

Abstract
Arsenic (As) contamination in drinking water is an epidemic in many areas of the world, especially Eastern Asian countries. Developing affordable and efficient procedures to remove arsenic from drinking water is critical to protect human health. In this study, the oxidation of aquifer solids through the use of sodium permanganate (NaMnO4), hydrogen peroxide (H2O2), and exposure to air, enhanced the adsorption of arsenic to the aquifer material resulting in treatment of the water. NaMnO4 was more effective than H2O2. NaMnO4 was tested at different loading rates (0.5, 1.5, 2.4, 3.4, and 4.9 g NaMnO4/kg aquifer material), and after 30 days contact time, arsenic removal ([As+3]INITIAL = 610 μg/L) was 77%, 88%, 93%, 95%, 97%, respectively, relative to un-oxidized aquifer material. Arsenic removal increased with increasing contact time (30, 60, 90 days) suggesting removal was not reversible under the conditions of these experiments. Oxidative treatment by exposing the aquifer solids to air for 68 days resulted in >99% removal of Arsenic ([As+3]INITIAL = 550 μg/L). Less arsenic removal (38.2%) was measured in the un-oxidized aquifer material. In-situ oxidation of aquifer materials using NaMnO4, or ex-situ oxidation of aquifer materials through exposure to air could be effective in the removal of arsenic in ground water and a potential treatment method to protect human health.
Keywords: Arsenic, Treatment, Permanganate, Oxidation, Ground water
Graphical Abstract
1. Introduction
1.1. Arsenic in ground water
Arsenic (As) contamination in drinking water is presently a worldwide epidemic (Vu et al., 2003). The World Health Organization current guideline value of As in drinking water is 10 μg/L, but in many regions of the world, As concentrations are much higher and have resulted in serious health consequences. In some Asian countries including Bangladesh, West Bengal, Vietnam, China, and Taiwan, the average concentrations of As range from 150 to 440 μg/L and concentrations greater than 3000 μg/L have been measured (Vu et al., 2003). The elevated concentrations of arsenic are generally associated with drinking water sourced from ground water in aquifer systems exhibiting low redox potential. Serious health illnesses related to As include melanosis, keratosis, cancer, and gangrene, and many deaths have been attributed to As contaminated ground water.
1.2. Arsenic chemistry
The two main forms of As are arsenite (As(III)) and arsenate (As(V)), and specific As species are dependent on redox conditions and pH (Table 1). It is apparent that numerous As species may occur in ground water under varying pH and redox conditions. The reduced form, arsenite, is more toxic, soluble, and mobile than the oxidized form, arsenate.
Table 1.
Prevalent Arsenic species under pH and redox conditions (Vu et al., 2003).
| Reducing Conditions | Oxidizing Conditions | ||
|---|---|---|---|
| pH | As(III) | pH | As(V) |
| 0–9 | H3AsO3 | 0–2 | H3AsO4 |
| 10–12 | H2AsO3− | 3–6 | H2AsO4− |
| 13 | HAsO32− | 7–11 | HAsO42− |
1.3. Arsenic removal in ground water
There are several mechanisms in which As removal from the ground water may occur as a result of the oxidation of aquifer materials. Complex As chemistry and variability in subsurface geochemical conditions contribute to the varying roles of removal mechanisms, and consequently the relative role of each mechanism may be difficult to quantify.
1.3.1. Adsorption onto aquifer solids
As(III) and As(V) can adsorb to iron (Fe) minerals found in aquifer materials through complexation reactions which form various As-Fe species (Sun et al., 1999, Vu et al., 2003, Akai et al., 2004, Xie et al., 2015). Under most environmental pH conditions (i.e., pH 6–9), As(V) present as H2AsO4−, adsorbs more readily than As(III) present as H3AsO3, and ferric iron (Fe(III)) complexes more As than ferrous iron (Fe(II)) (Table 1). Complexation and adsorption of As species involves the electrostatic attraction between the anionic form of As species (Table 1) and the positively charged sorption sites and surfaces, particularly those of oxidized iron oxides/hydroxides. Oxidized aquifer material containing greater quantities of ferric iron have greater potential for As adsorption than reduced aquifer materials.
1.3.2. Co-precipitation/coagulation
Arsenic removal may occur by co-precipitation with iron and other metals. Specifically, in ground water containing Fe(II) and As(III), a shift towards oxidative conditions would result in the oxidation of As(III) to As(V), Fe(II) to Fe(III), the precipitation of ferric iron (Fe(III)) and co-precipitation, or coagulation of As(V) (Johnston and Heijnen, 2001, Xie et al., 2015).
1.3.3. MnO2(s)
Once NaMnO4 or KMnO4 are applied to aquifer materials, the predominant manganese reaction byproduct is MnO2(s) (Huling and Pivetz, 2006). However, there are only a limited number of studies dealing with Mn oxides as stabilizing amendments in the scientific literature (Komarek et al., 2013). MnO2(s) readily oxidizes As(III) to As(V) (Driehaus et al., 1995) which becomes adsorbed to the hydroxyl group on the MnO2(s) surface (Oscarson et al., 1981, Sun et al., 1999) forming birnessite ((MnO)2AsOOH) (Manning et al., 2002). The reaction of Mn oxides can also be described as As(III) oxidation coupled with reductive dissolution of the Mn oxide (Manning et al., 2002, Komarek et al., 2013). The subsequent removal of Mn+2 cations from solution could involve adsorption onto sorbent surfaces comprised of solid phase iron and crystalline quartz media (Ocinski et al., 2016).
