

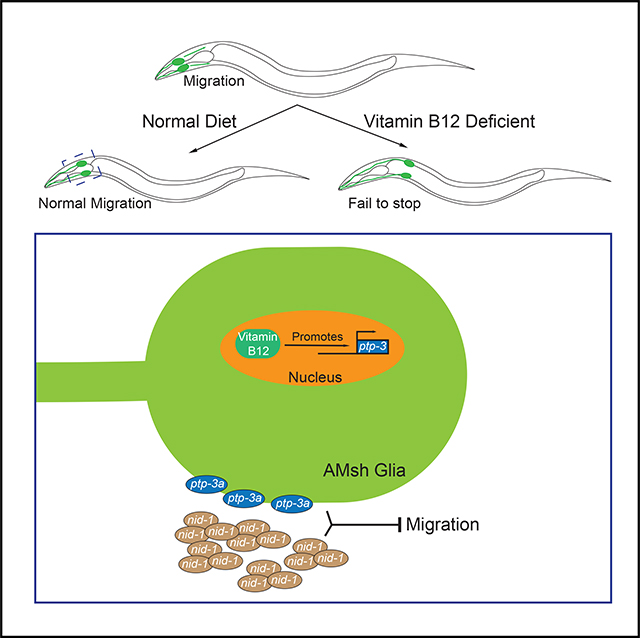
SUMMARY
Vitamin B12 is known to play critical roles during the development and aging of the brain, and vitamin B12 deficiency has been linked to neurodevelopmental and degenerative disorders. However, the underlying molecular mechanisms of how vitamin B12 affects the development and maintenance of the nervous system are still unclear. Here, we report that vitamin B12 can regulate glial migration and synapse formation through control of isoform-specific expression of PTP-3/LAR PRTP (leukocyte-common antigen-related receptor-type tyrosine-protein phosphatase). We found the uptake of diet-supplied vitamin B12 in the intestine to be critical for the expression of a long isoform of PTP-3 (PTP-3A) in neuronal and glial cells. The expression of PTP-3A cell autonomously regulates glial migration and synapse formation through interaction with an extracellular matrix protein NID-1/nidogen 1. Together, our findings demonstrate that isoform-specific regulation of PTP-3/ LAR PRTP expression is a key molecular mechanism that mediates vitamin-B12-dependent neuronal and glial development.
In Brief
Zhang et al. describe glial migration and synapse formation defects stemming from vitamin B12 deficiency. Their study identifies a cell-autonomous mechanism regulating glial migration, where vitamin B12 regulates expression of the cell adhesion molecule PTP-3/LAR PRTP in an isoform-specific manner.
Graphical Abstract
INTRODUCTION
Vitamin B12 deficiency has long been associated with a range of neurological complications, including neurodevelopmental disorders, subacute combined degeneration, behavioral changes, and cognitive decline (Black, 2008; Douaud et al., 2013; Healton et al., 1991; Lindenbaum et al., 1988; Reynolds, 2006). Vitamin B12 functions as a coenzyme in two important biochemical reactions that both may be relevant to the symptoms. The first is during the conversion methylmalonyl coenzyme A (CoA) to succinyl CoA, which is important for fatty acid metabolism, and reduced function may lead to incorporation of abnormal odd-numbered and branched fatty acids into the myelin, resulting in increased fragility (Cardinale et al., 1970; Dror and Allen, 2008; Green and Kinsella, 1995; Stollhoff and Schulte, 1987). The other reaction mediated by vitamin B12 is the conversion of homocysteine into methionine. This is important for the generation of S-adeno-sylmethionine, a methyl donor important for the methylation of DNA and the synthesis of lipids, proteins, and neurotransmitters in the nervous system (Briddon, 2003; Dror and Allen, 2008; Green et al., 2017; Reynolds, 2006). Although the coenzyme function of vitamin B12 has been well documented in the development and aging of the brain, other studies have also highlighted potential non-coenzyme functions of vitamin B12 in the nervous system (Scalabrino, 2009; Scalabrino et al., 1995), and there is still much to know about whether and how vitamin B12 affects the nervous system through its non-coenzyme functions.
Glial cells perform a wide range of vital roles in the nervous system, ranging from modulating development to actively playing a role in neuronal function (Barres, 2008; Fields et al., 2015; Mori et al., 2005). They need to migrate over what are often long distances from their birthplace to the appropriate regions to form functional units with neurons or perform other roles (Gilmour et al., 2002; Jarjour and Kennedy, 2004; Kinrade et al., 2001; Klämbt, 2009). The migration of glial cells can be divided into three steps: initiation; movement; and termination. Guidance molecules, including those from the netrin, slit, and semaphorin families can initiate and direct the movement of glial cells through interactions with their receptors that they express (Jarjour et al., 2003; Kinrade et al., 2001; Sasse and Klämbt, 2016; Spassky et al., 2002; Unni et al., 2012). However, it is still unclear what controls the termination of migration after glial cells reach their final positions.
C. elegans possess 56 glial cells in their body, which include 50 neuroepithelial glia that ensheath sensory neuron receptive endings (Oikonomou and Shaham, 2011; Shaham, 2015). The largest sensory organ of the worm, called the amphid sensilla, is a pair of sensilla composed of 12 neurons and 2 glial cells each (Ward et al., 1975). These glia, called the amphid sheath (AMsh) and amphid socket (AMso) cells, form channels that ensheath the dendrites of sensory neurons in the amphid sensilla (Oikonomou and Shaham, 2011; Perkins et al., 1986; Ward et al., 1975). Furthermore, these glial cells are vital for the proper functioning of the neurons that they ensheath, where they can modulate neural activity through secreted molecules at the receptive endings and control neuron receptive ending shape (Bacaj et al., 2008; Shaham, 2010; Singhvi et al., 2016).
Here, we describe a function of vitamin B12 in the regulation of glial migration and synapse formation. We found that either dietary vitamin B12 deficiency or reduced uptake in the intestine due to a mutation in the ABC transporter mrp-5 (multidrug resistance protein 5) leads to an over-migration of AMsh glia. Furthermore, vitamin B12 functions during glial migration and synapse formation by regulating the expression of the protein tyrosine phosphatase PTP-3/LAR PRTP in an isoform-specific manner. PTP-3A interacts with the extracellular matrix protein NID-1/nidogen 1 near the pharynx to regulate glial migration. Our findings reveal a vitamin-B12-dependent pathway regulating both glial and neuronal development that may inform our understanding of neuropathies derived from vitamin B12 deficiency.
