

Abstract
Inhibitory interneurons orchestrate information flow across the cortex and are implicated in psychiatric illness. Although interneuron classes have unique functional properties and spatial distributions, the influence of interneuron subtypes on brain function, cortical specialization, and illness risk remains elusive. Here, we demonstrate stereotyped negative correlation of somatostatin and parvalbumin transcripts within human and non-human primates. Cortical distributions of somatostatin and parvalbumin cell gene markers are strongly coupled to regional differences in functional MRI variability. In the general population (n = 9,713), parvalbumin-linked genes account for an enriched proportion of heritable variance in in-vivo functional MRI signal amplitude. Single-marker and polygenic cell deconvolution establish that this relationship is spatially dependent, following the topography of parvalbumin expression in post-mortem brain tissue. Finally, schizophrenia genetic risk is enriched among interneuron-linked genes and predicts cortical signal amplitude in parvalbumin-biased regions. These data indicate that the molecular-genetic basis of brain function is shaped by interneuron-related transcripts and may capture individual differences in schizophrenia risk.
Subject terms: Gene expression, Cognitive neuroscience, Schizophrenia
Interneuron subtypes have distinct properties and spatial distributions. Here, the authors show that the molecular-genetic basis of cortical resting-state brain function is shaped by distributions of interneuron-related transcripts and may capture individual differences in schizophrenia risk.
Introduction
Ramón y Cajal theorized that the functional diversity of the human brain arises, in part, from the vast assortment of neurons that pattern cortex1. Inhibitory interneurons are the most varied neuronal class2, exhibiting divergent morphological and physiological properties while coordinating information flow across the brain’s collective set of connections (functional connectome)3. Converging animal and human work provides evidence for the role of interneurons in healthy brain function as well as their dysregulation in psychiatric illnesses, including schizophrenia4 and major depressive disorder5. The development of dense spatial transcriptional atlases now enables the study of cellular and molecular associates of functional brain networks. Early work in this area identifies genes encoding ion channels6 and those enriched in supragranular layers of cortex7 as correlates of large-scale network organization. Other research indicates that cortical resting-state signal fluctuations follow the expression of neuron-enriched genes8. However, the transcriptional correlates of brain function have rarely been validated in-vivo6 and little is known about how the spatial distribution of specific interneuron subtypes shapes cortical function and associated risk for psychiatric illness in humans.
The spatial distribution of interneuron subtypes is theorized to contribute to regional specialization of cortex, partly by altering the relative balance of excitation and inhibition for a given cortical area9–11. Interneurons comprise 20–30% of cortical neurons12 and form stereotyped connections with excitatory projection neurons2,3. The majority of interneurons express one of a limited set of genetic markers: somatostatin (SST), parvalbumin (PVALB), and vasoactive-intestinal peptide (VIP; a subset of HTR3A interneurons)2. Each subtype possesses unique synaptic and functional characteristics, leading to the hypothesis that the ratio of interneuron classes underpins local differences in neural activity11,13. For example, SST interneurons preferentially target dendrites of cortical projection neurons to regulate their input, whereas PVALB interneurons synapse on perisomatic regions to regulate output2. Consequently, increased relative density of SST interneurons may facilitate filtering of noisy or task-irrelevant cortical signals and promote recurrent excitation required for higher-order cognition11,14. Conversely, relative increases in PVALB may produce stronger feedback inhibition on excitatory neurons, leading to shorter activation timescales suited for processing constantly changing sensorimotor stimuli11,15. Previous work documents cortical gradients of gene transcription that mirror the hierarchical organization of timescales from fast (unimodal cortex) to slow (multimodal cortex)8,10,16,17. Further, SST and PVALB interneuron markers are differentially expressed within distributed limbic and somato-motor cortico-striatal networks, respectively9. These observations suggest that spatial distributions of interneuron subtypes could underlie regional signaling differences across the cortical sheet, as indexed by blood oxygenation level-dependent (BOLD) functional magnetic resonance imaging (fMRI). However, the psychiatric and functional consequences of spatially variable SST- and PVALB-related transcription in cortex have yet to be fully characterized.
Establishing the principles by which cellular diversity influences brain function is a long-standing challenge in neuroscience and could reveal biological mechanisms of individual variability of the human brain. Consistent with this aim, cross-species evidence indicates the importance of PVALB interneurons for fMRI measures of brain function18. PVALB interneurons orchestrate gamma-band oscillations (30–80 Hz19), a frequency range that is tightly coupled to spontaneous BOLD fluctuations20. Experimental optogenetic stimulation of PVALB interneurons in rodents drives gamma-band rhythms, impacting information processing through the synchronization of excitatory neurons19. In psychiatric illness, decreased PVALB-mediated inhibition may be a core locus of disruption in schizophrenia, leading to altered gamma-band signals and working memory deficits that are a hallmark of the disorder21. However, a direct link between PVALB-related genetic variation and human brain activity has yet to be established. Linking cortical interneurons to individual differences in human brain function would yield deep biological insight into the hemodynamic BOLD signal, providing an engine for the discovery of functional connectome-linked genes and associated risk for illness onset.
Here, we bridge genetic, transcriptional, and neuroimaging data to advance three lines of inquiry linking interneurons to human brain function. First, we describe the organization of SST and PVALB expression in human and non-human primates, demonstrating a robust pattern of anti-correlation across cortex and subcortex. We perform single-cell polygenic deconvolution22 of bulk cortical tissue data to infer spatial distributions of inhibitory, excitatory, and non-neuronal cells across cortex, providing converging evidence with single-marker analyses. Second, we establish that the relative density of SST and PVALB tracks regional differences in cortical brain activity. In a large sample (N = 9713)23, genetic variation among PVALB-correlated genes explained an enriched proportion of heritable variance in resting-state signal amplitude (RSFA), in a manner that mirrors the spatial expression of PVALB measured in independent post-mortem brain tissue. These discoveries suggest that the molecular-genetic basis of cortical function is spatially nonuniform and that genes linked to PVALB interneurons explain heritable aspects of the BOLD signal. Third, we link PVALB interneurons and psychotic illness, demonstrating that genetic risk for schizophrenia is enriched among interneuron-linked genes and predicts reduced resting-state signal amplitude in a spatially heterogeneous manner that follows PVALB expression. These data help address a deep-rooted challenge in neuroscience to understand how cytoarchitecture shapes human brain function and related vulnerability for psychiatric illness.
Results
Anti-correlation of SST and PVALB interneuron markers across cortex
The properties of interneuron subtypes emerge early in development and are partly determined by their spatial origin in the embryonic ganglionic eminence24. SST and PVALB interneurons originate in the medial ganglionic eminence (MGE) along negatively correlated spatial gradients25. That is, PVALB- and SST-destined neurons differentially cluster within the dorsal and ventral MGE, respectively26. Evidence in humans9,10 and rodents11,17 suggests that SST and PVALB transcripts maintain a negative spatial correlation in adulthood, indicating that embryonic organization may constitute a “proto-map” of mature cortex. Although prior research suggests that SST and PVALB markers are differentially expressed across cortex9,10, the current work directly establishes transcriptional anti-correlation between these two cell types across multiple techniques, human datasets, non-human primate data, and human neurodevelopment. This comprehensive profiling of SST and PVALB interneuron expression is necessary for deep profiling of their relationship to in-vivo brain function and subsequent schizophrenia risk through statistical genetic approaches. The functional consequences of a negative spatial SST to PVALB relationship are not well understood, but the presence of replicable and evolutionarily conserved expression patterns may indicate the importance of interneuron gradients.
To characterize interneuron marker topography across human and non-human primate cortex, we analyzed gene expression data from the Allen Human Brain Atlas (AHBA)27 and NIH Blueprint Non-Human Primate (NHP) Atlas28. AHBA cortical samples from the left (n = 1265) and right (n = 418) hemispheres were analyzed. Microarrays do not provide absolute estimates of gene transcription, but can measure within-probe differences across samples. SST and PVALB expression were thus z-transformed across cortical samples, and subtracted (i.e., SST–PVALB) to reveal relative expression differences (Fig. 1a). Extending prior evidence9,10, SST and PVALB were anti-correlated across AHBA cortical samples (Fig. 1e) for both parametric (r(1681) = −0.45, p < 2.2e−16) and non-parametric tests (Spearman’s rho (rs) = −0.40, p < 2.2e−16). Relative to all possible gene-wise correlations, the SST to PVALB correlation was among the most negative (Fig. 1h, i; AUCsst = 0.009, AUCpvalb = 0.033). Further, the SST and PVALB relationship was among the most negative relative to all possible (n = 152,207,628) two-gene pairings (Fig. 1j; AUC = 0.001). SST and PVALB distributions were organized along an anterior to posterior gradient, with greatest relative SST in orbitofrontal and medial prefrontal cortex, anterior insula, and the temporal lobe (Fig. 1a, c and Supplementary Fig. 1). By contrast, relative PVALB expression was greatest within unimodal somato-motor, parietal, and visual cortices (Fig. 1a, c and Supplementary Fig. 1). Histologically defined anatomical categories were used to characterize regional differences of interneuron marker expression, reflected in Fig. 1c showing negatively correlated SST and PVALB median across cortical subregions (r(39) = −0.88, p = 2.4e−14).
Fig. 1. Cortical expression of SST and PVALB are negatively correlated across species and developmental stages.

a AHBA tissue samples mapped to the human cortical surface, and b an illustration of non-human primate tissue sample locations, colored by relative expression of SST (red) and PVALB (blue). Normalized expression difference reflects the sample-wise subtraction of z-transformed PVALB from SST. Relative SST–PVALB expression among anatomically defined groups from the c AHBA (n = 6 donors; n = 1683 samples; n = 41 regions) and d NIH Blueprint Non-Human Primate Atlas (n = 4 donors; n = 182 samples; n = 10 regions); circles = median, thick lines = interquartile range, thin lines = min and max values. e Sample-wise negative correlation of SST and PVALB in human cortex (r = −0.45, p < 2.2e−16; rs = −0.40, p < 2.2e−16) and f non-human primates (r(34) = −0.74, p = 2.2e−7; rs = −0.60, p = 0.0001). g Correlation of cortical SST and PVALB across nine developmental stages using data from the Brainspan Atlas of the Developing Human Brain (n = 42 donors; n = 362 samples; n = 9 developmental stages). h–j The SST to PVALB correlation is at the left tail of the distribution of all gene-wise correlations to SST (AUC = 0.009) and to PVALB (AUC = 0.033), as well as all possible two-gene spatial correlations (AUC = 0.001). *p ≤ 0.05, uncorrected; †p ≤ 0.10; error bars = standard error.