1.3.4. Organic materials
Organic-based material in aquifer material (i.e., wood, char, natural organic matter (NOM), carbonaceous solids, etc.) can be functionalized through oxidative treatments. Oxidative treatment increases surface oxide functional groups involved in various adsorption reactions. For example, hydrogen peroxide (H2O2) treatment of coal-based activated carbon increased the acidic surface oxide functional groups (i.e., phenolic, carboxylic, carbonyl, lactone groups) from 0.5 μeq/m2 to 1.25 μeq/m2, and changed the adsorptive properties of the activated carbon (Huling et al., 2005). Wood-based charcoal was efficient in the removal of As from arsenic-contaminated water (Hussain et al., 2001), however, a definitive mechanism was not provided for the removal of As. Alternatively, adsorbed NOM can significantly inhibit As(V) adsorption via competitive sorption processes (Ghosh et al., 2006, Li et al., 2017), suggesting that NOM oxidation could improve availability of sorption sites.
1.3.5. Microbial toxicity
Microbial mechanisms are responsible for the reduction of arsenate to arsenite resulting in the enrichment of arsenic in ground water (Akai et al., 2004). Oxidative conditions involving permanganate and H2O2 amendments would yield antiseptic conditions for microbial species responsible for these reductive processes. Therefore, oxidative treatment could inhibit, but not sterilize, microbial processes and limit the arsenic dissolution process.
Overall, the aquifer materials specifically used in this study contain elevated concentrations of organic matter (i.e., total organic carbon (TOC), and high concentrations of total iron ([TOC] 0.02–0.24%; Fe 2–10 g/kg) suggesting the mechanisms discussed above will play a role in arsenic fate and transport in this study.
1.4. Existing and developing treatment technologies
Various treatment technologies used to treat As contaminated water have different advantages and disadvantages. High performance technologies include reverse osmosis, ion exchange, and nano-filtration and achieve high levels of As removal but are costly, involve advanced technology, and require skilled workers to operate and maintain the treatment system (Ahmed, 2001, Bundschuh et al., 2010, van Halem et al., 2010). Other technologies include high pH precipitation and oxidation processes involving the addition of chemicals including alum, lime, iron salts, etc. These technologies involve significant removal of As, do not require highly skilled workers to perform the treatment, but involve a potentially toxic residual that requires safe handling and appropriate disposal, and are generally applicable to single households or small water treatment systems. Low-cost adsorptive media would provide the potential for low energy and water requirements, allow a decentralized operation, involve limited maintenance and cost, such as iron-amended, charred rice hulls (Cope et al., 2014).
In-situ remediation methods to lower arsenic concentrations may involve introducing O2-rich water and altering the pH to increase the anion adsorption capacity of aquifer materials, or some combination of these approaches (Welch et al., 2008). For example, the delivery of dissolved oxygen and HCl into iron-rich aquifer systems to lower As concentrations by adsorption or co-precipitation of As on Fe-oxides has been proposed (Welch et al., 2008). In-situ ground water treatment considered in the current study involves in-situ oxidation of aquifer solids through the use of permanganate (MnO4−) and hydrogen peroxide (H2O2); and ex-situ treatment would involve oxidation of aquifer solids or soils through passive air exposure. Criteria for evaluating each treatment process include low cost, simplicity, minimization of toxic residuals, production of high quality water, and applicability to small to mid-size water supply systems. Conceptually, an above-ground, ex-situ oxidative treatment process would utilize passively oxidized native soils and/or aquifer materials. Under this condition, purification of water is achieved by As immobilization on oxidized iron mineral surfaces. Arsenic-contaminated soil and/or aquifer material residuals would be projected, however, for the ex-situ application.
1.5. Objectives
The objectives of this study were to investigate the extent to which artificially oxidized aquifer material would enhance arsenic sorption and remove arsenic from water. Optimization of the treatment process was investigated by utilizing a range of oxidant loadings, and to quantify arsenic removal efficiency as a function of the oxidant loading.
2. Methods, materials, and analytical procedures
2.1. Oxidant selection
Three liquid-based oxidants were screened for use in this study; sodium persulfate (Na2S2O8), sodium permanganate (Na2MnO4), and hydrogen peroxide (H2O2). All three oxidants have high water solubility. Sodium persulfate was more expensive ($4.20/mole) and can be activated by either Fe2+, a base (KOH) or thermally (>30 °C) which would require additional cost, time, and complexity. Naturally occurring Fe(III) species (Wu et al., 2017) and reduce Fe species can also activate persulfate (Peluffo et al., 2016). The sulfate residual is non-toxic but there is a EPA secondary drinking water standard for sulfate (250 mg/L) involving aesthetic effects (EPA, 2017); and high sulfate leaves the water salty and is a mild laxative. For these reasons, sodium persulfate was eliminated from further investigation. MnO4− ($2.69/mol) (Carus Chemical Corporation, 2012) and H2O2 ($0.04/mol) are less costly. Residuals from NaMnO4 includes MnO2(s), which is non-toxic, highly stable, provides additional adsorptive surfaces for arsenic (Ocinski et al., 2016), and is able to oxidize As(III) to As(V) (Driehaus et al., 1995), the less mobile and less toxic form of As. An EPA secondary drinking water standard exists for manganese (Mn) (0.05 mg/L) involving aesthetics (i.e., black to brown color of water, black staining, bitter metallic taste) (EPA, 2017). O2(g), was also investigated in the study in the passive treatment of aquifer solids, both as a residual from H2O2 treatment and as a major component of air (20.9%). O2(g) is non-toxic and can oxidize reduced aquifer solids (Barcelona and Holm, 1991.
