

Abstract
The current pandemic has highlighted the need for methodologies that can quickly and reliably prioritize clinically approved compounds for their potential effectiveness for SARS-CoV-2 infections. In the past decade, network medicine has developed and validated multiple predictive algorithms for drug repurposing, exploiting the sub-cellular network-based relationship between a drug’s targets and disease genes. Here, we deployed algorithms relying on artificial intelligence, network diffusion, and network proximity, tasking each of them to rank 6,340 drugs for their expected efficacy against SARS-CoV-2. To test the predictions, we used as ground truth 918 drugs that had been experimentally screened in VeroE6 cells, and the list of drugs under clinical trial, that capture the medical community’s assessment of drugs with potential COVID-19 efficacy. We find that while most algorithms offer predictive power for these ground truth data, no single method offers consistently reliable outcomes across all datasets and metrics. This prompted us to develop a multimodal approach that fuses the predictions of all algorithms, showing that a consensus among the different predictive methods consistently exceeds the performance of the best individual pipelines. We find that 76 of the 77 drugs that successfully reduced viral infection do not bind the proteins targeted by SARS-CoV-2, indicating that these drugs rely on network-based actions that cannot be identified using docking-based strategies. These advances offer a methodological pathway to identify repurposable drugs for future pathogens and neglected diseases underserved by the costs and extended timeline of de novo drug development.
Introduction
The disruptive nature of the COVID-19 pandemic has unveiled the need for the rapid development, testing, and deployment of new drugs and cures. Given the compressed timescales, the de novo drug development process, that lasts a decade or longer, is not feasible. A time-efficient strategy must rely on drug repurposing (or repositioning), helping identify among the compounds approved for clinical use the few that may also have a therapeutic effect in patients with COVID-19. Yet, the lack of reliable repurposing methodologies has resulted in a winner-takes-all pattern, where more than one-third of registered clinical trials focus on hydroxychloroquine or chloroquine, siphoning away resources from testing a wider range of potentially effective drug candidates.
Drug repurposing algorithms rank drugs based on one or multiple streams of information, such as molecular profiles1, chemical structures2, adverse profiles3, molecular docking4, electronic health records5, pathway analysis6, genome wide association studies (GWAS)6, and network perturbations6–14. As typically only a small subset of the top candidates is validated experimentally, the true predictive power of the existing repurposing algorithms remains unknown. To quantify and compare their predictive power, different algorithms must make predictions for the same set of candidates, and the experimental validation must focus not only on the top candidates, but also on a wider list of drugs chosen independently of their predicted rank.
The COVID-19 pandemic presents both the societal imperative and the rationale to test drugs at a previously unseen scale. Hence, it offers a unique opportunity to quantify and improve the efficacy of the available predictive algorithms, while also identifying potential treatments for COVID-19. Here, we implement three recently developed network-medicine drug-repurposing algorithms that rely on artificial intelligence14,15, network diffusion16, and network proximity10 (Figure 1A, B). To test the validity of the predictions, we identified 918 drugs ranked by all predictive pipelines, that had been experimentally screened to inhibit viral infection and replication in cultured cells17. We also collected clinical trial data to capture the medical community’s collective assessment of promising drug candidates. We find that the predictive power varies for the different datasets and metrics, indicating that in the absence of a priori ground truth, it is impossible to decide which algorithm to trust. We propose, however, a multimodal ensemble forecasting approach that significantly improves the accuracy and the reliability of the predictions by seeking consensus among the predictive methods14,18.
Figure 1. Network Medicine Framework for Drug Repurposing.

(A) Study Design and Timeline. Following the publication of host-pathogen protein-protein interactions20 – March, 23rd, 2020 – we implemented three drug repurposing algorithms, relying on AI (A1-A4), network diffusion (D1-D5) and proximity (P1-P3), together resulting in 12 predictive ranking lists (pipelines, shown in (B)). Each pipeline offers predictions for a different number of drugs, what were frozen on April 15, 2020. We then identified 918 drugs for which all pipelines but P3 offered predictions, and experimentally validated their effect on the virus in VeroE6 cells17. The experimental (E918, E74) and clinical trial lists C415 offered the ground truth for validation and rank aggregation. (C) Direct target drugs bind either to a viral protein (D1) or to a host protein target of the viral proteins (D2). Network drugs (D3), in contrast, bind to the host proteins and limit viral activity by perturbing the host subcellular network.
