

Abstract
Mutations in the SLC13A5 gene, a sodium citrate cotransporter, cause a rare autosomal recessive epilepsy (EIEE25) that begins during the neonatal period and is associated with motor and cognitive impairment. Patient’s seizure burden, semiology, and electroencephalography (EEG) findings have not been well characterized. Data on 23 patients, 3 months to 29 years of age are reported. Seizures began during the neonatal period in 22 patients. Although seizures are quite severe in many patients later in life, seizure freedom was attainable in a minority of patients. Multiple patients’ chronic seizure management included a few common medications, phenobarbital and valproic acid in particular. Patients EEGs had a relatively well-preserved background for age, even in the face of frequent seizures, little slowing and multiple normal EEGs and do not support an epileptic encephalopathy. Other causes for the motor and cognitive delay beyond epilepsy warrant further study.
Keywords: antiseizure drugs, developmental disability, epileptic encephalopathy, inborn errors of metabolism, neonatal seizures, seizures, status epilepticus
Citrate transporter deficiency is an autosomal recessive disorder caused by mutations in the SLC13A5 gene, resulting in loss of NaCT (sodium chloride citrate cotransporter) activity.1-3 The protein is highly expressed in liver, teeth, testes, and brain though patients have only been noted to have brain and teeth enamel abnormalities.4,5 Point mutations and deletions have been identified throughout the gene in affected individuals.6 Several recurrent point mutations are present at low levels in the general population. SLC13A5 mutation carrier parents have not been reported to have a disease phenotype. Expression studies in heterologous expression systems have supported a loss of transporter function, with no mutations reported to have increased transporter activity.2,3 Interestingly, loss of function in animal models have not recapitulated the neurologic phenotype; mice and flies with NaCT deficiency have been described as having increased longevity and no neurologic phenotype has been reported.7,8
Epilepsy onset in the neonatal period has been a consistent feature in published reports of SLC13A5 citrate transporter deficient patients. However, the literature and our clinical experience supports a more varied epilepsy phenotype later in life, even among siblings, suggesting lack of a genotype–phenotype correlation. It has been hypothesized that the presence of elevated citrate may be contributing to the neurologic phenotype, but there is a lack of direct evidence. Additional support for abnormal citrate homeostasis is the finding of elevated citrate in the CSF and serum of patients with SLC13A5 disorder.9 Patients have cognitive delay and profoundly poor communication and motor skills; however, whether the developmental impairment is an epileptic encephalopathy remains unknown. One goal was to understand how ongoing seizures and interictal epileptiform activity relate to the disorder’s neurologic phenotype.
To further characterize the SLC13A5 epileptic phenotype, we decided to develop a caregiver survey to capture patients from around the world due to its rarity. Collaborating with the TESS Research Foundation (a nonprofit disease advocacy organization), we identified families with affected individuals and asked them to complete a survey to characterize seizure semiology, frequency, and treatment regimens. Caregivers were contacted through email addresses provided by the TESS Research Foundation. Caregivers were asked to fill out a questionnaire focused on the patient’s epilepsy phenotype. We also requested copies of medical records and electroencephalographies (EEGs) in an attempt to create the most robust phenotypic characterization to date of SLC13A5 disorder.
Methods
This research was carried out under a Stanford institutional review board extended approval protocol. Via caregiver submission, the TESS Research foundation has been collecting patients’ genotypes, and over 29 separate mutations have been identified to date (https://tessresearch.org/). Twenty-five caregivers identified by the TESS Research Foundation were sent an email and asked if they would volunteer to fill out 2 REDCap surveys, one for them to describe the epilepsy phenotype of the patient, and one to obtain medical records, EEGs and MRIs (Supplementary Questionnaires).
Results
A total of 50 EEGs were reviewed from 8 patients; 19 were by report only, but all 8 patients had at least one EEG tracing reviewed by BEP. In all cases, the reports (when available) were compared to impression of the EEG tracing(s), and agreement was present between BEP and the clinical EEG report. The EEGs were assessed for age of the patients at the time of the EEG, duration of the EEG, the awake and sleep background (if present), interictal epileptiform features, and seizures (Supplementary Table 1).