2.2. Aquifer materials
Aquifer core samples (8.5–16 ft bgs) were from the US Marine Corp Recruit Depot (MCRD) (Parris Island, S. Carolina) and from the Canadian River (Ada, OK) (6 ft bgs). The surficial aquifer underlying the MCRD site consists of the sandy Pliocene to Holocene sediments to an average depth of approximately 18 ft (Vroblesky et al., 2009). The water table is shallow and is typically encountered at a depth of 3–4 ft bgs at the site. The occurrence of reduced conditions is also evident from the presence of sulfides, ferrous Fe, hydrogen, and methane that have been measured in the ground water at depth (Vroblesky et al., 2009). The presence of total organic carbon TOC (0.11–0.28%) at the site represents the presence of an available source of substrate material to support biotic activity. Hydrogen (1.1–3.4 nM/L) measurements in 6 out of 7 wells is indicative of sulfate reducing conditions, and high sulfate concentrations (112–130 mg/L) in downgradient wells indicates that the sulfate reducing condition is outcompeting methanogenesis. The increase in acid-extractable Fe to a depth of 16 ft suggests that Fe reduction continues to be a predominant terminal electron acceptor process, assuming acid extractable Fe represents bioavailable ferric iron. The co-existence of ferrous iron, sulfide, and methane is a probable indication that multiple terminal electron accepting processes are occurring simultaneously either at different locations of the site, or at different micro-sites in the subsurface. Cores from the Canadian R. represent alluvial sands where the water table varies between 0 and 4 ft bgs. All aquifer cores collected at both sites were obtained using direct push technology and cores were extruded into transparent acetate sleeves which permitted the visual inspection of the core. The cores were immediately sealed at both ends, placed on ice/dry ice and transported to the research lab and frozen (−20 °C) until used.
2.3. General procedures
De-ionized (DI) water was de-aerated by sparging with N2 and used to prepare stock sodium arsenite (NaAsO2) solutions (520–610 μg/L as arsenite (As+3)). The stock solutions were analyzed (5×) for total and speciated As (As+5, As+3). Aquifer material handling, oxidation and As amendment was performed in an oxygen-free, anaerobic glove-box to prevent the oxidation of aquifer materials from exposure to air. Subsamples from the aquifer cores were collected and composited by hand-mixing in a glass beaker to assure consistency of aquifer material between test reactors. “Air exposed” (n = 2) and “oven dried” (n = 1) test conditions were evaluated as a form of passive oxidation where the aquifer material was air dried (24 h; ≈ 23 °C) and oven dried (24 h, 105 °C), respectively. These reactors were stored outside of the anaerobic glove box and were periodically opened and exposed to air. All of the test reactors were prepared in replicate and arsenic removal results were compared to control reactors containing un-oxidized aquifer material.
2.4. Oxidation and arsenic amendment
Reactors (250 mL glass vessel with air tight caps) containing wet aquifer material (20 g) were amended with NaMnO4 (10 mL; 10 g/L). An equimolar amount of H2O2 (2.4 g/L) was added in the H2O2-amended reactors. The test reactors were routinely turned to ensure a complete mix condition. The oxidant was allowed to react to completion (≥3 days). After oxidation, the arsenite solution (0.1 mL) was added to the oxidized aquifer material and routinely mixed. The temperature in the glove box was 23 ± 0.2 °C. After approximately 30 days contact time, aqueous samples were collected from the reactors, sampled in replicate, filtered (0.2 μm filter), frozen and stored in vials, and subsequently analyzed for total and As species. The pH was measured in the remaining solution after aqueous samples were collected. Reverse-order reactors were prepared in which the reactors first received the As solution followed by oxidant 48 h later. The reverse-order reactors contained saturated aquifer material from 16 ft bgs.
2.4.1. Control reactors
In oxidant-free control reactors, de-aerated DI water (10 mL) prepared by N2(g) sparging was added to the aquifer material (20 g) to achieve identical conditions as the test reactors. The arsenite stock solution (0.1 L) was added to the control reactors after oxidation was completed in the test reactors. The control reactors were handled and sampled in a similar manner as the test reactors.
2.4.2. Oxidation of aquifer material and arsenic immobilization feasibility studies
An “oxidant loading” study was conducted involving similar procedures as described above where aquifer material (20 g) in the test reactors was amended with 10 mL permanganate solutions (0, 1, 3, 5, 7, and 10 g/L NaMnO4). After the oxidant was fully reacted, an As solution (0.1 L; 610 μg/L As(III)) prepared with NaAsO2 was added to the oxidized aquifer material. After 30, 60, and 90 days reaction time, aqueous samples were collected, filtered, frozen and later analyzed for total As, As(III), and As(V). An “oxidant reaction rate” study was conducted where reactors were prepared containing aquifer material (20 g) and amended with an oxidant solution (50 mL; 5 g/L NaMnO4). The MnO4− concentration was measured with time using the spectrophotometer. The absorbance of the MnO4− solution, measured at a wavelength of 525 nm (λMAX = 525 nm), was used to calculate [MnO4−] from a calibration curve. The As uptake and oxidant reaction rate data from these experiments were used to assess the relationship between oxidant concentration and As immobilization, to quantify As immobilization in aquifer material, to define the behavior of the oxidant when injected into an aquifer, and to assess the long term behavior of As immobilization. A one dimensional reactive-transport model was used to conceptually estimate the radius of influence of the injected oxidant.
