP–caspase-8 heterodimer to FAS-associated protein with death domain (FADD) was also proposed to cleave receptor-interacting protein kinase 1 (RIPK1) and receptor-interacting protein kinase 3 (RIPK3), resulting in necroptosis inactivation29.
Considering the close relationship between hepatic cell death and TP/LPS-induced hepatotoxicity, we hypothesized that TP treatment might inhibit the transcriptional activity of NF-κB in hepatocytes, resulting in decreased expression of FLIP and an increase in the sensitivity of hepatocytes upon TNF-α stimulation. To test this hypothesis, the TNF-α antibody etanercept was injected prior the application of LPS to inhibit the effect of TNF-α induced by LPS. Additionally, the appearance of time-dependent NF-κB activity and NF-κB dependent pro-survival proteins were studied. Moreover, overexpression of FLIP in vivo and in vitro was carried out to interpret the relevance of FLIP in TP/LPS-induced hepatotoxicity.
2. Materials and methods
2.1. Material
TP (CAS number 38748-32-2, purity >98%) was obtained from Sanling Biotech (Guilin, China). LPS (L2755) and etanercept (Etan) were purchased from Sigma–Aldrich (St. Louis, MO, USA) and Pfizer (New York City, NY, USA), respectively. Mouse recombinant TNF-α (315-01A) and human recombinant TNF-α (300-01A) were obtained from Peprotech Inc. (Rocky Hill, NJ, USA). Z-VAD-FMK (A1902) and necrostatin-1 (Nec-1, A4213) were purchased from Apexbio (Houston, TX, USA). The reagents for qPCR, including Trizol reagent, SYBR Green Master Mix, and Reverse Transcription Kit, were obtained from Vazyme Biotech Co., Ltd. (Nanjing, China).
Primary antibodies against cleaved caspase-8 (8592) for mouse samples, cleaved caspase-8 (9496) for human samples, cleaved PARP (9532), BAX (2772), BCL-2 (2870), IκB-α (4814), FLIP (56,343), X-linked inhibitor of apoptosis protein (XIAP, 2042), and cleaved caspase-3 (9661) were purchased from Cell Signaling Technology (Boston, MA, USA). Antibodies against tubulin (sc-5286), GAPDH (sc-365,062), and NF-κB P65 (sc-372) were purchased from Santa Cruz Biotechnology (Dallas, CA, USA). Antibody against RIPK1 (17519-1-AP), mixed lineage kinase domain like pseudokinase (MLKL, 66675-1-Ig), myeloperoxidase (MPO, 22225-1-AP), and CIAP1 (10022-1-AP) were purchased from Proteintech (Chicago, IL, USA). Antibody against P-MLKL (Ser345) (ab196436) was purchased from Abcam (Cambridge, UK).
2.2. Animal and pharmacological treatments
Animal treatment of TP (500 μg/kg, i.g.) and LPS (0.1 mg/kg, i.p.) was described in our previous research paper8. Briefly, mice were treated daily with TP or the same volume of 0.5% CMC-Na for 7 days. LPS was administered 2 h after the last dose of TP and mice were sacrificed 8 h later. Doses of etanercept (10 mg/kg, i.p.) and mouse recombinant TNF-α (10 μg/kg, i.v.) were based on published literature30,31. AAV8-control and AAV8-FLIPL (Long form of FLIP) (1 × 1011 vg/mouse, i.v.) were prepared by Hanbio (Shanghai, China). FLIPL (NM_001289704.3) was vectored with AAV-CMV-MCS-GFP and tagged with 3 × Flag. All experiments and procedures involving mice were carried out on the basis of the guidelines from Ethical Committee of China Pharmaceutical University (Nanjing, China) and the Laboratory Animal Management Committee of Jiangsu Province.
To explore the function of anti-TNF-α antibody, etanercept, in TP/LPS-induced hepatotoxicity, mice were randomly divided into five groups (n = 10/group) and etanercept or the same volume of PBS was injected 1 h after the last dose of TP (Fig. 1A). To investigate whether TP-treatment increased the sensitivity of liver upon TNF-α stimulation, mice were divided into four groups (n = 6/group) and were injected intravenously with recombinant TNF-α or dilute PBS 2 h after the last dose of TP (Fig. 3A). The involvement of NF-κB in TP/LPS-induced hepatotoxicity was verified by the time-dependent experiment shown in Fig. 4A with six mice in each group. Moreover, the function of FLIPL in TP/LPS-induced hepatotoxicity was studied by the injection of AAV8-FLIPL or AAV8-NC in mice. Four weeks after AAV injection, mice were treated with TP and LPS as presented in Fig. 7A.
Figure 1.

Effects of etanercept administration on liver injury parameters disturbed by TP/LPS co-treatment. (A) Schematic presentation of the experimental procedure to testify the effects of etanercept upon TP/LPS-induced hepatotoxicity. (B)–(G) Levels of serum ALT, AST, TBA, Glucose, total protein, and ALB in TP/LPS treated mice, pretreated with etanercept (n = 10). (H) Pictures of liver tissue sections stained by H&E (top panels), analyzed by IHC for MPO (middle panels) or analyzed by PAS (bottom panels) (200 ×). Scale bar = 50 μm. Results were expressed as mean ± SEM and statistical analysis was performed using One-way ANOVA following by Tukey's multiple comparison test. *, #P < 0.05, ***, ##P < 0.01, ***, ###P < 0.001; ns, no statistical difference.
Figure 3.

Pretreatment with TP increased the sensitivity of mice and hepatocytes upon TNF-α. (A) Experimental design to prove hypersensitivity of TP-treated mice upon TNF-α. (B) and (C) Levels of serum ALT and AST in mice treated with TP, TNF-α, or both (n = 6). (D) Representative pictures of H&E staining of liver tissues obtained from mice treated with TP, TNF-α, or both (200 ×). Scale bar = 50 μm. (E) Cell viability of L-02 cells treated with TP (25 nmol/L) and TNF-α (50 ng/mL), 6, 12 or 24 h after TNF-α treatment (n = 3). (F)–(G) Cell viability of L-02 cells treated with TP (15, 20, or 25 nmol/L) and TNF-α (ranging from five to 50 ng/mL), 24 h after TNF-α treatment (n = 3). (H) and (I) Relative LDH release and representative images of the morphology of L-02 cells treated with TP (25 nmol/L) and TNF-α (50 ng/mL), 24 h after TNF-α treatment. Scale bar = 100 μm. (J)–(M) Representative Western blots and relative intensity of protein bands of cleaved caspase-3, cleaved caspase-8, and cleaved PARP from cells treated with TP (25 nmol/L) and TNF-α (50 ng/mL), 24 h after TNF-α application, with GAPDH as the loading control (n = 5–6). Results were expressed as mean ± SEM and statistical analysis was performed using One-way ANOVA or Two-way ANOVA following by Tukey's multiple comparison test. *, #P < 0.05, **, ##P < 0.01, ***, ###P < 0.001.
Figure 4.

Possible role of NF-κB and NF-κB-mediated pro-survival signals in TP/LPS-induced hepatotoxicity. (A) Experimental design to detect the time-dependent changes of NF-κB and its related pro-survival factors. (B)–(G) Relative mRNA levels of NF-κB target genes, including Ciap1, Ciap2, FlipL, Xiap, Tnfaip3, and Nfkbia, were detected by qPCR with tubulin as the internal control (n = 6). (H)–(L) Representative Western blots and relative intensity of protein bands of FLIPL, CIAP1, XIAP, and IκB-α after the treatment of TP and LPS with tubulin as the loading control (n = 4–6). (M) Representative photomicrographs of liver sections by IHC for P65 1 h after LPS application (200 × ). Scale bar = 50 μm. Results were expressed as mean ± SEM and statistical analysis was performed using Two-way ANOVA following by Tukey's multiple comparison test. *, #, $P<0.05, **, ##, $$P<0.01, ***, ###, $$$P<0.001; ns, no statistical difference.
Figure 7.