RESULTS
To study mechanisms regulating glial migration, we used C. elegans AMsh glial cells as a model. AMsh glia are born close to the nose of C. elegans and then their processes anchor to the surface of embryos while the cell bodies migrate toward the nerve ring (Heiman and Shaham, 2009). In day 1 (D1) young adults, AMsh cell bodies reside at the side of and align with the center of pharyngeal terminal bulbs (Figures 1A and 1B). In a genetic screen, we isolated a mutant yad138, whose AMsh cell body positions were indistinguishable from those of control animals in L1 larvae but possess AMsh cell bodies that fail to stop at the pharyngeal terminal bulbs and migrate further down the body in D1 adults (Figures 1A, 1B, and S1A–S1C). To further confirm these observations, we visualized single animals at different developmental stages and found all mutants examined in this study have similar AMsh cell body positions at the L1 stage when compared to control animals, but their AMsh cell bodies migrate further down the body and fail to stop at the correct place in the L4 and adult stages (Figure S1D). These results suggest that yad138 likely affects a molecule involved in terminating AMsh migration. yad138 causes a missense mutation in the C. elegans ABC transporter mrp-5, which has been shown to play essential roles in the export of vitamin B12 and heme from the intestine to other tissues (Korolnek et al., 2014; Na et al., 2018; Figure S1E). We found that driving expression of mrp-5 by its own promoter or an intestine-specific promoter was able to fully rescue yad138 phenotypes, although expression of mrp-5 in AMsh glia or pharyngeal muscles did not change the phenotypes (Figure 1C). These results show that yad138 is a loss-of-function allele of mrp-5, and mrp-5 functions in the intestine to affect AMsh migration.
Figure 1. Vitamin B12 Is Required for Terminating Glial Migration.

(A) yad138 causes over-migration of AMsh glia. Confocal images and schematic representation of AMsh glia in control and mrp-5(yad138) animals expressing Pf53f4.13::GFP (yadIs48) are shown. TB, pharyngeal terminal bulb. Scale bar, 10 μm.
(B) Quantification of Migration Index (MI) in control and mrp-5(yad138) animals. MI is calculated as (b − a)/a × 100%. As illustrated in (A), “a” represents the distance between the tip of nose and the center of the pharyngeal terminal bulb. “b” shows the distance between the tip of nose and the center of the AMsh cell bodies.
(C) mrp-5 functions in the intestine to regulate Amsh migration. White and gray bars show the percentage of animals with over-migration defects in one AMsh or both AMsh glia, respectively. Pvha-6 is an intestine-specific promoter, Pf53f4.13 drives expression only in AMsh glia, and Pmyo-2 is a pharyngeal-muscle-specific promoter.
(D) Injection of vitamin B12, but not heme, rescues mrp-5(lf) phenotypes.
(E) A schematic showing how vitamin B12 deficiency was induced.
(F) Quantification of MI of AMsh glia in animals cultured in control and vitamin-B12-deficient conditions.
(G) Dietary supplementation of vitamin B12, but not heme, rescued AMsh migration defects in vitamin-B12-deficient conditions. Data show the percentage of animals with AMsh migration defects.
In (B) and (F), data are represented as mean ± SEM. One-way ANOVA test; **p < 0.01; each point represents at least 30 worms. In (C), (D), and (G), data are represented as mean ± SEM. Two-way ANOVA test; **p < 0.01; ns, no significant difference. Each point represents three experiments of at least 50 worms each.
To determine whether the role of mrp-5 in AMsh glial migration is related to its function in the uptake of vitamin B12 or heme in the intestine, we directly injected vitamin B12 or heme to mrp-5(yad138) L1 larvae and observed full rescue of AMsh migration defects in vitamin B12-injected, but not heme-injected, animals (Figure 1D). Vitamin B12 also appears to function during early developmental stages, as injection of the same concentration of vitamin B12 in L4 larvae did not show any rescue ability on mrp-5(yad138) animals when they were scored as D1 adults (Figure 1D). To further confirm the importance of vitamin B12 in regulating AMsh glial migration, we cultured control animals in vitamin-B12-deficient conditions as outlined by Bito et al. (2013); Figure 1E. Similar to what they reported, we observed a reduction in brood size when animals were grown for five generations in vitamin-B12-deficient conditions (Bito et al., 2013; Figure S1F). Vitamin-B12-deficient animals displayed similar AMsh migration defects as those observed in mrp-5 mutants (Figure 1F). Dietary supplementation of vitamin B12, but not heme, was able to prevent the AMsh migration defects observed in vitamin-B12-deficient conditions (Figure 1G). We also found that feeding animals a vitamin-B12-enriched diet did not cause any observable abnormalities in AMsh development (Figure S1G).
In the same genetic screen, we isolated other mutants with similar phenotypes as those in mrp-5 mutants, and further analyses showed that two of them, yad122 and yad139, belong to a same complementation group and both affect cubn-1 (Figures S2A–S2D), the only C. elegans homolog of the intrinsic factor-vitamin B12 receptor cubilin (Kozyraki et al., 1998; Seetharam et al., 1997). Expression of cubn-1 under its own promoter fully rescued yad122 phenotypes, and yad122 exhibits a similar degree of AMsh migration defects as presumed null alleles (Figure S2C). These results support the conclusion that yad122 and yad139 are loss-of-function alleles of cubn-1, and yad122 is likely a null allele. Cubilin is a giant secreted protein and functions together with a receptor called megalin to uptake intrinsic factor-vitamin B12 (Ahuja et al., 2008; Moestrup et al., 1998; Nielsen et al., 2016). To understand how cubn-1 affects AMsh migration, we examined the function of C. elegans megalin lrp-1, and the results showed that knockdown of lrp-1 caused a similar degree of AMsh migration defects as was found in cubn-1 mutants, but knockdown of lrp-1 in a cubn-1(lf) background did not further enhance cubn-1 mutant phenotypes (Figures S3A–S3C). These results suggest that the function of cubn-1 likely relies on its binding with lrp-1 to uptake vitamin B12, similar to what was previously reported in mammalian systems (Ahuja et al., 2008; Moestrup et al., 1998; Nielsen et al., 2016). Further experiments showed that knockdown of lrp-1 expression only in AMsh glia caused the same degree of migration defects as those in global knockdown animals (Figure S3C), suggesting lrp-1 cell autonomously regulates AMsh migration. We also found that double mutants of cubn-1;mrp-5 exhibited similar defects as those in mrp-5 single mutants, and knockdown of lrp-1 did not further enhance mrp-5 mutant phenotypes (Figures S3D–S3F). These results suggest that CUBN-1 and LRP-1 likely function as co-receptors in AMsh glia to uptake vitamin B complex into AMsh glia to terminate AMsh migration. Taken together, these results demonstrate the important role of vitamin B12 in regulating glial migration. To understand how vitamin B12 regulates AMsh migration, we tested three important enzymes involved in C. elegans vitamin-B12-dependent metabolic pathways—pcca-1/propionyl-CoA carboxylase alpha chain, mmcm-1/methylmalonyl-CoA mutase, and metr-1/5-methyltetrahydrofolate-homocysteine methyltransferase (Watson et al., 2014), but we did not notice any abnormalities in AMsh migration in loss-of-function mutants of these three genes (Figure S3G), suggesting that vitamin B12 may regulate AMsh glial migration through other means.