The negative spatial relationship between SST and PVALB was evolutionarily conserved in non-human macaque primates (Fig. 1f; r(34) = −0.74, p = 2.2e−7; rs = −0.60, p = 0.0001), suggesting that interneuron marker gradients may reflect a core organizational feature of primate cortex. These data complement evidence for similar negatively correlated SST and PVALB gradients in rodents11,17. Given that SST and PVALB interneurons originate along stereotyped, anti-correlated gradients in the MGE26, we analyzed RNAseq data from the Brainspan Atlas of the Developing Human Brain to test whether the emergence of SST to PVALB negative correlations coincides with major waves of interneuron colonization, approximately 10–25 post-conception weeks (pcw)29,30. The negative correlation between SST and PVALB was absent in early-fetal (8–12 pcw; β = 0.32, p = 0.039) and early-midfetal (13–21 pcw; β = 0.02, p = 0.85) stages. Consistent with the hypothesis that mature interneuron distributions result from developmentally programmed migration patterns, we observed significant negative correlations between SST and PVALB emerge during late-fetal (24–37 pcw; β = −0.47, p = 0.012), early-infancy (4 months; β = −0.60, p = 0.0033), mid-late childhood (8–11 years; β = −0.52, p = 0.0038), and adult (18–40 years; β = −0.35, p = 0.0014) periods, as well as at a trend-level in adolescence (13–15 years; β = −0.54, p = 0.057). We did not observe a relationship in late infancy (10 months; b = −0.37, p = 0.36) or early childhood (1–4 years; b = −0.11, p = 0.48). These data provide developmental context as well as an external replication of the SST–PVALB cortical expression pattern observed in the AHBA (adult human) and NHP Atlas (adult macaque) samples.
Polygenic deconvolution of cell types across cortex
SST and PVALB are reliable genetic markers of their respective interneuron subtypes2, however single-cell transcriptomics show that cell classes possess polygenic signatures of expression31. To capture the molecular complexity of cell identity and quantify cell-level associates of brain function, we conducted polygenic cellular deconvolution of bulk AHBA expression data using CIBERSORTx (https://cibersortx.stanford.edu/)22. This method leverages transcriptomic “signatures” of cellular identity to estimate the relative abundance of cell types in bulk tissue data. Single-nucleus droplet-based sequencing (snDrop-seq) data from Lake and colleagues31 was used, providing cell-level expression data in dorsal frontal cortex (BA6/BA10; 10,319 cells) and visual cortex (BA17; 19,368 cells). Collinearity among transcriptionally similar cell types was reduced by using 18 superordinate cell identities defined by Lake and colleagues31. Cell signatures were created with CIBERSORTx (Supplementary Fig. 5) and cell abundance, expressed as a fraction, was estimated for each cortical tissue sample from AHBA donors (see Supplementary Data). AHBA tissue samples were projected to the cortical surface, allowing for vertex-level localization on a group-atlas (fsLR32k; see “Methods”). Fractional cell abundances were averaged for each of the 400 bi-hemispheric parcels from the atlas of Schaefer and colleagues32 (Supplementary Figs. 6–8).
We focus on imputed spatial distributions of SST and PVALB interneurons across cortex, estimated separately from visual and frontal cortex single-cell data (Fig. 2). Similar to single-marker analyses (Fig. 1a and Supplementary Fig. 1a), the inferred fractional abundance of SST interneurons was greatest in medial PFC, anterior insula, and temporal poles (Fig. 2a), which was highly consistent between frontal and visual cortex cell type derived maps (r(337) = 0.73, p < 2.2e−16). Conversely, PVALB fractional abundance was highest within visual, motor, and dorsal parietal cortex (Fig. 2b), which was also consistent across frontal and visual cortex derived estimates (r(337) = 0.76, p < 2.2e−16). Given the hypothesized importance of relative SST and PVALB abundance for cortical functional dynamics, Fig. 2c shows the spatial distribution of estimated SST- (red) and PVALB-dense (blue) areas of cortex. Figure 2d illustrates the spatial correlation of all deconvolved cell type fractions across cortex, recapitulating the negative spatial relationship between SST and PVALB single-gene markers (frontal cortex: r(337) = −0.62, p < 2.2e−16; visual cortex: r(337) = −0.37, p = 1.15e−12). Critically, cell fraction maps of PVALB and SST obtained through polygenic deconvolution were strongly correlated with the expression of their respective single-marker genes (Fig. 2f, g).
Fig. 2. Deconvolved cell type distributions are consistent with SST and PVALB single marker expression maps.

Using CibersortX22, frontal and visual cortex snDrop-Seq data from Lake and colleagues31 were used to deconvolve cell type fractions from bulk AHBA microarray expression data. Deconvolved cell fractions of a somatostatin and b parvalbumin interneurons across cortex using single-cell data from frontal (left) and visual (right) cortex. c Somatostatin and parvalbumin cell fraction maps were z-transformed and subtracted (SST–PVALB) to illustrate the relative density of each subtype across cortex. d Spatial correlations of each deconvolved cell type across cortex using frontal (top-left triangle) and visual (bottom-right triangle) cell signatures. e Example marker genes for each cell class. f Deconvolved parvalbumin fractions across cortex were positively spatially correlated (Pearson’s) to single-gene PVALB expression (frontal cortex: r(337) = 0.81, p < 2.2e−16; visual cortex: r(337) = 0.48, p < 2.2e−16). g Deconvolved somatostatin fractions across cortex are positively spatially correlated (Pearson’s) to single-gene SST expression (frontal cortex: r(337) = 0.72, p < 2.2e−16; visual cortex: r(337) = 0.60, p < 2.2e−16).
SST and PVALB anti-correlation in subcortical territories
We next examined whether the negative spatial relationship between SST and PVALB is also preserved across subcortex (see Supplementary Information for subcortical sample information). Although there is evidence for anti-correlated SST and PVALB interneuron gradients in striatum9 and hippocampus33, it remains unclear whether SST and PVALB inverse gradients are ubiquitous throughout the brain. Sample-wise expression was normalized and correlated separately for each of the seven areas: striatum, dorsal thalamus, hypothalamus, globus pallidus, amygdala, hippocampus, and combined substantia nigra/ventral tegmentum. The strength of the SST to PVALB correlation was benchmarked against all gene-wise correlations to SST and PVALB (Fig. 3a), and quantified as AUC of the distribution less than or equal to the SST–PVALB correlation. SST was significantly negatively correlated to PVALB in the hypothalamus (r(100) = −0.72, q = 1.9e−16, AUCsst = 0.002, AUCpvalb = 0.01), globus pallidus (r(37) = −0.39, q = 0.011, AUCsst = 0.03, AUCpvalb = 0.03), amygdala (r(65) = −0.30, q = 0.019, AUCsst = 0.06, AUCpvalb = 0.03), and thalamus (r(173) = −0.19, q = 0.019, AUCsst = 0.04, AUCpvalb = 0.07), but not the hippocampus (r(156) = −0.12, q = 0.14, AUCsst = 0.06, AUCpvalb = 0.21), ventral tegmentum/substantia nigra (r(63) = 0.09, q = 0.50, AUCsst = 0.39, AUCpvalb = 0.32), and striatum (r(168) = 0.23, q = 0.011, AUCsst = 0.65, AUCpvalb = 0.72).
Fig. 3. Subcortical SST and PVALB correlation and consistency in rodents.

a The correlation between SST and PVALB was estimated using AHBA data for each of seven subcortical territories. Red/blue circles denote Pearson correlations between SST and PVALB. Light and dark gray boxplots show the distribution of spatial correlations of all other genes (n = 17,447) to PVALB and SST, respectively (center = median, box = Q1–Q3, whiskers = 1.5*IQR, circles = outliers). The strength of the SST to PVALB relationship is quantified relative to gene-wide reference distributions. SST was more negatively correlated to PVALB than what is expected by chance in the hypothalamus (r(100) = −0.72, q = 1.9e−16, AUCsst = 0.002, AUCpvalb = 0.01), globus pallidus (r(37) = −0.39, q = 0.011), amygdala (r(65) = −0.30, q = 0.019), and thalamus (r(173) = −0.19, q = 0.019), but not the hippocampus (r(156) = −0.12, q = 0.14), ventral tegmentum/substantia nigra (r(63) = 0.09, q = 0.50), and striatum (r(168) = 0.23, q = 0.011). b Relative expression of SST and PVALB in human subcortical areas when compared to ground truth cell densities in rodent homologs from Kim et al.11 (Spearman’s rs = 0.41, p = 0.025).
These data identified subcortical territories with biased SST or PVALB expression in humans, which show striking correspondence to non-human animal literature11. Median z-transformed SST and PVALB was calculated for subdivisions of each subcortical region (Supplementary Fig. 9). Across 40 AHBA subcortical sub-regions, 31 were able to be mapped onto rodent homologs present in cell density (cells per mm3) data from Kim and colleagues11. The relative presence of human SST and PVALB across sub-regions was significantly positively correlated to ground truth cell density estimates in rodents (Fig. 3b; rs = 0.41, p = 0.025). Results were consistent when Spearman-rank correlation was used to limit the influence of outliers, and when the ratio of SST/PVALB rodent cell densities was used rather than the difference (rs = 0.49, p = 0.006).
In particular, the central nucleus (CeA) of the amygdala was enriched for SST in both rodents (SSTdensity = 24,342.60, PVALBdensity = 119.42) and humans (SSTexpr(z) = 1.27, PVALBexpr(z) = −0.76), paralleling observations in primates34. We also found greater relative SST in the ventral tegmental area (VTA) for both rodents (SSTdensity = 1953.98, PVALBdensity = 426.10) and humans (SSTexpr(z) = 1.12, PVALBexpr(z) = −0.42), but greater relative presence of PVALB in the substantia nigra pars reticulata (SNr) in humans (SSTexpr(z) = 0.22, PVALBexpr(z) = 0.72) and rodents (SSTdensity = 680.27, PVALBdensity = 4373.48). Supporting this VTA/SNr distinction, the VTA is connected to other SST-biased regions, including the nucleus accumbens (NAcc), anterior cingulate cortex, and mediodorsal thalamus35, whereas functional neuroimaging indicates a preferential coupling of the SNr to motor areas and sensorimotor striatum36. Building on the previous work9, the extent to which SST may be relatively increased within a distributed limbic cortico-striato-thalamic network is explored in Supplementary Fig. 10.