Overexpression of FLIPL protected mice from TP/LPS-induced hepatotoxicity. (A) Experimental procedure for the protective effects of FLIPL on TP/LPS-induced hepatotoxicity. (B) and (C) Changes of serum ALT and AST in mice treated with TP and LPS after the transfection of AAV-NC and AAV-FLIPL (n = 6). (D)–(H) Representative Western blots and relative intensity of the protein bands of RIPK1, P-MLKL, MLKL, and cleaved caspase-8 with GAPDH as the loading control (n = 4–6). (I) Pictures of liver sections analyzed by H&E staining (top panels), IHC staining for cleaved caspase-3 (middle panels) and MLKL (bottom panels) after the transfection of AAV-NC and AAV-FLIPL (200 ×). Scale bar = 50 μm. Results were expressed as mean ± SEM and statistical analysis was performed using One-way ANOVA following by Tukey's multiple comparison test. *, #P<0.05, **, ##P<0.01, ***, ###P<0.001; ns, no statistical difference.
2.3. Blood biochemistry analysis
Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), albumin (ALB), and total protein were detected using kits from Weiteman Biotech (Nanjing, China). Serum total bile acid (TBA) was measured using the kit from Jiancheng Bioengineering Institute (Nanjing, China). Serum glucose was measured with the kit from Rongsheng Biotech (Shanghai, China). All kits were used according to the manufacturer's instructions.
2.4. Histopathological examination
Fresh liver sections from mouse liver were washed with 4 °C PBS and fixed in 10% formaldehyde. Hematoxylin and eosin (H&E), immunohistochemistry (IHC) and periodic acid-Schiff (PAS) staining were performed by standard procedures.
2.5. Cell culture and cell viability assay
Human hepatic cell line L-02 was obtained from China Cell Culture Center (Shanghai, China). Cells were cultured in DMEM media with 10% fetal bovine serum (FBS) provided by Gibco (Grand Island, NY, USA) and maintained at 37 °C in a humidified atmosphere with 5% CO2. TP was added 1 h before human recombinant TNF-α in all in vitro experiments.
Cell viability was detected with the MTT assay in the 96-well plates. In brief, exponentially growing cells were plated at the density of 4 × 103 in each well. Twelve hours after seeding, old media was replaced with fresh DMEM media without FBS and different concentrations of TP as well as human recombinant TNF-α were added. Cell viability was detected at the indicated time.
Lactate dehydrogenase (LDH) leakage was detected according to the instructions of CytoTox 96® Non-Radioactive Cytotoxicity Assay Kit from Promega operation (Madison, WI, USA) and calculated using Eq. (1):
| (1) |
2.6. Cell transfection
For gene silencing or overexpression of human FLIPS (short form of FLIP) (NM_001127184.3), L-02 cells cultured in 24-well plates were transfected with scrambled control siRNA, FLIPS siRNA, pEX-1 vector, or pEX-1 vector carrying FLIPS sequence obtained from Genepharma (Shanghai, China). SiRNA or plasmid were transfected into the cells using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocols. The siRNA sequences are listed in Table 1. After 24 h transfection, fresh DMEM medium was added to the plates to replace the transfection medium for another 24 h before the treatment of TP (25 nmol/L) and TNF-α (50 ng/mL). Cell supernatant and lysate for LDH detection and Western Blot analysis were collected 24 h after TNF-α application.
Table 1.
Sequence of target gene siRNA.
| Gene | Gene sequence (5′–3′) |
|---|---|
| Negative control sense | UUCUCCGAACGUGUCACGUTT |
| Negative control antiense | ACGUGACACGUUCGGAGAATT |
| FLIPS sense | GGAGAAACUAAAUCUGGUUTT |
| FLIPS antisense | AACCAGAUUUAGUUUCUCCTT |
2.7. Immunocytochemistry
L-02 cells were plated in 24-well plates containing coverslips with the density of 2 × 104 in each well. Thirty minutes after pretreatment with TP and TNF-α, cells were fixed with 10% formaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 15 min, and blocked with 5% BSA. The fixed cells were then incubated with P65 primary antibody overnight at 4 °C followed by the incubation with Alexa Fluor 488 secondary antibody at room temperature for 1 h. The nucleus was labeled with DAPI (Beyotime Biotechnology, Shanghai, China) for 15 min and fluorescence images were obtained with FV-1000 (Olympus, TKY, Japan).
2.8. RNA extraction and qPCR
Total RNA was extracted from mouse liver with Trizol reagent. A total of 1 μg RNA for each sample was reversed to cDNA with Reverse Transcription Kit after the quantification of RNA concentration with Nanodrop2000 (Thermo Fisher Scientific, Waltham, MA, USA). The experiment was carried out on Stepone Plus using SYBR Green Master Mix and normalized with tubulin. The primer used for qPCR is listed in Table 2 and the relative mRNA expression was calculated using ΔΔCT method.
Table 2.
The primer sequences used for qPCR assay in mice.
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| Tubulin | GAGGAGATGACTCCTTCAACACC | TGATGAGCTGCTCAGGGTGGAA |
| Ciap1 | GATACGGATGAAGGGTCAGGAG | GGGTCAGCATTTTCTTCTCCTGG |
| Ciap2 | GGACATTAGGAGTCTTCCCACAG | GAACACGATGGATACCTCTCGG |
| Nfkbia | TGAAGGACGAGGAGTACGAGC | TTCGTGGATGATTGCCAAGTG |
| Tnfaip3 | AGCAAGTGCAGGAAAGCTGGCT | GCTTTCGCAGAGGCAGTAACAG |
| FlipL | GCTCTACAGAGTGAGGCGGTTT | CACCAATCTCCATCAGCAGGAC |
| Xiap | GGCAGAATATGAAGCACGGATCG | CACTTGGCTTCCAATCCGTGAG |
2.9. Western blot analysis
Total protein was extracted from mouse liver stored at −80 °C or cell samples using RIPA lysis buffer (Beyotime Biotechnology). Protein concentration was quantified by a BCA kit (Beyotime Biotechnology) and then protein was mixed with 4 × loading buffer from Bio-Rad laboratories (Hercules, CA, USA). Proteins were separated by SDS-PAGE with gels ranging from 8% to 12% and then transferred onto PVDF membranes. After blocking with 5% BSA at room temperature for 1 h, the membranes were incubated with primary antibodies overnight at 4 °C. The membranes were then incubated with second antibodies for 1 h and visualized at Tanon 4200 Chemiluminescent Imaging System (Shanghai, China) using ECL detection kit (Millipore, Danvers, MA, USA). The relative protein expression was normalized with Tubulin or GAPDH and analyzed with Image J (NIH, Bethesda, MA, USA).
2.10. Statistical analysis
Data were analyzed using GraphPad Prism 6 (La Jolla, CA, USA) and presented in the form of mean ± SEM. One-way analysis of variance (ANOVA) and two-way ANOVA followed by Tukey's multiple comparison test were performed to analyze the differences between groups. P-values <0.05 were considered to be statistically significant.
3. Results
3.1. Etanercept pretreatment protected mice from TP/LPS-induced hepatotoxicity
Previously, our results showed that apoptosis inhibitor, Z-VAD-FMK, together with necroptosis inhibitor, Nec-1, protected mice from TP/LPS induced hepatotoxicity, uncovering the essential role of apoptosis and necroptosis in TP/LPS-induced liver toxicity8. It is well established that the binding of TNF-α to tumor necrosis factor receptor type 1 (TNF-R1) on the surface of cell membrane leads to programmed cell death, including apoptosis and necroptosis, via promoting the formation of TNF-R1-bound complex (complex-I)32,33.
We supposed that LPS participated in TP/LPS-induced hepatotoxicity through stimulation of immune cells producing TNF-α. To test this, etanercept, a soluble decoy to the TNF-α receptor, was injected before LPS treatment to inhibit the binding of TNF-α to TNF-R1. Serum TNF-α levels were measured 1, 3, and 8 h post-LPS (Supporting Information Fig. S1A). Fig. S1B shows that LPS and TP/LPS-treated groups had significant increases in serum TNF-α, 1 h after LPS injection, whereas etanercept pretreatment decreased TP/LPS-induced TNF-α back to the normal, revealing efficient blockade of the binding of TNF-α to TNF-R1 in vivo.
