

Abstract
Lung cancer is the leading cause of cancer death worldwide, and the overall survival rate of advanced lung cancer patients is unsatisfactory. Ribosomal proteins (RPs) play important roles in carcinogenesis. However, the role of RPL32 in lung cancer has not been demonstrated. Here, we report that RPL32 is aberrantly, highly expressed in lung cancer tissues and that the overexpression of RPL32 is correlated with the poor prognosis of these patients. RPL32 silencing significantly inhibited the proliferation of lung cancer cells, with an observed p53 accumulation and cell-cycle arrest. Mechanistically, knockdown of RPL32 resulted in ribosomal stress and affected rRNA maturation. RPL5 and RPL11 sensed stress and translocated from the nucleus to the nucleoplasm, where they bound to murine double minute 2 (MDM2), an important p53 E3 ubiquitin ligase, which resulted in p53 accumulation and inhibition of cancer cell proliferation. As lung cancer cells usually express high levels of Toll-like receptor 9 (TLR9), we conjugated RPL32 small interfering RNA (siRNA) to the TLR9 ligand CpG to generate CpG-RPL32 siRNA, which could stabilize and guide RPL32 siRNA to lung cancer cells. Excitingly, CpG-RPL32 siRNA displayed strong anticancer abilities in lung cancer xenografts. Therefore, RPL32 is expected to be a potential target for lung cancer treatment.
Keywords: lung cancer, RPL32, p53, ribosomal stress
Graphical Abstract
Han and colleagues found knockdown of RPL32 resulted in ribosomal stress and RPL5-, RPL11-MDM2-p53-mediated inhibition of lung cancer cell proliferation. RPL32 is expected to be a potential target for lung cancer treatment.
Introduction
Lung cancer is the most common type of cancer and the leading cause of cancer death in men and women worldwide.1 Although the treatment of lung cancer has improved with the shift from using cytotoxic drugs to using precise individualized treatments, such as small molecule tyrosine kinase inhibitors and immune checkpoint blockades, in the past two decades, the 5-year survival rate is still very low (only approximately 15%). Most patients with advanced lung cancer continue to progress under anticancer treatment and eventually die due to treatment failure.2,3 Therefore, a comprehensive understanding of the molecular mechanism is of great significance for developing novel molecular targets for the treatment and prevention of lung cancer.
Rapidly proliferating cancer cells require higher rates of biological energy consumption and biosynthesis, which are partly achieved by an increase in the rate of ribosome production. As a major component of ribosomes, ribosomal proteins (RPs) play important roles in the tightly regulated protein synthesis process. In addition to their basic protein synthesis function, ribosomal proteins have important extraribosomal moonlighting functions in many cell physiological processes, such as the regulation of DNA damage repair, apoptosis, cell proliferation and differentiation, drug resistance, cell migration, and invasion.4,5 The dysregulation of ribosomal proteins has been reported to be closely related to the initiation and development of human cancer via tumor-promoting or -suppressing activities.6 The oncogenic activities include promoting cell-cycle progression and cell proliferation,7,8 participating in the regulation of genes essential for tumorigenesis,9, 10, 11 inhibiting cell death through reactive oxygen species (ROS) regulation12 and cell death regulatory gene alterations,13 and upregulating the expression of sirt1 post-transcriptionally14. The tumor-suppressive activities include induction of p53 family transcription factors;15,16 inactivation of non-p53 transcription factors, such as c-Myc17 and nuclear factor κB (NF-κB);18 control of gene expression at the post-transcriptional and translational levels;19 activation of a caspase-related apoptotic signaling pathway;20 promotion of cell-cycle arrest;21 and enhancement of DNA repair and genomic stability.22
Some ribosomal proteins are associated with the tumorigenesis and development of lung cancer. Chen et al.23 found that RPS6 and phosphorylated (phospho)-RPS6 are significantly overexpressed in non-small cell lung cancer (NSCLC) and that knockdown of RPS6 inhibits lung cancer cell growth by inducing cell-cycle arrest. RPL3 was reported to promote the apoptosis of p53-mutated lung cancer cells by downregulating cystathionine-β-synthase (CBS) and NF-κB upon fluorouracil (5-FU) treatment.24 Phosphorylated (phospho)-RPS3 was found to bind with the p65 subunit of NF-κB and confer radioresistance in lung cancer cells.25 However, the role of ribosomal proteins in lung cancer and their clinical significance deserve further study.
In a retrospective study of mutant genes in chronic B-lymphocytic leukemia (CLL) patients, it was found that RPL32 had high predictive accuracy toward SF3B1, one of the main recurrent mutated genes in CLL.26 In a lymph node carcinoma of the prostate (LNCaP) cell model, RPL32 was demonstrated to be upregulated in late-passage androgen-independent (LNCaP-C81) cells compared to early-passage androgen-sensitive (LNCaP-C33) cells, which suggests that RPL32 may positively correlate with the progression of human prostate cancer.27 In breast cancer patients, it has been reported that the expression of RPL32 in circulating tumor cell (CTC) clusters is higher than that in single CTC, which have greater metastatic potential.28 The above results suggest that RPL32 may be closely related to cancer proliferation and metastasis, but the function of RPL32 in lung cancer and its mechanism is still unclear.
In this study, we found that the expression of RPL32 in cancer tissues was significantly higher than that in adjacent tissues, and overexpression of RPL32 was associated with poor prognosis in lung cancer patients. RPL32 silencing significantly inhibited the proliferation of lung cancer cells. Mechanistically, RPL32 knockdown caused the release of RPL5 and RPL11 from the nucleus to the nucleoplasm, where they bound to murine double minute 2 (MDM2), resulting in accumulation of p53 and inhibition of cell proliferation. We also conjugated RPL32 small interfering RNA (siRNA) to CpG to guide RPL32 siRNA to the lung tumor tissue more efficiently and showed a strong antitumor effect in lung cancer xenografts. This study demonstrates that RPL32 may be a potential therapeutic target for lung cancer treatment.