2.5. Analytical methods
The quality of the data measurements and proper function of instruments was checked through analysis of quality control samples including method blanks, continuing calibration check standards, a second source quality control standard, lab duplicates, and matrix spikes.
pH. The pH was measured using an Orion Sure-Flow ROSS Combination pH probe. MnO4−. MnO4− concentration was determined using a spectrophotometer and measuring absorbance at 525 nm H2O2. H2O2 concentration was measured using a modified peroxytitanic acid colorimetric procedure (Boltz and Howell, 1978, Huling et al., 2001) with a detection limit of 2.9 μM. Aqueous samples collected from test reactors were filtered (0.2 μm) and analyzed in duplicate (standard error ranged between 2 and 3%). The TiSO4 reagent was from Pfaltz and Bauer Inc., and the H2O2 (30% wt. solution in water, reagent grade) was from Aldrich. Iron. Total iron was measured using EPA Phenanthroline Method No. 3500-Fe D (Clesceri et al., 1989). Dissolved oxygen (DO). DO was measured using a Thermo Scientific Orion 3 Star meter. Arsenic Aqueous Analysis. Total As was measured using inductively coupled plasma, optical emission spectroscopy using a Perkin Elmer Optima 3300 DV, ICP-OES. Arsenic speciation was performed using the liquid chromatography (LC)/inductively coupled plasma mass spectrometry (LC/ICPMS) system. The LC system separates the As species via a Dionex anion exchange column and gradient elution and is then pumped to the ICP-MS system, which quantifies the elemental species constituents. Due to a lower detection limit, calculations involving arsenic removal (%) used the results from the LC/ICP-MS results. Estimated numerical results reported as BQL (below quantitation limit) were used in the calculation, and the detection limit was used in the calculation when the results were reported as “non-detect”.
3. Results
The control and test reactors containing the aquifer material were amended with an arsenic solution (0.1 L; 520 μg/L NaAsO2 as As+3). Arsenic removal was based on the dry weight of aquifer material and results indicate that the oxidant-free control attenuated, or adsorbed an average of 0.36 μg arsenic/g of aquifer material (Fig. 1). The H2O2-amended reactors removed an average of 1.1 μg/g (∼3× the control), and the MnO4−-amended reactors removed an average of 2.5 μg/g (∼7× the control).
Fig. 1.

Arsenic removal in oxidized and un-oxidized aquifer material (20 g; 0.1 L [As+3]INITIAL = 520 μg/L). Oxidation of aquifer material was with NaMnO4 and H2O2 (10 mL; 0.071 M).
The aquifer samples collected at 6 ft and 8.5 ft had relatively low total organic carbon (TOC) contents (Fig. 2). These samples generally exhibited low As removal under the treatment conditions, with the exception of the H2O2-amended, 8.5 ft bgs sample. While it has been reported that organic-based materials were efficient in the removal of As from arsenic-contaminated water using wood charcoal (Hussain et al., 2001), natural organic matter (NOM) has been identified as an important parameter in controlling arsenic speciation and adsorption (Liu et al., 2008). Liu et al. (2008) reported adverse effects of NOM on arsenic adsorption at low to neutral pH values (pH range 4.0–9.4), and was attributed to NOM competition with As(III) for available binding sites. The aquifer material used in this study contained both fibrous wood particles and NOM indicating that biodegradation of either may have played a role. No firm explanation can be provided on the role of TOC regarding oxidation-related arsenic treatment and removal.
Fig. 2.

Total organic carbon content in aquifer material used in oxidation-impacted arsenic attenuation experiments.
The As species measured in the post-sorption solution was predominantly arsenate (As(V)) in the NaMnO4-amended reactors, and predominantly arsenite (As(III)) in the H2O2-amended and control reactors (Fig. 3). These results suggest that the manganese species, including MnO4− and MnO2(s), were more effective in oxidizing As(III) than H2O2, resulting in greater As removal.
Fig. 3.

Post-oxidation arsenic speciation in the test reactors. Refer to Table 1 for speciation of arsenic.
Regarding the reverse-order test reactors (i.e., As(III) amendment 48 h before oxidant amendment), relative to the baseline 16 ft bgs test reactor, a minor increase in As removal was measured in the NaMnO4-amended, and a significant increase was measured in the H2O2-amended reactors (Fig. 1). In both cases, the direct oxidation of As(III) to As(V) by the freshly-amended oxidant may have contributed to As(III) oxidation, and enhanced As(V) adsorption and immobilization. The oxidation of aquifer materials and/or NOM would have played a similar role in the reverse-order reactors as in the test reactors where oxidant amendment was followed by As(III) amendment. In both cases of reverse-order NaMnO4 and H2O2 amendment, greater As immobilization was measured than in the oxidant-free control.
The average post-oxidation pH was greater in the NaMnO4-amended reactors (pH = 8.8) relative to the H2O2-amended (pH = 6.6) and un-oxidized control (pH = 7.0) (Fig. 4). The form of As found in the NaMnO4-amended reactors was As(V) (Fig. 3). In this pH range, As(V) is primarily found in the anionic form (HAsO42−) (Table 1). In the H2O2-amended and un-oxidized control reactors, the main form of As was As(III), and was in the neutral form (H3AsO3). The di-valent anionic form of As(V) relative to the neutral form of As(III) helps explain the greater As removal in NaMnO4-amended aquifer material. Specifically, the positively charged forms of Fe on the surfaces of the aquifer solids (i.e., NaMnO4-amended) represent sorption sites for negatively charged As(V).