Network-based Drug Repurposing
Repurposing strategies often prioritize drugs approved for (other) diseases whose molecular manifestations are similar to those caused by the pathogen or disease of interest19. To search for diseases whose molecular mechanisms overlap with the COVID-19 disease, we first mapped the experimentally identified20 332 host protein targets of the SARS-CoV-2 proteins (Table S4) to the human interactome21–24 (Table S3), a collection of 332,749 pairwise binding interactions between 18,508 human proteins (SI Section 1.1). We find that 208 of the 332 viral targets form a large connected component (COVID-19 disease module hereafter, Figure 2B), indicating that the SARS-CoV-2 targets aggregate in the same network vicinity12,25. Next, we evaluated the network-based overlap between proteins associated with 299 diseases26 (d) and the host protein targets of SARS-CoV-2 (v) using the Svd metric26, finding Svd > 0 for all diseases, implying that the COVID-19 disease module does not directly overlap with the disease proteins associated with any single disease (Figure S1–2 and Table S7). In other words, a potential COVID-19 treatment cannot be derived from the arsenal of therapies approved for a specific disease, arguing for a network-based strategy that can identify repurposable drugs without regard for their established disease indication.
Figure 2. COVID-19 Disease Module.

(A) Proteins targeted by SARS-CoV-2 are not distributed randomly in the human interactome, but form a large connected component (LCC) consisting of 208 proteins, and multiple small subgraphs, shown in the figure. Almost all proteins in SARS-CoV-2 LCC are also expressed in the lung tissue, potentially explaining the effectiveness of the virus in causing pulmonary manifestations of the disease. (B) The random expectation of the LCC size indicates that the observed COVID-19 LCC, whose size is indicated by the red arrow, is larger than expected by chance (Z-score=1.65). (C) Heatmap of the Kendall τ statistic showing that the ranking list predicted by the different methods (A,D,P) are not correlated. We observe, however high correlations among the individual ranking list predicted by the same predictive method.
We implemented three competing network repurposing methodologies (Figure 1B and SI Section 1.4): i) The artificial intelligence-based algorithm14,15 maps drug protein targets and disease-associated proteins to points in a low-dimensional vector space, resulting in four predictive pipelines A1-A4, that rely on different drug-disease embeddings. ii) The diffusion algorithm16 is inspired by diffusion state distance, and ranks drugs based on capturing network similarity of a drug’s protein targets to the SARS-CoV-2 host protein targets. Powered by distinct statistical measures, the algorithm offers five ranking pipelines (D1-D5). iii) The proximity algorithm10 ranks drugs based on the distance between the host protein targets of SARS-CoV2 and the closest protein targets of drugs, resulting in three predictive pipelines of which P1 relies on all drug targets; P2 ignores targets identified as drug carriers, transporters, and drug-metabolizing enzymes; and P3 relies on differentially expressed genes identified by exposing each drug to cultured cells27. The low correlations across the three algorithms indicate that the methods extract complementary information from the network (Figure 2C, and SI Section 1.5).