Results
Upregulation of RPL32 in Lung Cancer and Its Correlation with Poor Clinical Outcomes
Through the analysis of a publicly available clinical database of lung cancer (http://kmplot.com/analysis/), we observed that the RPL32 expression level was associated with poor prognosis in patients with lung cancer (Figure 1A). To further confirm the protein levels of RPL32, we performed immunohistochemistry (IHC) to detect RPL32 in a large cohort of primary lung cancer patients (Table S1). For the 93 patients, 87 specimens contained both tumors and matched adjacent paracancerous tissues, whereas the remaining 6 specimens contained only tumors. In the 87 matched samples, we found that the RPL32 immunostaining intensity of tumors was significantly higher than that of adjacent normal tissues (Figures 1B and 1C). Clinically, higher RPL32 expression in tumors compared with paired tumor-adjacent normal tissues was significantly associated with shorter lung cancer patient survival (p = 0.0247) (Figure 1D). To confirm that RPL32 is an independent factor linked to clinical outcomes, we performed multivariate overall survival analysis by using a Cox proportional hazard model based on available clinical information. The results confirmed that RPL32 expression was an independent prognostic factor (Figure 1E). Together, our results confirm that the increased expression of RPL32 is positively correlated with the progression and survival rate of lung cancer patients.
Figure 1.

High Expression of RPL32 Is Associated with Adverse Clinical Outcomes in Patients with Lung Cancer
(A) Kaplan Meier (KM) Plotter analysis indicates that increased expression of RPL32 correlates with progression and poor survival in patients with lung cancer. (B) Representative IHC staining of RPL32 in lung cancer and paracancerous tissues. A total of 160 patient samples were stained and analyzed. (C) Quantitative analysis of RPL32 IHC staining intensity in 87 paired tumor/paratumor samples. **p < 0.01. (D) Kaplan-Meier survival curves of lung cancer patients based on the scores of RPL32 IHC staining. An IHC score in tumor tissue lower than or equal to that in its paired nontumor tissue was defined as “RPL32 Expression Low,” and the IHC score in tumor higher than its paired nontumor tissue was defined as “RPL32 Expression High.” A higher intensity of RPL32 immunostaining in tumors than in paired nontumor samples was strongly associated with poor patient survival, p = 0.0247. (E) Multivariate overall survival analysis of patients obtained by using a Cox proportion hazards model.
RPL32 Silencing Inhibits the Proliferation of Lung Cancer Cells with Significant p53 Accumulation and Cell-Cycle Arrest
To determine the potential biological function of RPL32, we employed a siRNA-based silencing strategy to transiently knock down RPL32 in several lung cancer cell lines. With the use of a cell proliferation colorimetric assay, we found that RPL32 silencing significantly suppressed the proliferation of lung cancer cell lines, except for p53-deficient H1299 cells (Figure 2A), indicating that the function of RPL32 might be p53 related. Subsequently, we found that RPL32 silencing resulted in significant accumulation of p53, accompanied by an increase in the p53 transcriptional target p21 (Figure 2B). Cell-cycle analysis by flow cytometry showed that RPL32 silencing resulted in G2/M arrest in A549 and NCI-H460 cells (Figure 2C). Accordingly, RPL32 silencing also dramatically reduced colony formation (Figure 2D).
Figure 2.

RPL32 Silencing Inhibits the Growth of Lung Cancer Cells in a p53-Dependent Manner
(A) Cell numbers were counted after transfection with scramble siRNA or RPL32 siRNA for 96 h (n = 3; mean ± SD). **p < 0.01. (B) Immunoblot analysis of RPL32, p53, and p21 in A549 and NCI-H460 cells transfected with scramble siRNA or RPL32 siRNA. (C) Cell-cycle analysis of A549 and NCI-H460 cells transfected with scrambled siRNA or RPL32 siRNA. (D) Colony formation assay of A549 and NCI-H460 cells transfected with scramble siRNA or RPL32 siRNA. (E) Quantitative real-time PCR analysis of p53 mRNA levels in A549 and NCI-H460 cells transfected with RPL32 siRNA and p53 siRNA alone or together (n = 3; mean ± SD). **p < 0.01. (F) Cells were counted 96 h after transfection with RPL32 siRNA and p53 siRNA alone or together (n = 3; mean ± SD). **p < 0.01. (G) Immunoblot analysis of RPL32, p53, and p21 in cells transfected with RPL32 siRNA and p53 siRNA alone or together. (H) Representative histograms of cell-cycle analysis of cells transfected with RPL32 siRNA and p53 siRNA alone or together.
The Biological Function of RPL32 in Lung Cancer Cells Is p53 Dependent
As mentioned above, siRNA-mediated silencing of RPL32 resulted in a decrease in the number of viable cells and an increase in the accumulation of p53 in both A549 and NCI-H460 cells. To investigate the relationship between RPL32 and p53, siRNA against p53 was transfected alone or together with RPL32 siRNA, and the knockdown efficiency was validated (Figure 2E). When RPL32 siRNA was cotransfected with p53 siRNA, the growth inhibitory effect of RPL32 silencing was significantly reduced (Figure 2F). In addition, the upregulation of p21 and G2/M cell-cycle arrest after knockdown of RPL32 was obviously reversed after cotransfection with p53 siRNA (Figures 2G and 2H). These results suggest that the effect of RPL32 knockdown on cell viability is p53 dependent.
RPL32 Silencing Inhibits MDM2-Mediated p53 Ubiquitination and Degradation
Next, we investigated how RPL32 silencing leads to the accumulation of p53. Quantitative real-time PCR results showed that there was no obvious change in the mRNA levels of p53 after RPL32 knockdown (Figures 3A and 3B), indicating that RPL32 silencing stabilized p53 at the protein level. To address this hypothesis, we used cycloheximide to block protein synthesis and found that the half-life of p53 increased in RPL32-knockdown A549 and NCI-H460 cells (Figure 3C). After treatment with the proteasome inhibitor MG132, the difference in p53 protein levels between RPL32-silenced and negative control cells was greatly reduced (Figure 3D), further indicating that the p53 accumulation caused by RPL32 knockdown is due to the reduction in p53 degradation.
Figure 3.