To further illuminate the molecular mechanisms underlying AMsh migration mediated by vitamin B12, we analyzed two other mutants with AMsh over-migration defects that were isolated in the initial screen, yad120 and yad121 (Figures 2A, 2B, and S1A). They failed to complement each other, and both alter the C. elegans homolog of LAR PRTP (leukocyte-common antigen-related receptor-type tyrosine-protein phosphatase) ptp-3 (Figure S4A). Interestingly, yad120 has stronger defects than yad121, but it only affects the long isoform ptp-3a, although yad121 affects both long and short isoforms ptp-3a and ptp-3b (Figures 2A, 2B, and S4A). Like what we observed in mrp-5 mutants, loss of function of ptp-3 did not affect the position of AMsh cells in L1 larvae, suggesting that it may play a role in terminating glial migration (Figure S1B). The two isoforms ptp-3a and ptp-3b have been shown to play distinct functions in synapse formation, axon guidance, and neuronal migration (Ackley et al., 2005; Sundararajan and Lundquist, 2012). To determine whether both isoforms are important for AMsh migration, we carried out rescue experiments and found that expression of ptp-3a under either its own promoter or an AMsh-specific promoter was able to fully rescue yad121 phenotypes, although expression of ptp-3b did not show any rescue ability (Figure 2B). Similar results were obtained from yad120 rescue experiments that ptp-3a, but not ptp-3b, can rescue AMsh migration defects (Figure 2B). We also examined a null allele of ptp-3a(ok244) and showed that it had the same degree of AMsh migration defects as observed in yad120 (Figure 2B). Using both transcription and translational reporters, we confirmed that ptp-3a is expressed in AMsh glial cells (Figures S4B and S4C). These results support the conclusion that ptp-3a, but not ptp-3b, cell autonomously regulates AMsh migration.
Figure 2. ptp-3a Functions in the Same Genetic Pathway as mrp-5 and Vitamin B12 in Regulating Glial Migration.

(A) Quantification of MI in control, ptp-3a(yad120), and ptp-3a,b(yad121) animals.
(B) ptp-3a, but not ptp-3b, regulates AMsh migration. White and gray bars show the percentage of animals with over-migration defects in one AMsh or both AMsh glia, respectively. Pf53f4.13 drives expression only in AMsh glia.
(C and D) Double mutants of ptp-3a;mrp-5 and ptp-3a;cubn-1 display similar defects as in ptp-3a single mutants. Quantification of MI of AMsh glia
(C) and the percentage of animals with over-migration defects (D) are shown.
In (A) and (C), data are represented as mean ± SEM. One-way ANOVA test; **p < 0.01; each point represents at least 30 worms. In (B) and (D), data are represented as mean ± SEM. Two-way ANOVA test; **p < 0.01; each point represents three experiments of at least 50 worms each.
Because both mrp-5(lf), cubn-1(lf), and ptp-3a(lf) cause over-migration of AMsh glia, we decided to test whether they might function in the same genetic pathway, and we found that double mutants of mrp-5;ptp-3a and cubn-1;ptp-3a showed a similar degree of phenotypes as was found in ptp-3a single mutants, supporting the conclusion that ptp-3a likely lies in the same pathway as mrp-5 and cubn-1 (Figures 2C, 2D, and S4D). Because ptp-3a functions in AMsh cells to affect migration, it is possible that its expression or functions are regulated by mrp-5 through vitamin B12. To test this hypothesis, we first examined whether loss of function of mrp-5 may affect ptp-3a expression or localization using a well-established ptp-3a translation reporter (Ackley et al., 2005). As shown in Figure 3A, in control animals, we observed puncta expression of PTP-3A in the nerve ring as well as in AMsh glial cells. Loss of function of mrp-5 reduced the overall expression of PTP-3A to less than 30% of that in control, and the PTP-3A expression reporter becomes invisible in the AMsh glia of mrp-5 mutants (Figures 3A and 3B). In contrast, mutations of mrp-5 did not alter PTP-3B expression (Figures 3A and 3C). Consistent with the observations in mrp-5(lf) animals, we showed that animals cultured in vitamin-B12-deficient conditions exhibited a similar decrease of PTP-3A expression, although the expression of PTP-3B was comparable to that in control conditions (Figures 3D–3F). The regulation of ptp-3a expression by mrp-5 and vitamin B12 has also been confirmed on the mRNA level by real-time PCR (Figures 3G and 3H). These results suggest that the uptake of vitamin B12 mediated by mrp-5 may regulate AMsh migration through transcriptional control of ptp-3a expression in AMsh cells. To further confirm this conclusion, we overex-pressed ptp-3a or ptp-3b only in AMsh cells and tested whether those transgenes were able to bypass the requirement of mrp-5 in the termination of AMsh migration. In support of our hypothesis, overexpression of ptp-3a, but not ptp-3b, in AMsh glia nearly completely suppressed AMsh migration defects in mrp-5 mutants (Figures 3I and S4E). We also found that overexpression of ptp-3a, but not ptp-3b, was able to eliminate the AMsh migration defects caused by vitamin B12 deficiency (Figure 3J). In conclusion, these results demonstrate that the uptake of vitamin B12 from the diet mediated by mrp-5 in the intestine can control the expression level of ptp-3a, and the expression of ptp-3a in AMsh glia has a critical role in terminating cell migration at the appropriate place. As AMsh glia are essential for the function of sensory neurons they ensheath, we used the classic dye filling assay to examine whether the AMsh migration defects we described in this study could affect sensory neuron function (Oikonomou and Shaham, 2011; Shaham, 2015). We found that neither mrp-5(lf) nor ptp-3a(lf) caused dye filling defects in D1 young adults (Figure S4F). However, we observed a significant reduction in the number of neurons that uptake Dil in D7 mrp-5(lf) and ptp-3a(lf) animals when compared with the same age control animals (Figure S4G). The dye filling defects displayed in aged mrp-5(lf) and ptp-3a(lf) animals were caused by abnormal migration of AMsh, as expression of ptp-3a only in AMsh glia could fully rescue their dye filling defects (Figure S4G). Taken together, these results suggest that the correct localization of AMsh cell bodies is critical for maintaining the function of sensory neurons they ensheath during aging.
Figure 3. mrp-5 and Vitamin B12 Regulates AMsh Migration through Isoform-Specific Control of ptp-3a, but Not ptp-3b, Expression.

(A–C) Loss offunction in mrp-5 decreases PTP-3A, but not PTP-3B, protein levels. Confocal images (A, left panel, GFP; right panel, differential interference contrast [DIC] merged with GFP; scale bar, 10 μm) and quantification (B and C) show the expression level of PTP-3A and PTP-3B in control and mrp-5(lf) animals. The red arrowheads point to AMsh cell bodies.