Experimental and Clinical Validation of Drug Repurposing Pipelines
We implemented the 12 pipelines to predict the expected efficacy of 6,340 drugs in Drugbank27 against SARS-CoV-2 and extracted and froze the predictions in the form of 12 ranked lists on April 15, 2020. All pipelines rely on the same input data and to maintain the prospective nature of the study, all subsequent analysis relies on this initial prediction list. As the different pipelines make successful predictions of a different subset of drugs, we identified 918 drugs for which all pipelines (except for P3, which predicts the smallest number of drugs) offer predictions and whose compounds were available in the Broad Institute drug repurposing library28 (Figure 1), and used two independent datasets to quantify the predictive power of each pipeline over the same set of drugs:
(1) As the first ground truth we used 918 compounds that had been experimentally screened for their efficacy against SARS-CoV-2 in VeroE6 cells, kidney epithelial cells derived from African green monkey17 (see SI Section 2). Briefly, the VeroE6 cells were pre-incubated with the drugs (from 8 μM down to 8 nM) and then challenged with wild type SARS-CoV-2 strain USA-WA1/2020. Of the 918 drugs, 806 had no detectable effect on viral infectivity (N drugs, 87.8% of the tested list); 35 were cytotoxic to the host cells (C drugs); 37 had a strong effect (S drugs), being active over a broad range of concentrations; and 40 had a weak effect (W drugs) on the virus (Figure 3A, Table S10). As the prediction pipelines offer no guidance on the magnitude of the in vivo effect, we consider as positive outcomes drugs that had a strong or a weak effect on the virus (S&W, 77 drugs, Table 2), and as negative outcomes the drugs without detectable effect (N, 806 drugs).
Figure 3. Experimental Outcomes and Network Origins.

(A) Examples of dose-response curves for eight of the 918 experimentally validated drugs17, illustrating the four observed outcomes (S, W, C and N). VeroE6 cells were challenged in vitro with SARS-CoV-2 virus and treated with the drug over a range of doses (from 8 nM to 8 μM). A two-steps drug-response model (see SI Section 2.3) was used to classify each drug into Strong, Weak, Cytotoxic or No-Effect categories, according to their response to the drug in different doses and cell and viral reduction. (B) The sub-network formed by the targets of the 77 S&W drugs within the interactome. The link corresponds to binding interactions. Purple proteins are targeted by S drugs only; orange by W drugs only; proteins targeted by both S&W drugs are shown as pie charts, proportional to the number of targets in each category. (C) The targets of N drugs have a positive proximity Z-Score to the COVID-19 module, meaning they are further from the COVID-19 module than random expectation. By contrast, the targets of S&W drugs are more proximal (closer) to the COVID-19 module than expected by change, suggesting that their COVID-19 vicinity contribute to their ability to alter the virus’s ability to infect the cells.
Table 2. Drugs with Positive Experimental Outcomes.
List of the 77 drugs with a positive outcome (S&W) from in vitro screen17. Drug response classification was obtained by a two-step model for drug response (see SI Section 2.3). Purple drugs show strong effect (S), and orange drugs showed weak effect (W).
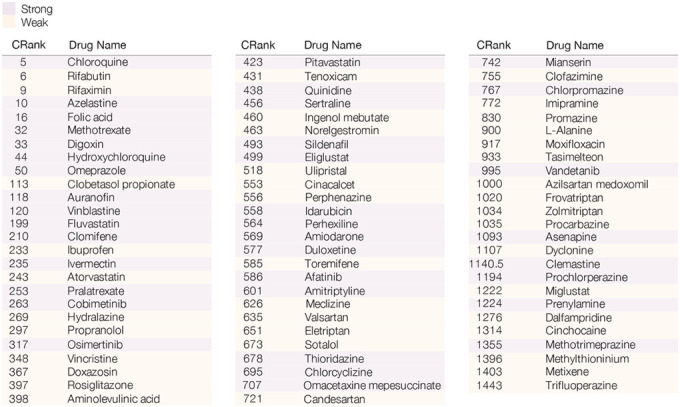
|
(2) On April 15, 2020 (prediction date), we scanned clinicaltrials.gov, identifying 67 drugs in 134 clinical trials for COVID-19 (CT415 dataset, Table S12). To compare outcomes across datasets, we limit our analysis to the experimentally tested 918 drugs, considering as positive the 37 drugs in clinical trial on the E918 list, and negative the remaining 881 drugs. As the outcomes of these trials are largely unknown, validation against CT415 dataset tests each pipeline’s ability to predict the pharmacological consensus of the medical community on drugs with expected potential efficacy for COVID-19 patients.