RPL32 Silencing Inhibits MDM2-Mediated p53 Ubiquitination and Degradation
(A and B) (A) A549 and (B) NCI-H460 cells were transfected with scramble siRNA or RPL32 siRNA and the RPL32 and p53 mRNA levels were determined with quantitative real-time PCR analysis. ***p < 0.001. (C) Immunoblot analysis of p53 and RPL32 protein levels in cells treated with cycloheximide (25 μg/mL) at the indicated time points. Densitometric analysis is also shown (n = 3; mean ± SD). (D) Immunoblot analysis of p53 protein levels in cells after treatment with MG132 (10 μM, 4 h). (E) Cell lysates of A549 and NCI-H460 cells were immunoprecipitated with anti-p53 antibody, followed by immunoblot with anti-p53, anti-MDM2, and anti-ubiquitin antibodies. The cells were treated with MG132 (10 μM) for 10 h before harvesting.
As MDM2 is an important ubiquitin E3 ligase that can interact with p53 to promote its ubiquitination and degradation, we wondered whether MDM2 is involved in the accumulation of p53 induced by RPL32 knockdown. We examined the interaction between MDM2 and p53 and p53 ubiquitination in A549 and NCI-H460 cells transfected with RPL32 siRNA. Before immunoprecipitation, MG132 was used to induce accumulation of ubiquitinated proteins. As shown in Figure 3E, the binding of MDM2 to p53 and the ubiquitination of p53 decreased after RPL32 knockdown, indicating that RPL32 silencing prevented MDM2-mediated ubiquitination and degradation of p53.
RPL32 Silencing Inhibits rRNA Maturation, Leads to RPL5 and RPL11 Translocation, and Prevents MDM2 from Binding to p53
Next, we investigated how RPL32 silencing inhibits MDM2-mediated p53 ubiquitination and degradation. Because RPL32 is a member of the ribosomal protein family, we first explored its effect on rRNA maturation. Primers that could distinguish between the precursor and mature human rRNAs were used to quantify the mature rRNAs (Figure 4A). We found that the levels of mature 18S and 28S rRNAs were significantly lower in RPL32-knockdown A549 and NCI-H460 cells than in their respective negative controls (Figure 4B), indicating that RPL32 silencing could result in ribosome biogenesis stress.
Figure 4.

RPL32 Silencing Inhibits rRNA Maturation and Results in RPL5 and RPL11 Translocation, Preventing MDM2 from Binding to p53
(A) Modular representation of the primary transcript of mammalian rRNA. Top: relative positions of regions absent (thin lines) and present (bold lines) in mature rRNAs. Bottom: relative positions of primer pairs used for analyzing rRNA processing. ETS, external transcribed spacer; ITS, internal transcribed spacer. (B) Quantitative real-time PCR analysis of rRNAs using primer pairs in cells transfected with scramble siRNA or RPL32 siRNA. The values represent the averages of primer pairs 4/3 (unprocessed) over 2/1 (total) for 18S rRNA and primer pairs d/c (unprocessed) over b/a (total) for 28S rRNA. (n = 3; mean ± SD). (C) Immunoblot analysis of p53, RPL32, RPL5, and RPL11 in cells after transfection with RPL32 siRNA, RPL5 siRNA, and RPL11 siRNA, alone or together. (D) Immunoblot analysis of MDM2, p53, RPL5, and RPL11 in cytoplasmic, nucleolar, and nucleoplasmic fractions after transfection with scramble siRNA or RPL32 siRNA. Actin, nucleophosmin (NPM), and histone H4 are the localization markers of the cytoplasm, nucleolus, and nucleoplasm, respectively. (E) Fluorescence microscopy of NCI-H460 cells after transfection with scramble siRNA or RPL32 siRNA and costained with anti-NPM (green) and anti-RPL5 (red) or anti-RPL11 (red) antibodies. Scale bars, 10 μm. (F) Cell lysates of A549 and NCI-H460 cells (+/− RPL32 siRNA) were immunoprecipitated with anti-RPL5 and anti-RPL11 antibodies, followed by immunoblot with anti-MDM2 antibody. **p < 0.01.
Under ribosome biogenesis stress, several well-studied RPs, including RPL529 and RPL11,30 are released from the nucleolus and then bind with MDM2 to inhibit its ubiquitin ligase activity on p53, resulting in the accumulation of p53. Whether these RPs also participate in the RPL32-mediated p53 response remains unclear. Therefore, we examined whether knockdown of RPL5 or RPL11 could affect the accumulation of p53 induced by RPL32 knockdown. As shown in Figure 4C, RPL5 or RPL11 siRNA transfection alone did not result in p53 accumulation, whereas cotransfection of RPL5 or RPL11 siRNA with RPL32 siRNA reduced the accumulation of p53, indicating that RPL5 and RPL11 may be involved in the RPL32-mediated p53 accumulation. Next, we investigated whether RPL32 knockdown interferes with the localization of RPL5 and RPL11 in cancer cells. When RPL32 was knocked down in A549 and NCI-H460 cells, we found that RPL5 and RPL11 levels decreased in the nucleolus but increased in the nucleoplasm, indicating their translocation from the nucleolus to the nucleoplasm (Figure 4D). This result was also confirmed by the immunostaining experiment in NCI-H460 cells (Figure 4E). Furthermore, the binding of endogenous MDM2 to RPL5 or RPL11 was significantly enhanced after knockdown of RPL32 in A549 and NCI-H460 cells (Figure 4F). Based on the above results, we speculate that RPL5 and RPL11 sense ribosomal biogenesis stress after RPL32 silencing and translocate from the nucleolus to the nucleoplasm, where they bind to MDM2 and prevent its binding to p53, leading to the accumulation of p53.