Of the caregivers approached, 9 did not fill out the forms at all or only in limited detail and were not included in this article. Seventeen caregivers filled the questionnaire out in full. A total of 23 patient’s data were included for analysis, 11 males and 12 females from 17 families and the age range was 3 months to 29 years (Table 1). The genetic findings are typical of the constellation of patients reported to date.1-3 The most common mutations—c.655G>A p.G219R and c.680 C>T, p.T227M—are present, along with unique and not previously reported heterozygous and homozygous mutations, including deletions.
Table 1.
Study Patients: Demographic, Genetic, Seizure Frequency, Seizure Semiology, Current Anti-Seizure Medications Data.
| Patient # | Age (months) | Sex | Gene mutation | How often does your child currently have seizures? | Seizure semiology | Current seizure medication(s) |
|---|---|---|---|---|---|---|
| 1 | 61 | Male | c.655G>A, p.G219R and c.1475T>C, p.L492P, compound heterozygous | 1 febrile seizure in the past year | Focal motor with retained awareness, Generalized tonic–clonic (GTC) | Acetazolamide, namenda, phenobarbital |
| 2 | 178 | Female | c.655G>A, p.G219R and c.1475T>C, p.L492P, compound heterozygous | 50-100 times per day | Focal motor with retained awareness, unresponsive with abnormal motor, GTC, myoclonic | Acetazolamide, diazepam, felbatol, namenda, perampanel, sertraline |
| 3 | 129 | Female | c644C>T, p.A215V, homozygous | Once every 6 months on an average | GTC | Phenobarbital, topiramate, valproic acid |
| 4 | 45 | Male | c644C>T, p.A215V, homozygous | Currently once every 2-3 months (related to fever and if missed a dose) | Focal motor with retained awareness, GTC, tonic | Carbamazepine and topiramate |
| 5 | 278 | Female | c.425C>T, p.T142M; 655G>A p.G219R, compound heterozygous | Since she turned 6 years she had no more seizures, normal EEG, no medication | GTC | No medications |
| 6 | 206 | Female | c.425C>T, p.T142M; 655G>A p.G219R, compound heterozygous | Nearly every day, sometimes a couple of seizures mainly during the night | GTC | Acetazolamide, clonazepam, phenobarbital, stiripentol, bromides |
| 7 | 185 | Male | c.425C>T, p.T142M; 655G>A p.G219R, compound heterozygous | Every 10 to 14 days | GTC, myoclonic | Vigabatrin |
| 8 | 151 | Female | c.511 delG amino acid P.E171Sfs* 16 frame shift due to single nucleotide deletion. Homozygous | 2 times per month | GTC | Acetazolamide, diazepam |
| 9 | 137 | Male | c.511 delG amino acid P.E171Sfs* 16 frame shift due to single nucleotide deletion. Homozygous | Once in every 3 to months or when he is severely sick | GTC | Acetazolamide, diazepam, lacosamide |
| 10 | 108 | Male | c.148T>C, p.Cys50Arg; homozygous | Only when ill with fever or missing medications | Focal motor with retained awareness, Unresponsive with abnormal motor | Levetiracetam, valproic acid, phenobarbital |
| 11 | 353 | Female | c.148T>C, p.Cys50Arg; homozygous | None since 5 years of age | GTC | Lacosamide |
| 12 | 9 | Female | c.425C>T, p.T142 M; Deletion of the entire gene, compound heterozygote | Several per day | GTC, myoclonic | Levetiracetam |
| 13 | 41 | Female | c.1022G>A, p.Trp341* and c.655G>A, p.Gly219Arg, compound heterozygous | It varies; can be 4 times a day or once every 2 to 5 weeks | GTC, tonic, myoclonic | Acetazolamide, carbamazepine, clobazam, diazepam, valproic acid |
| 14 | 110 | Female | c.425C>T, p.T142M; deletion of the entire gene, compound heterozygous | One time per year | GTC | Levetiracetam, phenobarbital, valproic acid |
| 15 | 156 | Male | c.997C>T P.R333* Nonsense truncated protein; C.680 C>T P.T227N, compound heterozygous | Not in about 3 years | GTC, myoclonic | Acetazolamide, carbamazepine, oxcarbazepine, phenobarbital |
| 16 | 60 | Male | CDNA C.655 G>A P.G219R; Deletion exon 1 to 5, compound heterozygous | No recent seizures | GTC | Valproic acid |
| 17 | 216 | Male | c.231+2T>G homozygous | 4 times per week | GTC, tonic, infantile spasms, | Clonazepam |