For the E918 experimental outcomes (Figure 4A), the best area under the curve (AUC) of 0.63 is provided by P1, followed by D2 (AUC = 0.58) and P3 (AUC = 0.58). For CT415 (Figure 4B), we observe particularly strong predictive power for the four AI-based pipelines (AUC of 0.73–0.76), followed by proximity P1 (AUC = 0.57) and P2 (AUC = 0.56).
Figure 4. Performance of the Predictive Pipelines.

(A,B) AUC (Area under the Curve), (C,D) Precision at 100, and (E,F) Recall at 100, for twelve pipelines tested for drug repurposing, each plot using as a gold standard the S&W drugs in E918 (left column) and drugs under clinical trials for treating COVID-19 as of April 15th, 2020 (CT415, right column). (G,H) The top K precision and recall for the different rank aggregation methods (connected points), compared to the individual pipelines (empty symbols) documenting the consistent predictive performance of CRank. Similar results are shown for two other datasets in Figure S8: the prospective expert curated E74 and the clinical trial data refreshed on 06/15/20 (CT615)
The goal of drug repurposing is to prioritize all available drugs, allowing the experimental efforts to limit their resources on the top-ranked ones. Thus, the most appropriate performance metric is the number of positive outcomes among top K ranked drugs (precision at K), and the fraction of all positive outcomes among the top K ranked drugs (recall at K). For the E918 dataset(Figure 4C) A2 ranks 9 S&W drugs among the top 100, followed by P1 (7 drugs) and A3 and A4 (6 drugs). We observe similar trends for recall (Figure 4E): the A2 pipeline ranks 11.7% of all positive drugs in the top 100, and P1 selects 9%. Finally, A1 ranks 12 drugs currently in clinical trials among the top 100 in CT415, followed by A3 (11 drugs) and A2 (10 drugs), trends that are similar for recall (Figure 4F).
Taken together, we find that while most algorithms show statistically significant predictive power (SI Section 3.1, Tables S1–2), they have different performance on the different ground truth datasets: the AI pipeline offers strong predictive power for the drugs selected for clinical trials, while proximity offers better predictive power for the E918 experimental outcomes. While together the twelve pipelines identify 22 positive drugs among the top 100, none of the pipelines offers consistent superior performance for all outcomes, prompting us to develop a multimodal approach that can extract the joint predictive power of all pipelines.
Multimodal Approach for Drug Repurposing
Predictive models for drug repurposing are driven by finite experimental resources that limit downstream experiments to those involving a finite number (K) of drugs. How do we identify these K drugs to maximize the positive outcomes of the tested list18? With no initial knowledge as to which of the Np = 12 predictive pipelines offer the best predictive power, we could place equal trust in all, by selecting the top K⁄Np drugs from each pipeline (Union list). We compare this scenario with an alternative strategy that combines the predictions of the different pipelines. A widely used approach is to calculate the average rank of each drug over the Np pipelines29 (Average Rank list). The alternative is to search for consensus ranking that maximizes the number of pairwise agreements between all pipelines15,18. As the optimal outcome, called the Kemeny consensus29, is NP-hard to compute, we implemented three heuristic rank aggregation algorithms (RAAs) that approximate the Kemeny consensus: Borda’s count30, the Dowdall method31, and CRank15. For example, if the resources allow us to test K = 120 drugs, we ask which ranked list offers the best precision and recall at 120: the Union list collecting the top 10 predictions from the 12 pipelines; or the top 120 predictions of Average Rank, Borda, Dowdall, or CRank; or the top 120 drugs ranked by an individual pipeline.