CpG-RPL32 siRNA Inhibits the Proliferation of Lung Cancer Cells In Vivo
Toll-like receptor 9 (TLR9)-positive cells can recognize unmethylated CpG oligodeoxynucleotides (ODNs).31 It has been demonstrated that the conjugates of CpG ODNs to siRNAs (CpG-siRNAs) can be actively internalized by TLR9-positive cells without any transfection reagents, achieving efficient gene silencing in vivo.32 We chemically linked RPL32 siRNA with CpG ODNs to produce CpG-RPL32 siRNA (Figure 5A), which was shown to be efficiently internalized by TLR9-positive A549 cells33 in vitro (Figure S1). To rule out the possible influence of TLR9-positive dendritic cells (DCs) and B cells,34 non-obese diabetic (NOD)-severe combined immunodeficiency (scid) gamma (NSG) mice were used in our xenograft experiments.
Figure 5.

RPL32 Silencing Inhibits the Growth of Lung Cancer In Vivo
(A) Sequence of the CpG-linked human RPL32 siRNA conjugate (CpG-RPL32 siRNA). CpG sequence (deoxyribonucleotides shown in black) were phosphorothioated and connected through a carbon linker (6 of C3 units) to the antisense strand of RPL32 siRNA (ribonucleotides shown in red). (B) Body weight of NSG mice inoculated with 5 × 106 A549 cells and randomly assigned to be injected peritumorally with PBS, CpG-scrambled RNA, or CpG-RPL32 siRNA. (C and D) Tumor volume (C) and weight (D) in each group. **p < 0.01. (E) Xenograft tumors in each group. (F) Tumor lysates from each group (n = 3) were subjected to immunoblot analysis to validate the RPL32 knockdown efficiency and p53 accumulation.
We inoculated 5 × 106 A549 cells into the right flanks of NSG mice. When the xenografted tumors were measurable, the mice were randomly divided into two groups that underwent peritumoral injection with 50 μL (80 μM) CpG-scrambled RNA or CpG-RPL32 siRNA, every 2 days for 5 consecutive times. As shown in Figures 5B–5D, CpG-RPL32 siRNA could significantly inhibit tumor growth compared with CpG-scrambled RNA. We also extracted protein lysates from xenografted tumors and confirmed the RPL32 silencing efficiency and p53 accumulation (Figure 5E).
Discussion
There are approximately 80 ribosomal proteins in eukaryotes, and recent studies have revealed that ribosomal proteins have additional extraribosomal functions.35 In this study, we found that high expression of RPL32 in lung cancer patients was associated with adverse clinical outcomes, and knockdown of RPL32 could significantly inhibit the proliferation of lung cancer cells. Furthermore, we found that knockdown of RPL32 resulted in significant accumulation of p53 and had less effect on the proliferation of p53 null H1299 lung cancer cells. In other words, the biological function of RPL32 in lung cancer cells is p53 dependent. To the best of our knowledge, the role of RPL32 in tumorigenesis and its relationship with p53 in lung cancer have not been reported elsewhere.
As p53 is related to drug sensitivity, we tested the cisplatin sensitivity, with or without the RPL32 knockdown in A549, NCI-H460, and H1299 lung cancer cell lines. We found knockdown of RPL32 could significantly increase the cisplatin sensitivity in p53 wild-type A549 and NCI-H460 cell lines but not in p53 null H1299 cells (data not shown). To accurately evaluate the relationship between RPL32 expression and the prognosis of lung cancer patients, the p53 status should also be also considered. Since there were no data on the p53 status in the tissue microarray analyzed in this study, we analyzed the relationship between the expression of the RPL32 gene and the prognosis of patients from The Cancer Genome Atlas (TCGA) lung adenocarcinoma dataset. Based on the p53 status, the patients were separated into two groups. It was found that in the mutant p53 group, the expression of RPL32 had no obvious relationship with the prognosis of patients (Figure S2A), whereas in the wild-type p53 group, patients with low expression of RPL32 had a longer survival, although the difference was not significant (p = 0.0524) (Figure S2B). These results further validate the relationship between RPL32 and p53 in lung cancer.
We also examined the effects of RPL32 silencing on the proliferation of the normal lung epithelial cell line BEAS-2B. As shown in Figure S3, the proliferation of BEAS-2B cells was slightly inhibited after RPL32 knockdown, with relatively little p53 accumulation and cell-cycle arrest. We have not yet fully clarified why RPL32 knockdown has different functions in normal cells and cancer cells. Besides, RPL32 overexpression was found to have a slight inhibition effect of the proliferation of A549 and NCI-H460 cells unexpectedly. We speculate that cancer cells have a high demand of biosynthesis, and they have stronger ribosomal function than normal cells, which makes them more susceptible to RPL32 silencing. Only overexpression of RPL32 may not increase the ribosomal biogenesis; conversely, it may also result in ribosomal stress. Although the mechanism is unclear, the above finding clearly suggests that RPL32 may be a specific therapeutic target for lung cancer.
Mechanically, we found that RPL32 silencing resulted in inhibition of rRNA maturation and ribosome biogenesis stress, which were further evidenced because the monosome and polysome levels were decreased after knockdown of RPL32 in A549 cells (data not shown). The nucleolus is the main component of the nucleus and the site of ribosome biosynthesis. It is considered a sensor of various stress conditions, which ultimately lead to the concept of “nucleolar stress.” It is noteworthy that many nucleolar proteins, such as NPM136 and GLTSCR2,37 translocate to the nucleoplasm during cellular stress responses. Ribosomal proteins, such as RPL5 and RPL11,38 have recently emerged as important factors in the activation of p53 in response to nucleolar stress. In our study, we found that both RPL5 and RPL11 also participated in the reduction of RPL32-induced nucleolar stress. These proteins sense and transmit the stress signal to p53, leading to the arrest of cancer cell proliferation. When we cut the “signaling bridge” through knockdown of RPL5 or RPL11, the RPL32 knockdown-induced p53 accumulation obviously decreased (Figure 4C).
We used mass spectrometry and a yeast two-hybrid system to detect the RPL32-interacting proteins. Nucleosome assembly protein 1-like 1 (NAP1L1) overlapped in both screening systems, and we further validated its interaction with RPL32 by immunoprecipitation experiments (Figures S4A–4C). NAP1L1 was recently reported as an oncogene that could activate the phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling pathway in hepatocellular carcinoma (HCC).39 NAP1L1 does not interact with p53 directly (Figure S4D), and knockdown of RPL32 had no influence on the expression of NAP1L1 (Figure S4E). However, knockdown of NAP1L1 affected the accumulation of p53 induced by RPL32 knockdown (Figure S4E). Interestingly, the expression of RPL5 and RPL11 increased after knockdown of NAP1L1 alone or together with RPL32, which implies that NAP1L1 participates in this process (Figure S4E). However, we have not yet fully elucidated the mechanism by which NAP1L1 is involved in the signaling transmitted from RPL32 to p53.