Relative SST and PVALB covaries with resting-state signal amplitude
Computational rodent work suggests the relative presence of SST and PVALB interneurons is a determinant of functional differences and hierarchical organization across cortex11. Spiking activity in cortex progresses from shorter timescales in sensory and unimodal cortices to longer timescales in association and integrative cortices15,37. This functional organization may be indexed by variability in the resting-state BOLD signal. Accordingly, we examined whether the difference of cortical SST and PVALB expression covaries with an in-vivo measurement of cortical signal variability, resting-state functional amplitude (RSFA). Voxel-wise RSFA was calculated using the UK Biobank sample (n = 9713) and averaged across the 400 parcel functional atlas of Schaefer and colleagues (Fig. 4a)32. Between-subject hierarchical clustering was conducted to reduce data dimensionality and identify cortical territories with similar patterns of signal amplitude across individuals (Fig. 4b). A seven-cluster solution was selected, corresponding to limbic A (light beige), limbic B (dark beige), cingulo-opercular (teal), temporo-parietal (orange), prefrontal (red), somato/motor (blue), and visual (purple) clusters. With the exception of Figs. 5a, b and 6b, clusters serve to aid in visualization and do not influence statistics. Consistent with recent work38, this data-driven dimensionality reduction broadly sorted association and unimodal aspects of cortex.
Fig. 4. SST–PVALB difference tracks inter-regional variation in cortical brain function.

a RSFA across each of the 400 Schaefer atlas parcels, averaged across 9713 UKB subjects. b Between-subject hierarchical clustering of residualized RSFA reveals 7-clusters cortical partitions with similar amplitude signatures; Light beige = limbic A, dark beige = limbic B, teal = cingulo-opercular, orange = temporal-parietal, red = prefrontal, blue = somato/motor, and purple = visual. c Relative presence of SST–PVALB is negatively correlated (Pearson’s) with cortical RSFA (r(337) = −0.53, p < 2.2e−16). d Across all deconvolved cell types, RSFA is most negatively spatially correlated (Pearson’s) to SST cell fractions (frontal cortex signature: r(337) = −0.48, p < 2.2e−16; visual cortex signature: r(337) = −0.36, p = 1.2e−11) and most positively correlated to PVALB cell fraction (frontal cortex signature: r(337) = 0.47, p < 2.2e−16; visual cortex signature: r(337) = 0.34, p = 1.7e−10). e Across frontal cortex (n = 272) and visual cortex (n = 240) cell type pairs, the relative difference of SST and PVALB cell fractions is most spatially associated with cortical RSFA.
Fig. 5. PVALB-linked genetic variation explains patterns of heritable brain function.

a RSFA was significantly heritable across the seven empirically defined spatial clusters (n = 9713 UKB subjects). b Partitioned heritability analyses reveal that the 500-gene PVALBSNP set accounted for a significant proportion of heritable variance in all seven clusters (n = 9713 UKB subjects). c Parcel-wise PVALBSNP partitioned heritability of RSFA (left panel) tracks the PVALB single-gene expression across cortex (Pearson’s r(326) = 0.35, p = 1.04e−10; rs = 0.40, p = 2.2e−14). d Across all genes, PVALB was the 115th most correlated gene to PVALBSNP partitioned heritability (AUC = 0.007). e Across all deconvolved cell types, inferred PVALB cell fraction was the most positively correlated to PVALBSNP partitioned heritability (frontal cortex: Pearson’s r(329) = 0.35, p = 2.27e−11; rs = 0.39, p = 2.20e−13; visual cortex: Pearson’s r(329) = 0.31, p = 6.67e−9; rs = 0.34, p = 1.50e−10). f SSTSNP partitioned heritability was not associated with SST single-marker expression (Pearson’s r(326) = −0.02, p = 0.75), a null finding that was consistent when put in context of all genes (g) and inferred cell types (h). Error bars = standard error.
Fig. 6. Schizophrenia polygenic risk predicts brain function and tracks PVALB expression.
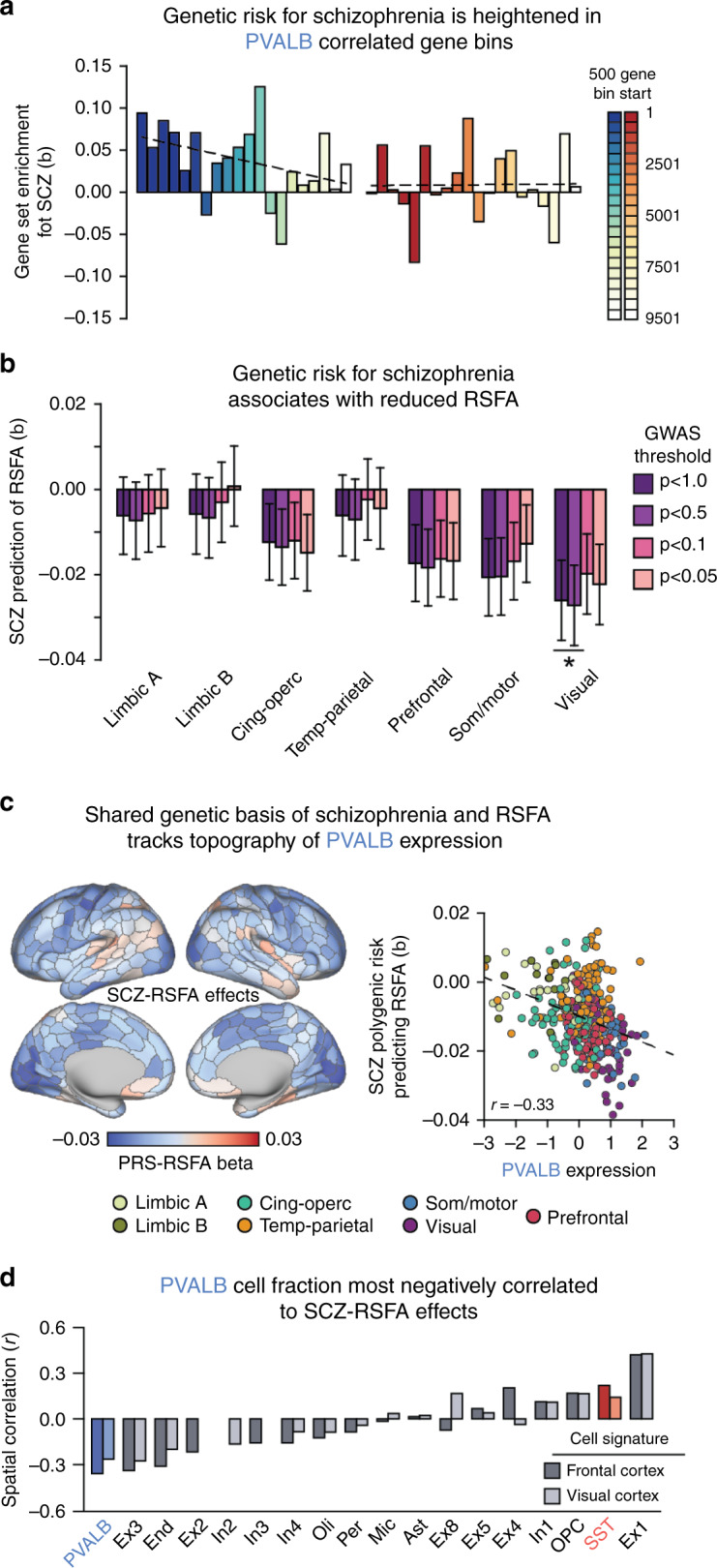
a Genes were rank-ordered by cortical spatial correlation to SST and PVALB, then divided into 500-gene bins. MAGMA competitive gene set analysis revealed enrichment of polygenic risk for schizophrenia in the top PVALB (p = 0.022), but not the top SST (p = 0.51) set. Enrichment decreased across ordered bins for PVALB (Spearman’s rs = −0.48, p = 0.03) but not for SST (Spearman’s rs = −0.001, p = 0.51). b Schizophrenia polygenic risk negatively predicts RSFA within the visual (q1.0 = 0.04) cluster, as well as somato/motor (q1.0 = 0.08) and prefrontal (q1.0 = 0.10) clusters at trend-levels (corrected for multiple-comparisons). c Parcel-wise prediction of RSFA by the schizophrenia PRS negatively correlated with cortical expression of PVALB (Pearson’s r = −0.33, p = 3.1e−10), which was also significant relative to all genes (PVALB = 145/17,448, AUC = 0.008). d Across all deconvolved cell type distributions, PVALB was the most negatively correlated to cortical SCZ-RSFA effects (frontal cortex: Pearson’s r(337) = −0.36, p = 9.5e−12; visual cortex: Pearson’s r(337) = −0.26, p = 9.0e−7). SCZ = schizophrenia; PRS = polygenic risk score; RSFA = resting state functional amplitude. *q ≤ 0.05. Error bars = standard error.
