

Abstract
Background
The coronavirus disease 2019 (COVID‐19) pandemic has caused a large surge of acute respiratory distress syndrome (ARDS). Prior phase I trials (non–COVID‐19) demonstrated improvement in pulmonary function in patients ARDS using fibrinolytic therapy. A follow‐up trial using the widely available tissue‐type plasminogen activator (t‐PA) alteplase is now needed to assess optimal dosing and safety in this critically ill patient population.
Objective
To describe the design and rationale of a phase IIa trial to evaluate the safety and efficacy of alteplase treatment for moderate/severe COVID‐19–induced ARDS.
Patients/Methods
A rapidly adaptive, pragmatic, open‐label, randomized, controlled, phase IIa clinical trial will be conducted with 3 groups: intravenous alteplase 50 mg, intravenous alteplase 100 mg, and control (standard‐of‐care). Inclusion criteria are known/suspected COVID‐19 infection with PaO2/FiO2 ratio <150 mm Hg for > 4 hours despite maximal mechanical ventilation management. Alteplase will be delivered through an initial bolus of 50 mg or 100 mg followed by heparin infusion for systemic anticoagulation, with alteplase redosing if there is a >20% PaO2/FiO2 improvement not sustained by 24 hours.
Results
The primary outcome is improvement in PaO2/FiO2 at 48 hours after randomization. Other outcomes include ventilator‐ and intensive care unit–free days, successful extubation (no reintubation ≤3 days after initial extubation), and mortality. Fifty eligible patients will be enrolled in a rapidly adaptive, modified stepped‐wedge design with 4 looks at the data.
Conclusion
Findings will provide timely information on the safety, efficacy, and optimal dosing of t‐PA to treat moderate/severe COVID‐19–induced ARDS, which can be rapidly adapted to a phase III trial (NCT04357730; FDA IND 149634).
Keywords: acute respiratory distress syndrome, clinical trial, coagulopathy, COVID‐19, fibrinolysis shutdown, tissue‐type plasminogen activator
Essentials.
Fibrinolytics have been used in phase I trials to treat acute respiratory distress syndrome (ARDS) with efficacy and low risk of bleeding complications.
Coronavirus disease 2019 (COVID‐19) is associated with microthrombi of the lungs and ARDS.
STARS is an adaptive, pragmatic, open‐label, phase IIa trial using tissue‐type plasminogen activator (t‐PA) to treat COVID‐19–related ARDS.
Two different dosing regimens of t‐PA will be used to test for efficacy in improving oxygenation at 48 hours from study enrollment.
1. INTRODUCTION
The worldwide incidence of coronavirus disease 2019 (COVID‐19) continues to rise, taxing the health care and economic resources of countries throughout the developed world. Based on the clinical experience in China and Italy, it is estimated that 5% to 27% of hospitalized patients with COVID‐19 will require prolonged intensive care, 1 , 2 , 3 , 4 , 5 , 6 with 50% to 99% requiring mechanical ventilation (MV) for viral‐induced pneumonitis progressing to acute hypoxemic respiratory failure and acute respiratory distress syndrome (ARDS). 2 , 7 , 8 In patients requiring MV, the reported mortality exceeds 50% 7 , 9 and approached 90% in a recent report from New York City. 10 There is no specific treatment for COVID‐19 ARDS other than routine mechanical ventilation, although prone positioning seems to be particularly effective in this population, 11 either as a consequence of enhanced alveolar drainage or redistribution of perfusion to better aerated portions of the lungs.
A remarkable feature of the pulmonary pathophysiology in COVID‐19 ARDS is the preservation of relatively normal lung compliance and a low incidence of barotrauma 12 suggesting extensive shunting, ventilation‐perfusion mismatch, and loss of regulation of alveolar perfusion. Autopsy and surgical specimens in these patients show a range of pathologic findings including diffuse alveolar damage, fibrin accumulation in the alveoli, the presence of mononuclear cell infiltrates and megakaryocytes, as well as fibrin‐platelet microthrombi in the pulmonary vasculature. 1 , 3 The concept of accumulation of pulmonary microthrombi leading to death dates back to 1845. 13 The angiographic appearance of filling defects of the pulmonary vasculature in patients with ARDS has been associated with a high mortality rate for decades. 14 , 15 , 16 , 17 Animal models of irreversible shock have demonstrated clots in organs driving organ failure. 18 , 19 This can be reversed with preemptive heparin 20 , 21 or postshock fibrinolytics. 22 Autopsies of critically ill patients also demonstrated clots in the organs of patients in the intensive care unit who died from organ failure. 19 , 23 These observations were eventually translated to 2 separate phase I human trials, 24 , 25 which were not followed up.
Given these vascular and hematologic findings and the distinct nature of the COVID‐19 ARDS, with preserved pulmonary mechanics, we postulate that this advanced ARDS is due to the microthrombosis and resistance to clot lysis in the pulmonary circulation. We believe these factors directly contribute to the high‐shunt type of hypoxemic respiratory failure seen in COVID‐19 ARDS. We hypothesized that administration of alteplase, a tissue‐plasminogen activator (t‐PA), followed by systemic anticoagulation will improve the PaO2/FiO2 ratio 48 hours after treatment.
1.1. Objective
We aimed to describe the design and rationale of a phase IIa trial (NCT04357730) that will evaluate the safety and efficacy of tPA (alteplase) treatment for moderate to severe ARDS in the setting of COVID‐19 infection.
2. METHODS
This is a phase IIa, open‐label clinical trial with a modified stepped‐wedge design, testing systemic administration of fibrinolytic therapy with alteplase (using Activase manufactured by Genentech, Inc) versus standard of care for patients infected with COVID‐19 resulting in severe ARDS. The study is registered at clinicaltrials.gov (NCT04357730), has received approval to proceed by the US Food and Drug Administration (FDA; IND 149634), and by all institutions’ institutional review boards (IRBs). The design is a rapidly adaptive, pragmatic clinical trial, with 3 interim analyses and 1 final look at the data. Preplanned adaptations described below will be contemplated at each interim analysis or earlier if recommended by the Data Safety Monitoring Board (DSMB).
2.1. Inclusion criteria
We will include patients ages 18‐75 years, with known or suspected COVID‐19 infection, with a normal neurological exam at time of enrollment (if patient is on paralytics, the patient has been awakened and showed no new neurological deficits in a complete neurological exam or had a magnetic resonance imaging (MRI)/computed tomography (CT) scan in the past 4.5 hours with no evidence of stroke), with a PaO2/FiO2 ratio <150 mm Hg (at sea level or adjusted for altitude) persisting for >4 hours despite maximal MV management according to each institution’s ventilation protocols (FiO2 ≥ 60% and positive end‐expiratory pressure [PEEP] ≥ 10 cm H2O). If obtaining arterial blood gases is not possible due to a surge‐related shortage of blood gas syringes, as we have experienced previously, we will infer the PaO2/FiO2 ratio from percent saturation of hemoglobin with oxygen as measured by pulse oximetry (SpO2), using the nonlinear imputation developed by the National Heart, Lung, and Blood Institute’s PETAL (Prevention and Early Treatment of Acute Lung Injury) Network Collaborators. 26 A normal neurological exam or CT/MRI scan to demonstrate no evidence of an acute stroke is needed due to recent reports of large‐vessel stroke as a presenting feature of COVID‐19 in young individuals. 27
Patients will be enrolled based on clinical characteristics, without consideration of language (using hospital interpreters and translated consent), race/ethnicity, or sex/gender. Patients are eligible to participate even if they are concurrently enrolled in other COVID‐19 therapeutic trials. Exclusion criteria are listed in Table 1.
TABLE 1.