Blood analysis was conducted 8 h after LPS injection to confirm the effects of etanercept on TP/LPS-induced hepatotoxicity. Consistent with our previous research, TP or LPS alone had little effect on serum ALT and AST, but TP/LPS co-treatment significantly increased ALT and AST levels (Fig. 1 B and C). Etanercept pretreatment inhibited TP/LPS-induced up-regulation of transaminases and protected mice from TP/LPS-induced hepatic cell apoptosis and necrosis. This was clear in the massive nuclear fragmentation and condensation of both cytoplasm and nucleus (see black arrows in Fig. 1B and C and H&E staining in Fig. 1H). TP/LPS co-treatment also up-regulated serum TBA level, an effect counteracted by Etanercept (Fig. 1D). Also, TP/LPS led to a dramatic decrease of serum glucose and almost completely depleted glycogen storage in the liver. Etanercept pretreatment partly enhanced serum glucose level and restored glycogen storage compared with TP/LPS co-treatment (Fig. 1E and PAS staining in Fig. 1H). Furthermore, ALB and total protein were inhibited by TP/LPS co-treatment which was alleviated by etanercept pretreatment (Fig. 1F and G). TP/LPS co-treatment also promoted the recruitment of neutrophils and increased serum TNF-α 8 h after LPS injection. Both effects were alleviated by etanercept (Fig. S1B and MPO staining in Fig. 1H).
To determine whether the protective effects of etanercept were related to cell death inhibition, the expressions of apoptosis and necroptosis related proteins were detected. The results (Fig. 2A, C–G) indicated that TP/LPS up-regulated cleaved caspase-8, cleaved caspase-3, and cleaved PARP, but had little effect on the expression of BAX and BCL-2. Likewise, etanercept pretreatment almost completely inhibited the up-regulation of cleaved caspase-8, cleaved caspase-3, and cleaved PARP. Moreover, etanercept weakened the up-regulation of the proteins related to necroptosis (RIPK1, MLKL, and P-MLKL [Ser345]) induced by TP/LPS co-treatment (Fig. 2B, H–J). The effect of etanercept on apoptosis and necroptosis was additionally confirmed by IHC staining of cleaved caspase-3 and MLKL, as etanercept decreased the positive areas of cleaved caspase-3 and MLKL induced by TP/LPS co-treatment (Fig. 2K).
Figure 2.

Pretreatment with etanercept protected mice from TP/LPS-induced apoptosis and necroptosis. (A) and (B) Representative protein bands related to apoptosis and necroptosis after etanercept treatment, with tubulin as the loading control (n = 3–6). (C)–(J) Densitometric analyses of the bands in Fig. 2A and B. (K) Pictures of liver tissue sections analyzed by IHC for cleaved caspase-3 (top panels) and MLKL (bottom panels) (200 × ). Scale bar = 50 μm. Results were expressed as mean ± SEM and statistical analysis was performed using One-way ANOVA following by Tukey's multiple comparison test. *, #P < 0.05, **, ##P < 0.01, ***,###P < 0.001.
These results show that anti-TNF-α antibody, etanercept, efficiently protected the mice from TP/LPS-induced bile acid and glucose metabolic disorder, inflammatory reactions, apoptosis, and necroptosis. LPS might participate in TP/LPS-induced apoptosis and necroptosis through promoting the release of TNF-α.
3.2. Pretreatment with TP increased the sensitivity of mice and hepatocytes upon TNF-α stimulation
The function of LPS-induced TNF-α was demonstrated by the injection of recombinant TNF-α in vivo (Fig. 3A). In normal mice, TNF-α (10 μg/kg) had little effect on serum ALT and AST levels vs. control, whereas TNF-α increased serum ALT and AST levels in mice pretreated with TP (Fig. 3B and C). H&E staining revealed that mice treated with TP or TNF-α alone showed little morphological abnormalities. However, TP/TNF-α co-treatment increased hepatic cellular injury, reflected by nuclear fragmentation and condensation, loss of cytoplasm, and increased inflammatory reactions (Fig. 3D). These results reveal the hypersensitivity of mice treated with TP upon TNF-α stimulation.
In vitro experiments were also carried out to discover the effects of TNF-α on TP-treated hepatocytes. In most circumstances, the cellular effect of TNF-α was pro-survival not pro-death. A 1 h pretreatment of L-02 cells with TP (25 nmol/L) followed by the application of TNF-α (50 ng/mL) for 24 h dramatically decreased cell viability. TP or TNF-α alone had insignificant effects on cell viability (Fig. 3E). Results also show that the TP/TNF-α-induced hepatic cell death was TP and TNF-α-dependent (Fig. 3F and G). The cytotoxicity of TP/TNF-α co-treatment was additionally confirmed by LDH release and histopathological observation. TP or TNF-α alone slightly increased LDH levels, but TP/TNF-α co-treatment significantly increased the release of LDH (Fig. 3H). Moreover, TP/TNF-α co-treatment impaired cellular morphology expressed by the shrinkage of hepatocytes, yet TP or TNF-α alone did not cause this kind of injury (Fig. 3I). Protein measurements revealed the apoptosis of hepatocytes induced by TP/TNF-α, reflected by an increase in the expression of cleaved caspase-3, cleaved caspase-8, and cleaved PARP (Fig. 3J–M).
In summary, TP treatment increased the sensitivity of mice and hepatocytes upon TNF-α stimulation. LPS participated in TP/LPS-induced hepatotoxicity through the release of TNF-α.
3.3. TP pretreatment counteracted NF-κB dependent pro-survival signals induced by LPS and TNF-α
As a vital pro-inflammatory factor in immune system, TNF-α participates in tissue homeostasis by regulating cytokine production, cell survival, and cell death34. However, under most circumstances, TNF-α is not able to promote cell death unless the checkpoints of TNF-α related pathways are disrupted. NF-κB and the relevant pro-survival proteins are important components of these pathways33, 34, 35. We suspected that the inhibition of NF-κB dependent pro-survival signals in hepatocytes by TP might be responsible for the sensitivity of hepatocytes following TNF-α stimulation. It has been reported that LPS activated NF-κB 1 h after LPS injection and this effect was gradually weakened with the passage of time31,36. Thus, a period of 1 h after LPS injection was suitable for the detection of NF-κB activity. Time-dependent changes in NF-κB induced by TP and LPS were detected 1, 3, and 8 h after LPS treatment (Fig. 4A). Two NF-κB target genes, Nfkbia encoding IκB-α and Tnfaip3 used to reflect the transcriptional activity of NF-κB, were increased 1 h after LPS injection and gradually returned to normal three and 8 h later (Fig. 4F and G). However, TP pretreatment, in TP/LPS co-treatment group, weakened NF-κB activity in comparison with LPS treatment alone. Western blots revealed that IκB-α (NF-κB inhibitor alpha) degraded 1 h after LPS administration and began to restore after 3 h, while TP pretreatment inhibited LPS induced IκB-α degradation 1 h after LPS injection (Fig. 4H and L). The effects of TP on LPS induced NF-κB activation were also confirmed by IHC staining of NF-κB P65. LPS alone promoted the translocation of P65 from cytoplasm to nucleus, especially in hepatocytes, while TP treatment decreased this effect (Fig. 4M). Thus, TP pretreatment inhibited LPS induced NF-κB activation in mice.