Fig. 4.

Post-oxidation pH of aquifer material in NaMnO4-, H2O2- and oxidant-free (control) reactors.
3.1. Oxidant loading study
Arsenic removal increased with increasing concentrations of NaMnO4 used to oxidize the aquifer material (Fig. 5). The average As concentration in the stock solution decreased 41% from 610 μg/L to 360 μg/L in the oxidant-free control reactor. Assuming 360 μg/L as the initial As concentration, reactors amended with NaMnO4 at concentrations of 1, 3, 5, 7, and 10 g/L removed an additional average of 72%, 85%, 91%, 94%, and 96%, respectively. The mass of As immobilized in aquifer material per mg NaMnO4 applied to the aquifer material decreased with an increase in NaMnO4 loading (i.e., NaMnO4 loading = mass of NaMnO4 applied per mass of aquifer material (mg NaMnO4/g aquifer material)) (Fig. 6). The lowest NaMnO4 loading to the aquifer material (0.49 mg NaMnO4/g aquifer material) using 1 g/L NaMnO4 resulted in the most efficient As removal (7.3 μg As/mg NaMnO4). This was 9 times greater than 0.82 μg As/mg NaMnO4 where the highest oxidant loading to the aquifer material (4.9 mg NaMnO4/g aquifer material) was applied. Although the efficiency of As removal was lower at 10 g/L NaMnO4, the extent of As removal was greatest. The concentration of NaMnO4 at 10 g/L achieved the lowest solution As concentration in water meeting the EPA primary drinking water standard for As of 10 μg/L.
Fig. 5.

Arsenic removal in NaMnO4-amended (10 mL; 0, 1, 3, 5, 7, 10 g/L) aquifer material (20 g), and oxidant-free control reactors; 30 days after the As(III) solution (0.1 L; 610 μg/L) was amended.
Fig. 6.

Arsenic concentration and immobilization efficiency (As immobilized per mass of NaMnO4 loaded to the aquifer material). Aquifer material (20 g) was amended with a NaMnO4 solution (10 mL; 0, 1, 3, 5, 7, 10 g/L) corresponding with a range in oxidant loading (0.49, 1.47, 2.4, 3.4, 4.9 mg NaMnO4/g soil).
The pH was 7.0 in the oxidant-free control, and increased to 8.1, 8.6, 8.7, 8.8, and 8.8 in reactors amended with 0.49, 1.47, 2.44, 3.44, and 4.87 mg NaMnO4/g aquifer material, respectively. The majority of the As in the oxidant-free control was As(III), and is likely un-ionized (H3AsO3) (Fig. 3, Table 1). In the oxidized aquifer material at pH 7–9, As is likely to be As(V), and present in the di-valent anionic form (HAsO42−) (Fig. 3, Table 1). The anionic form of As(V) relative to the neutral form of As(III), adsorbs to the positively charged sorption sites and helps explain the greater As removal in MnO4−-amended aquifer material.
3.2. Arsenic immobilization duration study
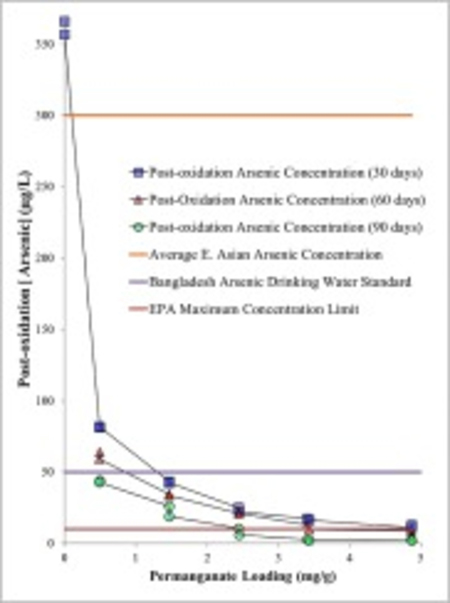
A decreasing trend in As concentration in the aqueous phase correlated with an increase in NaMnO4 loading over a 90 day reaction time period (Fig. 7). This result indicated the reaction was incomplete over the first 30 days, and arsenic removal continued for 90 days. Further, As concentrations did not rebound, and As removal was not reversible over the 90 day period (Fig. 7). The initial background concentration of As (i.e. 360 μg/L) was greater than the average E. Asian As concentration of 300 μg/L (Vu et al., 2003). Over a 30 day reaction period, the post-oxidation As concentrations were reduced from 360 μg/L to below the Bangladesh As drinking water standard (50 μg/L) using 3 g/L NaMnO4 (1.47 mg NaMnO4/g aquifer material) (Fig. 6, Fig. 7); and achieved the EPA maximum concentration limit (10 μg/L) for drinking water using 10 g/L NaMnO4 (4.9 mg NaMnO4/g aquifer material) (Fig. 6, Fig. 7). Results reported by others indicate that As(V) sorption on MnO2(s) is tenacious and desorption is weak (Lenoble et al., 2004).
Fig. 7.

The post-oxidation arsenic concentrations in NaMnO4-amended aquifer material measured 30, 60, and 90 days after the arsenic solution was amended to the oxidized aquifer materials.