| 18 | 24 | Male | No data | Monthly | Unresponsive with abnormal motor, GTC | Lacosamide, valproic acid |
| 19 | 18 | Male | c.997C>T, p.Arg333*, c.478G>T, p.Glu160*, compound heterozygous | No longer having seizures | Unresponsive with abnormal motor, GTC | Clobazam, topiramate, valproic acid |
| 20 | 3 | Male | c.997C>T p.R333*; c.1514C>T p.P505L, compound heterozygous | One time per week | Unresponsive with abnormal motor, GTC, myoclonic | Diazepam, levetiracetam, phenobarbital |
| 21 | 72 | Female | c680 C>T, p.T227M Homozygous | Big seizures—1 time per year | Absence, unresponsive with abnormal motor | Gabapentin, lamotrigine, lorazepam, leviteracetam, phenobarbital |
| 22 | 252 | Female | C.103-1 GtoA splicing mutation, intronic CCDX 11079.1; C.1276-1 G to A splicing mutation, compound heterozygous | Once a week | Absence, GTC, myoclonic | Acetazolamide, lacosamide, lamotragine, topiramate, valproic acid |
| 23 | 29 | Female | c.389G>A p.G130D; gene deletion exon 2 to 4, compound heterozygous | 3-5 times per day | Absence, unresponsive with abnormal motor, GTC, tonic | Diazepam, zarontin, phenobarbital, rufinamide |
Abbreviation: EEG, electroencephalography.
One question frequently asked about SLC13A5 disorder due to its potential metabolic phenotype is “are affected individuals growth impaired?.”9 The growth parameters for age were calculated using the 2000 CDC growth calculator. The mean weight was 32.7 percentile and height 37.9 percentile, though a number of patients (7 for weight, 4 for height) were less than 2 standard deviations for age.
All individuals developed epilepsy within the first year of life. Twenty-two of 23 patients developed epilepsy in the first week of life. This is in keeping with the literature, which says that seizures associated with SLC13A5 mutations start in the neonatal period.1-3,5
The description of seizure semiology varied. Multiple semiologies were reported in most individual patients, and varied seizure semiologies were noted within families with more than one affected individual. There was not a single semiology that occurred in all of the patients, though generalized tonic–clonic was most common, reported in 17 (∼74%) of the 23 patients. On average individuals had 1.7 seizure types (range: 0-4; Table 1 and Figure 1). We asked caregivers to describe the seizures in their own words. Seventeen of the caregivers describe prominent clonic activity, with body parts jerking as part of the seizures, often of one side or both sides of the body. Sometimes there is either a tonic component at the beginning or a brief lead in with a change in appearance, suggesting a focal onset. One EEG tracing we had access to captured multiple myoclonic seizures. On video, there is a head turn to the left and jerking of the limbs without loss of tone associated with high-voltage bifrontal spike and wave at 3 HZ frequency with a modest, less than a second in duration right-sided lead-in (Figure 2D).
Figure 1.

Seizure semiology. The number of patients reporting each seizure semiology.
Figure 2.

Electroencephalography (EEG): Normal EEG background, awake, asleep, interictal EEG, hemiclonic seizure onset. A, Normal awake background at 7 years 6 months, patient 9. B, Normal sleep architecture at 4 years 2 months, patient 10. C, Multifocal spikes with otherwise normal background at 6 years 5 months, patient 8. D, Hemiclonic seizure with head turn to the left and left more than right body jerks, with brief right-sided lead to a more generalized high-voltage spike and wave, otherwise normal awake background at 11 year 2 months, patient 2.
Seizure frequency varied widely, with one 5-year-old described as seizure-free off antiseizure medication, but all other patients reported to be taking antiseizure medication (Figure 3). Seven of the patients have not had seizures in over a year or only when ill. However, 10 had daily or weekly seizures.
Figure 3.

Seizure frequency. Current seizure burden.