Inclusion and exclusion criteria for the STARS trial
| Inclusion criteria: age 18‐75 y old with known or suspected COVID‐19 infection with a PaO2/FiO2 ratio <150 (at sea level) or (inferred PaO2/FiO2 ratio from SpO2 if an arterial blood gas is unavailable) persisting for >4 h despite maximal mechanical ventilation management according to each institution's ventilation protocols. |
| Absolute exclusion criteria (documented at the time of enrollment): |
| Stroke or inability to demonstrate a normal neurological exam unless a CT scan within 4.5 h of enrollment excludes a cerebral vascular event |
| Active bleeding |
| Acute myocardial infarction or history of myocardial infarction within the past 3 wk or cardiac arrest during hospitalization |
| Hemodynamic instability with noradrenaline >0.2 µg/kg/min |
| Acute renal failure requiring dialysis |
| Liver failure (escalating liver failure with total bilirubin > 3 mg/dL) |
| Cardiac tamponade |
| Bacterial endocarditis |
| Severe uncontrolled hypertension defined as SBP >185 mm Hg or DBP >110 mm Hg |
| History of severe head injury within prior 3 mo, or prior history of intracranial hemorrhage |
| Seizure during prehospital course or during hospitalization for COVID‐19 |
| Diagnosis of brain tumor, arteriovenous malformation (AVM), or ruptured aneurysm |
| Currently on ECMO |
| Major surgery or major trauma within the past 2 wk |
| GI or GU bleed within the past 3 wk |
| Known bleeding disorder |
| Arterial puncture at a noncompressible site within the past 7 d |
| Lumbar puncture within past 7 d |
| Pregnancy |
| INR > 1.7 (with or without concurrent use of warfarin) |
| Platelet count <100 × 109/L or history of HITT |
| Fibrinogen <300 mg/dL |
| Known abdominal or thoracic aneurysm |
| History of CNS malignancy or CNS metastasis within past 5 y |
| History of non‐CNS malignancy within the past 5 y that commonly metastasizes to the brain (lung, breast, melanoma) |
| Prisoner status |
There are 3 treatment arms:
Group tPA50 (n = 20) will receive 50 mg of alteplase intravenous bolus administration over 2 hours, given as a 10‐mg push followed by the remaining 40 mg over a total time of 2 hours. Immediately following the alteplase infusion, 5000 U of unfractionated heparin (UFH) will be delivered; the heparin drip will be continued to maintain the activated partial thromboplastin time (aPTT) at 60 to 80 seconds (2.0‐2.5 times the upper limit of normal). This t‐PA protocol is a modification of the GUSTO (Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries) I to III trials. 28 , 29
Group tPA100 (n = 20) will receive 100 mg of t‐PA intravenous bolus administration over 2 hours, given as a 10‐mg push followed by the remaining 90 mg over a total time of 2 hours. Immediately following the t‐PA infusion, 5000 U of UFH will be delivered, and the heparin drip will be continued to maintain the aPTT at 60 to 80 seconds (2.0‐2.5 times the upper limit of normal). This t‐PA protocol is similar to that used by Konstantinides et al. 30
Control: institution’s standard‐of‐care protocol for ARDS.
Rebolusing of tPA is permitted in the first two intervention groups, particularly in those patients who show an initial transient response, but is not sustained (<50% PaO2/FiO2 improvement by 24 hours). All exclusion criteria (Table 1) also apply to the second t‐PA (alteplase) bolus.
Other modifications of the alteplase dosing are as follows:
Fibrinogen monitoring: For all t‐PA administration groups, fibrinogen levels will be measured before and after t‐PA intravenous bolus, 6 hours after the start of the infusion, then every 6 hours for first 24 hours, and once a day for 6 days following treatment intervention in all the groups (see detailed lab testing schedule below). If fibrinogen levels fall below 300 mg/dL, the second bolus of t‐PA (alteplase) will not be given.
Heparin dosing: An infusion of unfractionated heparin will be continued for up to 7 days or until the patient is extubated and has an O2 requirement of ≤4 L/min by nasal cannula, and titrated to maintain the activated partial thromboplastin time to 60 to 80 seconds (2.0‐2.5 times the upper limit of normal). The goal of this treatment is to prevent recurrent microvascular thrombotic hypoxemia or macrovascular complications (stroke, myocardial infarction, or venous thromboembolism) due to possible rebound t‐PA effects causing hypercoagulability. If necessary, an infusion of antithrombin concentrate will be administered in heparin‐resistant patients.
2.2. Diverse positioning and/or paralytic agents for ventilation
If the position or use of paralytics must be changed before the 24‐ and 48‐hours post‐randomization, the PaO2/FiO2 measured immediately before these changes (within <6 hours of the 48‐hour postrandomization end point) will be used as primary outcome.
2.3. Outcomes
The primary outcome of interest is change in PaO2/FiO2 at 48 hours from randomization. Secondary outcomes are listed in Table 2.
TABLE 2.
Primary and secondary outcomes for the STARS trial
| Outcome | Timing |
|---|---|
| Primary outcome: PaO2/FiO2 improvement from pre‐to‐post intervention | 48 h after randomization. |
| Secondary outcomes | |
| Achievement of PaO2/FiO2 ≥200 or 50% increase in PaO2/FiO2 (whichever is lower) | 48 h after randomization |
| National Early Warning Score (NEWS) a |
48 h after randomization 14 d |
| NIAID ordinal scale b | |
| Reduction of FiO2 <80% (if started on higher concentration) | |
| Return to supine or lateral position (if started in prone position) | |
| Reduction of positive end expiratory pressure | |
| Reduction of inhaled prostonoids (if started before t‐PA therapy) | |
| Reduction of inhaled nitric oxide (if started before t‐PA therapy) | |
| Reduction of paralytic (if on before t‐PA therapy) | |
| Reciever operating characteristic curve of coagulation variables associated with achievement a PaO2/FiO2 >50% | |
| Reciever operating characteristic curve of coagulation variables associated with bleeding complications | |
| 48 h in‐hospital mortality | |
| 14 d in‐hospital mortality | 14 d |
| 28 d in‐hospital mortality | 28 d |
| ICU‐free days (up to 28 d) | 28 d |
| In‐hospital coagulation‐related event‐free (arterial and venous) days (up to 28 d) | 28 d |
| Ventilator‐free days (up to 28 d) | 28 d |
| Successful extubation (no reintubation <3 d after initial extubation) | 28 d |
| Survival to discharge | Discharge |
National Early Warning Score (NEWS2): based on 7 clinical parameters (respiration rate, oxygen saturation, any supplemental oxygen, temperature, systolic blood pressure, heart rate, level of consciousness); bNIAID ordinal scale: The ordinal scale is an assessment of the clinical status as follows: (i) death; (ii) hospitalized, on invasive mechanical ventilation or extracorporeal membrane oxygenation; (iii) hospitalized, on noninvasive ventilation or high‐flow oxygen devices; (iv) hospitalized, requiring supplemental oxygen; (v) hospitalized, not requiring supplemental oxygen–requiring ongoing medical care (COVID‐19 related or otherwise); (vi) hospitalized, not requiring supplemental oxygen—no longer requires ongoing medical care; (vii) not hospitalized, limitation on activities and/or requiring home oxygen; (viii) not hospitalized, no limitations on activities (we will combine 7 and 8 as discharge from hospital to home, as the trial is limited to in‐hospital morbidity/mortality).
2.4. Rapidly adaptive design (Figure 1)
The design is a rapidly adaptive, pragmatic clinical trial, with 3 interim analyses and 1 final look at the data, with test boundaries determined by the Pocock method to maintain overall experiment error at <0.05. Preplanned adaptations described below will be contemplated at each interim analysis or earlier if recommended by the DSMB. For rapid efficacy assessment to isolate the arm(s) with the highest likelihood of success and lowest bleeding risk, we will deploy each intervention arm sequentially up to each interim analysis, in a modified stepped‐wedge fashion 31 , with preplanned adaptations(below) at each interim analysis Figure 1.
FIGURE 1.