Computational models suggest that increased PVALB density may correspond to greater recurrent inhibition of excitatory neurons and faster timescales (i.e., higher RSFA). Likewise, increased relative presence of SST is predicted to correspond to longer timescales of activity (i.e., lower RSFA)11. Earlier work documents a correlation of interneuron marker expression with fractional Amplitude of Low-Frequency Fluctuations (fALFF)8, a metric closely tied to RSFA, within a circumscribed set of cortical areas. Consistent with these predictions, a negative spatial correlation was observed between RSFA and the relative difference of SST and PVALB expression (Fig. 4c; r(337) = −0.53, p < 2.2e−16; rs = −0.60, p < 2.2e−16). Across 152,207,628 two-gene pairings, the SST–PVALB to RSFA correlation (r = −0.53; Fig. 4c) was the 54,303rd most negatively associated pair (top 0.04%). Analysis of deconvolved cell fractions across cortex revealed that SST had the strongest negative spatial association to RSFA (Fig. 4d; frontal cortex signature: r(337) = −0.48, p < 2.2e−16; visual cortex signature: r(337) = −0.36, p = 1.2e-11), while PVALB cell fractions were the most positively correlated (frontal cortex signature: r(337) = 0.47, p < 2.2e−16; visual cortex signature: r(337) = 0.34, p = 1.7e−10), relative to all other cell types. Relative difference of SST and PVALB cell fractions was the most associated with RSFA, relative to all pairwise cell combinations from frontal (n = 272) and visual (n = 240) cortex (Fig. 4e). Overall, these analyses use ex-vivo transcriptional data to identify SST and PVALB interneurons as cellular correlates of in-vivo measures of cortical signal variability (i.e., RSFA).
Polygenic variation among PVALB-correlated genes underlies cortical brain function
Genome-wide association studies (GWAS) demonstrate that the genetic bases of many complex traits are due to the cumulative weight of genetic variants spread across the entire genome, each with a subtle effect39. Brain phenotypes such as resting-state functional amplitude likely possess a similar polygenic architecture. However, functionally-relevant polymorphisms can cluster in genes expressed within associated tissue and cell types40. We tested whether single-nucleotide polymorphisms (SNPs) that explain heritable variance in brain activity (i.e., RSFA) are enriched within genes linked to PVALB and SST, which would provide insight into the molecular basis of the resting-state BOLD fluctuations.
We established the SNP heritability of RSFA. A significant proportion of between-subject variation in cluster-wise RSFA was due to common genetic variants [Fig. 5a; : limbic A = 0.25 (SE 0.04), limbic B = 0.30 (SE 0.04), cingulo-opercular = 0.21 (SE 0.04), temporo-parietal = 0.29 (SE 0.04), prefrontal = 0.33 (SE 0.04), somato/motor = 0.21 (SE 0.04), visual = 0.07 (SE 0.04)]41. See Supplementary Fig. 11 for parcel-wise heritability estimates. Interneuron-correlated gene sets were nominated using a “guilt-by-association” logic. That is, genes that were spatially correlated to interneuron markers (i.e., SST, PVALB) were assumed to relate to each interneuron subtype. Using cortical AHBA data, genes were rank-ordered by their spatial correlation to each interneuron marker and the top 500 most-correlated genes were selected. Interneuron-related SNP lists were generated for each gene set by identifying variants within ±5000 base pairs from transcription start and stop site of each gene. PVALB and SST SNP sets were non-overlapping. The eQTL variants for each gene set were included, defined using cortical data from the CommonMind consortium42 and NIH GTEx43. We denote the SNP lists for each interneuron gene set as PVALBSNP and SSTSNP (see Supplementary Data). Genetic relatedness matrices were calculated for the UKB sample using each SNP set, and heritability was estimated using GCTA-GREML simultaneously across three partitions: PVALBSNP, SSTSNP, and a partition containing all remaining genotyped variants41.
Indicating that genetic variance in RSFA, a measure of in-vivo functional variability, is explained by genes linked to PVALB interneurons, the PVALBSNP set accounted for a significant proportion of heritable variance of the temporo-parietal [ (SE 0.010), q = 0.0007], prefrontal [ (SE 0.0091), q = 0.015], and somato/motor [ (SE 0.0090), q = 0.015], visual [ (SE 0.0089), q = 0.021], limbic A [ (SE 0.009), q = 0.04], limbic B [ (SE 0.009), q = 0.007] RSFA clusters, but at only trend-level in cingulo-opercular cluster [ (SE = 0.0090), q = 0.053]. Conversely, the SSTSNP set did not explain a significant proportion of heritable variance across any partition (’s < 0.011, q’s > 0.44). We next tested whether the PVALBSNP set explains more genetic variance in RSFA than would be expected by chance. Enrichment was calculated as the proportion of heritability explained by the SNP partition, divided by the fraction of SNPs in that partition (>1 denotes enrichment). We observed enrichment greater than 1 for visual (enrich = 8.10, SE = 0.31), somato/motor (enrich = 3.56, SE = 0.32), temporo-parietal (enrich = 4.10, SE = 0.34), prefrontal (enrich = 2.36, SE = 0.32), cingulo-opercular (enrich = 2.04, SE = 0.31), limbic A (enrich = 1.98, SE = 0.32), and limbic B (enrich = 3.41, SE = 0.33) clusters. The PVALBSNP list (N = 9571 variants) constituted 2.8% of total analyzable genotyped SNPs (N = 337,501 variants), but accounted for 5.6–22.9% (M = 10.4, SD = 6.0) of total genetic variance across each of the RSFA clusters (Fig. 5a). The SSTSNP partition (2.5% of available variants) only explained an enriched proportion of variance within the cingulo-opercular (enrich = 1.86, SE = 0.33) RSFA cluster.
An important unanswered question is whether the genetic determinants of RSFA are uniform across cortex, or whether they vary according to underlying transcriptional patterns. We tested whether the PVALBSNP and SSTSNP partitions explain a greater percentage of heritable RSFA variance in regions where the respective marker is most expressed. Partitioned heritability was calculated across 400 Schaefer cortical parcels. Across all parcels with AHBA expression data, normalized genetic variance explained by the PVALBSNP partition was positively correlated to PVALB expression (Fig. 5c; r(326) = 0.35, p = 1.04e−10; rs = 0.40, p = 2.2e−14), corresponding to visual, superior temporal, and parietal cortex. Across all genes, PVALB was the 115th most positively correlated gene (top 0.66% of 17,448 transcripts; Spearman = 29th/17,448 = 0.17%) to the PVALBSNP partition map, indicating that this positive relationship is not obligated by global statistical properties (Fig. 5d). This relationship remained significant after controlling for the overall SNP heritability of each parcel (β = 0.22, t(336) = −4.70, p = 3.9e−6). Conversely, PVALBSNP heritability was negatively correlated to SST expression (r(326) = −0.39, p = 6.32e−14; rs = −0.37, p = 6.5e−12). There was not a significant parcel-wise relationship between SSTSNP partitioned heritability and SST gene expression (Fig. 5f; r(326) = −0.017, p = 0.75; rs = −0.021, p = 0.71). Converging with single-gene analyses, deconvolved estimates of PVALB cell fractions were the most positively correlated to PVALB partitioned heritability (Fig. 5e; frontal cortex: r(329) = 0.35, p = 2.27e−11; rs = 0.39, p = 2.2e−13; visual cortex: r(329) = 0.31, p = 6.67e−9; rs = 0.34, p = 1.5e−10). These results were highly consistent across parallel techniques using linkage-disequilibrium score regression (LDSC) and estimates of heritability from GWAS analyses of RSFA (Supplementary Fig. 12)44. Cumulatively, these data indicate that the heritable basis of resting-state functional amplitude is spatially heterogeneous, demonstrating a particularly important role of genes co-expressed with PVALB.
Functional associates of schizophrenia genetic risk correlate with PVALB expression
Convergent evidence from animal models and post-mortem tissue analyses suggests that interneuron dysfunction is a core pathophysiological feature of schizophrenia21. To determine whether interneuron-related genetic variation is tied to disease liability, we tested whether polygenic risk for schizophrenia45 is greater among PVALBSNP and SSTSNP variants, relative to the rest of the genome. Using a partitioned MAGMA analysis46, we divided rank-ordered PVALB and SST gene lists into bins of 500. We observed significant enrichment of schizophrenia polygenic risk for the top PVALB gene set (β = 0.094, p = 0.022), but not the top SST set (β = −0.001, p = 0.51). Suggesting that polygenic schizophrenia risk is greater among interneuron-related genes, we examined 20 gene bins and found that the enrichment of schizophrenia genetic risk decreased as gene sets became less spatially correlated with PVALB (rs = −0.48, p = 0.03), but not SST (rs = 0.027, p = 0.91; Fig. 6a).
To test whether polygenic risk for schizophrenia relates to cortical RSFA, we calculated schizophrenia polygenic risk scores (SCZ-PRS)47 using genotyped variants from individuals in the UK Biobank imaging sample. Across the seven RSFA clusters, SCZ-PRS negatively predicted RSFA in the visual cluster (Benjamini–Hochberg corrected q = 0.04; Fig. 6b; GWAS threshold p < 1.0), as well at trend-levels in somato/motor (q = 0.08) and prefrontal (q = 0.10) clusters. Consistent with the hypothesized link between PVALB interneurons and psychotic illness, the relationship between RSFA and polygenic schizophrenia risk was negatively correlated to PVALB expression across cortex (Fig. 6c; r(337) = −0.33, p = 3.1e−10; rs = −0.35, p = 5.5e−11). That is, regions with greater PVALB expression (e.g., motor and visual parcels) showed stronger negative relationships between SCZ-PRS and RSFA. This relationship remained significant after controlling for overall SNP heritability of each parcel (β = −0.28, t(336) = −5.32, p = 1.95e−7), indicating that the effect isn’t driven by parcel-wise explainable genetic variance. Comparing the RSFA-schizophrenia polygenic risk map to all genes, PVALB was the among the top 0.83% negatively correlated expression profiles (145 out of 17,448; Spearman = 105/17,448 = 0.60%), showing that this relationship is not statistically obligated. Across all deconvolved cell types, parcel-wise PVALB cell fractions were on average the most negatively spatially correlated to schizophrenia risk RSFA effects (Fig. 6d; frontal cortex: r(337) = −0.36, p = 9.5e−12; rs = −0.35, p = 2.4e−11; visual cortex: r(337) = −0.26, p = 9.0e−7; rs = −0.26, p = 1.3e−6). Ontological enrichment analysis further revealed that the top 500 genes correlated with PVALB in the AHBA data contained genes associated to schizophrenia, neuronal signaling, and gated channel activity (Table 1). Together, these data suggest that schizophrenia-related genetic variants are associated with cell types, particularly parvalbumin interneurons, and could explain functional disruptions across cortex.
Table 1.