(D–F) Vitamin B12 deficiency suppresses PTP-3A, but not PTP-3B, expression. Confocal images (D, left panel, GFP; right panel, DIC merged with GFP; scale bar, 10 μm) and quantification (E and F) show the expression level of PTP-3A and PTP-3B in control vitamin-B12-deficient and vitamin-B12- supplemented conditions.
(G and H) Real-time PCR results show that loss of function in mrp-5 (G) or culturing animals in vitamin-B12-deficient conditions (H) decrease ptp-3a mRNA levels. A housekeeping gene, cdc-42, was used as an internal control.
(I) Overexpression of ptp-3a, but not ptp-3b, in AMsh cells suppresses migration defects in mrp-5(lf) mutants.
(J) Overexpression of ptp-3a, but not ptp-3b, in AMsh cells suppresses migration defects caused by vitamin B12 deficiency.
In (B), (C), (E), and (F), data are represented as mean ± SEM. One-way ANOVA test; **p < 0.01; each point represents at least 20 worms. In (G)–(H), data are represented as mean ± SEM. One-way ANOVA test; **p < 0.01; each point represents three experimental replicates. In (I)–(J), data are represented as mean ± SEM. Two-way ANOVA test; **p < 0.01; each point represents three experiments of at least 50 worms each.
Because ptp-3a plays a critical role in regulating synapse formation (Ackley et al., 2005), we tested whether mrp-5 and vitamin B12 have a role in synapse formation as well. As shown in Figures 4A and 4B, loss of function of mrp-5 caused enlarged synapses and decreased synapse number, similar to what has been shown in ptp-3a mutants. mrp-5(lf) phenotypes were caused by defects in vitamin B12 uptake, as injection of vitamin B12 was able to completely rescue mrp-5(lf) phenotypes (Figures 4A and 4B). We also found that overexpression of ptp-3a can cell autonomously suppress mrp-5(lf) defects, supporting the conclusion that lack of vitamin B12 caused by a mutant form of mrp-5 disrupts synapse formation through downregulating ptp-3a expression. We further confirmed this by showing that culturing animals in vitamin-B12-deficient conditions caused similar synapse formation defects as those displayed in mrp-5(lf) or ptp-3a(lf) animals, and overexpression of ptp-3a prevented synapse formation defects caused by vitamin B12 deficiency (Figures 4C and 4D).
Figure 4. Vitamin B12 and mrp-5 Regulate Synapse Formation through ptp-3a, and the Interactions of nid-1 with ptp-3a Play an Important Role in Both Synapse Formation and Glial Migration.

(A and B) mrp-5 regulates synapse formation through vitamin B12 and ptp-3a. Confocal images (A) show GABA neuron synapses visualized by SNB-1::GFP (juIs1) at the dorsal cord (scale bar, 10 μm), and quantification data (B) show the total SNB-1::GFP puncta number in the dorsal cord.
(C and D) Vitamin B12 regulates synapse formation through ptp-3a. Confocal images (C) show GABA neuron synapses visualized by SNB-1::GFP (juIs1) at the dorsal cord (scale bar, 10 μm), and quantification data (D) show the total SNB-1::GFP puncta number in the dorsal cord.
(E and F) nid-1 functions together with ptp-3a to regulate AMsh migration.
(E) Quantification of MI in control, nid-1, ptp-3a(yad120), and nid-1;ptp-3a(yad120) animals.
(F) The percentage of animals with over-migration defects. Pmyo-2 is a pharyngeal-muscle-specific promoter.
In (B), (D), and (E), data are represented as mean ± SEM. One-way ANOVA test; **p < 0.01; each point represents at least 20 worms. In (F), data are represented as mean ± SEM. Two-way ANOVA test; **p < 0.01; each point represents three experiments of at least 50 worms each.
In synapse formation, PTP-3A functions through interactions with an extracellular matrix protein NID-1, an ortholog of human nidogen 1 and nidogen 2 (Ackley et al., 2005). We found that loss of function of nid-1 caused AMsh over-migration defects, and double mutants of nid-1;ptp-3a exhibited a similar degree of defects as those found in ptp-3a single mutants (Figures 4E, 4F, and S1A–S1D), confirming that nid-1 is likely also a functional partner of ptp-3a in regulating glial migration. More interestingly, expression of nid-1 only in the pharyngeal muscle was able to fully rescue nid-1(lf) phenotypes, which is consistent with the strong expression of nid-1 in the pharynx, as previously shown (Figure 4F; Ackley et al., 2005; Kang and Kramer, 2000). Expression of NID-1::GFP suggested potential accumulation of NID-1 on the surface of AMsh cell bodies (Figure S4H). Therefore, it is likely that the interactions between AMsh-expressed PTP-3A and pharynx-expressed NID-1 provide the signals to terminate AMsh migration at the pharyngeal terminal bulbs. In conclusion, our study provides a common mechanism for regulation of both neuronal and glial development by vitamin B12, in which the uptake of vitamin B12 by the intestine can regulate the expression of PTP-3 in an isoform-specific manner, and this regulation also relies on the interactions between PTP-3 and the extracellular matrix protein NID-1 (Figure S5).
DISCUSSION
In our study, we uncover a role of vitamin B12 in glial migration and synapse formation, and we show that vitamin B12 functions by regulating the expression of ptp-3a, the C. elegans homolog of mammalian LAR PRTP. C. elegans ptp-3a and mammalian LAR PRTPs are highly expressed in the nervous system and have been shown to be important for synapse formation, axon development, and axon regeneration in neurons (Ackley et al., 2005; Chagnon et al., 2004; Chen et al., 2011; Stoker, 2015; Takahashi and Craig, 2013). Our findings further reveal a function of LAR PRTPs in regulating glial migration and show ptp-3a/LAR PRTP expression to be connected to vitamin B12 uptake in the intestine. As vitamin B12 deficiency causes many neurodevelopmental and degenerative disorders that are associated with abnormality in both neurons and glial cells (Cardinale et al., 1970; Douaud et al., 2013; Healton et al., 1991; Reynolds, 2006; Scalabrino et al., 1995; Stollhoff and Schulte, 1987), it is possible that ptp-3a/LAR PRTP as a target for vitamin B12 is associated with multiple neurological symptoms of vitamin B12 deficiency.