We find that Average Rank offers the worst performance, trailing the predictive power of most individual pipelines (Figure 4G–H). The Union List and Dowdall offer better outcomes, but trail behind the best performing individual pipelines (E918, CT415). Borda has a strong predictive performance for E918, but not for CT415. In contrast, CRank, that relies on Bayesian factors, offers a consistently high predictive performance for all datasets and most K values. CRank performs equally well for two other datasets: a manually curated prospective list E74 (described below) and the list of clinical trials updated on 06/15/20 (C615, Figure S8). In other words, we find that CRank extracts the cumulative predictive power of all methods, matching or exceeding the predictive power of the individual pipelines across all datasets. Its persistent performance indicates that an unsupervised multimodal approach can significantly improve the hit rate over individual prediction algorithms. It also suggests that in the absence of a ground truth, the Kemeny consensus, which seeks a ranking with the smallest number of pairwise disagreements between the individual pipelines, represents an effective and theoretically principled strategy when each pipeline carries some predictive power.
Network Effects
Most computationally informed drug repurposing methods rely on chemical binding energy minimization and docking patterns, limited to compounds that bind either to viral proteins or to the host targets of the viral proteins20 (Figure 1C). A good example is remdesivir, a direct-acting antiviral that inhibits viral RNA polymerase32,33. In contrast, our pipelines identify drugs that target host proteins to induce network-based perturbations that alter the virus’s ability to enter the cell or to replicate within it. An example of such host-targeting drug34 is dexamethasone, which reduces mortality in COVID-19 patients by modulating the host immune system35. We find that only one of the 77 S&W drugs are known to directly target a viral protein binding target: amitriptyline, which targets SIGMAR1, the target of the NSP6 SARS-CoV-2 protein. In other words, 76 of the 77 drugs that show efficacy in our experimental screen are “network drugs”, achieving their effect indirectly, by perturbing the host subcellular network. As these drugs do not target viral proteins or their host targets target, they cannot be identified using traditional binding-based methods yet are successfully prioritized by network-based methods.
Searching for common mechanistic or structural patterns that could account for the efficacy of the 77 S&W drugs, we explored their target and pathway enrichment profiles (Figures S6–7), as well as their reported mechanisms of action, failing to identify statistically significant features shared by most S&W drugs. This failure is partly explained by the diversity of the S&W drugs (Table S10), containing antipsychotics (9S & 4W), serotonin receptor agonists (3W), non-steroidal anti-inflammatory drugs (2W), angiotensin receptor blockers (2W), tyrosine kinase inhibitors (5S), statins (1W & 2S), and others.
As CRank extracts its predictive power from the network, we hypothesized that network-based patterns may help distinguish the S&W drugs from the N drugs. Indeed, we find that the targets of the 37 S drugs form a statistically significant large connected component (Z-Score=2.05), indicating that these targets agglomerate in the same network neighborhood. We observe the same pattern for the targets of the 40 W drugs (Z-Score=3.42). The negative network separation between the S and W drug targets (SSW = −0.69) indicates that, in fact, the S and the W drugs target the same network neighborhood. To characterize this neighborhood, we measured the network-based proximity of the targets of the S, W, and N drug classes to the SARS-CoV-2 targets. We find that compared to random expectation, the N drug targets are far from the COVID-19 module (Figure 3C), while the S and W drug targets are closer to the COVID-19 disease module than expected by chance. The magnitude of the effect is also revealing: the S drug targets are closer than the W drug targets, suggesting that network proximity is a positive predictor of a drug’s efficacy.
Taken together, our analyses suggest that S&W drugs are diverse, and lack pathway-based or mechanistic signatures that distinguish them. We do find, however, that S&W drug target the same interactome neighborhood, located in the network vicinity of the COVID-19 disease module, potentially explaining their ability to influence viral activity, and the effectiveness of network-based methodologies to identify them.