Subsequently, NF-κB-dependent pro-survival genes and proteins were detected. The mRNA levels of the pro-survival genes, including Xiap, FlipL, Ciap1, and Ciap2, were significantly elevated by LPS, especially 1 h after LPS administration and subsequently fell back to normal. However, TP treatment repressed the transcription of those genes being induced by LPS (compared TP/LPS- vs. LPS-treated groups, Fig. 4B–E). Additionally, TP alone was able to inhibit the mRNA levels of Ciap1, Ciap2, Xiap, and FlipL (Fig. 4B–E). The effects of TP on the pro-survival proteins were also detected. In contrast to the gene expression, LPS alone had little effect on CIAP1 and XIAP protein expressions (Fig. 4H, J and K). Previous work has revealed that CIAP1, CIAP2, and XIAP cooperated with each other to maintain embryonic development and protected cells from TNF-α-induced cell death37. Although TP significantly suppressed the expression of XIAP, it had little effect on CIAP1 expression. However, suppression of the expression of CIAP1 sensitized cells to TNF-α while repression of XIAP or CIAP2 had no effect38. Thus, we excluded the possibility of XIAP inhibition on TP/TNF-α-induced liver hypersensitivity. Likewise, Fig. 4H and I shows that TP treatment not only blocked the up-regulation of FLIPL 1 h after the injection of LPS, but also inhibited the expression of FLIPL 8 h after LPS administration. Published work revealed that FLIPL induced by TNF-α was indispensable for cell survival and cells deficient of FLIPL were prone to death in the presence of TNF-α29. We speculated that inhibition of NF-κB-mediated FLIPL expression by TP might be responsible for hypersensitivity of TP-treated mice upon LPS stimulation.
The effects of TP on NF-κB activity and the expression of FLIP were also confirmed in vitro. L-02 cells treated with TNF-α showed NF-κB P65 aggregation in nucleus (especially in partially magnified images), while TP pretreatment suppressed this process (Fig. 5A). However, in contrast to the results in mice, TP increased the expression of FLIPL in L-02 cells and TNF-α seemed to have no effect on it, indicated that FLIPL may not participate in cytoprotective effect on L-02 cells against TNF-α-induced cell death (Fig. 5B and C). Nevertheless, TNF-α alone increased the expression of the FLIPS, which was not present in mouse, implied that FLIPS instead of FLIPL may protect L-02 cells against TNF-α. We also discovered that TP pre-treatment counteracted the up-regulation of FLIPS induced by TNF-α and TP treatment reduced the FLIPS protein level, which was consistent with the effects of TP on the expression of FLIPL in vivo (Fig. 5B and D).
Figure 5.

Possible role of NF-κB and NF-κB-mediated pro-survival signals in TP/TNF-α-induced hepatic cell apoptosis. (A) L-02 cells were treated with TP (25 nmol/L) plus TNF-α (50 ng/mL) and were collected 30 min after TNF-α treatment for immunocytochemistry. Scale bar = 50 μm. (B)–(D) L-02 cells were treated with TP (25 nmol/L) plus TNF-α (50 ng/mL) and were collected 24 h after TNF-α application. Representative Western blots and relative intensity of protein bands of FLIPL and FLIPS with GAPDH as the loading control (n = 3–4). Results were expressed as mean ± SEM and statistical analysis was performed using One-way ANOVA following by Tukey's multiple comparison test. *, #P<0.05, **, ##P<0.01, ***, ###P<0.001; ns, no statistical difference.
In this part, we found that TP inhibited NF-κB-dependent transcriptional activity as well as the pro-survival protein FLIPL in vivo and FLIPS in vitro, which were induced by LPS or TNF-α. However, whether inhibition of NF-κB dependent FLIP expression involved in TP/LPS-induced hepatotoxicity or TP/TNF-α-induced hepatic cell apoptosis remained elusive.
3.4. Overexpression of FLIPs alleviated TP/TNF-α-induced apoptosis in hepatocytes
Next, the function of FLIPS on L-02 cells was studied by FLIPS knockdown. L-02 cells were transfected with FLIPS siRNA or negative control siRNA and the efficiency of siRNA was verified in Supporting Information Figs. S2A and S2B. We found that transfection with FLIPS siRNA promoted hepatic cell apoptosis in the presence of TNF-α, revealing the role of FLIPS in protecting cells against TNF-α-induced cell death (Fig. 6A, C, and D). However, the results of LDH release and the protein level of cleaved PARP indicated that there was no difference between cells treated with siRNA-NC plus TP/TNF-α and siRNA-FLIPS plus TP/TNF-α. We propose the following reason behind these results: although siRNA-FLIPS was enough to inhibit the expression of FLIPS, cells transfected with siRNA-FLIPS plus TP/TNF-α co-treatment were not able to further inhibit the expression of FLIPS, compared with siRNA-NC plus TP/TNF-α co-treatment (Fig. 6E and F).
Figure 6.

Protective role of FLIPS in TP/TNF-α-induced hepatic cell apoptosis. (A) and (B) Relative LDH release in L-02 cells transfected with siRNA or plasmid before the treatment of TP (25 nmol/L) and TNF-α (50 ng/mL) (n = 4). (C)–(F) Representative Western blots and relative intensity of protein bands of FLIPS and cleaved PARP in L-02 cells transfected with siRNA-NC or siRNA-FLIPS before the treatment of TP and TNF-α with GAPDH as the loading control (n = 4–5). (G)–(I) Representative Western blots and relative intensity of protein bands of FLIPS and cleaved PARP in L-02 cells transfected with pEX-1-NC or pEX-1-FLIPS before the treatment of TP and TNF-α with GAPDH as the loading control (n = 3). Results were expressed as mean ± SEM and statistical analysis was performed using One-way ANOVA following by Tukey's multiple comparison test. *P < 0.05, **P < 0.01, ***P < 0.001; ns, no statistical difference.
The role of FLIPS in TP/TNF-α-induced hepatic cell apoptosis was further clarified by FLIPS overexpression and the efficiency of FLIPS plasmid was confirmed by Western blot (Figs. S2C and S2D). As expected, cells transfected with FLIPS plasmid expressed high level of FLIPS even after TP and TNF-α treatment (Fig. 6I). Furthermore, compared with cells transfected with NC, hepatocytes transfected with FLIPS protected cells from TP/TNF-α-induced apoptosis and decreased the LDH release induced by TP/TNF-α (Fig. 6B, G, and H). Thus, the inhibitory effect of TP on NF-κB-dependent FLIPS up-regulation sensitized hepatocytes upon TNF-α exposure. Overexpression of FLIPS protected hepatocytes from TP/TNF-α-induced apoptosis.
3.5. Overexpression of FLIPL protected the mice from TP/LPS induced hepatotoxicity
Lastly, the protective effect of FLIPL on TP/LPS-induced hepatotoxicity was confirmed in vivo. We generated AAV vectors carrying FLIPL plasmid to overexpress FLIPL in hepatocytes (Fig. 7A). Fluorescence microscopy revealed GFP expression in mice injected with AAV-NC or AAV-FLIPL (Fig. S2E). Measurement of serum ALT and AST revealed that overexpression of FLIPL partly alleviated the liver injury induced by TP/LPS, as shown in comparison between AAV-FLIPL/TP/LPS and AAV-NC/TP/LPS groups (Fig. 7B and C). This was supported by H&E staining, where the apoptosis and necrosis areas were attenuated by FLIPL overexpression (Fig. 7I). To identify the effect of FLIPL overexpression on TP/LPS-induced apoptosis, we detected the protein level of cleaved caspase-8 and positive areas of cleaved caspase-3 in liver tissues (Fig. 7D, H, and I). FLIPL overexpression inhibited TP/LPS-induced caspase-8 cleavage and attenuated the positive areas of cleaved caspase-3, implying that the protective effect of FLIPL overexpression was partly relied on apoptosis inhibition. In addition, FLIPL overexpression inhibited necroptosis related proteins, including RIPK1, MLKL, and P-MLKL, activated by TP/LPS co-treatment (Fig. 7D–G). This was supported by IHC staining of MLKL (Fig. 7I). Thus, FLIPL protected mice from TP/LPS-induced hepatotoxicity through the inhibition of both apoptosis and necroptosis in vivo.