Besides the nervous system, LAR PRTPs are also expressed in a wide range of cell types, including T lymphocytes, kidney cells, prostate cells, and intestinal epithelial cells (Murphy et al., 2005; Pulido et al., 1995; Streuli et al., 1988) and are known to be involved in the regulation of insulin signaling, epithelial integrity, and cancer progression (Mooney and LeVea, 2003; Müller et al., 1999; Murchie et al., 2014; Xu and Fisher, 2012). Furthermore, mutations in LAR PRTPs have been linked to hyperinsulinism and obesity in human patients (Alliance of Genome Resources). A major symptom of vitamin B12 deficiency is anemia, which may or may not coincide with neurological symptoms (Green et al., 2017; Lindenbaum et al., 1988; Reynolds, 2006). Other symptoms may include fatigue, decreased appetite, glossitis, and reduced gastrointestinal function (Briani et al., 2013; Green et al., 2017; Hunt et al., 2014). As some of these symptoms appear to relate to either insulin signaling or epithelial integrity, where LAR PRTPs are known to play important roles, the regulation of ptp-3a by vitamin B12 may also be relevant.
In this study, we found that vitamin B12 controls glial migration and synapse formation through regulating ptp-3a transcription. However, the question of how this regulation happens is still unclear. Different levels of vitamin B12/folic acid have been shown to affect specific DNA methylation patterns, and maternal vitamin B12/folic acid levels during pregnancy were also shown to affect DNA methylation patterns in infants (Hoyo et al., 2011; Kok et al., 2015; McKay et al., 2012; Rampersaud et al., 2000). In our experiments, we found that injection of vitamin B12 at the early, but not late, larval stages is sufficient for normal glial and neuronal development, suggesting that this regulation of ptp-3a by vitamin B12 occurs during early developmental stages. Therefore, one possibility is that, during development, vitamin B12 can affect the methylation of DNA in the regulatory elements of the ptp-3a promoter to facilitate its transcription. A second possibility is that vitamin B12 may regulate ptp-3a on the mRNA level, as previous studies show that vitamin B12 can bind to regulatory elements of messenger RNAs to control gene expression (Mandal and Breaker, 2004; Nahvi et al., 2004; Padmanabhan et al., 2017). It is also possible that vitamin B12 may regulate the expression or activation of certain transcription factors that are important for ptp-3a expression. Further studies are needed to uncover the detailed mechanism of how vitamin B12 regulates ptp-3a transcription.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Dong Yan (dong.yan@duke.edu). C. elegans strains and plasmids generated in this study are available from the lead contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans genetics
C. elegans strains were maintained on nematode growth media (NGM) plates using E. coli OP50 as a food source. Animals were grown according to standard methods (Brenner, 1974) at 20°C unless otherwise stated. Wild-type worms were of the Bristol N2 strain. Only hermaphrodite animals were used for experiments. All transgenes and strains are described in Table S1. yadIs48 (Pf53f4.13::GFP) was used to visualize AMsh cells. juIs1 (Punc-25::SNB-1::GFP) was used to visualize GABA synapses. The alleles used in genetic analyses were mrp-5(yad138), cubn-1(yad122), ptp-3a(yad120), and nid-1(cg119) unless specific alleles were mentioned.
METHOD DETAILS
Cloning and constructs
All DNA expression constructs were generated using Gateway cloning technology (Invitrogen, Carlsbad, CA) and subsequently sequenced. mrp-5 and nid-1 were amplified from a homemade genomic pool. A 2kb fragment before the start codon of mrp-5 was used as its promoter. A 2.5 kb fragment before the start codon of nid-1 was used as its promoter. A complete list of DNA constructs used is included in Table S1. In general, plasmid and PCR DNA used in this study were injected at a concentration of 1–50ng/μL with a Pttx-3::RFP co-injection marker injected at a concentration of 50ng/μL.
RNA Interference
RNA interference (RNAi) experiments were carried out by feeding anaimls with E. coli strain HT115 (DE3) expressing double-stranded RNA against target genes for at least two generations. All lrp-1 RNAi clones were made by inserting 1kb cDNA fragments into the L4440 vector. RNAi clones were first cultured overnight using LB broth containing ampicillin (100 μg/ml) at 37°C, and then concentrated bacteria were seeded on NGM plates containing 100μg/ml ampicillin and 5mM isopropyl 1-thio-β-D-galactopyranoside (IPTG) and cultured overnight at 37°C. Animals were grown on RNAi plates for at least two generations. In all experiments, either GFP RNAi or unc-22 RNAi was included as a control to account for RNAi efficiency. Tissue-specific RNAi was carrired out as described (Calixto et al., 2010; Feinberg and Hunter, 2003). In brief, to knock down gene expression only in pharyngeal muscles, we expressed the sid-1(cDNA) under the control of the Pf53f4.13 promoter, a AMsh specific promoter in an sid-1(pk3321) mutant background. sid-1 mutants are resistant to RNAi, and transgenic animals (yadEx760;sid- 1(pk3321)) are resistant to RNAi in all tissues except in the AMsh glial cells which express sid-1 cDNA.
Microscopy
Representative images were acquired with a Zeiss LSM700 confocal microscope using a Plan-Apochromat 40×/1.4 objective. Worms were immobilized using 1.5% 1-phenoxy-2-propanol (TCI America, Portland, OR) in M9 buffer and mounted on 5% agar slides. 3D reconstructions were done using Zeiss Zen software. Images of GABA neuron synapses show the dorsal cord at the middle part of animals. A Zeiss Axio Imager 2 microscope equipped with Chroma HQ filters was used to score AMsh migration defects and take images to measure Migration Index. For GABA neuron synapse number quantification, a Zeiss Axio Imager 2 microscope equipped with Chroma HQ filters was used to quantify the total number of SNB-1::GFP puncta at the dorsal cord. Each condition represented 3 experiments of at least 50 Day 1 adult animals each unless otherwise noted.
Brood size assays
Brood size assays were performed by allowing gravid adults to lay eggs for 3 days on plates seeded with vitamin B12-deficient OP50 before the adults were removed. One day later, the total number of embryos or hatched animals were counted. Three experiments of at least 20 animals were quantified for each condition.
Vitamin B12 deficiency assays
Vitamin B12 deficiency assays were performed as previously described (Bito et al., 2013). In brief, vitamin B12-deficient OP 50 was cultured as a food source: OP50 was grown in M9 medium at 37°C for 3 days. Animals were then cultured using M9 agar (2%) plates seeded with the vitamin B12-deficient OP50 as their food source. All animals were grown in this condition for five generations with or without vitamin B12 (cyanocobalamin) (Sigma) (100 μg/L) or heme (Sigma) (500 μM) dietary supplementation.
Vitamin B12/heme injection
Vitamin B12 (3.2 mM) or heme (500 μM) were prepared using autoclaved water and were injected into the middle part of animals. We followed the standard protocol for all injections. For L1 injections, we cultured newly hatched animals in 20°C for 12 hours to reach the late L1 stage. The injection was performed by placing animals on 10% agar pads, which were newly made and air-dried for 30–45 mins, with lower injection air pressure (1/3 of pressure for L4 animal injection). The L1 injection needle tips also had to be thinner than regular L4 injection ones. When injections were performed in L1 animals, about 40%–60% animals failed to recover, and all data were collected from day 1 adults. Injected L4 animals had over a 90% recovery rate, and data were also collected in day 1 adults
Dye filling
Dil is purchased from Molecular Probes (Eugene, OR), and the stock solution is 2 mg/ml in dimethylformamide. We performed the experiments following the standard protocol. In brief, D1 and D7 adult animals were collected and washed three times in M9 buffer, and then incubated in 10 ng/ml Dil solution (in M9) for 2 hours at 20°C. After 2 hours incubation with Dil solution, animals were washed 3 times (20 min/each) in M9 buffer before visualization using 63X lens.