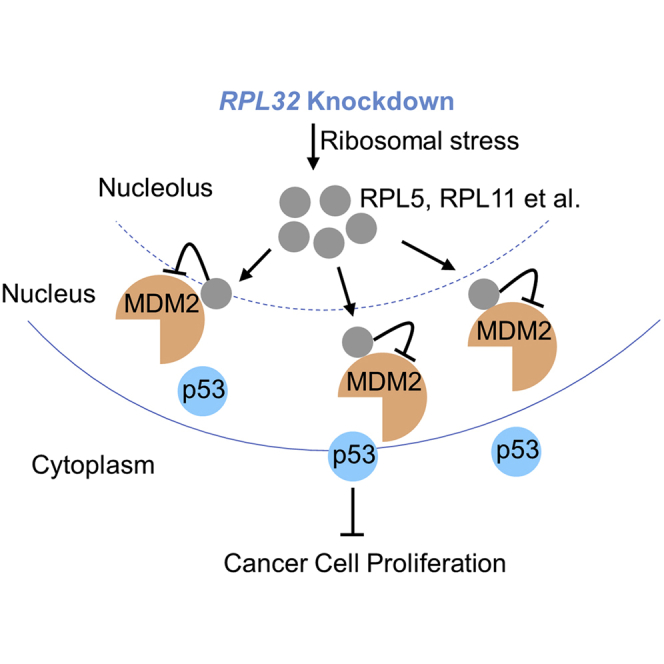
In summary, as illustrated in Figure 6, we discovered that RPL32 silencing resulted in ribosome biogenesis stress, leading to the translocation of RPL5 and RPL11 from the nucleus to the nucleoplasm, where they competitively bound to MDM2, resulting in the accumulation of p53 and inhibition of lung cancer proliferation. Our findings demonstrate that RPL32 might be a promising therapeutic target for lung cancer.
Figure 6.

Proposed Model of the Process after RPL32 Silencing in Lung Cancer Cells
RPL32 silencing results in ribosome biogenesis stress, inducing RPL5 and RPL11 translocation from the nucleus to the nucleoplasm, where they competitively bind to MDM2, which results in p53 accumulation and inhibition of lung cancer cell proliferation.
Materials and Methods
Patient Samples and IHC Assay
To assess the correlation between RPL32 IHC scores and clinical stages of lung cancer patients, a 160-spot, paraffin-embedded tissue array chip (HBre-Duc170Sur-01), which included 87 paired lung tumor and normal tissues and 6 tumor tissues with 10 years of follow-up information, was purchased from Shanghai Outdo Biotech (Shanghai, China). RPL32 IHC scores were calculated from RPL32 IHC staining intensity multiplied by the positive staining rate. Staining intensity is generally divided into four levels: 0 indicates negative staining, 0.5 indicates weakly positive staining, 1 indicates light-yellow or light-brown staining, and 2 indicates yellow or brown staining. The ratio of the number of positive cells to the total number of cells in the tissue was defined as the positive staining rate. Detailed clinical features of the lung cancer patients are summarized in Table S1.
For survival analysis of patients with p53 information, primary data were obtained from TCGA lung adenocarcinoma dataset. A total of 473 samples with mutant p53 (n = 256) and wild-type p53 (n = 217) were included. In each group, the patients were divided into two groups, according to the relative expression level of the RPL32 gene.
Reagents and Antibodies
Cycloheximide and MG132 were purchased from Sigma-Aldrich. Primary antibodies against p53, p21, and Myc-tag were purchased from Cell Signaling Technology. Antibodies against RPL32, MDM2, and NAP1L1 were obtained from Abcam. Antibodies against RPL11 and NPM1 were from Proteintech. Anti-RPL5 antibody was obtained from GeneTex. The secondary antibodies were horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin G (IgG) and anti-mouse IgG (Jackson ImmunoResearch).
Cell-Cycle Assay
The cell cycle was determined by flow cytometric (FCM) analysis with the Cell Cycle Staining Kit (MultiSciences Biotech, China). Briefly, cells were collected, washed, and resuspended in 500 μL DNA staining solution mixed with 5 μL permeabilization solution and incubated for 30 min. The samples were then analyzed by FCM. The results were analyzed with the BD FACSCalibur system.
Cell Lines and Animals
HEK293 cell and the lung cancer cell lines A549, NCI-H460, PC-9, H1792, and H1299 were purchased from the Cell Bank of the Chinese Academy of Science and were cultured in DMEM (Gibco), supplemented with 10% fetal bovine serum (Gibco) at 37°C under a 5% CO2 and 90% humidified atmosphere.
Female NSG mice (Shanghai Slac Laboratory Animal) were maintained in a specific pathogen-free facility and were treated with humane care after approval from the Animal Care and Use Committee of Zhejiang University.
Colony Formation Assay
To conduct a colony formation assay, 1 × 103 A549 or NCI-H460 cells that were in the logarithmic phase were seeded into a 6-well plate and cultured at 37°C under a 5% CO2 and 90% humidified atmosphere until visible clones appeared (approximately 2 weeks). Cell clones were washed twice with PBS, fixed with methanol for 10 min, stained with 0.1% crystal violet for 30 min, and washed with PBS.