4. Discussion
Drugs containing TP had been reported to induce severe liver injury according to the data from the National Center for Adverse Drug Reaction Monitoring of China. However, mice or rats treated with TP even at high doses ranging from 400 to 1000 μg/kg did not completely express the toxic reactions which were observed clinically1,3,4,6. We proposed that animals maintained in barrier systems cannot fully imitate the conditions faced by the patients and that TP treatment might disrupt immune homeostasis, leading to the liver hypersensitivity upon the stimulation of pathogens. Thus, we chose LPS as the stimulant and found that TP/LPS co-treatment caused serious liver injury together with massive hepatic cell death8. However, the role of LPS was unclear in TP/LPS-induced hepatotoxicity and liver hypersensitivity. TNF-α, the pro-inflammatory factor produced by LPS-activated immune cells, is the main cytokine for apoptosis and necroptosis activation. We proposed that decreased TNF-α level facilitated by LPS might counteract TP/LPS-induced cell death and found that etanercept abolished the toxic reactions induced by TP/LPS, and especially alleviated TP/LPS-induced apoptosis and necroptosis (Figure 1, Figure 2). Although etanercept greatly protected mice from TP/LPS-induced hepatotoxicity, it did not completely inhibit TP/LPS-induced transaminase up-regulation. Thus, it is rational to speculate that other cytokines might also involve in TP/LPS-induced hepatotoxicity. For example, it has been reported that another two TNF family members, FasL and TRAIL expressed on activated immune cells, can participate in the development of liver disease and promote cell death in the condition of pro-survival function deficiency39,40. Thus, it is necessary to learn the role of other cytokines involved in the mechanism of hypersensitivity.
In most cases, cell death of parenchymal cells (i.e., hepatocytes) vs. non-parenchymal cells (including Kupffer cells, endothelial cells, lymphocytes, and stellate cells) is responsible for the progression of liver diseases and drug-induced liver injury41, 42, 43. Therefore, the human hepatic cell line L-02 was used in the in vitro experiments. It is known that monocytes and macrophages express the highest levels of TLR4 and facilitate the production of TNF-α in response to LPS44,45. Although low levels of TLR4 are expressed on hepatocytes, the expression of TLR4 on hepatocytes is mostly related to hepatic cell metabolism rather than LPS-induced cell death46,47. However, TNF-R1, expressed on almost every mammalian cell type, including hepatocytes, regulates a number of biological responses ranging from NF-κB activation to cell death when it is being activated by TNF-α. Thus, TNF-α was used as a stimulant rather than LPS in vitro. We found that TP-treatment increased the sensitivity of hepatocytes upon TNF-α stimulation and promoted apoptosis of hepatocytes (Fig. 3E–M). However, only Z-VAD-FMK protected hepatocytes from TP/TNF-α-induced cell death and decreased LDH release, while Nec-1 had little protective effect (Supporting Information Figs. S3A and S3B). This suggested that necroptosis may not participate in TP/TNF-α induced cell death in vitro. These results were inconsistent with our in vivo results in which both Z-VAD-FMK and Nec-1 alleviated TP/LPS-induced hepatotoxicity8. Some research groups reported that RIPK3 is only expressed in liver non-parenchymal cells, but not in hepatocytes, while others reported the RIPK3-independent necroptosis during hepatitis and RIPK3-dependent necroptosis in hepatocellular carcinoma cell line48, 49, 50, 51. Differences between our in vivo and in vitro results may be due to the RIPK3 deficiency in hepatocytes and the in vivo activation of both apoptosis and necroptosis might not be restricted to hepatocytes. Thus, only apoptotic proteins were detected in vitro (Fig. 3J–M). Additionally, mouse recombinant TNF-α was used in our in vivo experiments to replace LPS as the stimulant, confirming the role of LPS in TP/LPS-induced hepatotoxicity. These results also support our hypothesis that TP-treated mice were sensitized to TNF-α (Fig. 3A–D).
Subsequent experiments were designed to explain why TP-treatment sensitized mice or hepatocytes to TNF-α. In vitro results revealed that TNF-α did not promote hepatic cell death, which was consistent with the published research26,52. It was reported that the binding of TNF-α to TNF-R1 led to the receptor trimerization and the formation of complex-I33. Complex I, closely associated with plasma membrane, is known to additionally recruit caspase-8 to complex IIa (containing TNF receptor-associated death domain (TRADD), FADD, caspase-8, and FLIP) and complex IIb (containing RIPK1, FADD, caspase-8, and RIPK3) in the cytoplasm, which potentiate apoptosis and necroptosis33. There are several essential checkpoints controlling the formation of complex II. The ubiquitin-dependent checkpoint hinders RIPK1 to complex II and activates kinases upstream of NF-κB. The kinase-dependent checkpoint is mediated mainly by IKKα, IKKβ, and MAPK-activated protein kinase 2. Finally, the transcription-dependent and caspase-dependent checkpoint is dependent on the NF-κB-mediated pro-survival function and the formation of FLIP–caspase-8 heterodimer34. Among these, transcription-dependent mechanisms have been extensively studied. The inhibition of NF-κB by TP, especially in immune cells, is closely related to its pharmacodynamic effects53, 54, 55. Our results indicated that TP inhibited LPS induced NF-κB activation in vivo together with NF-κB-dependent pro-survival genes. In particular, NF-κB-mediated FLIPL expression was inhibited by TP (Fig. 4). In vitro experiments demonstrated that NF-κB and its related pro-survival protein, FLIPS, which was especially expressed in human cells, were inhibited by TP in hepatocytes (Fig. 5). Thus, it was rational to speculate that the deficiency of NF-κB-mediated pro-survival function in TP-treated hepatocytes might be the reason for its sensitization.
It has been reported that three isoforms of FLIP, have been identified: FLIPL, FLIPS, and FLIPR56. As the structural analogue of caspase-8, all isoforms of FLIP combine with caspase-8 to form FLIP-caspase-8 complex and inhibit the formation of caspase-8 homodimer, which is essential for external apoptosis activation57. Additionally, FLIPL–caspase-8 complex functions to inhibit necroptosis via RIPK1 and RIPK3 proteolytic cleavage32. Importantly, FLIPL is expressed in human and mice, while FLIPS is expressed in human and FLIPR is specially expressed in some specific cell lines57. We found that LPS administration was able to induce the expression of FLIPL in vivo, while TNF-α induced the specific up-regulation of FLIPS without affecting the physiological levels of FLIPL in L-02 cells (Figure 4, Figure 5B). We supposed that FLIPS rather than FLIPL might be the regulator of caspase-8 processing for TNF-α-mediated sensitization in L-02 cells, which was similar to HeLa cells58. The results in Fig. 6 uncovered that knockdown of FLIPS sensitized L-02 cells to TNF-α by activating apoptosis and overexpression of FLIPS rescued TP/TNF-α-induced apoptosis, implying the cytoprotective function of FLIPS upon TP/TNF-α application. It has been reported that deletion of FLIPL in intestinal epithelial cells or hepatocytes resulted in perinatal lethality as a consequence of apoptosis and programmed necrosis while knockout FLIPL in T lymphocytes promoted apoptosis and necroptosis in the presence of anti-CD359,60. We supposed that apoptosis and necroptosis activation in TP/LPS-induced hepatotoxicity might be the consequence of FLIPL inhibition by TP and AAV carrying FLIPL plasmid was injected in vivo to prove the function of FLIPL in apoptosis and necroptosis regulation. The results in Fig. 7 reflected that overexpression of FLIPL alleviated TP/LPS-induced hepatotoxicity as well as apoptosis and necroptosis.
Although the results in Figure 4, Figure 5 show that TP-treatment inhibited LPS induced NF-κB activation in whole liver homogenate with mixed cell types and TNF-α induced NF-κB activation in L-02 cells, we were surprised that TP treatment enhanced serum TNF-α level one and 8 h after LPS administration, compared between TP/LPS and LPS treatments (Fig. S1B). It is widely accepted that cell necrosis is able to destroy cellular integrity and triggers a massive inflammatory response, which can be the reason for an increase in serum TNF-α level 8 h after LPS administration. However, we found that TP/LPS co-treatment did not induce any obvious liver injury 1 h after LPS injection by monitoring the time-dependent changes of serum ALT and AST, which excluded the possibility of necrosis in increasing serum TNF-α level in TP/LPS co-treatment group (Figs. S1C and S1D). One possible reason for this phenomenon might associate with the effect of TP on the number of hepatic macrophages and their phagocytic function. Our group showed that TP treatment inhibited phagocytic function of macrophages, resulting in the up-regulation of serum LPS and increased the number of hepatic macrophages, which was mainly responsible for the production of inflammatory factors upon LPS stimulation61. Moreover, we cannot rule out the possibility that TP has different effects on NF-κB activation between macrophages and hepatocytes. Thus, reasons for massive TNF-α production by TP-treated immune cells upon LPS stimulation needs further explanation.