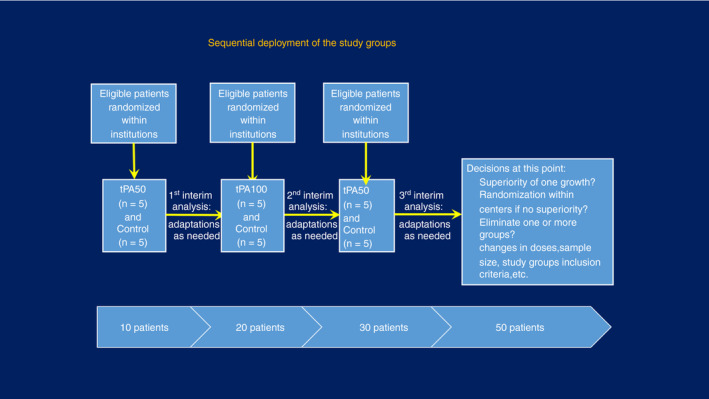
Study design of the STARS trial
2.5. Data collection and storage
Study data will be collected and managed using REDCap (Research Electronic Data Capture) electronic data capture tools hosted by the University of Colorado Anschutz Medical Campus. 32
2.6. Randomization
All randomizations will be conducted intrahospital (ie, no cluster randomization) to avoid the confounding effect of practice variation, in blocks of 10 to allow better distribution between groups at each interim analysis. It will be done by the Data Coordinating Center and automated in a REDCap instrument. Upon confirmed eligibility and consent, the REDCap instrument will reveal the assignment (Group tPA50, Group tPA100, Control) to the pharmacy of the enrolling institution, which will then release the drug if the patient was assigned to one of the intervention groups. Time 0 is assigned as the time of randomization. We anticipate that each of the 5 centers will enroll 5 to 10 patients.
2.7. Sample size rationale
The sample size was fixed at n = 50 (with 20 patients in each intervention group and 10 patients in the control group) due to budgetary and feasibility constraints. The minimum detectable difference was then calculated using PASS version 14.0 (NCSS LLC, Kaysville, UT, USA), focusing on the primary outcome (PaO2/FiO2 improvement) and assuming (i) power = 80%, confidence = 95%, and four sequential tests (3 interim + 1 final), using the Pocock method to determine test boundaries; (ii) potential improvement assumptions based on a previous study 25 as well as a more favorable scenario with mean baseline PaO2/FiO2 = 149 with an overestimated standard deviation of 100, (iii) design effect = 1.12 due to the study’s multicenter nature (intraclass correlation coefficient = 0.03, 33 , 34 average cluster = 5); and (iv) 20% inflation to account for premature death or withdrawal for any reasons.
A sample size of 50 (20 in each intervention group and 10 in the control group) patients would detect a ≥68% improvement in PaO2/FiO2 between the 2 intervention groups and ≥73% improvement between intervention groups and controls. While balanced group sizes will maximize a study’s statistical power, unequal randomization ratios will only significantly reduce the power of a study if the ratio is 3:1 or more. Reasons for the unequal randomization include (i) more safety information, an essential component of a phase IIa study; (ii) experience with dosing of t‐PA; and (iii) to allow 3 equal sequential phases that would inform the remainder of the trial. 35 The initial 2 phases (t‐PA 50 mg vs control; t‐PA 100 mg vs control) will provide a signal that allows the termination of the control arm.
Recruitment will assume at least a 30% increase to account for refusal or inability to consent. We anticipate enrolling enough individuals to result in a sample of 50 eligible patients, to be reevaluated during each of the interim analyses. A legally authorized representative, as defined by each state and each institution’s legislation and policies, will be able to consent.
2.8. Criteria for stopping the clinical trial early for efficacy or harm
Criteria include reaching adjusted P value for the primary outcome and at least 1 of the secondary outcomes at all follow‐up time points, or DSMB deemed the harm profile unacceptable.
2.9. Criteria for stopping for futility
We will follow the guidelines established by Jitlal et al 36 .These criteria are (i) low conditional power (<15%), calculated using bootstrapping simulations, based on the target minimum differences for all primary and secondary outcomes; (ii) observed difference size in the primary or secondary outcomes favor the control group (<5%); (iii) the DSMB and trial team agree that enough patients and events have been observed so far to produce a reliable effect; (iv) only 1 center interested in continuing enrollment; and (v) no evidence of an effect in any prespecified subgroups. If the DSMB deemed the adverse events profile acceptable, we may wish to continue to ensure that a modest effect is not missed.
2.10. Preplanned adaptations at each interim analysis
The study interim analyses will be used to propose preplanned modifications based on observed effects, recruitment, eligibility, and other aspects of the study as determined below.
Drop/add study arms: Deploying study arms sequentially (vs in parallel) allows sufficient sample sizes in each arm to assess outcomes and adverse events. Study arms that show significant improvement may ethically preclude the deployment of other arms. Similarly, study arms that show adverse events (as listed) attributable to the intervention (per trial team with DSMB/IRB determination) or minimal/no improvement may be eliminated. Study arms may be added if concurrent trials demonstrate significant evidence of benefit of a different route, dose, or mode of administration of the study drugs.
Inclusion criteria: Although currently the trial entry criteria are based on age and PaO2/FiO2, we recognize the potential role of coagulation assays (eg, D‐dimer, fibrinogen, fibrinolysis) in better defining the group most likely to benefit from the fibrinolytic intervention. Thus, such assays may be added as entry criteria if identified as predictors of good results during interim analyses or in other clinical trials. In addition, if the stratified analysis on initial PaO2/FiO2 shows benefit or harm in low and moderate PaO2/FiO2, the PaO2/FiO2 level for entry in the study may be modified to increase the probability of benefit.
Sample size: The current sample size is defined by budget and feasibility constraints and may prove insufficient if the effect detected is substantial but there is low power to detect it. A larger sample size may be recommended by the trial team and the DSMB, in which case we will pursue additional resources to increase enrollment.
Cessation rules: Based on interim analyses, coagulation and oxygenation variables may become important determinants of benefit/risk for the subjects as explained above; thus, these variables may be proposed as further determinants for cessation rules.
Enrollment/refusal rates: Modifications on enrollment and consent procedure may be proposed to remedy low enrollment and high refusal rate. One potential alternative is the addition of an observational arm as done by Pieracci et al. 37
Crossover: If 1 treatment arm shows a signal of benefit (as defined in our proposed outcomes), we are under the ethical mandate to offer it to patients who were enrolled in the other arms but did not show improvement. These patients “cross over” to the alternative arm. The analysis will be conducted as an intent‐to‐treat approach (patients are analyzed according to their initial assigned group) and subsequently in a separate as‐treated analysis considering the combination of the 2 treatments.
Comparison of prone/supine position: Additional arms or change in entry criteria may have to added if the prone position for ventilation is demonstrated to have a major benefit (eg, criteria for entry may be modified to PaO2/FiO2 <150 in prone position).
Doses/duration/administration mode of t‐PA and heparin: As more is learned during this trial as well as other clinical trials about the administration of t‐PA in relation to other ventilation techniques (prone position, PEEP, pulmonary vasodilators, etc) and the risk/benefit associated with the doses, duration, and model of administration (eg, bolus vs continuous drip), it may be beneficial for study subjects to modify the study arms.
2.11. Laboratory measurements
Laboratory tests obtained on all patients will include:
Arterial blood gases, fibrinogen and D‐dimer levels, prothrombin time/International Normalized Ratio, aPTT, C‐reactive protein, and complete blood count with platelet count: pre and post alteplase intravenous bolus (only baseline for controls), then every 6 hours for the first 24 hours, and once a day for 7 days (or earlier if patient is extubated) following treatment intervention in all the groups.
Thrombelastography (TEG) or rotational thromboelastometry (ROTEM) (where available): pre and post alteplase intravenous bolus (baseline for controls), then every 6 hours for the first 24 hours, and once a day for 7 days (or earlier if patient is extubated) following treatment intervention in all the groups.
Troponin, creatinine, bilirubin, alanine aminotransferase, and creatine kinase pre‐alteplase (baseline for controls) and at 24 and 72 hours after infusion.