3.3. Air exposed/oven dried aquifer material
Two aggressive oxidants, NaMnO4 and H2O2 were tested and successfully demonstrated the ability to oxidize aquifer solids and to enhance the removal of arsenic from solution. These experiments were conducted in an anaerobic glove box to allow for independent investigation of the effects of these oxidants (i.e. in the absence of air exposure). A final set of experiments were conducted to investigate ex-situ, passive oxidation of aquifer solids and arsenic immobilization by exposing the aquifer material to air for a sustained period of time. Ideally, exposure to oxygen, a less aggressive oxidant than NaMnO4 or H2O2, but abundant and readily available, may potentially lead to similar results and lower treatment cost for arsenic removal in an ex-situ application. In an ex-situ reactor configuration using excavated aquifer and/or soil material, exposure of the solids to air would be expected to result in the oxidation of reduced surface mineral species. Subsequently, contact between oxidized mineral surfaces and arsenite-contaminated water would result in the oxidation of arsenite to arsenate. Through this process, enhanced arsenic sorption and immobilization may lead to arsenic removal from the ground water and a potential cost effective arsenic treatment method.
Results indicated >99.0% removal of arsenic was achieved in all reactors both exposing the aquifer solids to air (n = 2), and by oven drying the aquifer solids (n = 1), and 38.2% (n = 2) arsenic removal occurred in the un-oxidized aquifer material (Fig. 8). Given that the aquifer solids were exposed to air, oxidation of sulfur and iron may have occurred through chemolithotrophy, a microbiotic process carried out by autotrophic soil microorganisms (Kappler et al., 2000). An autoclaved aquifer material sample subsequently exposed to air for 30 days had similar pH as the aquifer material stored under anaerobic conditions (i.e., un-oxidized) suggesting a biotic influence. The practical implication of these results indicate that the pH may need to be buffered.
Fig. 8.

Arsenic removal in water by oxidized aquifer material achieved through passive air exposure (AE) and oven-dried (OD) aquifer samples. Arsenic loss (%) was calculated ([As]INITIAL - [As]FINAL)/[As]INITIAL) × 100) and based on samples collected 68 days after the arsenic solution ([As]INITIAL = 550 μg/L) was amended to the test reactors. All samples (replicates) in AE and OD reactors were non-detect (detection limit = 5 μg/L).
4. Discussion
Enhanced arsenic immobilization in the soil was attributed to oxidative treatment and collective influence of the following mechanisms, (1) oxidation of reduced ferrous iron (Fe(II) to ferric iron (Fe(III)), a well-known sorbent of arsenic species, (2) co-precipitation of As with oxidized iron (Fe(III)) and possibly other metals, (3) oxidation of As(III) to As(V), by the oxidants (i.e., NaMnO4, H2O2), their residuals (i.e., MnO2(s), dissolved oxygen, O2(g)), and by O2(g) in air, and (4) complexation and adsorption of HAsO42− with cationic sorption sites on mineral surfaces. Other potential mechanisms include, the oxidative activation of natural organic matter (i.e., functionalization) known to be present in the aquifer material resulting in an increase in the functional groups (i.e., sorption sites); and the oxidation of NOM occupying sorption sites that free up sorption sites for arsenic sorption.
NaMnO4 amendments resulted in a general increase in the average pH (8.8) relative to the un-oxidized aquifer materials (pH 7.0) (Fig. 4). Various iron species (i.e., Fe3O4, FeOOH, Fe2O3, amorphous Fe(OH)3) and MnO2(s) exhibit a pH point of zero charge (pHPZC) in the pH range of 6.5–8.5. An increase in the pH would reduce the number of positively charged surface sites, but this does not appear to negatively impact As removal (i.e., Fig. 1, Fig. 5, Fig. 6, Fig. 7). However, the acidic pH in the air-exposed and oven-dried aquifer solids decreased below this range in pHPZC (Fig. 8). This likely increased the number of positively charged surface sites, and contributed to the significant removal of As. Multiple, undifferentiated As removal mechanisms played a role in these treatment systems, but specific mechanisms were not independently investigated.
Results indicate that the artificial oxidation of aquifer materials or soils may be an effective method of reducing As concentrations in ground water. The treatment of ground water using these artificial oxidation treatment processes could occur in either in-situ or ex-situ treatment systems. Historically, there have been limited applications and development of in-situ treatment methods. Long term studies are needed to advance the technology, and to quantify the sustainability of treatment performance. In general, the feasibility of in-situ treatment would involve greater uncertainty due to limited characterization of subsurface hydrogeology and geochemical characteristics, and the impact these may have on treatment effectiveness. An EPA secondary drinking water standard exists for manganese (Mn) (0.05 mg/L) is based on aesthetics (i.e., black to brown color of water, black staining, bitter metallic taste) (EPA, 2017). Pilot-scale data is needed to further assess the technical and economic feasibility of the technology, including the extent to which manganese residuals are present in the treated ground water. More frequent applications of engineered systems involving ex-situ treatment provides greater certainty in geochemistry, system operations, and treatment methods. The results of this study represent long-term contact conditions between the aerated (oxidized) soil and arsenic-contaminated water. Pilot-studies are needed to assess arsenic removal in systems designed with site-specific soil material, and with potentially shorter contact times reflecting practical operational conditions.