In this article, we demonstrated that the inhibition of FLIP in hepatocytes by TP is one reason for TNF-α/LPS induced hypersensitivity and the possible role of NF-κB in FLIP regulation. It has been postulated that the combination of LPS and certain drugs aggravate drug-induced adverse reactions62, 63, 64. For instance, the application of LPS aggravated the liver injury induced by amiodarone, trovafloxacin, sulindac, or ranitidine, while anti-TNF-α therapy restrained this liver damage43,65, 66, 67. However, none of these researchers focused on the function of TNF-α-related checkpoints in cell death or NF-κB-mediated transcriptional activity in their hypersensitivity models. Some of the drugs mentioned above, such as isoniazid and trovafloxacin, had been reported to inhibit RNA transcription, uncovering the possibility between their transcriptional inhibition (including NF-κB-mediated transcriptional inactivation) and hypersensitivity68,69. Nevertheless, whether there is an existence of any common pathways, such as transcriptional, translational, or NF-κB inactivation, for drug hypersensitivity or whether the drugs suppress NF-κB-mediated survival functions in hepatocytes, hypersensitivity upon LPS or TNF-α stimulation needs further investigation. In addition, the disruption of ubiquitin-dependent and kinases-dependent checkpoints lying in the upstream of NF-κB, can modify its pro-survival function. There is a great need to discover the actual target of TP in order to explain whether NF-κB inactivation is the secondary effect of TP on ubiquitination and kinase-mediated pathways or not. Overall, a deeper understanding of the checkpoints in TNF-α-induced cell death and their relationship to drug-induced adverse reactions is needed to provide a better theoretical basis for clinically safe medications.
5. Conclusions
This report showed that LPS participates in TP/LPS-induced hepatotoxicity through the induction of TNF-α. Mice or hepatic cells treated with TP were sensitized to TNF-α, while inactivation of NF-κB and NF-κB-mediated FLIP expression in hepatocytes may be responsible for this sensitization. Exogenous overexpression of FLIP protected mice or hepatocytes from TP/LPS-induced hepatotoxicity or TP/TNF-α-induced apoptosis. These results are critical in the search for the toxic targets of TP and they broaden our understanding of drug-induced hypersensitivity. A comprehensive mechanism for TP/LPS-induced hepatotoxicity is interpreted shown in Fig. 8.
Figure 8.

Schematic presentation indicated the suggested mechanisms by which TP/LPS-induced hepatotoxicity and the possible mechanistic intervention of etanercept, Z-VAD-FMK, Nec-1, as well as FLIP overexpression for liver protection in TP/LPS-induced liver injury. Under physiological conditions, the binding of LPS on the surface of TLR4 on immune cells triggers the production of TNF-α. On one hand, the attachment of TNF-α to TNF-R1 on hepatocytes facilitated the formation of cell death Complex IIa and Complex IIb, thus promoting TNF-α treated hepatic cell death. On the other hand, stimulation of TNF-R1 activated NF-κB and up-regulated the expression of pro-survival proteins (including FLIP) to inhibit cell death. Thus, TNF-α alone cannot induce hepatic cell death due to the balance between pro-survival and pro-death signals (indicated as black arrows). However, TP treatment inhibited the NF-κB-dependent transcriptional activity and the subsequent pro-survival protein FLIP in hepatocytes. TP-treated hepatocytes cannot counteract TNF-α induced cell death, ultimately promoting hepatic cell death in the presence of TNF-α (indicated as orange arrows). Z-VAD-FMK and Nec-1 inactivated apoptosis and necroptosis, thus alleviated TP/LPS-induced hepatotoxicity. Etanercept treatment inhibited the binding of TNF-α to TNF-R1 and protected mice from TP/LPS-induced hepatotoxicity. FLIP overexpression increased the formulation of FLIP–caspase-8 complex, ultimately inhibited TP/LPS-induced cell death and hepatotoxicity.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81973562, 81773995, 81773827, 81320108029, 81573690, 81573514, and 81673684), the National “Major Scientific and Technological Special Project for Significant New Drugs” project (2015ZX09501004-002-004, China), Specific Fund for Public Interest Research of Traditional Chinese Medicine, Ministry of Finance (201507004–002, China), and "Double First-Class” University project (CPU2018GY33, China).
Author contributions
Ziqiao Yuan, Zihang Yuan, and Luyong Zhang participated in the research design. Ziqiao Yuan, Zihang Yuan, Muhammad Hasnat, Haoran Zhang, and Peishi Liang carried out the experiments. Ziqiao Yuan, Peishi Liang and Lixin Sun performed the data analysis. Ziqiao Yuan and Zhenzhou Jiang wrote the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.02.009.
Contributor Information
Zhenzhou Jiang, Email: beaglejiang@cpu.edu.cn.
Luyong Zhang, Email: lyzhangchina@hotmail.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Li X.J., Jiang Z.Z., Zhang L.Y. Triptolide: progress on research in pharmacodynamics and toxicology. J Ethnopharmacol. 2014;155:67–79. doi: 10.1016/j.jep.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Zhao X., Liu X., Zhang P., Liu Y., Ran W., Cai Y., Wang J., Zhai Y., Wang G., Ding Y., Li Y. Injectable peptide hydrogel as intraperitoneal triptolide depot for the treatment of orthotopic hepatocellular carcinoma. Acta Pharm Sin B. 2019;9:1050–1060. doi: 10.1016/j.apsb.2019.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang J., Sun L., Wang L., Hassan H.M., Wang X., Hylemon P.B., Wang T., Zhou H., Zhang L., Jiang Z. Activation of Sirt1/FXR signaling pathway attenuates triptolide-induced hepatotoxicity in rats. Front Pharmacol. 2017;8:1–11. doi: 10.3389/fphar.2017.00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang J., Jiang Z., Ji J., Wang X., Wang T., Zhang Y., Tai T., Chen M., Sun L., Li X., Zhang L. Gene expression profiling and pathway analysis of hepatotoxicity induced by triptolide in Wistar rats. Food Chem Toxicol. 2013;58:495–505. doi: 10.1016/j.fct.2013.04.039. [DOI] [PubMed] [Google Scholar]
- 5.Hasnat M., Yuan Z., Naveed M., Khan A., Raza F., Xu D., Ullah A., Sun L., Zhang L., Jiang Z. Drp1-associated mitochondrial dysfunction and mitochondrial autophagy: a novel mechanism in triptolide-induced hepatotoxicity. Cell Biol Toxicol. 2019;35:267–280. doi: 10.1007/s10565-018-9447-8. [DOI] [PubMed] [Google Scholar]
- 6.Wang X., Jiang Z., Cao W., Yuan Z., Sun L., Zhang L. Th17/Treg imbalance in triptolide-induced liver injury. Fitoterapia. 2014;93:245–251. doi: 10.1016/j.fitote.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Tan Q.Y., Hu Q., Zhu S.N., Jia L.L., Xiao J., Su H.Z., Huang S.Y., Zhang J., Jin J. Licorice root extract and magnesium isoglycyrrhizinate protect against triptolide-induced hepatotoxicity via up-regulation of the Nrf2 pathway. Drug Deliv. 2018;25:1213–1223. doi: 10.1080/10717544.2018.1472676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan Z., Zhang H., Hasnat M., Ding J., Chen X., Liang P., Sun L., Zhang L., Jiang Z. A new perspective of triptolide-associated hepatotoxicity: liver hypersensitivity upon LPS stimulation. Toxicology. 2019;414:45–56. doi: 10.1016/j.tox.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 9.Thomson A.W., Knolle P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol. 2010;10:753–766. doi: 10.1038/nri2858. [DOI] [PubMed] [Google Scholar]