Real-time Reverse Transcription PCR
To measure changes in ptp-3a mRNA levels in mrp-5 mutants and N2 animals with or without vitamin B12, total RNA was isolated from mixed population plates. Animals were washed with M9 buffer three times for 90 minutes in total, and TRI Reagent® (Invitrogen) was subsequently added to isolate the RNA. Animals were independently cultured and collected three times for each condition. Reverse transcription was performed on total RNA (1 μg) using an iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad Laboratories). Quantitative real-time, reverse-transcription PCR (qPCR) was performed using iTaq Universal Probes Supermix (Bio-Rad Laboratories) and ready-to-use TaqMan Gene Expression Assays (Invitrogen), and mRNA levels of ptp-3a and cdc-42 were quantified, with cdc-42/cell division control protein 42 serving as an internal control. qPCR amplifications were performed using an Applied Biosystems StepOnePlus Real-Time PCR System (Applied Biosystems). Relative mRNA levels were quantified using the StepOnePlus Software v2.1 based on the comparative ΔΔCT method.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Data was analyzed using one-way or two-way ANOVA followed by Tukey’s post hoc test in Graphpad Prism (Graphpad Software, La Jolla, CA). Statistical details are included in the figure legends. In general, a p value cutoff of 0.05 was considered statistically significant. The data are represented by mean ± SEM. For phenotype penetrance experiments, three biological replicates of at least 50 animals each were used unless otherwise stated.
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or code.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Escherichia coli OP50 | http://www.wormbook.org/chapters/www_strainmaintain/strainmaintain.html | N/A |
| Escherichia coli DA1977 | Caenorhabditis Genetics Center (CGC) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1-phenoxy-2-propanol | TCI America, Portland, OR | Cat#P0118, CAS RN:770-35-4 |
| Vitamin B12 (Cyanocobalamin) | Sigma | Cat#V6629, CAS RN:68-19-9 |
| Hemin | Sigma | Cat#H9039, CAS RN:16009-13-5 |
| Dil Stain | Molecular Probes, Eugene, OR | Cat#:D282, CAS RN:41085-99-8 |
| Critical Commercial Assays | ||
| iScript Reverse Transcription Supermix for RT-qPCR | Bio-Rad Laboratories | Cat#1708840 |
| iTaq Universal Probes Supermix | Bio-Rad Laboratories | Cat#1725131 |
| TaqMan Gene Expression Assays – cdc-42 | Thermo Scientific | Cat#:4331182 Assay ID:Ce02435136_g1 |
| TaqMan Gene Expression Assays – ptp-3 | Thermo Scientific | Cat#:4448892 Assay ID: Ce02406216_g1 |
| Experimental Models: Organisms/Strains | ||
| C. elegans strains, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| Pmyo-2::mrp-5 | This paper | PNYL1218 |
| Pf53f4.13::mrp-5 | This paper | PNYL1219 |
| Pf53f4.13::ptp-3a | This paper | PNYL1215 |
| Pf53f4.13::ptp-3b | This paper | PNYL1216 |
| Pptp-3A::ptp-3a::HA | This paper | PBH21 |
| Punc-25::ptp-3a | This paper | PNYL1217 |
| Pf16f9.3::mCherry::H2B | This paper | PNYL1142 |
| Fosmid WRM0627BF04 | Yishi Jin | WRM0627BF04 |
| lrp-1 RNAi 1 | This paper | PNYL1280 |
| lrp-1 RNAi 1 | This paper | PNYL1281 |
| Pf53f4.13::sid-1(cDNA) | This paper | PNYL1279 |
| Pf53f4.13::mCherry | This paper | PNYL696 |
| Pnid-1::nid-1::GFP | Brian Ackley/ Jim Kramer Lab | pEVL54 |
| Software and Algorithms | ||
| Graphpad Prism 6 | Graphpad Software | N/A |
| Zeiss Zen Black | Zeiss | N/A |
| StepOnePlus Software v2.1 | Applied Biosystems | N/A |
Highlights.
Vitamin B12 plays a role in regulating glial migration through PTP-3/LAR PRTP
PTP-3 functions cell autonomously in an isoform-specific manner
Vitamin B12 deficiency leads disruption of synapse formation
PTP-3 functions by interacting with the extracellular matrix protein NID-1
ACKNOWLEDGMENTS
We thank Dr. Yuji Kohara for cDNAs. Some strains used in this study were provided by the National BioResource Project (NBRP, Japan) as well as the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). We also thank Dr. Yishi Jin for kindly providing us with the fosmid used in this study. This project is supported by the Holland Trice Awards. A.Z. and D.Y. are supported by NIH R01 (NS094171 and NS105638 to D.Y.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.113.