5. Conclusions
The oxidation of reduced aquifer solids was achieved through the addition of sodium permanganate and hydrogen peroxide, and by contact with air. Through these oxidative treatments, arsenic adsorption onto the aquifer material was enhanced relative to un-oxidized controls resulting in an increase in the removal of arsenic from solution. The 30-day, un-oxidized control attenuated an average of 0.36 μg/g of As from the water arsenic solution. The H2O2-amended and NaMnO4-amended reactors removed 1.1 μg/g (∼3× the control) and 2.5 μg/g (∼7× the control), respectively. Arsenic immobilization was directly correlated with NaMnO4 concentration applied to the aquifer material where 1, 3, 5, 7, and 10 g/L NaMnO4 removed 86%, 93%, 96%, 97%, and 98%, respectively. The efficiency of As removal (i.e., μg As/g NaMnO4) decreased as NaMnO4 concentration increased; however, greater As removal and lower As concentrations were achieved with increasing concentration. Arsenic removal from water continued over the 90 day treatment period indicating that the reaction was incomplete in the first 30 days after arsenic amendment, and that As removal was not reversible over the 90 day testing period. The initial concentration of As (360 μg/L) was greater than the average E. Asian As concentration of 300 μg/L, was reduced to below the Bangladesh As drinking water standard (50 μg/L) in 30 days using 3 g/L NaMnO4 (1.47 mg NaMnO4/g aquifer material), and achieved the EPA maximum concentration limit (10 μg/L) for drinking water using 5 g/L NaMnO4 (2.44 mg NaMnO4/g aquifer material) in 90 days. Oxidative treatment of the aquifer solids by exposure to air for 68 days resulted in >99% removal of Arsenic ([As+3]INITIAL = 550 μg/L); less arsenic removal (37%) was measured in the un-oxidized aquifer material. The pH of the soil slurry increased over background (pH = 7.0) to pH 8.8 as the concentration of NaMnO4 amended to the aquifer material increased. Under oxidized conditions in this pH range, As(III) was oxidized to As(V), an anionic form of arsenic (HAsO42−) that is highly amenable to electrostatic adsorption on the positively charged surfaces of iron and manganese oxides. Results suggest that the artificial oxidation of aquifer materials or soils could be an effective method of reducing As concentrations in ground water.
Acknowledgments
The Authors acknowledge Shauna Bennett, Andrew Greenwood, and Steve Markham (Shaw Environmental and Infrastructure, Inc.) for arsenic analyses; Cherrie Adair and Dr. John Wilson at the US Environmental Protection Agency, R. S. Kerr Environmental Research Center (US EPA, RSKERC) (Ada, OK) for allowing the use of laboratory equipment.
Notice
The U.S. Environmental Protection Agency, through its Office of Research and Development, funded and managed the research described here. It has not been subjected to Agency review and therefore does not necessarily reflect the views of the Agency, and no official endorsement should be inferred.
References
- Ahmed MF An overview of arsenic removal technologies in Bangladesh and India Proceedings of BUET-UNU International Workshop on Technologies for Arsenic Removal in Drinking Water, Dhaka, 5–7 May, 2001, Bangladesh University of Engineering and Technology and United Nations University, Bangladesh (2001), pp. 251–269 [Google Scholar]
- Akai J, Izumi K, Fukuhara H, et al. Mineralogic and geomicrobiological investigations on groundwater arsenic enrichment in Bangladesh Appl. Geochem, 19 (2004), pp. 215–230 [Google Scholar]
- Barcelona MJ, Holm TR Oxidation-reduction capacity of aquifer solids Environ. Sci. Technol, 25 (9) (1991), pp. 1565–1572 [Google Scholar]
- Boltz DF, Howell JA (Eds.), Colorimetric Determination of Non-metals. Wiley- Interscience Publications, John Wiley and Sons; (1978) [Google Scholar]
- Bundschuh J, Litter M, Ciminelli V, Morgada M, et al. Emerging mitigation needs and Sustainable options for solving the arsenic problems of rural and Isolated urban areas in Latin America- A critical analysis Wat. Res, 44 (19) (2010), pp. 5328–5345 [DOI] [PubMed] [Google Scholar]
- Carus Chemical Corporation, 2012. http://www.Caruscorporation.com.