RNA Interference, Plasmids, and Transfections
Cells were transfected with scrambled or siRNAs using RNAiMAX (Thermo Fisher Scientific), according to the manufacturer’s protocol. siRNA oligonucleotides were purchased from RiboBio (Guangzhou, China). A nonspecific oligo that is not complementary to any human gene was used as a negative control. siRNA sequences against RPL32 were as follows: 5′-UGGCCAUCAGAGUCACCAA deoxythymidine dinucleotide (dTdT)-3′ and 5′-CGAGAUCGCUCACAAUGUU dTdT-3′. siRNA sequences against p53 were as follows: 5′-GUACCACCAUCCACUACAA dTdT-3′ and 5′-AGAGAAUCUCCGCAAGAAA dTdT-3′. siRNA sequence against NAP1L1 was as follows: 5′-GGCAGACAUUGACAACAAA dTdT-3′.
cDNA encoding human RPL32 was cloned from a human lung tissue sample and inserted into the pcDNA3.1-Flag backbone plasmid that was purchased from Addgene to generate pcDNA3.1-Flag-RPL32. Cells were transfected with plasmids by Lipofectamine 3000 (Thermo Fisher Scientific), according to the manufacturer’s protocol.
Quantitative Real-Time PCR Analysis
Total RNA was isolated from cells or tissues, and first-strand cDNA was generated from total RNA using oligo-dT primers and Reverse Transcriptase II (Life Technologies). Quantitative real-time PCR was performed using specific primers and the LightCycler 480 II analyzer (Roche) with the SYBR GreenER qPCR SuperMix Universal Kit (Life Technologies). Target gene expression values were normalized to human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) values. Sequences of the primers used for quantitative real-time PCR were as follows: human RPL32: forward primer 5′-TCTCCTTCTCGGCATCATGG-3′, reverse primer 5′-CGAACCCTGTTGTCAATGCC-3′; human GAPDH: forward primer 5′-ACAGCCTCAAGATCATCAGCAA-3′, reverse primer 5′-ACCACTGACACGTTGGCAGT-3′; and human p53: forward primer 5′-GGACGGAACAGCTTTGAGGT-3′, reverse primer 5′-CCCACGGATCTGAAGGGTGA-3′.
Oligonucleotide Design and Synthesis
CpG-conjugated siRNA was synthesized as previously described.40,41 D19 phosphorothioate ODNs were linked to the antisense strand (AS) of RPL32 siRNA, using 6 units of the C3 carbon chain linker (CH2)3. The resulting construct was hybridized to the complementary sense strand (SS) of siRNAs to generate the chimeric ODN-siRNA constructs used in the study (deoxynucleotides are shown underlined). Sequences of single-stranded constructs are listed below, and the target RPL32 sequence is from bases 261 to 283 of the coding sequence (CDS) of the human RPL32 gene (GenBank: NM_000994): RPL32 siRNA (SS): 5′-GCUGCUGAUGUGCAACAAAUCUUAC-3′; CpG-RPL32 siRNA (AS): 5′-GGTGCATCGATGCAGGGGGG-linker-GUAAGAUUUGUUGCACAUCAGCAGCAC-3′; scrambled RNA (SS): 5′-UCCAAGUAGAUUCGACGGCGAAGTG-3′; ανδ CpG-scrambled RNA (AS): 5′-GGTGCATCGATGCAGGGGGG-linker-CACUUCGCCGUCGAAUCUACUUGGAUU-3′.
Human Lung Cancer Xenograft Model
Human lung cancer xenograft models were established according to our previous study.42 6- to 8-week-old female NSG mice were utilized. Briefly, 5 × 106 A549 cells were injected subcutaneously into the flank regions of the mice. When the diameter of the tumors reached approximately 0.4 cm, the mice were randomly assigned to three groups: PBS, CpG-scrambled RNA, and CpG-RPL32 siRNA. The CpG-siRNA doses were given peritumorally every 2 days for 5 consecutive times. The tumor volume was determined using the formula V = LW2/2, where L is the largest diameter and W is the smallest diameter.
Immunofluorescence
Cells were fixed in 4% paraformaldehyde for 10 min and permeabilized with 0.5% Triton X-100 for 10 min. After blocking with 5% BSA, cells were incubated overnight with antibody in PBS containing 5% BSA at 37°C. Staining was detected using DyLight 488- or DyLight 549-labeled secondary antibodies (MultiScience). Nuclei were costained with 4′,6-diamidino-2-phenylindole (DAPI; Roche). Stained cells were imaged using a fluorescence microscope.
Immunoprecipitation Analysis
Cells were lysed in NP-40 lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 0.5% NP-40, 10 mM PMSF, 20 mM NaF, 1 mM Na3VO4, and protease inhibitor cocktail. Lysates were immunoprecipitated with the appropriate antibodies overnight at 4°C. Protein A/G Sepharose beads (GE Healthcare) were then added and incubated for 4 h at 4°C. Alternatively, lysates were immunoprecipitated with anti-Flag M2 Magnetic Beads (Sigma) overnight at 4°C. After five washes with lysis buffer, samples were resolved on SDS-PAGE gels and blotted.
Cell Fractionation
Cytoplasmic, nuclear, and nucleolar fractions were obtained, as previously described, with minor modifications.43 Briefly, 5 × 106 cells were transfected with negative control or RPL32 siRNA. After 48 h, cells were collected and resuspended in 500 μL hypotonic buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT). After centrifugation (500 × g) for 10 min, the supernatant was recovered to serve as the cytoplasmic fraction. The pellet was resuspended in 300 μL S1 buffer (0.25 M sucrose, 10 mM MgCl2), overlaid onto 300 μL S2 buffer (0.35 M sucrose, 0.5 mM MgCl2), and centrifuged (1,500 × g) for 5 min. The pellet was resuspended in 300 μL S2 buffer to serve as the nuclear fraction. Then, the nuclear fraction was sonicated (30 s sonication, 60 s intervals for 5 cycles), overlaid onto 300 μL S3 buffer (0.88 M sucrose, 0.05 mM MgCl2), and centrifuged (3,000 × g) for 10 min. The supernatant was collected as the nucleoplasmic fraction. The pellet was resuspended in radioimmunoprecipitation assay (RIPA) buffer to serve as the nucleolar fraction. Protease inhibitor cocktail was added to all fraction samples. The fractions were resolved on SDS-PAGE gels and blotted.
Statistical Analysis
Unless otherwise noted, quantitative data are expressed as the mean ± standard error. Statistical significance was determined with paired Student’s t test; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001, when compared with control.