2.12. Safety assessment
Safety considerations and monitoring with plan during the intervention administration are listed in Table 3. The estimated risk of the most feared complication, intracranial hemorrhage, is estimated to be 0.72% 28 if total t‐PA dose is <1.4 mg/kg over total infusion. Estimated risk of severe or life‐threatening bleeding other than intracranial is 0.4% 38 if total t‐PA dose is <1.4 mg/kg over total infusion. Given the emergence of data on the increased rate of thrombotic strokes in young patients with COVID‐19, 27 exclusion of stroke prior to fibrinolytic therapy is essential. The inability to demonstrate a normal neurological exam unless a CT scan within 4.5 hours 39 of enrollment excludes a cerebral vascular event represents an absolute exclusion to study enrollment. Safety assessments will consist of monitoring and reporting adverse events and serious adverse events per protocol. This includes any adverse event, defined as any unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease temporally associated with the use of alteplase or other protocol‐imposed intervention (eg, heparin, blood sampling). Given alteplase and heparin relative short half‐life (<72 minutes and <90 minutes, respectively), any adverse event manifesting within 3 hours of administration of the intervention drugs would be considered potentially temporally related to the intervention drugs. Adverse events associated with blood sampling are considered temporally related if happening within 30 minutes of the sampling. Methods and timing of safety check assessments are listed in Table 3.
TABLE 3.
Safety check assessments: methods and timing
| Serious adverse events | Method for safety check | Safety check frequency | Cessation rule a |
|---|---|---|---|
| Death | NA | NA | NA |
| Cardiopulmonary arrest | NA | NA | Any cardiopulmonary arrest |
| Allergic reactions including angioedema | Clinical exam | Clinical exam before, during, and immediately after alteplase infusion; every 6 h after alteplase infusion up to 24 h; at least every 24 h after alteplase infusion during heparin infusion or more frequently if any abnormality detected | Any allergic reaction |
| Worsening of neurological function | Clinical neurological exam and imaging if applicable per care provider’s decision. Most patients will use GCS without verbal component | Clinical exam before, during, and immediately after alteplase infusion; every 6 h post alteplase infusion up to 24 h; at least every 24 h after alteplase infusion during heparin infusion or more frequently if any abnormality detected Imaging per attending’s discretion | GCS decrease of >2 points or focal deficit within 24 h of study drug infusion or new hemorrhage on CT scan or MRI |
| Worsening of pulmonary function | Arterial blood gas and ventilation indices | Every 6 h in the first 24 h and every 12 until 48 h; if second alteplase dose, every 6 h until 48 h | >30% PaO2/FiO2 baseline reduction |
| External bleeding | Clinical exam | Clinical exam before, during, and immediately after alteplase infusion; every 6 h post alteplase infusion up to 24 h; at least every 24 h after alteplase infusion during heparin infusion or more frequently if any abnormality detected | Unresponsive to compression |
| Gastrointestinal bleeding | Clinical exam and hemoglobin | Clinical exam before, during, and immediately after alteplase infusion; every 6 h post alteplase infusion up to 24 h; at least every 24 h after alteplase infusion during heparin infusion or more frequently if any abnormality detected. Endoscopic exam per attending's discretion. | Hemoglobin reduction >3 g/dL within 24 hours of study drug intervention or requiring RBC transfusion with suspected gastrointestinal bleeding |
| Hemoptysis | Clinical exam | Clinical exam before, during, and immediately after alteplase infusion; every 6 h after alteplase infusion up to 24 h; at least every 24 h after alteplase infusion during heparin infusion or more frequently if any abnormality detected. Endoscopic exam per attending’s discretion. | Persistent hemoptysis for ≥4 h or compromising airway |
| Hematuria | Clinical exam | Clinical exam before, during, and immediately after alteplase infusion; every 6 h post alteplase infusion up to 24 h; at least every 24 h after alteplase infusion during heparin infusion or more frequently if any abnormality detected. Endoscopic exam per attending’s discretion. | Persistent hematuria for ≥4 h or urinary obstruction |
| Retroperitoneal bleeding | Clinical exam pre, during and immediately post alteplase infusion; every 6 h post alteplase infusion up to 24 h; at least every 24 h after alteplase infusion during heparin infusion or more frequently if any abnormality detected. Endoscopic exam per attending's discretion. | Hemoglobin reduction >3 g/dL within 24 h of infusion of study drug infusion or requiring RBC transfusion | |
| Tube thoracotomy | Clinical exam and Hgb | Clinical exam before, during, and immediately after alteplase infusion; every 6 h post alteplase infusion up to 24 h; at least every 24 h after alteplase infusion during heparin infusion or more frequently if any abnormality detected | Hemoglobin reduction >3 g/dL within 24 h of infusion of study drug infusion or requiring RBC transfusion |
| Any of the below listed criteria developing during or up to 3 h after alteplase or heparin infusion b | Clinical exam and laboratory | Any of listed exclusion criteria developing during or up to 3 h after alteplase or heparin infusion, except for fibrinogen, for which we will set cessation cutoff at 100 mg/dL |
Criteria or attending’s decision.
Criteria: Acute myocardial infarction; acute renal failure (escalating renal failure with creatinine >3 times baseline); liver failure (escalating liver failure with ALT >3 times baseline); cardiac tamponade; bacterial endocarditis; severe uncontrolled hypertension defined as SBP >185 mm Hg or DBP >110 mm Hg; seizure; placement on ECMO; major surgery required; requirement of lumbar puncture; INR >1.7; platelet count <100 × 109/L or history of HITT; fibrinogen <100 mg/dL.
2.13. Follow‐up
Patients will be followed until death or discharge up to 28 days. Laboratory measurements related to the research study, however, will end at day 7 after randomization.
2.14. Statistical analysis
The statistical analysis plan followed the recently published guidelines 40 and is available in the Supplemental Material. All outcome variables will be examined for distribution. If very skewed, we will attempt log and Box‐Cox power transformations to approximate normality. If those are unsuccessful, the outcomes will be categorized using the median or previously defined cutoff. All outcomes will also be analyzed as relative change from baseline. Effectiveness of the randomization to determine baseline comparability of the groups will be done using the absolute standardized mean difference (SMD <0.20 defined as acceptable balance). Any differences deemed clinically relevant or with absolute SMD >0.2 will be adjusted for using inverse probability weighting methods as described below.
All outcome comparison analyses will be conducted initially as an intent‐to‐treat (patients are analyzed in the group they were randomized to), followed by an as‐treated analysis. The primary outcome will be assessed within groups and between groups. Differences in the primary outcome will be evaluated using linear mixed models, with appropriate transformations if normality departure of residuals is detected. Linear mixed models allow (i) adjustment for potential confounders detected in the comparison of the groups at baseline using inverse probability weighting by a propensity score; and (ii) change in the covariance structure to account for repeated measures and the intrahospital correlation (as this is a multicenter study). In addition, it tolerates missing observations. We will also compare percent change over baseline, using t tests with the appropriate adjustment for heteroscedasticity if needed. Categorical outcomes will be compared using generalized estimating equations to account for confounders (as above), covariance structure and intrahospital correlation. In addition, we will compare the “dose” of the intervention (ie, how much of the treatment the patient received) as an effect of interest, as premature death and withdrawals are expected. Survival analysis with inverse probability weighted Cox proportional hazards model and robust sandwich variance estimate to account for clustering for hospitals will be used for mortality as well as for survivor‐bias subject outcomes (eg, pulmonary embolism) censoring for death. As all outcomes are in‐hospital, loss to follow‐up is not likely. The preplanned comparisons include within group (improvement over baseline) and between groups, all 2‐tailed with significance declared as defined by the Pocock spending method.