REFERENCES
- Ackley BD, Harrington RJ, Hudson ML, Williams L, Kenyon CJ, Chisholm AD, and Jin Y (2005). The two isoforms of the Caenorhabditis elegans leukocyte-common antigen related receptor tyrosine phosphatase PTP-3 function independently in axon guidance and synapse formation. J. Neurosci 25, 7517–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja R, Yammani R, Bauer JA, Kalra S, Seetharam S, and Seetharam B (2008). Interactions of cubilin with megalin and the product of the amnion-less gene (AMN): effect on its stability. Biochem. J 410, 301–308. [DOI] [PubMed] [Google Scholar]
- Bacaj T, Tevlin M, Lu Y, and Shaham S (2008). Glia are essential for sensory organ function in C. elegans. Science 322, 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440. [DOI] [PubMed] [Google Scholar]
- Bito T, Matsunaga Y, Yabuta Y, Kawano T, and Watanabe F (2013). Vitamin B12 deficiency in Caenorhabditis elegans results in loss of fertility, extended life cycle, and reduced lifespan. FEBS Open Bio 3, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black MM (2008). Effects of vitamin B12 and folate deficiency on brain development in children. Food Nutr. Bull 29 (2, Suppl), S126–S131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briani C, Dalla Torre C, Citton V, Manara R, Pompanin S, Binotto G, and Adami F (2013). Cobalamin deficiency: clinical picture and radiological findings. Nutrients 5, 4521–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briddon A (2003). Homocysteine in the context of cobalamin metabolism and deficiency states. Amino Acids 24, 1–12. [DOI] [PubMed] [Google Scholar]
- Calixto A, Chelur D, Topalidou I, Chen X, and Chalfie M (2010). Enhanced neuronal RNAi in C. elegans using SID-1. Nat. Methods 7, 554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinale GJ, Carty TJ, and Abeles RH (1970). Effect of methylmalonyl coenzyme A, a metabolite which accumulates in vitamin B 12 deficiency, on fatty acid synthesis. J. Biol. Chem 245, 3771–3775. [PubMed] [Google Scholar]
- Chagnon MJ, Uetani N, and Tremblay ML (2004). Functional significance of the LAR receptor protein tyrosine phosphatase family in development and diseases. Biochem. Cell Biol 82, 664–675. [DOI] [PubMed] [Google Scholar]
- Chen L, Wang Z, Ghosh-Roy A, Hubert T, Yan D, O’Rourke S, Bowerman B, Wu Z, Jin Y, and Chisholm AD (2011). Axon regeneration pathways identified by systematic genetic screening in C. elegans. Neuron 71, 1043–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douaud G, Refsum H, de Jager CA, Jacoby R, Nichols TE, Smith SM, and Smith AD (2013). Preventing Alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. Proc. Natl. Acad. Sci. USA 110, 9523–9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror DK, and Allen LH (2008). Effect of vitamin B12 deficiency on neurodevelopment in infants: current knowledge and possible mechanisms. Nutr. Rev 66, 250–255. [DOI] [PubMed] [Google Scholar]
- Feinberg EH, and Hunter CP (2003). Transport of dsRNA into cells by the transmembrane protein SID-1. Science 301, 1545–1547. [DOI] [PubMed] [Google Scholar]
- Fields RD, Woo DH, and Basser PJ (2015). Glial regulation of the neuronal connectome through local and long-distant communication. Neuron 86, 374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour DT, Maischein HM, and Nüsslein-Volhard C (2002). Migration and function of a glial subtype in the vertebrate peripheral nervous system. Neuron 34, 577–588. [DOI] [PubMed] [Google Scholar]
- Green R, and Kinsella LJ (1995). Current concepts in the diagnosis of cobalamin deficiency. Neurology 45, 1435–1440. [DOI] [PubMed] [Google Scholar]
- Green R, Allen LH, Bjørke-Monsen AL, Brito A, Guéant JL, Miller JW, Molloy AM, Nexo E, Stabler S, Toh BH, et al. (2017). Vitamin B12 deficiency. Nat. Rev. Dis. Primers 3, 17040. [DOI] [PubMed] [Google Scholar]
- Healton EB, Savage DG, Brust JC, Garrett TJ, and Lindenbaum J (1991). Neurologic aspects of cobalamin deficiency. Medicine (Baltimore) 70, 229–245. [DOI] [PubMed] [Google Scholar]
- Heiman MG, and Shaham S (2009). DEX-1 and DYF-7 establish sensory dendrite length by anchoring dendritic tips during cell migration. Cell 137, 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyo C, Murtha AP, Schildkraut JM, Jirtle RL, Demark-Wahnefried W, Forman MR, Iversen ES, Kurtzberg J, Overcash F, Huang Z, and Murphy SK (2011). Methylation variation at IGF2 differentially methylated regions and maternal folic acid use before and during pregnancy. Epigenetics 6, 928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt A, Harrington D, and Robinson S (2014). Vitamin B12 deficiency. BMJ 349, g5226. [DOI] [PubMed] [Google Scholar]
- Jarjour AA, and Kennedy TE (2004). Oligodendrocyte precursors on the move: mechanisms directing migration. Neuroscientist 10, 99–105. [DOI] [PubMed] [Google Scholar]
- Jarjour AA, Manitt C, Moore SW, Thompson KM, Yuh SJ, and Kennedy TE (2003). Netrin-1 is a chemorepellent for oligodendrocyte precursor cells in the embryonic spinal cord. J. Neurosci 23, 3735–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, and Kramer JM (2000). Nidogen is nonessential and not required for normal type IV collagen localization in Caenorhabditis elegans. Mol. Biol. Cell 11, 3911–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinrade EF, Brates T, Tear G, and Hidalgo A (2001). Roundabout signalling, cell contact and trophic support confine longitudinal glia and axons in the Drosophila CNS. Development 128, 207–216. [DOI] [PubMed] [Google Scholar]
- Klämbt C (2009). Modes and regulation of glial migration in vertebrates and invertebrates. Nat. Rev. Neurosci 10, 769–779. [DOI] [PubMed] [Google Scholar]
- Kok DE, Dhonukshe-Rutten RA, Lute C, Heil SG, Uitterlinden AG, van der Velde N, van Meurs JB, van Schoor NM, Hooiveld GJ, de Groot LC, et al. (2015). The effects of long-term daily folic acid and vitamin B12 supplementation on genome-wide DNA methylation in elderly subjects. Clin. Epigenetics 7, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolnek T, Zhang J, Beardsley S, Scheffer GL, and Hamza I (2014). Control of metazoan heme homeostasis by a conserved multidrug resistance protein. Cell Metab. 19, 1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozyraki R, Kristiansen M, Silahtaroglu A, Hansen C, Jacobsen C, Tommerup N, Verroust PJ, and Moestrup SK (1998). The human intrinsic factor-vitamin B12 receptor, cubilin: molecular characterization and chromosomal mapping of the gene to 10p within the autosomal recessive megaloblastic anemia (MGA1) region. Blood 91, 3593–3600. [PubMed] [Google Scholar]
- Lindenbaum J, Healton EB, Savage DG, Brust JC, Garrett TJ, Podell ER, Marcell PD, Stabler SP, and Allen RH (1988). Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N. Engl. J. Med 318, 1720–1728. [DOI] [PubMed] [Google Scholar]
- Mandal M, and Breaker RR (2004). Gene regulation by riboswitches. Nat. Rev. Mol. Cell Biol 5, 451–463. [DOI] [PubMed] [Google Scholar]