Author Contributions
W.H. and Y.Y. conceived and designed the study. J.X., H.P., W.Z., X.L., and C.S. performed the experiments. J.X. and H.P. analyzed the data and prepared the figures. W.H. and J.X. wrote the paper. Y.Z. modified the paper. All authors reviewed the manuscript, and the manuscript is approved by all authors for publication.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81872238 and 81972745), Ten Thousand Plan Youth Talent Support Program of Zhejiang Province (ZJWR0108009), and Zhejiang Medical Innovative Discipline Construction Project-2016.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.05.019.
Contributor Information
Hongming Pan, Email: panhongming@zju.edu.cn.
Yunhai Yang, Email: docyyh@163.com.
Weidong Han, Email: hanwd@zju.edu.cn.
Supplemental Information
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Herbst R.S., Morgensztern D., Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. doi: 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 3.Testa U., Castelli G., Pelosi E. Lung Cancers: Molecular Characterization, Clonal Heterogeneity and Evolution, and Cancer Stem Cells. Cancers (Basel) 2018;10:248. doi: 10.3390/cancers10080248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou X., Liao W.J., Liao J.M., Liao P., Lu H. Ribosomal proteins: functions beyond the ribosome. J. Mol. Cell Biol. 2015;7:92–104. doi: 10.1093/jmcb/mjv014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiong X., Liu X., Li H., He H., Sun Y., Zhao Y. Ribosomal protein S27-like regulates autophagy via the β-TrCP-DEPTOR-mTORC1 axis. Cell Death Dis. 2018;9:1131. doi: 10.1038/s41419-018-1168-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molavi G., Samadi N., Hosseingholi E.Z. The roles of moonlight ribosomal proteins in the development of human cancers. J. Cell. Physiol. 2019;234:8327–8341. doi: 10.1002/jcp.27722. [DOI] [PubMed] [Google Scholar]
- 7.Guo X., Shi Y., Gou Y., Li J., Han S., Zhang Y., Huo J., Ning X., Sun L., Chen Y. Human ribosomal protein S13 promotes gastric cancer growth through down-regulating p27(Kip1) J. Cell. Mol. Med. 2011;15:296–306. doi: 10.1111/j.1582-4934.2009.00969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kummalue T., Inoue T., Miura Y., Narusawa M., Inoue H., Komatsu N., Wanachiwanawin W., Sugiyama D., Tani K. Ribosomal protein L11- and retinol dehydrogenase 11-induced erythroid proliferation without erythropoietin in UT-7/Epo erythroleukemic cells. Exp. Hematol. 2015;43:414–423.e1. doi: 10.1016/j.exphem.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Lim K.H., Kim K.H., Choi S.I., Park E.S., Park S.H., Ryu K., Park Y.K., Kwon S.Y., Yang S.I., Lee H.C. RPS3a over-expressed in HBV-associated hepatocellular carcinoma enhances the HBx-induced NF-κB signaling via its novel chaperoning function. PLoS ONE. 2011;6:e22258. doi: 10.1371/journal.pone.0022258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Z.Y., Jiang H., Qu Y., Wei M., Yan M., Zhu Z.G., Liu B.Y., Chen G.Q., Wu Y.L., Gu Q.L. Metallopanstimulin-1 regulates invasion and migration of gastric cancer cells partially through integrin β4. Carcinogenesis. 2013;34:2851–2860. doi: 10.1093/carcin/bgt226. [DOI] [PubMed] [Google Scholar]
- 11.Zhu X., Yuan C., Tian C., Li C., Nie F., Song X., Zeng R., Wu D., Hao X., Li L. The plant sesquiterpene lactone parthenolide inhibits Wnt/β-catenin signaling by blocking synthesis of the transcriptional regulators TCF4/LEF1. J. Biol. Chem. 2018;293:5335–5344. doi: 10.1074/jbc.M117.819300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J., Chen Z., Liu N., Chen Y. Ribosomal protein L10 in mitochondria serves as a regulator for ROS level in pancreatic cancer cells. Redox Biol. 2018;19:158–165. doi: 10.1016/j.redox.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez C.D., Martinovsky G., Naumovski L. Inhibition of cell death by ribosomal protein L35a. Cancer Lett. 2002;180:195–202. doi: 10.1016/s0304-3835(02)00024-1. [DOI] [PubMed] [Google Scholar]
- 14.Zhao L., Cao J., Hu K., Wang P., Li G., He X., Tong T., Han L. RNA-binding protein RPS3 contributes to hepatocarcinogenesis by post-transcriptionally up-regulating SIRT1. Nucleic Acids Res. 2019;47:2011–2028. doi: 10.1093/nar/gky1209. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Fumagalli S., Ivanenkov V.V., Teng T., Thomas G. Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev. 2012;26:1028–1040. doi: 10.1101/gad.189951.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daftuar L., Zhu Y., Jacq X., Prives C. Ribosomal proteins RPL37, RPS15 and RPS20 regulate the Mdm2-p53-MdmX network. PLoS ONE. 2013;8:e68667. doi: 10.1371/journal.pone.0068667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou X., Hao Q., Liao J.M., Liao P., Lu H. Ribosomal protein S14 negatively regulates c-Myc activity. J. Biol. Chem. 2013;288:21793–21801. doi: 10.1074/jbc.M112.445122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russo A., Esposito D., Catillo M., Pietropaolo C., Crescenzi E., Russo G. Human rpL3 induces G1/S arrest or apoptosis by modulating p21 (waf1/cip1) levels in a p53-independent manner. Cell Cycle. 2013;12:76–87. doi: 10.4161/cc.22963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mukhopadhyay R., Ray P.S., Arif A., Brady A.K., Kinter M., Fox P.L. DAPK-ZIPK-L13a axis constitutes a negative-feedback module regulating inflammatory gene expression. Mol. Cell. 2008;32:371–382. doi: 10.1016/j.molcel.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jang C.Y., Kim H.D., Kim J. Ribosomal protein S3 interacts with TRADD to induce apoptosis through caspase dependent JNK activation. Biochem. Biophys. Res. Commun. 2012;421:474–478. doi: 10.1016/j.bbrc.