There will be no adjustment for multiple outcomes, as all were preplanned. Adjustments for multiple comparisons in preplanned hypotheses leads to more type II errors. 41 , 42
2.15. Preplanned subgroup analyses
We anticipate the following subgroup analyses, which will assist in determining whether there is a subgroup of patients for whom the intervention is more beneficial/harmful: (i) baseline PaO2/FiO2 < 100 and < 50; (ii) hemodynamic instability with vasopressors; (iii) age <35, 36‐50, 51‐65, 66‐75 years; (iv) D‐dimers median levels; (v) fibrinolysis shutdown (by TEG or ROTEM); (vi) fibrinogen median levels; (vii) prone/supine positioning; (viii) requirement of rebolusing of lteplase; (ix) received dose of alteplase as premature death or adverse event or other reasons may preclude the administration of complete treatment; and (x) elimination of centers contributing <2 cases.
Additional subgroup analyses may be defined at an interim analysis and will be added for the subsequent interim analyses. This will be documented by filing another version of this statistical analysis plan with the IRBs, DSMB, funder, and FDA.
2.16. Missing data
Missing data are expected to be minimal. If <15% and nondifferential between study groups, we will proceed with analyses of complete data. If >15% or differential between groups (possibly missing not at random), we will add 2 strategies to the complete data set analyses: (i) multiple imputation by chained equations (MICE), recognizing that MICE is better for missing at random data; and (ii) sensitivity analyses: we will assume worst and best clinical scenarios and compare the results with the complete data set.
3. DISCUSSION
The mechanistic rationale for the administration of t‐PA (alteplase) is based on (i) autopsy findings of patients who succumbed to COVID‐19 ARDS showing presence of microthrombi in the lungs and other organs 1 , 3 ; (ii) COVID‐19 poor outcomes associated with hypercoagulability/hypofibrinolysis 43 , 44 , 45 ; (iii) the high rate of pulmonary thrombotic complications while on prophylactic heparin dosing 46 ; (iv) and previous demonstrations of improved pulmonary function in ARDS with plasminogen activators with no bleeding complications. 25 The pathophysiology of COVID can be conceptualized into an early phase, dominated by symptoms directly attributable to the virus, and a later phase manifesting the consequences of the inflammatory response to the invading pathogen. Much of the therapy currently being investigated focuses of reducing the viral load and attenuating cell entry lacking and reducing inflammation. 47 , 48 , 49 , 50 , 51 While the conspicuous hypercoagulable state has been widely documented, the role of suppressed fibrinolysis remains largely overlooked.
COVID‐19 has a clear association with thrombotic complications, which predominantly occur in the lungs. 46 Coagulation biomarkers have been associated with poor prognosis. 52 Functional coagulation measurements have further supported the hypercoagulable state of these patients. 43 While the mechanism of thrombosis and hypercoagulability remains unclear, inflammation driving cytokine release is believed to be the initiator of coagulation changes based on prior work in sepsis. 53 Cytokine production is believed to drive tissue factor product, resulting in systemic activation of coagulation. 54 Tissue factor expression is upregulated on macrophages and endothelial cells in response to elevated tumor necrosis factor‐α (TNF‐α), interleukin (IL)‐6 and IL‐1. 55 At the same time, the cytokine storm damages the endothelium, reducing the antithrombic capacity of the systemic circulation via suppression of protein C, protein S, antithrombin, and tissue factor pathway inhibitor. 56 This is compounded by the severe acute respiratory syndrome coronavirus 2 directly infecting the endothelium of the lungs, heart, and small bowel. 57 COVID also is commonly associated with a high fibrinogen level that correlates with IL‐6 levels. 45 IL‐6 has previously been reported to be the main stimulator of fibrinogen synthesis. 58 With the combination of coagulation activation and hyperfibrinogenemia it is not surprising that this population is prone to thrombosis.
Endotoxin leading to cytokine production has also been demonstrated to activate of the fibrinolytic system 2 hours after infusion, followed by a shutdown of fibrinolysis within the following hour due to elevated plasminogen activator inhibitor −1 (PAI‐1) levels. 59 Fibrinolysis activation with rapid suppression from PAI‐1 was appreciated with endotoxin infusion in nonhuman primates, with concurrent increases in thrombin generation. 60 Pentoxifylline attenuates these fibrinolytic changes in this animal model, whereas IL‐6 and TNF‐α inhibitors have no effect. 61 These experiments were followed up with the hypothesis that an antifibrinolytic (eg, tranexamic acid) would prevent progression to disseminated intravascular coagulation by blocking plasmin activation; however, tranexamic acid had no impact on the prothrombic component of endotoxin infusion in healthy subjects and did not alter cytokine production. 62 Our group’s recent work has demonstrated that COVID‐19 patients with a thrombelastography Lysis 30 (LY30) of 0% and D‐dimer level >2600 ng/mL have a venous thrombosis rate of 50%. 63 Due to D‐dimers having a half‐life that exceeds 12 hours, 64 this value is reflective of the cumulative amount of polymerized fibrin present over the past day or longer, while low fibrinolytic activity measures the current fibrinolytic systemic state of the patient. Therefore, an elevated D‐dimer with low fibrinolytic activity is consistent with prior activation of fibrinolysis with current low fibrinolytic activity, fulfilling the definition of fibrinolysis shutdown, which has been described for the past half‐century. 65 , 66 This fibrinolytic phenotype has been associated with poor outcomes in trauma. 67 , 68 , 69 , 70 , 71 Elevated D‐dimer and low fibrinolytic state has previously been mistermed overt hyperfibrinolysis 72 ; however, the overt hyperfibrinolytic phenotype in trauma does not commonly bleed to death and receives significantly fewer transfusions compared to patients with elevated D‐dimer and elevated LY30s that represents true hyperfibrinolysis. Hyperfibrinolytic trauma patients have the hallmark signs of excessive fibrinolysis with excessive bleeding and low fibrinogen levels that can be reversed with an antifibrinolytic medication. 73 Patients with COVID‐19 have elevated fibrinogen but are not bleeding to death and should not be classified as hyperfibrinolytic, which has already been proposed. 74 The combination of prothrombotic and lack of fibrinolysis poses a major logistical challenge in treating COVID‐19, as both ends of coagulation likely require treatment for effective outcomes. There is a potential that a combination of IL‐6 blockage to attenuate hyperfibrinogenemia in combination with t‐PA could provide a more durable response, which can be adapted from this phase II trial.
The 48‐hour assessment of PaO2/FiO2 in patients after t‐PA treatment will also have limitations in quantifying pulmonary dysfunction improvement. There a numerous ventilator adjuncts to improve oxygenation in the setting of severe ARDS, including proning the patient, 11 paralytics, 75 nitric oxide, 76 and prostonoids. 77 Prolonged use of these interventions is associated with adverse events. 78 Therefore, getting the patients off of these medications or prone positioning would be considered a beneficial outcome with t‐PA regardless of change in PaO2/FiO2 over 48 hours. This also includes reducing toxic levels of oxygen (FiO2 > 80%) and reducing PEEP. In addition to assessing improvements in each of these individual variables, a composite score of each adjunctive measure will also be conducted to represent a global change in requirement of adjuncts for improving oxygenation at 48 hours.
Due to different treatment practices at the 5 enrolling centers, we anticipate that there will be variability in the techniques used before patient enrollment to optimize oxygenation, an acknowledged limitation. This is in line with the pragmatic nature of the trial. Moreover, the crisis created by the pandemic without available treatments precluded the development of agreed‐upon standard operating procedures for ventilation as well as other intensive care procedures.