Enrichment terms for interneuron-correlated genes.
| Gene list | Category | ID | Name | p | q FDR-BH | Hits | Genes in GO |
|---|---|---|---|---|---|---|---|
| SST | GO: BP | GO:0099536 | Synaptic signaling | 1.37e−5 | 3.04e−2 | 37 | 687 |
| SST | GO: BP | GO:0099537 | Trans-synaptic signaling | 2.43e−5 | 3.04e−2 | 36 | 678 |
| SST | GO: CC | GO:0097458 | Neuron part | 1.26e−6 | 6.83e−4 | 66 | 1545 |
| SST | GO: CC | GO:0098793 | Presynapse | 4.03e−4 | 3.64e−2 | 20 | 341 |
| PVALB | GO: MF | GO:0005261 | Cation channel activity | 3.33e−12 | 1.74e−9 | 33 | 306 |
| PVALB | GO: MF | GO:0005249 | Voltage-gated potassium channel activity | 1.33e−10 | 1.69e−8 | 17 | 91 |
| PVALB | GO: BP | GO:0071805 | Potassium ion transmembrane transport | 1.53e−9 | 2.43e−6 | 21 | 181 |
| PVALB | GO: BP | GO:0098655 | Cation transmembrane transport | 3.23e−9 | 3.09e−6 | 48 | 738 |
| PVALB | GO: CC | GO:0034703 | Cation channel complex | 1.04e−10 | 5.07e−8 | 23 | 176 |
| PVALB | GO: CC | GO:0034702 | Ion channel complex | 3.03e−10 | 5.07e−8 | 29 | 291 |
| PVALB | Disease | C0036341 | Schizophrenia | 5.85e−7 | 1.6e−3 | 68 | 1561 |
Ontological enrichment analyses were conducted with ToppGene on the 500 genes used to generate the PVALBSNP and SSTSNP lists. See Supplementary Data for full enrichment tables.
Discussion
Integrating genetic, transcriptional, and neuroimaging data, we demonstrate that spatial distributions of interneurons are stereotyped across species and development, and explain a substantial portion of the heritable variation in RSFA, a measure of in-vivo brain activity. Somatostatin and parvalbumin interneuron markers were negatively spatially correlated across cortex, a relationship that was robust in early developmental periods in humans and evolutionarily conserved in non-human primates (Fig. 1). Stereotyped patterns of SST and PVALB expression were observed in subcortex and were consistent with cell density estimates in rodents (Fig. 3). Computational models theorize that interneuron ratios underlie regional differences in cortical brain function11. Providing empirical support for this hypothesis, relative SST and PVALB expression in post-mortem brain tissue aligned with spatial RSFA in the general population (Fig. 4). Indicating the functional relevance of this spatial relationship, genetic polymorphisms linked to PVALB correlated genes accounted for an enriched proportion of heritable variance underlying cortical signal amplitude (Fig. 5). Critically, the amount of in-vivo variance explained by PVALB-linked SNPs positively tracked spatial expression of PVALB in independent post-mortem brain tissue, suggesting that common genetic polymorphisms influence brain function in a cell-preferential and regionally variable manner. Implicating genetic differences among interneurons in schizophrenia, schizophrenia-related polygenic risk was enriched among genes co-expressed with interneurons, and predicted reduced resting-state functional amplitude across cortex, following the spatial landscape of PVALB gene expression (Fig. 6).
Adaptive functioning depends on the integration of information across timescales. Higher-order cognition often requires information accumulation over time, whereas sensorimotor processing entails rapid adaption to changing external stimuli15,18. These computational demands are met, in part, through the hierarchical organization of anatomic and functional connections in cortex, as well as cytoarchitectural differences across brain regions10,48. Our data indicate that interneuron ratios, as indexed by SST and PVALB expression, are an important feature of regionally variable brain function. Due to unique functional and synaptic properties of somatostatin and parvalbumin interneurons, relative shifts in their density can alter the balance of inhibitory control11. SST interneurons generally synapse onto dendrites of pyramidal neurons to gate incoming cortical signals, whereas PVALB interneurons generally provide perisomatic inhibition that is well-suited for feedback inhibition and output regulation2. Increased dendritic (i.e., SST) over perisomatic (i.e., PVALB) inhibition could lead to more robust filtering of task-irrelevant information, allowing for greater recurrent excitation and integration of information over time19. Conversely, parvalbumin-dense sensorimotor regions may benefit from fast responses and lower recurrent excitation to adapt to rapidly changing inputs15.
Our analyses provide molecular genetic support for a relationship between parvalbumin interneurons and the hemodynamic signal. BOLD signals are preferentially coupled to gamma-band oscillations (30–80 Hz) relative to other frequency domains20. Importantly, fast-spiking parvalbumin interneurons have been experimentally linked to gamma oscillations49. Here, we provide initial evidence in humans for the influence of parvalbumin interneurons on fMRI signal variability. For instance, polygenic variation among parvalbumin correlated genes explained upwards of 23% of the heritable variance in RSFA in visual cortex.
Schizophrenia is one of the most heritable forms of psychiatric illnesses (h2 ~81%)50, with converging lines of evidence pointing toward GABAergic and parvalbumin interneuron abnormalities as cardinal features of the disorder21,51. Patients with schizophrenia exhibit reduced levels of GAD67, an enzymatic precursor of GABA52, and are characterized by PVALB interneurons with atypical perineuronal nets53. These abnormalities are thought to underlie disorder-related disruption of gamma-band oscillations and working memory51. Linking these observations, we demonstrate that polygenic schizophrenia risk is increased among genes that are spatially correlated to PVALB (Fig. 6a), expanding upon cell transcriptomic work implicating cortical interneurons as an illness marker40. We further show a negative association between individual polygenic schizophrenia risk and RSFA in a large population-based sample (Fig. 6b). The topography of these effects follows the spatial profile of PVALB expression across cortex (Fig. 6c), highlighting the potential role of parvalbumin interneurons in mediating brain-based intermediate phenotypes associated with psychotic illness. This report also compliments evidence that schizophrenia risk gene expression is spatially correlated to disorder-related changes in brain morphology54,55.
Disruption of excitatory/inhibitory balance may reflect a cross-diagnostic marker of psychiatric illness56. For instance, decreased expression of parvalbumin cell markers is evident in both schizophrenia and bipolar disorder57, while major depressive disorder is marked by preferential reductions in somatostatin interneurons5. Delineating the region-specific roles of cortical interneuron subtypes will provide insight into cross-diagnostic patterns of both behavior and brain function. With regard to depressed mood, modulation of cortical somatostatin interneurons causally influences anxiety- and depression-like behavioral phenotypes in rodents5,58. In line with this observation, we observed preferential expression of somatostatin within mPFC, NAcc, mediodorsal thalamus, and VTA (Supplementary Fig. 9), a distributed set of regions implicated in reward and affective information processing35. Somatostatin-biased regions (ACC, mPFC, and insula) also correspond to areas of cortical thinning in patients with MDD and individuals reporting elevated negative affect59–61.
The present findings should be interpreted in light of several limitations. First, we use single molecular markers to infer the relative presence of SST and PVALB interneurons, which are not sensitive to morphological and physiological differences among interneuron subgroups2, a point we sought to address through the analyses of deconvolved cell type distributions. Second, we employ a “guilt-by-association” logic to nominate interneuron related gene sets. While we cannot conclude that genes within each identified interneuron group directly influence interneuron function, similar correlation-based nomination approaches have been shown to correspond well with a priori defined gene groups62. However, the examination of enrichment terms (Table 1 and Supplementary Information) allows for a more precise understanding of the biological processes contributing to these results. Third, our measure of brain signal amplitude (RSFA) is likely explained by a mixture of neural and vascular signals. However, effects related to BOLD variability have been shown to be stable even after controlling for measures of vasculature63, and vascular confounds are a caveat of many BOLD measures of brain function. Last, our in-vivo imaging and genetic analyses focus on an aging population of white British individuals. Future work should examine the stability of these results across diverse populations64,65.
Inherited genetic variation shapes brain function within and across individuals66. Analyses of spatially-dense, whole-genome, expression atlases increasingly reveal transcriptional correlates of brain function8, structure67, functional connectivity6,7,9, and psychiatric illness54. With the parallel emergence of large-scale imaging genetic data23, it is now possible to bridge structural genetic, transcriptional, and large-scale neuroimaging brain phenotypes. Here, we leverage these data to show that interneuron marker distributions are consistent across species, correlate with cortical signal amplitude, explain regional differences in heritable brain function, and associate with genetic risk for schizophrenia in the general population.
Methods
Allen Human Brain Atlas
Publicly available human gene expression data from six postmortem donors (1 female), aged 24–57 years (42.5 ± 13.4) were obtained from the Allen Institute27. Data reflect the microarray normalization pipeline implemented in March 2013 (http://human.brain-map.org) and analyses were conducted according to the guidelines of the Yale University Human Subjects Committee. Microarray probes from eight overarching ontological categories were selected: cortex, dorsal thalamus, striatum, globus pallidus, hypothalamus, hippocampus, amygdala, and the combined substantia nigra and ventral tegmentum (see Supplementary Information). Probes without Entrez IDs were removed. Probe-wise noise for each donor was quantified as the number of above-threshold samples in cortex, divided by total cortical sample count. A probe-wise average was computed across all six donors, which was used to remove probes expressed in fewer than 20% of cortical samples68. If more than one probe existed for a given gene, the one with the highest mean expression level was selected for further analysis, resulting in 17,448 brain-expressed genes.
Individual cortical tissue samples were mapped to each AHBA donor’s Freesurfer derived cortical surfaces, downloaded from Romero-Garcia and colleagues69. Native space midthickness surfaces were transformed to a common fsLR32k group space while maintaining the native cortical geometry of each individual donor. The native voxel coordinate of each tissue sample was mapped to the closest surface vertex using tools from the HCP workbench70. A cortical tissue sample was not analyzed if it was greater than 4 mm from the nearest surface vertex, resulting in 1683 analyzable cortical samples. Microarray expression of each gene was mean- and variance-normalized separately for each of the eight analyzed regions, revealing relative expression differences within cortical and subcortical territories. For region-wise expression analyses (e.g., Fig. 1c), ontological categories from the AHBA were used to calculate the median, min–max, and interquartile range of relative expression in each region. Detailed information about the analyzed regions is provided in the Supplementary Information. Cortical data visualization was carried out using wb_view from the HCP workbench70. The MNI locations of striatal and thalamic samples were cross-referenced to functional atlases of Choi and colleagues71 and Hwang and colleagues72. With AFNI, a single voxel (1 mm3) region of interest (ROI) was generated at the MNI location of each sample. A functional network label was assigned if the ROI fell within a volumetric parcel. If the sample did not overlap with the functional atlas, the associated ROI was expanded to 2 mm3 and the network with the most overlapping voxels in the ROI was assigned. If the expanded 2 mm3 ROI did not overlap, the process was repeated using a 3 mm3 ROI. A sample was omitted from analysis if the 3 mm3 ROI did not overlap with the associated functional atlas. Functional sub-regions with 3 or fewer samples were excluded from analyses. This process was repeated for coordinates aligned to MNI152 1 mm space using ANTs registration tools (https://github.com/chrisgorgo/alleninf/tree/master/alleninf/data).