- 10.Crispe I.N. The liver as a lymphoid organ. Annu Rev Immunol. 2009;27:147–163. doi: 10.1146/annurev.immunol.021908.132629. [DOI] [PubMed] [Google Scholar]
- 11.Mencin A., Kluwe J., Schwabe R.F. Toll-like receptors as targets in chronic liver diseases. Gut. 2009;58:704–720. doi: 10.1136/gut.2008.156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao E., Ilyas G., Cingolani F., Choi J.H., Ravenelle F., Tanaka K.E., Czaja M.J. Pentamidine blocks hepatotoxic injury in mice. Hepatology. 2017;66:922–935. doi: 10.1002/hep.29244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olleros M.L., Vesin D., Fotio A.L., Santiago-Raber M.L., Tauzin S., Szymkowski D.E., Garcia I. Soluble TNF, but not membrane TNF, is critical in LPS-induced hepatitis. J Hepatol. 2010;53:1059–1068. doi: 10.1016/j.jhep.2010.05.029. [DOI] [PubMed] [Google Scholar]
- 14.Liedtke C., Trautwein C. The role of JNK2 in toxic liver injury. J Hepatol. 2006;45:762–764. doi: 10.1016/j.jhep.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J., Huang N., Guo Y., Cui S., Ge C., He Q., Pan X., Wang G., Wang H., Hao H. Combined obeticholic acid and apoptosis inhibitor treatment alleviates liver fibrosis. Acta Pharm Sin B. 2019;9:526–536. doi: 10.1016/j.apsb.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim A.H.., Song W.Y. TNF-alpha-mediated apoptosis in chondrocytes sensitized by MG132 or actinomycin D. Biochem Biophys Res Commun. 2002;295:937–944. doi: 10.1016/s0006-291x(02)00789-1. [DOI] [PubMed] [Google Scholar]
- 17.Karahashi H., Amano F. Lipopolysaccharide (LPS)-induced cell death of C3H mouse peritoneal macrophages in the presence of cycloheximide: different susceptibilities of C3H/HeN and C3H/HeJ mice macrophages. J Endotoxin Res. 2000;6:33–39. doi: 10.1177/09680519000060010501. [DOI] [PubMed] [Google Scholar]
- 18.Luo P., Zhao Y., Li D., Chen T., Li S., Chao X., Liu W., Zhang L., Qu Y., Jiang X., Lu G., Poon W., Fei Z. Protective effect of Homer 1a on tumor necrosis factor-alpha with cycloheximide-induced apoptosis is mediated by mitogen-activated protein kinase pathways. Apoptosis. 2012;17:975–988. doi: 10.1007/s10495-012-0736-z. [DOI] [PubMed] [Google Scholar]
- 19.Wang L., Du F., Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 20.Baldwin A.S., Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 21.Oeckinghaus A., Hayden M.S., Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 22.Micheau O., Lens S., Gaide O., Alevizopoulos K., Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21:5299–5305. doi: 10.1128/MCB.21.16.5299-5305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karin M., Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka M., Fuentes M.E., Yamaguchi K., Durnin M.H., Dalrymple S.A., Hardy K.L., Goeddel D.V. Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity. 1999;10:421–429. doi: 10.1016/s1074-7613(00)80042-4. [DOI] [PubMed] [Google Scholar]
- 25.Rudolph D., Yeh W.C., Wakeham A., Rudolph B., Nallainathan D., Potter J., Elia A.J., Mak T.W. Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev. 2000;14:854–862. [PMC free article] [PubMed] [Google Scholar]
- 26.Geisler F., Algul H., Paxian S., Schmid R.M. Genetic inactivation of RelA/p65 sensitizes adult mouse hepatocytes to TNF-induced apoptosis in vivo and in vitro. Gastroenterology. 2007;132:2489–2503. doi: 10.1053/j.gastro.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 27.Schattenberg J.M., Zimmermann T., Worns M., Sprinzl M.F., Kreft A., Kohl T., Nagel M., Siebler J., Schulze Bergkamen H., He Y.W., Galle P.R., Schuchmann M. Ablation of c-FLIP in hepatocytes enhances death-receptor mediated apoptosis and toxic liver injury in vivo. J Hepatol. 2011;55:1272–1280. doi: 10.1016/j.jhep.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Hughes M.A., Powley I.R., Jukes-Jones R., Horn S., Feoktistova M., Fairall L., Schwabe J.W., Leverkus M., Cain K., MacFarlane M. Co-operative and hierarchical binding of c-FLIP and caspase-8: a unified model defines how c-FLIP isoforms differentially control cell fate. Mol Cell. 2016;61:834–849. doi: 10.1016/j.molcel.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oberst A., Dillon C.P., Weinlich R., McCormick L.L., Fitzgerald P., Pop C., Hakem R., Salvesen G.S., Green D.R. Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filliol A., Piquet-Pellorce C., Raguenes-Nicol C., Dion S., Farooq M., Lucas-Clerc C., Vandenabeele P., Bertrand M.J.M., Le Seyec J., Samson M. RIPK1 protects hepatocytes from Kupffer cells-mediated TNF-induced apoptosis in mouse models of PAMP-induced hepatitis. J Hepatol. 2017;66:1205–1213. doi: 10.1016/j.jhep.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 31.Filliol A., Piquet-Pellorce C., Le Seyec J., Farooq M., Genet V., Lucas-Clerc C., Bertin J., Gough P.J., Dimanche-Boitrel M.T., Vandenabeele P., Bertrand M.J., Samson M. RIPK1 protects from TNF-alpha-mediated liver damage during hepatitis. Cell Death Dis. 2016;7 doi: 10.1038/cddis.2016.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tummers B., Green D.R. Caspase-8: regulating life and death. Immunol Rev. 2017;277:76–89. doi: 10.1111/imr.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ting A.T., Bertrand M.J.M. More to life than NF-kappaB in TNFR1 signaling. Trends Immunol. 2016;37:535–545. doi: 10.1016/j.it.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Annibaldi A., Meier P. Checkpoints in TNF-induced cell death: implications in inflammation and cancer. Trends Mol Med. 2018;24:49–65. doi: 10.1016/j.molmed.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 35.Peltzer N., Darding M., Walczak H. Holding RIPK1 on the ubiquitin leash in TNFR1 signaling. Trends Cell Biol. 2016;26:445–461. doi: 10.1016/j.tcb.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 36.Schneider A.T., Gautheron J., Feoktistova M., Roderburg C., Loosen S.H., Roy S., Benz F., Schemmer P., Buchler M.W., Nachbur U., Neumann U.P., Tolba R., Luedde M., Zucman-Rossi J., Panayotova-Dimitrova D., Leverkus M., Preisinger C., Tacke F., Trautwein C., Longerich T., Vucur M., Luedde T. RIPK1 Suppresses a TRAF2-dependent pathway to liver cancer. Canc Cell. 2017;31:94–109. doi: 10.1016/j.ccell.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 37.Moulin M., Anderton H., Voss A.K., Thomas T., Wong W.W., Bankovacki A., Feltham R., Chau D., Cook W.D., Silke J., Vaux D.L. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 2012;31:1679–1691. doi: 10.1038/emboj.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vanlangenakker N., Vanden Berghe T., Bogaert P., Laukens B., Zobel K., Deshayes K., Vucic D., Fulda S., Vandenabeele P., Bertrand M.J. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2011;18:656–665. doi: 10.1038/cdd.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geserick P., Hupe M., Moulin M., Wong W.W., Feoktistova M., Kellert B., Gollnick H., Silke J., Leverkus M. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J Cell Biol. 2009;187:1037–1054. doi: 10.1083/jcb.200904158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haag C., Stadel D., Zhou S., Bachem M.G., Moller P., Debatin K.M., Fulda S. Identification of c-FLIPL and c-FLIPS as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut. 2011;60:225–237. doi: 10.1136/gut.2009.202325. [DOI] [PubMed] [Google Scholar]