Discussion
A recent in vitro screen36 of 12,000 compounds in VeroE6 cells identified 100 compounds that inhibit viral infectivity. Yet, only 2 of the 12,000 compounds tested are FDA approved, the rest being in the preclinical or experimental phase, years from reaching patients. In contrast, 96% of the 918 drugs prioritized and screened here are FDA approved, hence should they also show efficacy in human cell lines, could be moved immediately to rapid clinical trials. Brute force screening does, however, offer an important benchmark: Its low hit rate of 0.8% highlights the value in prioritizing resources towards the most promising compounds. Indeed, the unsupervised CRank offers an order of magnitude higher (9%) hit rate among the top 100 drugs, and the top 800 of the 6,340 drugs prioritized by CRank contains 58 of the 77 S&W drugs (Figure 4G–H). The hit rate can be further increased by expert knowledge. To demonstrate this point, we mimicked the traditional drug repurposing process whereby a physician-scientist manually inspected the top 10% of the CRank consensus ranking on April 15, removing drugs with known significant toxicities in vivo and lower-ranked members of the same drug class, and arrived at 74 drugs available for testing. Using the experimental design as described above, but over a wider range of doses (0.625 – 20μM, 0.2 MOI), we screened these 74 compounds separately from the E918 list, and found 39 N, 10 W, and 11 S outcomes (Table S11). The resulting 28% enrichment of S&W drugs suggests that in the case of limited resources, outcomes are maximized by combining algorithmic consensus ranking with expert knowledge.
Inspecting the CRank list and the experimental outcomes, we find two highly ranked drugs with strong outcomes, but not yet in clinical trials (Table 1): azelastine (CRank #10, S), an antihistamine used to treat upper airway symptoms of allergies, and digoxin (CRank #33, S), used to treat heart failure. Our findings, coupled with extensive experience in their use in the clinical community, argue for their consideration in clinical trials. Other highly ranked candidates include folic acid (CRank #16, S), or methotrexate (CRank #32, S), which impairs folate metabolism and attenuates host inflammatory response in autoimmune diseases. This latter mechanism argues that methotrexate is likely to be effective at the other end of the disease spectrum, i.e., in the face of profound hyperimmune response to the infection. Omeprazole (CRank #50, S), used to suppress gastric acid production, alters lysosome acidification and, along with other benzimidazoles, binds to nonstructural protein 3 (nsp3). Blocking this protein, which enhances the virus’s ability to evade the immune system37, was found to interfere in viral formation of SARS-CoV-238. The combination of CRank and strong outcomes highlighted a few other drugs that may be considered for clinical trial based on knowledge of the general pharmacology, including fluvastatin (CRank #199, S), an HMG-CoA reductase inhibitor used to lower cholesterol, but with pleiotropic effects, including anti-inflammatory effects (likely a class effect, as atorvastatin and pitavastatin also had similar effects); ivermectin (CRank #235, S), an anti-parasitic agent; and sildenafil (CRank #493, S), a phosphodiesterase-5 inhibitor.
Table 1. CRank Predictions for Drug Repurposing.
Top 100 consensus predictions of the drug repurposing pipelines aggregated using the CRank algorithm. The top 100 drugs contain 9 drugs with positive experimental outcomes (S&W), 3 of which are among the top 10 drugs. Drugs highlighted in purple correspond to strong outcomes (S), in orange weak outcomes (W), in green to cytotoxic drugs, while non-highlighted drugs have shown no effect (N) in VeroE6 cells.
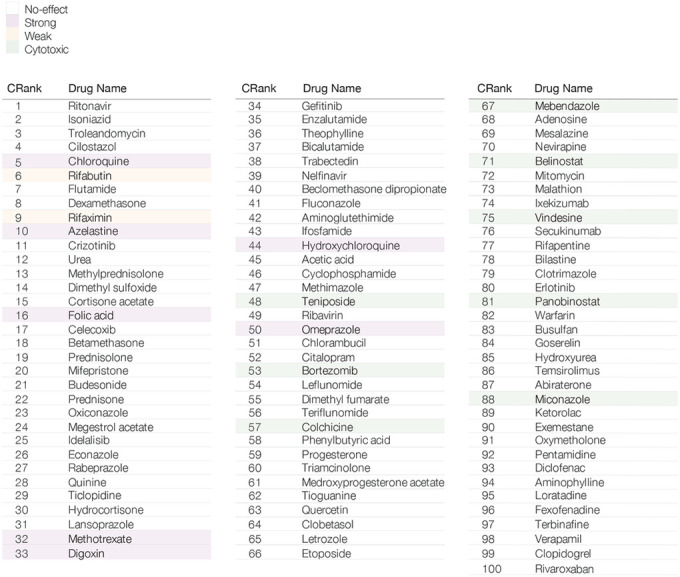
|
Taken together, the methodological advances presented here not only suggest potential drug candidates for COVID-19, but offer a principled algorithmic toolset to identify future treatments for diseases underserved by the cost and the timelines of conventional de novo drug discovery processes. As only 918 of the 6,340 drugs prioritized by CRank were screened, a selection driven by compound availability, many potentially efficacious FDA-approved drugs remain to be tested. Finally, it is also possible that some drugs that lacked activity in VeroE6 cells may nevertheless show efficacy in human cells, like loratadine (rank #95, N), which inhibited viral activity in the human cell line Caco-239. Ritonavir, our top-ranked drug, also showed no effect in our screen, despite the fact that over 42 clinical trials are exploring its potential efficacy in patients. In other words, some of the drugs ranked high by CRank may show efficacy, even if they are not among the 77 S&W drugs with positive outcomes.