- McKay JA, Groom A, Potter C, Coneyworth LJ, Ford D, Mathers JC, and Relton CL (2012). Genetic and non-genetic influences during pregnancy on infant global and site specific DNA methylation: role for folate gene variants and vitamin B12. PLoS ONE 7, e33290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moestrup SK, Kozyraki R, Kristiansen M, Kaysen JH, Rasmussen HH, Brault D, Pontillon F, Goda FO, Christensen EI, Hammond TG, and Verroust PJ (1998). The intrinsic factor-vitamin B12 receptor and target of teratogenic antibodies is a megalin-binding peripheral membrane protein with homology to developmental proteins. J. Biol. Chem 273, 5235–5242. [DOI] [PubMed] [Google Scholar]
- Mooney RA, and LeVea CM (2003). The leukocyte common antigen-related protein LAR: candidate PTP for inhibitory targeting. Curr. Top. Med. Chem 3, 809–819. [DOI] [PubMed] [Google Scholar]
- Mori T, Buffo A, and Götz M (2005). The novel roles of glial cells revisited: the contribution of radial glia and astrocytes to neurogenesis. Curr. Top. Dev. Biol 69, 67–99. [DOI] [PubMed] [Google Scholar]
- Müller T, Choidas A, Reichmann E, and Ullrich A (1999). Phosphorylation and free pool of beta-catenin are regulated by tyrosine kinases and tyrosine phosphatases during epithelial cell migration. J. Biol. Chem 274, 10173–10183. [DOI] [PubMed] [Google Scholar]
- Murchie R, Guo CH, Persaud A, Muise A, and Rotin D (2014). Protein tyrosine phosphatase σ targets apical junction complex proteins in the intestine and regulates epithelial permeability. Proc. Natl. Acad. Sci. USA 111, 693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AM, Sheils OM, McDonald GS, and Kelleher DP (2005). Detection of a tyrosine phosphatase LAR on intestinal epithelial cells and intraepithelial lymphocytes in the human duodenum. Mediators Inflamm. 2005, 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na H, Ponomarova O, Giese GE, and Walhout AJM (2018). C. elegans MRP-5 exports vitamin B12 from mother to offspring to support embryonic development. Cell Rep. 22, 3126–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahvi A, Barrick JE, and Breaker RR (2004). Coenzyme B12 riboswitches are widespread genetic control elements in prokaryotes. Nucleic Acids Res. 32, 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Christensen EI, and Birn H (2016). Megalin and cubilin in proximal tubule protein reabsorption: from experimental models to human disease. Kidney Int. 89, 58–67. [DOI] [PubMed] [Google Scholar]
- Oikonomou G, and Shaham S (2011). The glia of Caenorhabditis elegans. Glia 59, 1253–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan S, Jost M, Drennan CL, and Elias-Arnanz M (2017). A new facet of vitamin B12: gene regulation by cobalamin-based photoreceptors. Annu. Rev. Biochem. 86, 485–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins LA, Hedgecock EM, Thomson JN, and Culotti JG (1986). Mutant sensory cilia in the nematode Caenorhabditis elegans. Dev. Biol 117, 456–87. [DOI] [PubMed] [Google Scholar]
- Pulido R, Serra-Pagès C, Tang M, and Streuli M (1995). The LAR/PTP delta/PTP sigma subfamily of transmembrane protein-tyrosine-phosphatases: multiple human LAR, PTP delta, and PTP sigma isoforms are expressed in a tissue-specific manner and associate with the LAR-interacting protein LIP.1. Proc. Natl. Acad. Sci. USA 92, 11686–11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampersaud GC, Kauwell GP, Hutson AD, Cerda JJ, and Bailey LB (2000). Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am. J. Clin. Nutr 72, 998–1003. [DOI] [PubMed] [Google Scholar]
- Reynolds E (2006). Vitamin B12, folic acid, and the nervous system. Lancet Neurol. 5, 949–960. [DOI] [PubMed] [Google Scholar]
- Sasse S, and Klämbt C (2016). Repulsive epithelial dues direct glial migration along the nerve. Dev. Cell 39, 696–707. [DOI] [PubMed] [Google Scholar]
- Scalabrino G (2009). The multi-faceted basis of vitamin B12 (cobalamin) neurotrophism in adult central nervous system: Lessons learned from its deficiency. Prog. Neurobiol 88, 203–220. [DOI] [PubMed] [Google Scholar]
- Scalabrino G, Lorenzini EC, Monzio-Compagnoni B, Colombi RP, Chiodini E, and Buccellato FR (1995). Subacute combined degeneration in the spinal cords of totally gastrectomized rats. Ornithine decarboxylase induction, cobalamin status, and astroglial reaction. Lab. Invest 72, 114–123. [PubMed] [Google Scholar]
- Seetharam B, Christensen EI, Moestrup SK, Hammond TG, and Verroust PJ (1997). Identification of rat yolk sac target protein of teratogenic antibodies, gp280, as intrinsic factor-cobalamin receptor. J. Clin. Invest 99, 2317–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham S (2010). Chemosensory organs as models of neuronal synapses. Nat. Rev. Neurosci 11, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham S (2015). Glial development and function in the nervous system of Caenorhabditis elegans. Cold Spring Harb. Perspect. Biol 7, a020578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhvi A, Liu B, Friedman CJ, Fong J, Lu Y, Huang XY, and Shaham S (2016). A glial K/Cl transporter controls neuronal receptive ending shape by chloride inhibition of an rGC. Cell 165, 936–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassky N, de Castro F, Le Bras B, Heydon K, Quéraud-LeSaux F, Bloch-Gallego E, Chédotal A, Zalc B, and Thomas JL (2002). Directional guidance of oligodendroglial migration by class 3 semaphorins and netrin-1. J. Neurosci 22, 5992–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoker AW (2015). RPTPs in axons, synapses and neurology. Semin. Cell Dev. Biol 37, 90–97. [DOI] [PubMed] [Google Scholar]
- Stollhoff K, and Schulte FJ (1987). Vitamin B12 and brain development. Eur. J. Pediatr 146, 201–205. [DOI] [PubMed] [Google Scholar]
- Streuli M, Krueger NX, Hall LR, Schlossman SF, and Saito H (1988). A new member of the immunoglobulin superfamily that has a cytoplasmic region homologous to the leukocyte common antigen. J. Exp. Med 168, 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararajan L, and Lundquist EA (2012). Transmembrane proteins UNC-40/DCC, PTP-3/LAR, and MIG-21 control anterior-posterior neuroblast migration with left-right functional asymmetry in Caenorhabditis elegans. Genetics 192, 1373–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, and Craig AM (2013). Protein tyrosine phosphatases PTPδ, PTPσ, and LAR: presynaptic hubs for synapse organization. Trends Neurosci. 36, 522–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unni DK, Piper M, Moldrich RX, Gobius I, Liu S, Fothergill T, Donahoo AL, Baisden JM, Cooper HM, and Richards LJ (2012). Multiple slits regulate the development of midline glial populations and the corpus callosum. Dev. Biol 365, 36–49. [DOI] [PubMed] [Google Scholar]
- Ward S, Thomson N, White JG, and Brenner S (1975). Electron microscopical reconstruction of the anterior sensory anatomy of the nematode Caenorhabditis elegans.?2UU. J. Comp. Neurol 160, 313–337. [DOI] [PubMed] [Google Scholar]
- Watson E, MacNeil LT, Ritter AD, Yilmaz LS, Rosebrock AP, Caudy AA, and Walhout AJM (2014). Interspecies systems biology uncovers metabolites affecting C. elegans gene expression and life history traits. Cell 156, 1336–1337. [DOI] [PubMed] [Google Scholar]
- Xu Y, and Fisher GJ (2012). Receptor type protein tyrosine phosphatases (RPTPs) - roles in signal transduction and human disease. J. Cell Commun. Signal 6, 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.