During the current pandemic, there has been a call for rigorous trials with concurrent control groups. 79 However, randomized controlled trials bring ethical dilemmas, especially when no current treatment exists. Thus, it is imperative that creativity and the rigorous application of the scientific method are combined to produce an innovative, efficient study design. Our design uses the adaptive framework, 80 , 81 which allows preplanned modifications to improve the efficiency of the trial and detect effect or harm more promptly, and a modified stepped‐wedge design. More recently, the stepped‐wedge randomized trials have gained popularity have been proposed. 82 , 83 The modified stepped‐wedge pragmatic design is different than the usual parallel randomized controlled trials, in which the intervention and control groups run, as the name implies, in parallel. The traditional stepped‐wedged approach involves a sequential rollout of an intervention to participants (individuals or clusters) over a number of time periods, such that at the end of the study, all participants will have received the intervention. The name of the design (stepped‐wedged) comes from the schematic illustration of the design. The 1987 Gambia hepatitis intervention study 84 was the pioneer stepped‐wedge study, and tested the effectiveness of a hepatitis B vaccine. We modified the stepped‐wedge design to deploy the intervention groups sequentially to more quickly accrue the sample size with one of the intervention groups with a parallel control. In a traditional parallel design, the first interim analyses would require sufficient number in 3 (as opposed to 2) groups, thus increasing the efficiency of the trial and increasing the likelihood of isolating the more successful arm.
It should be noted that using the traditional (yet arbitrary) confidence level of 95% (α = 0.05) is overly stringent for the current circumstances. The rigid cutoff of 95% level of certainty, as eloquently summarized by Nuzzo 85 in one of the most cited Nature articles should not be applied without regard to the conditions in which the study is conducted. Is it appropriate or necessary to be 95% certain when the condition in question has high lethality and morbidity, threatening to exceed the health care system’s capacity, and has no known prevention (except for isolation) or treatment? While we do not believe this is the precise time to reignite a discussion about the traditional probability (P < .05), we will, in our study, for all comparisons present the effect size with appropriate confidence intervals depicting the uncertainty surrounding our estimation. The clinical experience of the investigators working together with the independent DSMB and IRBs will produce the appropriate interpretation of the results, which can then inform current medical decisions and a subsequent phase III trial if appropriate. Peer review and the readers can then assess the results applying their own tolerance for uncertainty.
Two phase I clinical trials support the potential beneficial effect of thrombolytics in severe ARDS with no reported bleeding complications 24 , 25 and prompted our group’s interest in the potential use of t‐PA in COVID‐19. 86 An important observation was that patients can have transient responders to lytic therapy, with autopsy confirmation of subsequent pulmonary rethrombosis. 24 Our early off‐label use of t‐PA in COVID‐19–related ARDS demonstrated similar transient or long‐lasting improvement on patients using lower doses of this medication. 87 Redosing of fibrinolytics in the Hardaway et al 25 study when indicated demonstrated an overall improvement in oxygenation in 80%. These observations stress the importance of continued heparin anticoagulation following fibrinolytic therapy, as proposed in our trial, together with redosing t‐PA as needed. In both prior phase I clinical trials using fibrinolytics in the setting of ARDS, 24 , 25 the conclusions made clear that future follow‐up studies were required. Our proposal for a phase II study is the natural progression to expand upon those preliminary findings to identify the optimal dosing of a fibrinolytic in the setting of ARDS and ensure safety in regards to treatment strategies using t‐PA, which is a commonly used fibrinolytic agent in clinical practice.
RELATIONSHIP DISCLOSURE
HBM, CDB, EEM, and MBY have patents pending related to both coagulation/fibrinolysis diagnostics and therapeutic fibrinolytics, and are passive cofounders and hold stock options in Thrombo Therapeutics, Inc. HBM and EEM have received grant support from Haemonetics and Instrumentation Laboratories. MBY has previously received a gift of alteplase (t‐PA) from Genentech and owns stock options as a cofounder of Merrimack Pharmaceuticals. RJ, RCM, PKM, JW, NH, DST, and AS declare nothing to report.
AUTHOR CONTRIBUTIONS
HBM, CDB, EEM, AS, and MBY prepared the manuscript, with critical input and revisions from RJ, RCM, PKM, JW, NH, and DST.
Moore HB, Barrett CD, Moore EE, et al. Study of alteplase for respiratory failure in severe acute respiratory syndrome coronavirus 2/COVID‐19: Study design of the phase IIa STARS trial. Res Pract Thromb Haemost. 2020;4:984–996. 10.1002/rth2.12395
Hunter B. Moore and Christopher D. Barrett denotes co‐first authors.
Funding information
The STARS trial is funded by an investigator‐initiated grant from Genentech, the manufacturer of alteplase (Activase). Genentech will have no participation in the conduction of the trial, statistical analysis, interim adaptations, and publications. Data collection and management is supported by NIH/NCRR Colorado CTSI Grant Number UL1 RR025780. Its contents are the authors’ sole responsibility and do not necessarily represent official views of the National Institutes of Health.
Handling Editor: Cihan Ay
Contributor Information
Hunter B. Moore, @hbumoore.
Ernest E. Moore, Email: ernest.moore@dhha.org.
Michael B. Yaffe, Email: myaffe@mit.edu.
REFERENCES
- 1. Yao XH, Li TY, He ZC, Ping YF, Liu HW, Yu SC, et al. A pathological report of three COVID‐19 cases by minimally invasive autopsies. Zhonghua Bing Li Xue Za Zhi. 2020;49:E009. [DOI] [PubMed] [Google Scholar]
- 2. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS‐CoV‐2 pneumonia in Wuhan, China: a single‐centered, retrospective, observational study. Lancet Respir Med. 2020;8(5):475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wiedermann CJ. Anticoagulant therapy for septic coagulopathy and disseminated intravascular coagulation: where do KyberSept and SCARLET leave us? Acute Med Surg. 2020;7(1):e477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grasselli G, Pesenti A, Cecconi M. Critical care utilization for the COVID‐19 outbreak in Lombardy, Italy: early experience and forecast during an emergency response. JAMA. 2020;323(16):1545. [DOI] [PubMed] [Google Scholar]
- 6. Livingston E, Bucher K. Coronavirus disease 2019 (COVID‐19) in Italy. JAMA. 2020;323(14):1335. [DOI] [PubMed] [Google Scholar]
- 7. Grasselli G, Zangrillo A, Zanella A, Antonelli M, Cabrini L, Castelli A, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS‐CoV‐2 admitted to ICUs of the Lombardy region, Italy. JAMA. 2020;323(16):1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bhatraju PK, Ghassemieh BJ, Nichols M, Kim R, Jerome KR, Nalla AK, et al. Covid‐19 in critically ill patients in the Seattle region ‐ case series. N Engl J Med. 2020;382(21):2012–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID‐19 in the New York City area. JAMA. 2020;323(20):2052–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pan C, Chen L, Lu C, Zhang W, Xia JA, Sklar MC, et al. Lung Recruitability in SARS‐CoV‐2 associated acute respiratory distress syndrome: a single‐center, observational study. Am J Respir Crit Care Med. 2020;201(10):1294–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gattinoni L, Chiumello D, Caironi P, Busana M, Romitti F, Brazzi L, et al. COVID‐19 pneumonia: different respiratory treatments for different phenotypes? Intensive Care Med. 2020;46(6):1099–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paget J. Additional observations on obstructions of the pulmonary arteries. Med Chir Trans. 1845;28:353–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vesconi S, Rossi GP, Pesenti A, Fumagalli R, Gattinoni L. Pulmonary microthrombosis in severe adult respiratory distress syndrome. Crit Care Med. 1988;16(2):111–3. [DOI] [PubMed] [Google Scholar]