Supplementary Material
Acknowledgments
This work was supported, in part, by NIH grants HG007690, HL108630, and HL119145, and by American Heart Association grants D700382 and CV-19 to J.L; A.L.B is supported by NIH grant 1P01HL132825, American Heart Association grant 151708, and ERC grant 810115-DYNASET. M.Z. is supported, in part, by NSF grants IIS-2030459 and IIS-2033384, and by Harvard Data Science Initiative. J.J.P and R.A.D are supported by NIH grants PO1AI120943, RO1AI128364, RO1Ai125453 and from the Massachusetts Consortium on Pathogen Readiness. We wish to thank Nicolette Lee and Grecia Morales for providing support, Helia Sanchez for helping curate the list of drugs in clinical trials for COVID-19, Marc Santolini for suggestions in the diffusion-based methods and Raj S Dattani for comments on the manuscript.
Footnotes
Declaration of interests
J.L. and A.L.B are co-scientific founder of Scipher Medicine, Inc., which applies network medicine strategies to biomarker development and personalized drug selection. A.L.B is the founder of Nomix Inc. and Foodome, Inc. that apply data science to health; O.V and D.M.G are scientific consultants for Nomix Inc. I.D.V. is a scientific consultant for Foodome Inc.
References
- 1.Dudley J. T. et al. Computational Repositioning of the Anticonvulsant Topiramate for Inflammatory Bowel Disease. Sci. Transl. Med. 3, 96ra76–96ra76 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keiser M. J. et al. Predicting new molecular targets for known drugs. Nature 462, 175–181 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campillos M., Kuhn M., Gavin A. C., Jensen L. J. & Bork P. Drug target identification using side-effect similarity. Science 321, 263–266 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Dakshanamurthy S. et al. Predicting New Indications for Approved Drugs Using a Proteochemometric Method. J. Med. Chem. 55, 6832–6848 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paik H. et al. Repurpose terbutaline sulfate for amyotrophic lateral sclerosis using electronic medical records. Sci. Rep. 5, 8580 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greene C. S. & Voight B. F. Pathway and network-based strategies to translate genetic discoveries into effective therapies. Hum. Mol. Genet. 25, R94–R98 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casas A. I. et al. From single drug targets to synergistic network pharmacology in ischemic stroke. Proc. Natl. Acad. Sci. U. S. A. 116, 7129–7136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng F. et al. Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat. Commun. 9, 1–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng F., Kovács I. A. & Barabási A. L. Network-based prediction of drug combinations. Nat. Commun. 10, 1197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guney E., Menche J., Vidal M. & Barábasi A. L. Network-based in silico drug efficacy screening. Nat. Commun. 7, 10331 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadegh S. et al. Exploring the SARS-CoV-2 virus-host-drug interactome for drug repurposing. Nat. Commun. 11, 1–9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Y. et al. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 6, 1–18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zitnik M., Agrawal M. & Leskovec J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 34, i457–i466 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zitnik M. et al. Machine Learning for Integrating Data in Biology and Medicine: Principles, Practice, and Opportunities. Inf. Fusion 50, 71–91 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zitnik M., Sosic R. & Leskovec J. Prioritizing Network Communities. Nat. Commun. 9, 2544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao M. et al. Going the Distance for Protein Function Prediction: A New Distance Metric for Protein Interaction Networks. PLoS One (2013). doi: 10.1371/journal.pone.0076339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patten J. J. & et al. BU Drug Screening. Under Prep. (2020). [Google Scholar]