- Clesceri LS, Greenberg AE, Trussel RR Standard Methods for the Examination of Water and Wastewater Port City Press, Baltimore: (1989) [Google Scholar]
- Cope CO, Webster DS, Sabatini DA Arsenate adsorption onto iron oxide amended rice husk char Sci. Total Environ, 488–489 (2014), pp. 554–561 [DOI] [PubMed] [Google Scholar]
- Driehaus W, Seith R, Jekel M Oxidation of arsenate(III) with manganese oxides in water treatment Wat. Res, 29 (1) (1995), pp. 297–305 [Google Scholar]
- EPA Secondary Drinking Water Standards: Guidance for Nuisance Chemicals (2017) https://www.epa.gov/dwstandardsregulations/secondary-drinking-water-standards-guidance-nuisance-chemicals
- Ghosh A, Saez E, Ela W Effect of pH, competitive anions and NOM on the leaching of arsenic from solid residuals Sci. Total Environ, 363 (2006), pp. 46–59 [DOI] [PubMed] [Google Scholar]
- Huling SG, Arnold RG, Sierka RA, Miller MA Influence of peat on Fenton oxidation Wat. Res, 35 (7) (2001), pp. 1687–1694 [DOI] [PubMed] [Google Scholar]
- Huling SG, Jones PK, Ela WP, Arnold RG Repeated reductive and oxidation treatments of granular activated carbon J. Environ. Eng, 131 (2) (2005), pp. 287–297 [Google Scholar]
- Huling SG, Pivetz B In-Situ Chemical Oxidation – Engineering Issue US Environmental Protection Agency, National Risk Management Research Laboratory, R.S. Kerr Environmental Research Center, Ada, OK: (2006) EPA/600/R-06/072 [Google Scholar]
- Hussain MD, Haque MA, Islam MM, Hossen MA Approaches for removal of arsenic from tubewell water for drinking purpose Feroze Ahmed M, et al. (Eds.), Technologies for Arsenic Removal from Drinking Water, A Compilation of Papers Presented at the Intl. Workshop on Technol. For Arsenic Removal from Drinking Water (2001) Bangladesh Univ. of Engineering and Technol., Dhaka, Bangladesh and the United Nations Univ., Tokyo: May 2001 [Google Scholar]
- Johnston R, Heijnen H Safe water technology for arsenic removal Ahmed MF, et al. (Eds.), Technologies for Arsenic Removal from Drinking Water, Bangladesh University of Engineering/Technology, Dhaka, Bangladesh: (2001) [Google Scholar]
- Kappler U, et al. Sulfite: cytochrome c oxidoreductase from thiobacillus noveelus J. Biol. Chem, 275 (18) (2000), pp. 13202–13212 [DOI] [PubMed] [Google Scholar]
- Komarek M, Vanek A, Ettler V Chemical stabilization of metals and arsenic in contaminated soils using oxides - a review Environ. Poll, 172 (2013), pp. 9–22 [DOI] [PubMed] [Google Scholar]
- Lenoble V, Laclautre C, Serpaud B, Deluchat V, Bollinger VC As(V) retention and As(III) simultaneous oxidation and removal on a MnO2-loaded polystyrene resin Sci. Total Environ, 326 (2004), pp. 197–207 [DOI] [PubMed] [Google Scholar]
- Li F, Guo H, Zhou X, Zhao K, Shen J, Liu F, Wei J Impact of natural organic matter on arsenic removal by modified granular natural siderite: evidence of ternary complex formation by HPSEC-UV-ICP-MS Chemosphere, 168 (2017), pp. 777–785 [DOI] [PubMed] [Google Scholar]
- Liu G, Zhang X, Talley JW, Neal CR, Wang H Effect of NOM on arsenic adsorption by TiO2 in simulated As(III)-contaminated raw waters Wat. Res, 42 (2008), pp. 2309–2319 [DOI] [PubMed] [Google Scholar]
- Manning AB, Fendorf ES, Bostick B, Suarez LD Arsenic (III) oxidation and arsenic (V) adsorption reactions on synthetic birnessite Environ. Sci. Technol, 36 (5) (2002), pp. 976–981 [DOI] [PubMed] [Google Scholar]
- Ocinski D, Jacukowicz-Sobala I, Mazur P, Raczyk J, Kociołek-Balawejder E Water treatment residuals containing iron and manganese oxides for arsenic removal from water– Characterization of physicochemical properties and adsorption studies Chem. Eng. J, 294 (2016), pp. 210–221 [Google Scholar]
- Oscarson DW, Huang PM, Liaw WK Role of manganese in the oxidation of arsenite by freshwater lake sediments Clay Clay Mineral, 29 (3) (1981), pp. 219–225 [Google Scholar]
- Peluffo M, Pardo F, Santos A, Romero A Use of different kinds of persulfate activation with iron for the remediation of PAH-contaminated soil Sci. Total Env, 563–564 (2016), pp. 649–656 [DOI] [PubMed] [Google Scholar]
- Sun X, Doner HE, Zavarin M Spectroscopy study of arsenire [As(III0] oxidation on Mn- substituted goethite Clay Clay Mineral., 47 (4) (1999), pp. 474–480 [Google Scholar]
- van Halem D, Heijman SGJ, Johnston R, Huq IM, Ghosh SK, Verbek JQJC, Amy GL, van Dijk JC Subsurfave iron and arsenic removal: low cost technology for community-based water supply in Bangladesh Water Sci. Technol, 62 (11) (2010), pp. 2702–2709 [DOI] [PubMed] [Google Scholar]
- Vroblesky DA, Petkewich MD, Landmeyer JE, Lowery MA Source, Transport, and Fate of Groundwater Contamination at Site 45, Marine Corps Recruit Depot, Parris Island, South Carolina U.S. Geological Survey Scientific Investigations Report 2009–5161; (2009), p. 80 [Google Scholar]
- Vu KB, Kaminski MD, Nunez L Review of Technologies for Contaminated Groundwater ANL-CMT-03/2Argonne Natl. Laboratories, Argonne, IL: (2003) 60439 [Google Scholar]
- Welch AH, Stollenwerk KG, Paul AP, Maurer DK, Halford KJ In situ arsenic removal in an alkaline elastic aquifer Appl. Geochem, 23 (8) (2008), pp. 2477–2495 [Google Scholar]
- Wu Y, Prulho R, Brigante M, Dong W, Hanna K, Mailhot G Activation of persulfate by Fe(III) species: implications for 4-tert-butylphenol degradation J. Haz. Mat, 322 (2017), pp. 380–386 [DOI] [PubMed] [Google Scholar]
- Xie X, Wang Y, Pi K, Liu C, Li J, Liu Y, Wang Z, Duan M In situ treatment of arsenic contaminated groundwater by aquifer iron coating: experimental study Sci. Total Environ, 527–528 (2015), pp. 38–46 [DOI] [PubMed] [Google Scholar]