Subcortical AHBA expression data was compared to rodent cell density counts published by Kim et al.11. Z-transformed AHBA expression values were summarized across major subdivisions of the seven subcortical regions analyzed (e.g., CA1, NAcc, etc.; Supplementary Data). Median AHBA expression values were used for analyses in Fig. 3b. Rodent homologs of each sub-region were manually identified and PVALB and SST cell densities were averaged across male and female samples.
UKB imaging processing
Minimally preprocessed resting-state fMRI data from the UK Biobank were analyzed, reflecting the following preprocessing steps: motion correction with MCFLIRT73, grand-mean intensity normalization, highpass temporal filtering, fieldmap unwarping, and gradient distortion correction. Noise terms were identified and removed using FSL ICA+FIX. Full information on the UKB preprocessing is published23. Additional processing was conducted in AFNI74 and consisted of 3dDespike, resampling to MNI152 space using the UKB generated linear and nonlinear transforms, FWHM blur of 4.0 mm, regression of CSF, WM, and global resting state signals, and first- and second-order trend removal. Voxel-wise RSFA maps were generated with 3dRSFC and then averaged within each of the approximately symmetrical 400 volumetric parcels from the 7-network parcellation of Schaefer and colleagues32. Due to signal blurring between lateral striatum and insular cortex, supplemental resting-state functional connectivity analyses reflect an additional local white matter regression against gray matter using AFNI anaticor. Imaging analyses were conducted in volume, but visualized on the cortical surface. Resting-state functional connectivity between striatum, thalamus, and cortex was estimated using AFNI’s 3dNetCorr, which calculated the Fisher-Z transformed correlation values of timeseries across the Choi 7-region striatal atlas71, the Hwang 9-region thalamic atlas72, and the Schaefer 400-region cortical atlas32.
A total of 13,236 UKB subjects were processed through the imaging pipeline. Subjects with mean run-wise frame-to-frame head motion greater than 0.20 mm, and inverted resting-state SNR greater than three standard deviations above the mean were removed. After filtering for white British subjects with usable genetic data, cryptic relatedness <0.025, and conducting row-wise deletion for the variables age, sex, height, weight, BMI, three head position coordinates (X, Y, Z), combined gray/white matter volume, combined ventricular/CSF volume, diastolic and systolic blood pressure, run-wise resting state motion, resting state inverse SNR, T1 inverse SNR, and UK Biobank assessment center, 9713 subjects remained for analyses (percent female = 54.33, mean age = 63.67, SD = 7.45, min/max age = 45–80). We also included the anthropometric measures of height, BMI, weight, and blood pressure. Analyses were conducted according to the guidelines of the Yale University IRB.
UKB genetics
UK Biobank genotype data was filtered to include only white British subjects with imaging data passing the quality control thresholds described above. Plink v1.9 was used to remove samples with missingness >0.10, SNPs with minor-allele frequency <0.05, Hardy–Weinberg equilibrium p < 1 × 10−6, and call rate <0.02, resulting in 337,501 autosomal variants75. GCTA software was used to calculate a genetic relatedness matrix to remove individuals with cryptic relatedness more than 0.025, leaving N = 9713 subjects for analysis41. Ten genetic principal components were then calculated for use as covariates in polygenic risk score and heritability analyses. When calculating polygenic risk for schizophrenia, SNPs from the major histocompatibility complex were censored except for the most significantly associated variant from the region.
RSFA between-subject clustering and heritability
Voxel-wise RSFA data from the (N = 9713) UK Biobank sample was averaged within each of 400 roughly symmetric volumetric ROIs from the 7-Network cortical parcellation of Schaefer and colleagues32. Parcel-wise RSFA values were residualized for the effect of age, sex, age2, age × sex, age2 × sex, height, weight, BMI, combined gray/white matter volume (normed for head size), combined ventricular/CSF volume (normed for head size), diastolic and systolic blood pressure, run-wise rsfMRI motion, rsfMRI inverse SNR, T1 inverse SNR, three head position coordinates (X, Y, Z), and UK Biobank assessment center. Hierarchical clustering of residualized RSFA estimates was conducted using R in order to group regions with similar between-subject patterns of covariation. A 7-parcel RSFA clustering was selected. Raw RSFA values were then averaged across parcels falling within the same data-derived between-subject cluster for use in heritability analyses. SNP heritability of RSFA was estimated with genotyped data using GCTA-REML software. Age, sex, age2, height, weight, BMI, combined normed gray/white matter volume, combined normed ventricular/CSF volume, diastolic and systolic blood pressure, run-wise rsfMRI motion, rsfMRI inverse SNR, T1 inverse SNR, head coordinates (X, Y, Z), UK Biobank assessment center, and 10 genetic principal components were included as covariates.
Partitioned heritability analyses were conducted for the seven RSFA clusters and for each of the 400 individual cortical parcels. Using AHBA expression data, genes were rank ordered by their spatial cortical correlation to SST and PVALB. Genes without Entrez IDs were removed. The BioMart package76 was used to identify each gene’s transcription start and stop sites (±5000 base pairs) according to the GRCh37-hg19 genome assembly. Otherwise, the gene was cross-referenced to cortical eQTL databases from the NIH GTEx project43 and CommonMind consortium42. Intragenic (±5000 base pairs) and eQTL SNPs associated with the top 500 SST (NSNP = 8308) and PVALB (NSNP = 9571) correlated genes were used for partitioned heritability analyses, respectively denoted SSTSNP and PVALBSNP. A small subset of genes in each bin did not have analyzable variants using these criteria and thus did not contribute to results. Genetic-relatedness matrices for the SSTSNP and PVALBSNP partitions were generated, as well as one for all remaining genotyped SNPs. RSFA heritability accounted for by each genetic relatedness matrix was estimated simultaneously for each of the three partitions using GCTA41. Partitioned heritability was then defined as the phenotypic variance explained by either SSTSNP or PVALBSNP, divided by the total phenotypic variance. To calculate the significance of individual partitions, we consider the Wald test statistic against the null of , which follows a half-half mixture of (a χ2 distribution with a probability mass at zero) and (a χ2 distribution with 1 degree of freedom). Enrichment values were calculated to determine if the proportion of variability explained by a partition was greater than the proportion of variants within the partition, defined as:
| 1 |
where is the heritable variance explained by the SNP partition (e.g., PVALBSNP), is the heritable variance explained by all partitions, gpart is the number of variants within the SNP partition, and gtotal is the total number of genotyped SNPs. Standard error for SNP partitions were similarly scaled by the genome partition denominator. When calculating RSFA partitioned heritability across individual parcels (i.e., Fig. 5), those with outlier partitioned heritability (i.e., PVALBPART, SSTPART) and expression (i.e., PVALB, SST) greater than 4 standard deviations from the mean were excluded, resulting in 328 observations across cortex. The spatial relationship between partitioned heritability estimates and ex-vivo AHBA gene expression patterns was then quantified using correlation (Fig. 5).
GCTA heritability results were replicated using LDSC regression44. Preprocessing of UKB imputed genetic data included censoring of SNPs with imputation accuracy estimate less than 90% (i.e., INFO < 0.9), minor allele frequency <0.01, Hardy–Weinberg equilibrium p < 1 × 10−6, call rate <0.1, genotype rate <0.1, and removal of non-biallelic variants using Plink v2.0. Genome-wide association analyses were conducted using the linear regression form of GCTA’s fastGWA utility77. The same quantitative and categorical covariates were used across GCTA and GWAS analyses. LDSC based estimates of RSFA heritability were conducted on 1,158,800 HapMap3 variants that overlapped with variants in the UKB GWAS summary statistics, using precomputed LD scores from 1000 Genomes European data (i.e., “eur_w_ld_chr”). Partitioned RSFA heritability analyses were conducted by examining SNPs occurring near the coding regions of the 500 genes most spatially correlated to PVALB and SST (i.e., two non-overlapping 500-gene bins). SNP locations were mapped to genes using the biomaRt package in R and AHBA Entrez IDs (i.e., GRCh37 build)76. SNP annotation files encompassed genetic variants occurring ±10,000 base pairs from the start–stop positions of each gene. A small subset of genes in each bin did not have analyzable variants using these criteria and thus did not contribute to results. The LDSC “make_annot.py” tool was used to create annotation files, filtering on HapMap3 variants and using 1000 Genomes Phase 1 genetic data to estimate LD. Partitioned heritability of the SST and PVALB SNP sets was estimated in conjunction with a full baseline model of 53 annotations (i.e., “1000G_Phase1_baseline_ldscores”), using precomputed allele frequencies (i.e., 1000G_frq) and weights (i.e., “weights_hm3_no_hla”). These analyses provide a measure of genetic variance explained by the SST and PVALB SNP bins, conditioned on the baseline annotation model to prevent non-specific genetic signals from inflating stratified heritability estimates.
To assess whether schizophrenia polygenic risk was enriched among SST and PVALB correlated gene sets, competitive gene-set analysis was conducted using MAGMA46. Rank-ordered SST and PVALB genes were divided into twenty non-overlapping 500-gene bins. Schizophrenia summary statistics from the GWAS of Ripke and colleagues45 were used. Intragenic variants were defined using a ±5000 base pair window, and gene set enrichment was estimated simultaneously across all 40 gene bins, revealing whether a particular bin is more associated with polygenic risk for schizophrenia than all other genes. Polygenic risk for schizophrenia45 was calculated using PRSice47. Only the top-SNP from the major histocompatibility complex was used for the generation of individual risk scores. Benjamini–Hochberg false-discovery rate correction was conducted separately for each GWAS p-value threshold examined (e.g., correction for seven tests at the GWAS p < 1.0 threshold).