- 18.Guala D. & Sonnhammer E. L. A large-scale benchmark of gene prioritization methods. Sci. Rep. 7, 1–10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gulbahce N. et al. Viral perturbations of host networks reflect disease etiology. PLoS Comput. Biol. 8, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gordon D. E. et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luck K., Sheynkman G. M., Zhang I. & Vidal M. Proteome-Scale Human Interactomics. Trends in Biochemical Sciences 42, 342–354 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caldera M., Buphamalai P., Müller F. & Menche J. Interactome-based approaches to human disease. Curr. Opin. Syst. Biol. 3, 88–94 (2017). [Google Scholar]
- 23.Silverman E. K. et al. Molecular networks in Network Medicine: Development and applications. Wiley Interdiscip. Rev. Syst. Biol. Med. e1489 (2020). doi: 10.1002/wsbm.1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buchanan M., Caldarelli G., De Los Rios P., Rao F. & Vendruscolo M. Networks in Cell Biology. (Cambridge University Press, 2010). doi: 10.1017/CBO9780511845086 [DOI] [Google Scholar]
- 25.Gulbahce N. et al. Viral perturbations of host networks reflect disease etiology. PLoS Comput. Biol. 8, 1002531 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Menche J. et al. Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 347, 1257601 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wishart D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corsello S. M. et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat. Med. 23, 405–408 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dwork C., Kumar R., Naor M. & Sivakumar D. Rank aggregation methods for the web. in Proceedings of the 10th International Conference on World Wide Web, WWW 2001 613–622 (Association for Computing Machinery, Inc, 2001). doi: 10.1145/371920.372165 [DOI] [Google Scholar]
- 30.Borda J. C. Memoire sur les elections au scrutin. Mémoires de l’académie royale 657–664 (1781). [Google Scholar]
- 31.Reilly B. Social choice in the south seas: Electoral innovation and the Borda count in the Pacific Island countries. Int. Polit. Sci. Rev. (2002). doi: 10.1177/0192512102023004002 [DOI] [Google Scholar]
- 32.Wang M. et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 30, 269–271 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beigel J. H. et al. Remdesivir for the Treatment of Covid-19 — Preliminary Report. N. Engl. J. Med. NEJMoa2007764 (2020). doi: 10.1056/NEJMoa2007764 [DOI] [PubMed] [Google Scholar]
- 34.Lin K. & Gallay P. Curing a viral infection by targeting the host: The example of cyclophilin inhibitors. Antiviral Res. 99, 68–77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horby P. et al. Effect of Dexamethasone in Hospitalized Patients with COVID-19: Preliminary Report. medRxiv (2020). doi: 10.1101/2020.06.22.20137273 [DOI] [Google Scholar]
- 36.Riva L. et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 1–11 (2020). doi: 10.1038/s41586-020-2577-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Virdi R. S., Bavisotto R. V., Hopper N. C. & Frick D. N. Discovery of Drug-like Ligands for the Mac1 Domain of SARS-CoV-2 Nsp3. bioRxiv 2020.07.06.190413 (2020). doi: 10.1101/2020.07.06.190413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aguila E. J. T. & Cua I. H. Y. Repurposed GI Drugs in the Treatment of COVID-19. Dig. Dis. Sci. 1, 1–2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ellinger B. et al. Identification of inhibitors of SARS-CoV-2 in-vitro cellular toxicity in human (Caco-2) cells using a large scale drug repurposing collection. Res. Sq. 1–19 (2020). doi: 10.21203/RS.3.RS-23951/V1 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.