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 21.Russo A., Pagliara V., Albano F., Esposito D., Sagar V., Loreni F., Irace C., Santamaria R., Russo G. Regulatory role of rpL3 in cell response to nucleolar stress induced by Act D in tumor cells lacking functional p53. Cell Cycle. 2016;15:41–51. doi: 10.1080/15384101.2015.1120926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim T.H., Leslie P., Zhang Y. Ribosomal proteins as unrevealed caretakers for cellular stress and genomic instability. Oncotarget. 2014;5:860–871. doi: 10.18632/oncotarget.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen B., Zhang W., Gao J., Chen H., Jiang L., Liu D., Cao Y., Zhao S., Qiu Z., Zeng J. Downregulation of ribosomal protein S6 inhibits the growth of non-small cell lung cancer by inducing cell cycle arrest, rather than apoptosis. Cancer Lett. 2014;354:378–389. doi: 10.1016/j.canlet.2014.08.045. [DOI] [PubMed] [Google Scholar]
- 24.Russo A., Saide A., Cagliani R., Cantile M., Botti G., Russo G. rpL3 promotes the apoptosis of p53 mutated lung cancer cells by down-regulating CBS and NFκB upon 5-FU treatment. Sci. Rep. 2016;6:38369. doi: 10.1038/srep38369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H.J., Youn H., Seong K.M., Jin Y.W., Kim J., Youn B. Phosphorylation of ribosomal protein S3 and antiapoptotic TRAF2 protein mediates radioresistance in non-small cell lung cancer cells. J. Biol. Chem. 2013;288:2965–2975. doi: 10.1074/jbc.M112.385989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernández-Martínez J.L., deAndrés-Galiana E.J., Sonis S.T. Genomic data integration in chronic lymphocytic leukemia. J. Gene Med. 2017;19:e2936. doi: 10.1002/jgm.2936. [DOI] [PubMed] [Google Scholar]
- 27.Karan D., Kelly D.L., Rizzino A., Lin M.F., Batra S.K. Expression profile of differentially-regulated genes during progression of androgen-independent growth in human prostate cancer cells. Carcinogenesis. 2002;23:967–975. doi: 10.1093/carcin/23.6.967. [DOI] [PubMed] [Google Scholar]
- 28.Aceto N., Bardia A., Miyamoto D.T., Donaldson M.C., Wittner B.S., Spencer J.A., Yu M., Pely A., Engstrom A., Zhu H. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110–1122. doi: 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai M.S., Lu H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem. 2004;279:44475–44482. doi: 10.1074/jbc.M403722200. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y., Wolf G.W., Bhat K., Jin A., Allio T., Burkhart W.A., Xiong Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 2003;23:8902–8912. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krieg A.M. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene. 2008;27:161–167. doi: 10.1038/sj.onc.1210911. [DOI] [PubMed] [Google Scholar]
- 32.Hossain D.M., Moreira D., Zhang Q., Nechaev S., Swiderski P., Kortylewski M. TLR9-Targeted SiRNA Delivery In Vivo. Methods Mol. Biol. 2016;1364:183–196. doi: 10.1007/978-1-4939-3112-5_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barnie P.A., Zhang P., Lu P., Chen X., Su Z., Wang S., Xu H. CpG-oligodeoxynucleotides suppress the proliferation of A549 lung adenocarcinoma cells via toll-like receptor 9 signaling and upregulation of Runt-related transcription factor 3 expression. Biomed. Rep. 2014;2:374–377. doi: 10.3892/br.2014.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kortylewski M., Swiderski P., Herrmann A., Wang L., Kowolik C., Kujawski M., Lee H., Scuto A., Liu Y., Yang C. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat. Biotechnol. 2009;27:925–932. doi: 10.1038/nbt.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Warner J.R., McIntosh K.B. How common are extraribosomal functions of ribosomal proteins? Mol. Cell. 2009;34:3–11. doi: 10.1016/j.molcel.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurki S., Peltonen K., Latonen L., Kiviharju T.M., Ojala P.M., Meek D., Laiho M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell. 2004;5:465–475. doi: 10.1016/s1535-6108(04)00110-2. [DOI] [PubMed] [Google Scholar]
- 37.Lee S., Kim J.Y., Kim Y.J., Seok K.O., Kim J.H., Chang Y.J., Kang H.Y., Park J.H. Nucleolar protein GLTSCR2 stabilizes p53 in response to ribosomal stresses. Cell Death Differ. 2012;19:1613–1622. doi: 10.1038/cdd.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dai M.S., Zeng S.X., Jin Y., Sun X.X., David L., Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol. Cell. Biol. 2004;24:7654–7668. doi: 10.1128/MCB.24.17.7654-7668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z., Gao W., Pu L., Zhang L., Han G., Zuo X., Zhang Y., Li X., Shen H., Wu J., Wang X. PRDM8 exhibits antitumor activities toward hepatocellular carcinoma by targeting NAP1L1. Hepatology. 2018;68:994–1009. doi: 10.1002/hep.29890. [DOI] [PubMed] [Google Scholar]
- 40.Iwasaki A., Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Q., Hossain D.M., Nechaev S., Kozlowska A., Zhang W., Liu Y., Kowolik C.M., Swiderski P., Rossi J.J., Forman S. TLR9-mediated siRNA delivery for targeting of normal and malignant human hematopoietic cells in vivo. Blood. 2013;121:1304–1315. doi: 10.1182/blood-2012-07-442590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shou J., You L., Yao J., Xie J., Jing J., Jing Z., Jiang L., Sui X., Pan H., Han W. Cyclosporine A sensitizes human non-small cell lung cancer cells to gefitinib through inhibition of STAT3. Cancer Lett. 2016;379:124–133. doi: 10.1016/j.canlet.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 43.Sasaki M., Kawahara K., Nishio M., Mimori K., Kogo R., Hamada K., Itoh B., Wang J., Komatsu Y., Yang Y.R. Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat. Med. 2011;17:944–951. doi: 10.1038/nm.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.