NIH Blueprint processing
Publicly available microarray data from six adult macaque primates (three female) were downloaded from the Gene Expression Omnibus website (https://www.ncbi.nlm.nih.gov/geo; accession number GSE31613)78. Expression values were converted from log10 to log2. Data from two macaques (one female) were excluded due to sparse sampling across cortex. Samples from the following 10 cortical regions were included in our analyses: OFC, ACC, medial temporal lobe, temporal area, DLPFC, A1C, S1C, M1C, V1, and V2. The collapseRows function was used in R to select the probe with the highest mean expression and ComBat was used to remove residual donor effects. SST and PVALB expression were mean and variance-normalized to reveal relative expression differences across cortex.
BrainSpan processing
Publicly available RNAseq reads per kilobase per million (RPKM) data from the Brainspan atlas were used to characterize patterns of interneuron-marker gene expression across development. Cortical tissue samples were analyzed from early fetal [8–12 post-conception weeks (pcw), donors = 10, samples = 88], early/mid fetal (13–21 pcw, donors = 10, samples = 88), late fetal (24–37 pcw; donors = 5, samples = 27), early infancy (4 months; donors = 3, samples = 22), late infancy (10 months; donors = 1, samples = 8), early childhood (1–4 years; donors = 5, samples = 41), mid/late childhood (8–11 years; donors = 2, samples = 30), adolescence (13–15 years; donors = 2, samples = 14), and adulthood (18–40 years; donors = 8, samples = 85) developmental stages. RNAseq probes without Entrez IDs were excluded and duplicated probes were removed by selecting the probe with the highest mean expression. Data was log2 transformed and the effect of donor was removed separately for each age group using ComBat79. Gene expression was then mean- and variance-normalized across cortical tissue samples separately for each developmental stage. When multiple ages were present in a development stage, age was included as a covariate in a linear regression predicting normalized SST expression from normalized PVALB expression.
Single-cell analysis and deconvolution
Single-nucleus droplet-based sequencing (snDrop-seq) data from Lake and colleagues31 was obtained from the Gene Expression Omnibus website (“GSE97930” [https://www.ncbi.nlm.nih.gov/geo]). Count matrices derived from unique molecular identifier (UMI) were analyzed, reflecting 19,368 cells from visual cortex (BA17) and 10,319 cells from frontal cortex (BA10 and BA6) across six postmortem adult brains. Collinearity among transcriptionally similar cell types was reduced by labeling cells according to 18 superordinate cell identities defined by Lake and colleagues31. Single-cell data were preprocessed using Seurat80. After checking for outlier cells and minimally expressed genes, default global-scaling normalization was applied (i.e., “LogNormalize”). Entrez IDs and gene symbols were used to cross-reference single-cell and AHBA data, and genes lacking matches or with more than one match were removed. Non-log transformed data were then analyzed using CIBERSORTx22 to impute cell type fractions present in the AHBA data. Independent gene signature matrices were defined with visual cortex and frontal cortex cell data. Cell type fractions were then estimated separately for each AHBA donor, once using a visual cell signature and once using a frontal cortex cell signature. Cell type fractions for each AHBA cortical sample were mapped to the cortical surface and summarized among Schaefer32 atlas ROIs in the same manner as single-gene expression data.
Replication analyses
In this paper, we sought to replicate findings with alternative methodologies and independent datasets whenever possible. In Fig. 1, we document a negative spatial relationship between SST and PVALB interneuron markers in cortex, using microarray data from the AHBA. This effect was replicated using RNASeq data from the AHBA (Supplementary Fig. 4), in an independent sample from the Brainspan Atlas (Fig. 1g), in cortex of non-human macaque primates (Fig. 1d, f), and with a complementary polygenic technique implemented with single-cell data and CibersortX (Fig. 2). We also identify stereotyped patterns of SST and PVALB expression in subcortical territories, which were replicated with independent measures of cell density in rodents (Fig. 3). Spatial associations between cortical function (i.e., RSFA) and markers of interneurons were also established with two techniques. The first approach examined highly cell-specific individual gene markers and the second approach utilized polygenic cellular deconvolution based on multivariate transcriptional signatures of cell types (Fig. 4). Further, heritability and partitioned heritability analyses of cortical RSFA were replicated with two techniques, GCTA and LD score regression (Fig. 5 and Supplementary Fig. 12). These methods yielded highly convergent results that were robust to genetic preprocessing choices (e.g., analysis of genotyped versus imputed variants) and model assumptions of underlying genetic architecture.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This work was supported by the National Institute of Mental Health (Grants K01MH099232 and R01MH120080 to A.J.H.), the National Science Foundation (DGE-1122492 to K.M.A.), and the National Institute on Aging (K99AG054573 to T.G.). Analyses were made possible by the high-performance computing facilities provided through the Yale Center for Research Computing. We thank B. J. Casey, Danielle Gerhard, Lauren Patrick, and Erica Ho for their feedback on early versions of the project. This work used data from the Allen Institute for Brain Science, NIH Blueprint Non-Human Primate Atlas, CommonMind Consortium, Brainspan Atlas of the Developing Human Brain, and has been conducted using the UK Biobank Resource under Application Number 25163. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 01/16/2018.
Author contributions
K.M.A. and A.J.H. designed the research. K.M.A. conducted the research. M.A.C., R.C., M.D.R., and T.G. provided analytic support. K.M.A. and A.J.H. wrote the manuscript and made figures. All authors edited the manuscript.
Data availability
The data that support these findings are either publicly available, provided with the paper, or are under third party restrictions. Publicly available data are accessible at the following locations: Allen Human Brain Atlas (https://human.brain-map.org/), UK Biobank (https://www.ukbiobank.ac.uk/), Brainspan Atlas of the Developing Human Brain (https://www.brainspan.org/), NIH Blueprint NHP Atlas (https://www.blueprintnhpatlas.org/), NIH GTEx (https://commonfund.nih.gov/gtex), CommonMind (https://www.nimhgenetics.org/resources/commonmind), snDrop-seq data at Gene Expression Omnibus “GSE97942” (https://www.ncbi.nlm.nih.gov/geo/), and GWAS data from the Psychiatric Genomics Consortium (https://www.med.unc.edu/pgc/).
Data not under third-party restrictions are available at https://github.com/HolmesLab/2020_NatComm_interneurons_cortical_function_schizophrenia.
Code availability
Code not under third-party restrictions are available at https://github.com/HolmesLab/2020_NatComm_interneurons_cortical_function_schizophrenia.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41467-020-16710-x.
References
- 1.Ramón y Cajal, S. Recollections of My Life (The MIT Press, 1989).
- 2.Kepecs A, Fishell G. Interneuron cell types are fit to function. Nature. 2014;505:318–326. doi: 10.1038/nature12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Markram H, et al. Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Burgos G, Cho RY, Lewis DA. Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol. Psychiatry. 2015;77:1031–1040. doi: 10.1016/j.biopsych.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fee C, Banasr M, Sibille E. Somatostatin-positive gamma-aminobutyric acid interneuron deficits in depression: cortical microcircuit and therapeutic perspectives. Biol. Psychiatry. 2017;82:549–559. doi: 10.1016/j.biopsych.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richiardi J, et al. Correlated gene expression supports synchronous activity in brain networks. Science. 2015;348:1241–1244. doi: 10.1126/science.1255905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krienen FM, Yeo BTT, Ge T, Buckner RL, Sherwood CC. Transcriptional profiles of supragranular-enriched genes associate with corticocortical network architecture in the human brain. Proc. Natl. Acad. Sci. USA. 2016;113:E469–E478. doi: 10.1073/pnas.1510903113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang G-Z, et al. Correspondence between resting-state activity and brain gene expression. Neuron. 2015;88:659–666. doi: 10.1016/j.neuron.2015.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson KM, et al. Gene expression links functional networks across cortex and striatum. Nat. Commun. 2018;9:1428. doi: 10.1038/s41467-018-03811-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burt JB, et al. Hierarchy of transcriptomic specialization across human cortex captured by structural neuroimaging topography. Nat. Neurosci. 2018;21:1251–1259. doi: 10.1038/s41593-018-0195-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim Y, et al. Brain-wide maps reveal stereotyped cell-type-based cortical architecture and subcortical sexual dimorphism. Cell. 2017;171:456–459. doi: 10.1016/j.cell.2017.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hendry SHC, Schwark HD, Jones EG, Yan J. Numbers and proportions of GABA-immunoreactive neurons in different areas of monkey cerebral cortex. J. Neurosci. 1987;7:1503–1519. doi: 10.1523/JNEUROSCI.07-05-01503.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawaguchi Y, Kubota Y. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb. Cortex. 1997;7:476–486. doi: 10.1093/cercor/7.6.476. [DOI] [PubMed] [Google Scholar]
- 14.Wang XJ, Tegner J, Constantinidis C, Goldman-Rakic PS. Division of labor among distinct subtypes of inhibitory neurons in a cortical microcircuit of working memory. Proc. Natl. Acad. Sci. USA. 2004;101:1368–1373. doi: 10.1073/pnas.0305337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaudhuri R, Knoblauch K, Gariel M-A, Kennedy H, Wang X-J. A large-scale circuit mechanism for hierarchical dynamical processing in the primate cortex. Neuron. 2015;88:419–431. doi: 10.1016/j.neuron.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cioli C, Abdi H, Beaton D, Burnod Y, Mesmoudi S. Differences in human cortical gene expression match the temporal properties of large-scale functional networks. PLoS ONE. 2014;9:e115913. doi: 10.1371/journal.pone.0115913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fulcher BD, Murray JD, Zerbi V, Wang X-J. Multimodal gradients across mouse cortex. Proc. Natl. Acad. Sci. U.S.A. 2019;116:4689–4695. doi: 10.1073/pnas.1814144116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JH, et al. Global and local fMRI signals driven by neurons defined optogenetically by type and wiring. Nature. 2010;465:788–792. doi: 10.1038/nature09108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cardin JA, et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niessing J, et al. Hemodynamic signals correlate tightly with synchronized gamma oscillations. Science. 2005;309:948–951. doi: 10.1126/science.1110948. [DOI] [PubMed] [Google Scholar]