- 15. Mergoni M, Caberti P, Vergallo A, Pagliari S, Salvadori A, Gulli E. Vascular obstruction in patients with ARDS. A study with selective pulmonary angiography with a wedged catheter. Minerva Anestesiol. 1991;57(10):932–3. [PubMed] [Google Scholar]
- 16. Greene R. Pulmonary vascular obstruction in the adult respiratory distress syndrome. J Thorac Imaging. 1986;1(3):31–8. [DOI] [PubMed] [Google Scholar]
- 17. Zapol WM, Jones R. Vascular components of ARDS. Clinical pulmonary hemodynamics and morphology. Am Rev Respir Dis. 1987;136(2):471–4. [DOI] [PubMed] [Google Scholar]
- 18. Hardaway RM 3rd, Mc KD. Disseminated intravascular coagulation: a cause of shock. Ann Surg. 1959;149(4):462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cafferata HT, Aggeler PM, Robinson AJ, Blaisdell FW. Intravascular coagulation in the surgical patient: its significance and diagnosis. Am J Surg. 1969;118(2):281–91. [DOI] [PubMed] [Google Scholar]
- 20. Turpini R, Stefanini M. The nature and mechanism of the hemostatic breakdown in the course of experimental hemorrhagic shock. J Clin Invest. 1959;38(1, Part 1):53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hardaway RM, Brune WH, Geever EF, Burns JW, Mock HP. Studies on the role of intravascular coagulation in irreversible hemorrhagic shock. Ann Surg. 1962;155:241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hardaway RM, Drake DC. Prevention of “irreversible” hemorrhagic shock with fibrinolysin. Ann Surg. 1963;157:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hardaway RM. The significance of coagulative and thrombotic changes after haemorrhage and injury. J Clin Pathol. 1970;4 (suppl):110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Greene R, Lind S, Jantsch H, Wilson R, Lynch K, Jones R, et al. Pulmonary vascular obstruction in severe ARDS: angiographic alterations after i.v. fibrinolytic therapy. AJR Am J Roentgenol. 1987;148(3):501–8. [DOI] [PubMed] [Google Scholar]
- 25. Hardaway RM, Harke H, Tyroch AH, Williams CH, Vazquez Y, Krause GF. Treatment of severe acute respiratory distress syndrome: a final report on a phase I study. Am Surg. 2001;67(4):377–82. [PubMed] [Google Scholar]
- 26. Etchill E, Sperry J, Zuckerbraun B, Alarcon L, Brown J, Schuster K, et al. The confusion continues: results from an American association for the surgery of trauma survey on massive transfusion practices among United States trauma centers. Transfusion. 2016;56(10):2478–86. [DOI] [PubMed] [Google Scholar]
- 27. Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, et al. Large‐vessel stroke as a presenting feature of Covid‐19 in the young. N Engl J Med. 2020;382(20):e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Esmon CT. Molecular events that control the protein C anticoagulant pathway. Thromb Haemost. 1993;70(1):29–35. [PubMed] [Google Scholar]
- 29. World Medical Association Declaration of Helsinki . Recommendations guiding physicians in biomedical research involving human subjects. Cardiovasc Res. 1997;35(1):2–3. [PubMed] [Google Scholar]
- 30. Konstantinides S, Geibel A, Heusel G, Heinrich F, Kasper W, Management S, et al. Heparin plus alteplase compared with heparin alone in patients with submassive pulmonary embolism. N Engl J Med. 2002;347(15):1143–50. [DOI] [PubMed] [Google Scholar]
- 31. Carless PA, Henry DA, Moxey AJ, O'Connell DL, Brown T, Fergusson DA. Cell salvage for minimising perioperative allogeneic blood transfusion. Cochrane Database Syst Rev. 2006;4 10.1002/14651858.CD001888.pub2 [DOI] [PubMed] [Google Scholar]
- 32. Spanos A, Jhanji S, Vivian‐Smith A, Harris T, Pearse RM. Early microvascular changes in sepsis and severe sepsis. Shock. 2010;33(4):387–91. [DOI] [PubMed] [Google Scholar]
- 33. Driessen A, Schafer N, Albrecht V, Schenk M, Frohlich M, Sturmer EK, et al. Infrastructure and clinical practice for the detection and management of trauma‐associated haemorrhage and coagulopathy. Eur J Trauma Emerg Surg. 2015;41(4):413–20. [DOI] [PubMed] [Google Scholar]
- 34. Geddes AE, Burlew CC, Wagenaar AE, Biffl WL, Johnson JL, Pieracci FM, et al. Expanded screening criteria for blunt cerebrovascular injury: a bigger impact than anticipated. Am J Surg. 2016;212(6):1167–74. [DOI] [PubMed] [Google Scholar]
- 35. Dumville JC, Hahn S, Miles JNV, Torgerson DJ. The use of unequal randomisation ratios in clinical trials: a review. Contemp Clin Trials. 2006;27(1):1–12. [DOI] [PubMed] [Google Scholar]
- 36. Jitlal M, Khan I, Lee SM, Hackshaw A. Stopping clinical trials early for futility: retrospective analysis of several randomised clinical studies. Br J Cancer. 2012;107(6):910–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pieracci FM, Leasia K, Bauman Z, Eriksson EA, Lottenberg L, Majercik S, et al. A multicenter, prospective, controlled clinical trial of surgical stabilization of rib fractures in patients with severe, nonflail fracture patterns (Chest Wall Injury Society NONFLAIL). J Trauma Acute Care Surg. 2020;88(2):249–57. [DOI] [PubMed] [Google Scholar]
- 38. investigators G . An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med. 1993;329(10):673–82. [DOI] [PubMed] [Google Scholar]
- 39. Lees KR, Emberson J, Blackwell L, Bluhmki E, Davis SM, Donnan GA, et al. Effects of alteplase for acute stroke on the distribution of functional outcomes: a pooled analysis of 9 trials. Stroke. 2016;47(9):2373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gamble C, Krishan A, Stocken D, Lewis S, Juszczak E, Dore C, et al. Guidelines for the content of statistical analysis plans in clinical trials. JAMA. 2017;318(23):2337–43. [DOI] [PubMed] [Google Scholar]
- 41. Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology (Cambridge, Mass). 1990;1(1):43–6. [PubMed] [Google Scholar]
- 42. Perneger TV. What’s wrong with Bonferroni adjustments. BMJ. 1998;316(7139):1236–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Panigada M, Bottino N, Tagliabue P, Grasselli G, Novembrino C, Chantarangkul V, et al. Hypercoagulability of COVID‐19 patients in intensive care unit. a report of thromboelastography findings and other parameters of hemostasis. J Thromb Haemost. 2020;18(7):1738–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(4):844–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ranucci M, Ballotta A, Di Dedda U, Bayshnikova E, Dei Poli M, Resta M, et al. The procoagulant pattern of patients with COVID‐19 acute respiratory distress syndrome. J Thromb Haemost. 2020;18(7):1747–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID‐19. Thromb Res. 2020;191:145–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. He XL, Liu Z, Xia SY. Vascular endothelial injuries and changes of blood coagulation and fibrinolysis indexes in patients with acute respiratory distress syndrome. Chin Med Sci. 2004;19(4):252–6. [PubMed] [Google Scholar]
- 48. Hofstra JJ, Haitsma JJ, Juffermans NP, Levi M, Schultz MJ. The role of bronchoalveolar hemostasis in the pathogenesis of acute lung injury. Semin Thromb Hemost. 2008;34(5):475–84. [DOI] [PubMed] [Google Scholar]
- 49. Idell S. Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit Care Med. 2003;31(4 Suppl):S213–20. [DOI] [PubMed] [Google Scholar]
- 50. Idell S, Koenig KB, Fair DS, Martin TR, McLarty J, Maunder RJ. Serial abnormalities of fibrin turnover in evolving adult respiratory distress syndrome. Am J Physiol. 1991;261(4 Pt 1):L240–8. [DOI] [PubMed] [Google Scholar]
- 51. Olson TM, Driscoll DJ, Edwards WD, Puga FJ, Danielson GK. Pulmonary microthrombi. Caveat for successful modified Fontan operation. J Thorac Cardiovasc Surg. 1993;106(4):739–44. [PubMed] [Google Scholar]