- 41.Schwabe R.F., Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol. 2018;15:738–752. doi: 10.1038/s41575-018-0065-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woolbright B.L., Jaeschke H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J Hepatol. 2017;66:836–848. doi: 10.1016/j.jhep.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaw P.J., Hopfensperger M.J., Ganey P.E., Roth R.A. Lipopolysaccharide and trovafloxacin coexposure in mice causes idiosyncrasy-like liver injury dependent on tumor necrosis factor-alpha. Toxicol Sci. 2007;100:259–266. doi: 10.1093/toxsci/kfm218. [DOI] [PubMed] [Google Scholar]
- 44.Iwasaki A., Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 45.Wang Z., Xu G., Gao Y., Zhan X., Qin N., Fu S., Li R., Niu M., Wang J., Liu Y., Xiao X., Bai Z. Cardamonin from a medicinal herb protects against LPS-induced septic shock by suppressing NLRP3 inflammasome. Acta Pharm Sin B. 2019;9:734–744. doi: 10.1016/j.apsb.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li L., Chen L., Hu L., Liu Y., Sun H.Y., Tang J., Hou Y.J., Chang Y.X., Tu Q.Q., Feng G.S., Shen F., Wu M.C., Wang H.Y. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology. 2011;54:1620–1630. doi: 10.1002/hep.24552. [DOI] [PubMed] [Google Scholar]
- 47.Jia L., Vianna C.R., Fukuda M., Berglund E.D., Liu C., Tao C., Sun K., Liu T., Harper M.J., Lee C.E., Lee S., Scherer P.E., Elmquist J.K. Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat Commun. 2014;5:1–26. doi: 10.1038/ncomms4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin C.Y., Chang T.W., Hsieh W.H., Hung M.C., Lin I.H., Lai S.C., Tzeng Y.J. Simultaneous induction of apoptosis and necroptosis by Tanshinone IIA in human hepatocellular carcinoma HepG2 cells. Cell Death Dis. 2016;2:1–11. doi: 10.1038/cddiscovery.2016.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roychowdhury S., McMullen M.R., Pisano S.G., Liu X., Nagy L.E. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57:1773–1783. doi: 10.1002/hep.26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gunther C., He G.W., Kremer A.E., Murphy J.M., Petrie E.J., Amann K., Vandenabeele P., Linkermann A., Poremba C., Schleicher U., Dewitz C., Krautwald S., Neurath M.F., Becker C., Wirtz S. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J Clin Invest. 2016;126:4346–4360. doi: 10.1172/JCI87545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dara L., Liu Z.X., Kaplowitz N. Questions and controversies: the role of necroptosis in liver disease. Cell Death Dis. 2016;2:1–10. doi: 10.1038/cddiscovery.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suda J., Dara L., Yang L., Aghajan M., Song Y., Kaplowitz N., Liu Z.X. Knockdown of RIPK1 markedly exacerbates murine immune-mediated liver injury through massive apoptosis of hepatocytes, independent of necroptosis and inhibition of NF-kappaB. J Immunol. 2016;197:3120–3129. doi: 10.4049/jimmunol.1600690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geng Y., Fang M., Wang J., Yu H., Hu Z., Yew D.T., Chen W. Triptolide down-regulates COX-2 expression and PGE2 release by suppressing the activity of NF-kappaB and MAP kinases in lipopolysaccharide-treated PC12 cells. Phytother Res. 2012;26:337–343. doi: 10.1002/ptr.3538. [DOI] [PubMed] [Google Scholar]
- 54.Park S.W., Kim Y.I. Triptolide induces apoptosis of PMA-treated THP-1 cells through activation of caspases, inhibition of NF-kappaB and activation of MAPKs. Int J Oncol. 2013;43:1169–1175. doi: 10.3892/ijo.2013.2033. [DOI] [PubMed] [Google Scholar]
- 55.Wang X., Zhang L., Duan W., Liu B., Gong P., Ding Y., Wu X. Anti-inflammatory effects of triptolide by inhibiting the NF-kappaB signalling pathway in LPS-induced acute lung injury in a murine model. Mol Med Rep. 2014;10:447–452. doi: 10.3892/mmr.2014.2191. [DOI] [PubMed] [Google Scholar]
- 56.Krueger A., Baumann S., Krammer P.H., Kirchhoff S. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol. 2001;21:8247–8254. doi: 10.1128/MCB.21.24.8247-8254.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsuchiya Y., Nakabayashi O., Nakano H. FLIP the switch: regulation of apoptosis and necroptosis by cFLIP. Int J Mol Sci. 2015;16:30321–30341. doi: 10.3390/ijms161226232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perez D., White E. E1A sensitizes cells to tumor necrosis factor alpha by downregulating c-FLIPS. J Virol. 2003;77:2651–2662. doi: 10.1128/JVI.77.4.2651-2662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He M.X., He Y.W. A role for c-FLIPL in the regulation of apoptosis, autophagy, and necroptosis in T lymphocytes. Cell Death Differ. 2013;20:188–197. doi: 10.1038/cdd.2012.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Piao X., Komazawa-Sakon S., Nishina T., Koike M., Piao J.H., Ehlken H., Kurihara H., Hara M., Van Rooijen N., Schutz G., Ohmuraya M., Uchiyama Y., Yagita H., Okumura K., He Y.W., Nakano H. c-FLIP maintains tissue homeostasis by preventing apoptosis and programmed necrosis. Sci Signal. 2012;5:1–18. doi: 10.1126/scisignal.2003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang L., Xu D., Li L., Xing X., Liu L., Ismail Abdelmotalab M., Xiao L., Pang T., Huang X., Wang X., Wang T., Jiang Z., Zhang L., Sun L. Possible role of hepatic macrophage recruitment and activation in triptolide-induced hepatotoxicity. Toxicol Lett. 2018;299:32–39. doi: 10.1016/j.toxlet.2018.08.017. [DOI] [PubMed] [Google Scholar]
- 62.Luyendyk J.P., Maddox J.F., Cosma G.N., Ganey P.E., Cockerell G.L., Roth R.A. Ranitidine treatment during a modest inflammatory response precipitates idiosyncrasy-like liver injury in rats. J Pharmacol Exp Therapeut. 2003;307:9–16. doi: 10.1124/jpet.103.054288. [DOI] [PubMed] [Google Scholar]
- 63.Uetrecht J., Naisbitt D.J. Idiosyncratic adverse drug reactions: current concepts. Pharmacol Rev. 2013;65:779–808. doi: 10.1124/pr.113.007450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hassan H.M., Guo H., Yousef B.A., Guerram M., Hamdi A.M., Zhang L., Jiang Z. Role of inflammatory and oxidative stress, cytochrome P450 2E1, and bile acid disturbance in rat liver injury induced by isoniazid and lipopolysaccharide cotreatment. Antimicrob Agents Chemother. 2016;60:5285–5293. doi: 10.1128/AAC.00854-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tukov F.F., Luyendyk J.P., Ganey P.E., Roth R.A. The role of tumor necrosis factor alpha in lipopolysaccharide/ranitidine-induced inflammatory liver injury. Toxicol Sci. 2007;100:267–280. doi: 10.1093/toxsci/kfm209. [DOI] [PubMed] [Google Scholar]
- 66.Zou W., Beggs K.M., Sparkenbaugh E.M., Jones A.D., Younis H.S., Roth R.A., Ganey P.E. Sulindac metabolism and synergy with tumor necrosis factor-alpha in a drug-inflammation interaction model of idiosyncratic liver injury. J Pharmacol Exp Therapeut. 2009;331:114–121. doi: 10.1124/jpet.109.156331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lu J., Jones A.D., Harkema J.R., Roth R.A., Ganey P.E. Amiodarone exposure during modest inflammation induces idiosyncrasy-like liver injury in rats: role of tumor necrosis factor-alpha. Toxicol Sci. 2012;125:126–133. doi: 10.1093/toxsci/kfr266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klamerth O.L. Inhibition of transcription by isonicotinic acid hydrazide. Mutat Res. 1976;35:53–64. doi: 10.1016/0027-5107(76)90168-8. [DOI] [PubMed] [Google Scholar]
- 69.Liguori M.J., Anderson M.G., Bukofzer S., McKim J., Pregenzer J.F., Retief J., Spear B.B., Waring J.F. Microarray analysis in human hepatocytes suggests a mechanism for hepatotoxicity induced by trovafloxacin. Hepatology. 2005;41:177–186. doi: 10.1002/hep.20514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.