- 21.Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newman AM, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller KL, et al. Multimodal population brain imaging in the UK Biobank prospective epidemiological study. Nat. Neurosci. 2016;19:1523–1536. doi: 10.1038/nn.4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wamsley B, Fishell G. Genetic and activity-dependent mechanisms underlying interneuron diversity. Nat. Rev. Neurosci. 2017;18:299–309. doi: 10.1038/nrn.2017.30. [DOI] [PubMed] [Google Scholar]
- 25.Wonders CP, Anderson SA. The origin and specification of cortical interneurons. Nat. Rev. Neurosci. 2006;7:687–696. doi: 10.1038/nrn1954. [DOI] [PubMed] [Google Scholar]
- 26.Wonders CP, et al. A spatial bias for the origins of interneuron subgroups within the medial ganglionic eminence. Dev. Biol. 2008;314:127–136. doi: 10.1016/j.ydbio.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hawrylycz MJ, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–399. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bakken TE, et al. A comprehensive transcriptional map of primate brain development. Nature. 2016;535:367–375. doi: 10.1038/nature18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Letinic K, Zoncu R, Rakic P. Origin of GABAergic neurons in the human neocortex. Nature. 2002;417:645–649. doi: 10.1038/nature00779. [DOI] [PubMed] [Google Scholar]
- 30.Sidman RL, Rakic P. Neuronal migration, with special reference to developing human brain: a review. Brain Res. 1973;62:1–35. doi: 10.1016/0006-8993(73)90617-3. [DOI] [PubMed] [Google Scholar]
- 31.Lake BB, et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018;36:70–80. doi: 10.1038/nbt.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaefer A, et al. Local-global parcellation of the human cerebral cortex from intrinsic functional connectivity MRI. Cereb. Cortex. 2018;28:3095–3114. doi: 10.1093/cercor/bhx179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogel JW, et al. A molecular gradient along the longitudinal axis of the human hippocampus informs large-scale behavioral systems. Nat. Commun. 2020;11:960. doi: 10.1038/s41467-020-14518-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kovner R, et al. Somatostatin gene and protein expression in the non-human primate central extended amygdala. Neuroscience. 2019;400:157–168. doi: 10.1016/j.neuroscience.2018.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haber SN, Knutson B. The reward circuit: linking primate anatomy and human imaging. Neuropsychopharmacology. 2009;35:4–26. doi: 10.1038/npp.2009.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murty VP, et al. Resting state networks distinguish human ventral tegmental area from substantia nigra. NeuroImage. 2014;100:580–589. doi: 10.1016/j.neuroimage.2014.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murray JD, et al. A hierarchy of intrinsic timescales across primate cortex. Nat. Neurosci. 2014;17:1661–1663. doi: 10.1038/nn.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bijsterbosch J, et al. Investigations into within- and between-subject resting-state amplitude variations. NeuroImage. 2017;159:57–69. doi: 10.1016/j.neuroimage.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Visscher PM, et al. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 2017;101:5–22. doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skene NG, et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 2018;50:825–833. doi: 10.1038/s41588-018-0129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fromer M, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016;19:1442–1453. doi: 10.1038/nn.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lonsdale J, et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bulik-Sullivan BK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Euesden J, Lewis CM, O’Reilly PF. PRSice: Polygenic Risk Score software. Bioinformatics. 2015;31:1466–1468. doi: 10.1093/bioinformatics/btu848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Felleman DJ, Van Essen DC. Distributed hierarchical processing in the primate cerebral cortex. Cereb. Cortex. 1991;1:1–47. doi: 10.1093/cercor/1.1.1-a. [DOI] [PubMed] [Google Scholar]
- 49.Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry. 2003;60:1187–1192. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- 51.Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci. 2010;11:100–113. doi: 10.1038/nrn2774. [DOI] [PubMed] [Google Scholar]
- 52.Akbarian S, et al. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch. Gen. Psychiatry. 1995;52:258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- 53.Mauney SA, et al. Developmental pattern of perineuronal nets in the human prefrontal cortex and their deficit in schizophrenia. Biol. Psychiatry. 2013;74:427–435. doi: 10.1016/j.biopsych.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morgan SE, et al. Cortical patterning of abnormal morphometric similarity in psychosis is associated with brain expression of schizophrenia-related genes. Proc. Natl. Acad. Sci. USA. 2019;116:9604–9609. doi: 10.1073/pnas.1820754116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Romme IAC, de Reus MA, Ophoff RA, Kahn RS, van den Heuvel MP. Connectome disconnectivity and cortical gene expression in patients with schizophrenia. Biol. Psychiatry. 2017;81:495–502. doi: 10.1016/j.biopsych.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 56.Marín O. Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 2012;13:107–120. doi: 10.1038/nrn3155. [DOI] [PubMed] [Google Scholar]
- 57.Toker L, Mancarci BO, Tripathy S, Pavlidis P. Transcriptomic evidence for alterations in astrocytes and parvalbumin interneurons in subjects with bipolar disorder and schizophrenia. Biol. Psychiatry. 2018;84:787–796. doi: 10.1016/j.biopsych.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fuchs T, et al. Disinhibition of somatostatin-positive GABAergic interneurons results in an anxiolytic and antidepressant-like brain state. Mol. Psychiatry. 2017;22:920–930. doi: 10.1038/mp.2016.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schmaal L, et al. Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the ENIGMA Major Depressive Disorder Working Group. Mol. Psychiatry. 2017;22:900–909. doi: 10.1038/mp.2016.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anderson, K. M. et al. Convergent molecular, cellular, and neural signatures of major depressive disorder. Preprint at 10.1101/2020.02.10.942227 (2020). [DOI] [PMC free article] [PubMed]
- 61.Holmes AJ, et al. Individual differences in amygdala-medial prefrontal anatomy link negative affect, impaired social functioning, and polygenic depression risk. J. Neurosci. 2012;32:18087–18100. doi: 10.1523/JNEUROSCI.2531-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wolfe CJ, Kohane IS, Butte AJ. Systematic survey reveals general applicability of ‘guilt-by-association’ within gene coexpression networks. BMC Bioinformatics. 2005;6:227. doi: 10.1186/1471-2105-6-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garrett DD, Lindenberger U, Hoge RD, Gauthier CJ. Age differences in brain signal variability are robust to multiple vascular controls. Sci. Rep. 2017;7:10149. doi: 10.1038/s41598-017-09752-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duncan L, et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun. 2019;10:3328. doi: 10.1038/s41467-019-11112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Elliott ML, et al. A polygenic score for higher educational attainment is associated with larger brains. Cereb. Cortex. 2018;491:56–59. doi: 10.1093/cercor/bhy219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ge T, Holmes AJ, Buckner RL, Smoller JW, Sabuncu MR. Heritability analysis with repeat measurements and its application to resting-state functional connectivity. Proc. Natl. Acad. Sci. USA. 2017;114:5521–5526. doi: 10.1073/pnas.1700765114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reardon PK, et al. Normative brain size variation and brain shape diversity in humans. Science. 2018;360:1222–1227. doi: 10.1126/science.aar2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arnatkevic̆iūtė A, Fulcher BD, Fornito A. A practical guide to linking brain-wide gene expression and neuroimaging data. NeuroImage. 2019;189:353–367. doi: 10.1016/j.neuroimage.2019.01.011. [DOI] [PubMed] [Google Scholar]
- 69.Romero-Garcia R, et al. Structural covariance networks are coupled to expression of genes enriched in supragranular layers of the human cortex. NeuroImage. 2018;171:256–267. doi: 10.1016/j.neuroimage.2017.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marcus DS, et al. Informatics and data mining tools and strategies for the Human Connectome Project. Front. Neuroinformatics. 2011;5:1–12. doi: 10.3389/fninf.2011.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Choi EY, Yeo BTT, Buckner RL. The organization of the human striatum estimated by intrinsic functional connectivity. J. Neurophysiol. 2012;108:2242–2263. doi: 10.1152/jn.00270.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hwang K, Bertolero MA, Liu WB, D’Esposito M. The human thalamus is an integrative hub for functional brain networks. J. Neurosci. 2017;37:5594–5607. doi: 10.1523/JNEUROSCI.0067-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jenkinson M, Bannister P, Brady M, Smith S. Improved optimization for the robust and accurate linear registration and motion correction of brain images. NeuroImage. 2002;17:825–841. doi: 10.1016/s1053-8119(02)91132-8. [DOI] [PubMed] [Google Scholar]
- 74.Cox RW. AFNI: software for analysis and visualization of functional magnetic resonance neuroimages. Comput. Biomed. Res. 1996;29:162–173. doi: 10.1006/cbmr.1996.0014. [DOI] [PubMed] [Google Scholar]
- 75.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Durinck S, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- 77.Jiang L, et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nat. Genet. 2019;51:1749–1755. doi: 10.1038/s41588-019-0530-8. [DOI] [PubMed] [Google Scholar]
- 78.Bakken TE, et al. A comprehensive transcriptional map of primate brain development. Nature. 2016;535:367–375. doi: 10.1038/nature18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2006;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 80.Stuart T, et al. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The data that support these findings are either publicly available, provided with the paper, or are under third party restrictions. Publicly available data are accessible at the following locations: Allen Human Brain Atlas (https://human.brain-map.org/), UK Biobank (https://www.ukbiobank.ac.uk/), Brainspan Atlas of the Developing Human Brain (https://www.brainspan.org/), NIH Blueprint NHP Atlas (https://www.blueprintnhpatlas.org/), NIH GTEx (https://commonfund.nih.gov/gtex), CommonMind (https://www.nimhgenetics.org/resources/commonmind), snDrop-seq data at Gene Expression Omnibus “GSE97942” (https://www.ncbi.nlm.nih.gov/geo/), and GWAS data from the Psychiatric Genomics Consortium (https://www.med.unc.edu/pgc/).
Data not under third-party restrictions are available at https://github.com/HolmesLab/2020_NatComm_interneurons_cortical_function_schizophrenia.
Code not under third-party restrictions are available at https://github.com/HolmesLab/2020_NatComm_interneurons_cortical_function_schizophrenia.