- 52. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID‐19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spero JA, Lewis JH, Hasiba U. Disseminated intravascular coagulation. Findings in 346 patients. Thromb Haemost. 1980;43(1):28–33. [PubMed] [Google Scholar]
- 54. Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers. 2016;2:16037. [DOI] [PubMed] [Google Scholar]
- 55. Doshi SN, Marmur JD. Evolving role of tissue factor and its pathway inhibitor. Crit Care Med. 2002;30(5 Suppl):S241–S250. [DOI] [PubMed] [Google Scholar]
- 56. Sungurlu S, Kuppy J, Balk RA. Role of antithrombin iii and tissue factor pathway in the pathogenesis of sepsis. Crit Care Clin. 2020;36(2):255–65. [DOI] [PubMed] [Google Scholar]
- 57. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID‐19. Lancet. 2020;395(10234):1417–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Carty CL, Heagerty P, Heckbert SR, Jarvik GP, Lange LA, Cushman M, et al. Interaction between fibrinogen and IL‐6 genetic variants and associations with cardiovascular disease risk in the cardiovascular health study. Ann Hum Genet. 2010;74(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spiel AO, Mayr FB, Firbas C, Quehenberger P, Jilma B. Validation of rotation thrombelastography in a model of systemic activation of fibrinolysis and coagulation in humans. J Thromb Haemost. 2006;4(2):411–6. [DOI] [PubMed] [Google Scholar]
- 60. Biemond BJ, Levi M, Ten Cate H, Van der Poll T, Buller HR, Hack CE, et al. Plasminogen activator and plasminogen activator inhibitor I release during experimental endotoxaemia in chimpanzees: effect of interventions in the cytokine and coagulation cascades. Clin Sci (Lond). 1995;88(5):587–94. [DOI] [PubMed] [Google Scholar]
- 61. Levi M, ten Cate H, Bauer KA, van der Poll T, Edgington TS, Buller HR, et al. Inhibition of endotoxin‐induced activation of coagulation and fibrinolysis by pentoxifylline or by a monoclonal anti‐tissue factor antibody in chimpanzees. J Clin Invest. 1994;93(1):114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Renckens R, Weijer S, de Vos AF, Pater JM, Meijers JC, Hack CE, et al. Inhibition of plasmin activity by tranexamic acid does not influence inflammatory pathways during human endotoxemia. Arterioscler Thromb Vasc Biol. 2004;24(3):483–8. [DOI] [PubMed] [Google Scholar]
- 63. Wright FLVT, Moore EE, Moore HB, Wohlauer MV, Urban S, Nydam TL, et al. Fibrinolysis shutdown correlates to thromboembolic events in severe COVID‐19 infection. JACS. 2020;S1072‐7515(20):30400‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ruhl H, Berens C, Winterhagen A, Muller J, Oldenburg J, Potzsch B. Label‐free kinetic studies of hemostasis‐related biomarkers including d‐dimer using autologous serum transfusion. PLoS ONE. 2015;10(12):e0145012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chakrabarti R, Hocking ED, Fearnley GR. Reaction pattern to three stresses–electroplexy, surgery, and myocardial infarction–of fibrinolysis and plasma fibrinogen. J Clin Pathol. 1969;22(6):659–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Moore HB, Moore EE, Neal MD, Sheppard FR, Kornblith LZ, Draxler DF, et al. Fibrinolysis shutdown in trauma: historical review and clinical implications. Anesth Analg. 2019;129(3):762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Moore HB, Moore EE, Gonzalez E, Chapman MP, Chin TL, Silliman CC, et al. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J Trauma Acute Care Surg. 2014;77(6):811–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Moore HB, Moore EE, Liras IN, Gonzalez E, Harvin JA, Holcomb JB, et al. Acute fibrinolysis shutdown after injury occurs frequently and increases mortality: a multicenter evaluation of 2540 severely injured patients. J Am Coll Surg. 2016;222(4):347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Leeper CM, Neal MD, McKenna CJ, Gaines BA. Trending fibrinolytic dysregulation: fibrinolysis shutdown in the days after injury is associated with poor outcome in severely injured children. Ann Surg. 2017;266(3):508–15. [DOI] [PubMed] [Google Scholar]
- 70. Meizoso JP, Karcutskie CA, Ray JJ, Namias N, Schulman CI, Proctor KG. Persistent fibrinolysis shutdown is associated with increased mortality in severely injured trauma patients. J Am Coll Surg. 2017;224(4):575–82. [DOI] [PubMed] [Google Scholar]
- 71. Roberts DJ, Kalkwarf KJ, Moore HB, Cohen MJ, Fox EE, Wade CE, et al. Time course and outcomes associated with transient versus persistent fibrinolytic phenotypes after injury: a nested, prospective, multicenter cohort study. J Trauma Acute Care Surg. 2019;86(2):206–13. [DOI] [PubMed] [Google Scholar]
- 72. Gall LS, Vulliamy P, Gillespie S, Jones TF, Pierre RSJ, Breukers SE, et al. The S100A10 pathway mediates an occult hyperfibrinolytic subtype in trauma patients. Ann Surg. 2018;269(6):1184–91. [DOI] [PubMed] [Google Scholar]
- 73. Moore HB, Moore EE, Chapman MP, Hansen KC, Cohen MJ, Pieracci FM, et al. Does tranexamic acid improve clot strength in severely injured patients who have elevated fibrin degradation products and low fibrinolytic activity, measured by thrombelastography? J Am Coll Surg. 2019;229(1):92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ji HL, Zhao R, Matalon S, Matthay MA. Elevated plasmin(ogen) as a common risk factor for COVID‐19 susceptibility. Physiol Rev. 2020;100(3):1065–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Slutsky AS, Villar J. Early paralytic agents for ARDS? Yes, No, and sometimes. N Engl J Med. 2019;380(21):2061–3. [DOI] [PubMed] [Google Scholar]
- 76. Gebistorf F, Karam O, Wetterslev J, Afshari A. Inhaled nitric oxide for acute respiratory distress syndrome (ARDS) in children and adults. Cochrane Database Syst Rev. 2016;6 10.1002/14651858.CD002787.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fuller BM, Mohr NM, Skrupky L, Fowler S, Kollef MH, Carpenter CR. The use of inhaled prostaglandins in patients with ARDS: a systematic review and meta‐analysis. Chest. 2015;147(6):1510–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fan E, Brodie D, Slutsky AS. Acute respiratory distress syndrome: advances in diagnosis and treatment. JAMA. 2018;319(7):698–710. [DOI] [PubMed] [Google Scholar]
- 79. Coccolini F, Sartelli M, Kluger Y, Pikoulis E, Karamagioli E, Moore EE, et al. COVID‐19 the showdown for mass casualty preparedness and management: the Cassandra syndrome. World J Emerg Surg. 2020;15(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Adaptive designs for medical device clinical studies DRAFT GUIDANCE2015. [Accessed 2020 May 15] Available from http://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm446729.pdf
- 81. Pallmann P, Bedding AW, Choodari‐Oskooei B, Dimairo M, Flight L, Hampson LV, et al. Adaptive designs in clinical trials: why use them, and how to run and report them. BMC Med. 2018;16(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hemming K, Haines TP, Chilton PJ, Girling AJ, Lilford RJ. The stepped wedge cluster randomised trial: rationale, design, analysis, and reporting. BMJ. BMJ. 2015;350:h391. [DOI] [PubMed] [Google Scholar]
- 83. Brown CA, Lilford RJ. The stepped wedge trial design: a systematic review. BMC Med Res Methodol. 2006;6(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Group TGHS . The Gambia hepatitis intervention study. Can Res. 1987;47(21):5782–7. [PubMed] [Google Scholar]
- 85. Nuzzo R. Scientific method: statistical errors. Nature. 2014;506(7487):150–2. [DOI] [PubMed] [Google Scholar]
- 86. Moore HB, Barrett CD, Moore EE, McIntyre RC, Moore PK, Talmor DS, et al. Is there a role for tissue plasminogen activator (tPA) as a novel treatment for refractory COVID‐19 associated acute respiratory distress syndrome (ARDS)? J Trauma Acute Care Surg. 2020;88(6):713–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang J, Hajizadeh N, Moore EE, McIntyre RC, Moore PK, Veress LA, et al. Tissue plasminogen activator (tPA) treatment for COVID‐19 associated acute respiratory distress syndrome (ARDS): a case series. J Thromb Haemost. 202018(7):1752–5. [DOI] [PMC free article] [PubMed] [Google Scholar]