Twenty-two medications were listed by caregivers as being taken for antiseizure purposes (Figure 4). The most common medications include phenobarbital and valproic acid 9 patients each. Two atypical antiseizure medications include acetazolamide 8 patients and memantine 2 patients. No patients were reported to be taking pregabalin, phenytoin, tigabine, or zonisamide. The number of patients who were having less than a seizure per year or seizures only with illness are in the column next to each of the medications. Medications with more than one patient in the infrequent seizure category include phenobarbital (5 of the 9) and valproic acid (4 of the 9).
Figure 4.

Current medication usage in all patients, including the seizure-free patient subset. The current number of patients using each medication is shown in blue. The medications of those patients who are seizure free or rare yearly seizures are shown in orange.
When asked what medications they felt worked the best for their child; 5 caregivers said phenobarbital, 4 said valproic acid, 2 vigabatrin, 2 lacosamide, 1 felbatol, 1 clonazepam, 1 clobazam, 1 acetazolamide, 1 levetiracetam, 1 carbamazepine or phenytoin, and 1 oxcarbazepine. While mixed in mechanism of action, multiple GABAA receptor agonist medications appear in both the common current medications being consumed and the medications they felt worked best for their child.
Fifty EEGs or reports were available for review in 8 of the patients (Supplementary Table 1). Electroencephalographies were evaluated by BEP, who is board certified in Epilepsy and Clinical Neurophysiology. A checklist was used to assess the background, interictal, and ictal features.
Electroencephalography tracings were available from 3 patients with EEGs during the first week of life. All appeared to be in status epilepticus with frequent “typical” neonatal seizures, with focal onset from multiple central or temporal locations, occurring up to several times per hour, all with a short duration of less than 2 minutes. The EEG background appeared abnormal during the periods with frequent seizures, with excessive discontinuity and frequent multifocal spikes and sharps (Figure 5B). However, after several days, the patients had improvement in the background with continuous background, normal features for age, state changes, and multifocal sharp transients (Figure 5A).
Figure 5.
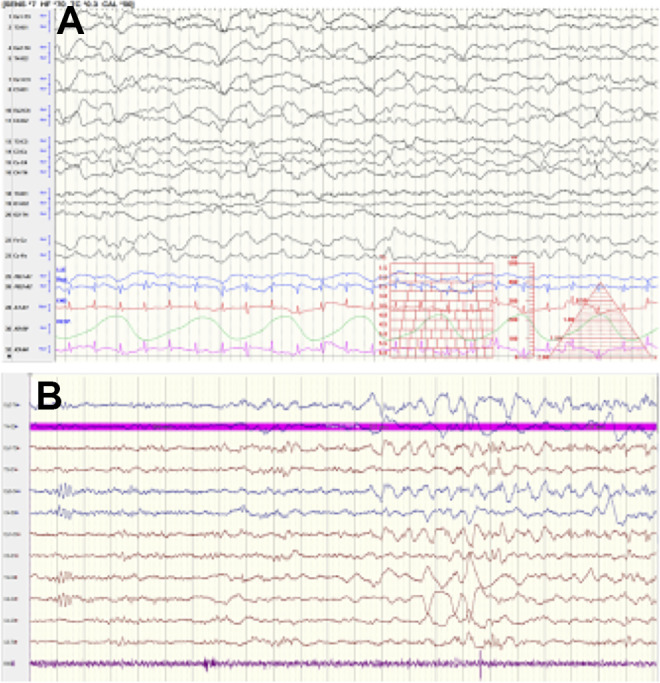
Electroencephalography (EEG): Neonatal EEG background and neonatal seizure. A, Normal background on day of life 8 in patient 1, with multifocal sharps. B, Seizure onset and excessive discontinuous background on day of life 3 in patient 12.
In older patients outside the neonatal period and up to 24 years of age (Figure 2A-D), the EEG background appeared normal or had mild theta range slowing. Interictal spiking was multifocal and/or appeared generalized with bilateral high-voltage spikes when present though some EEGs were normal. Patient 2 had 5 brief seizures, less than 15 seconds in a 30-minute EEG, with consistent head and eyes to the left and had more left body jerking, the EEG correlate appeared as a generalized spike and a wave discharge, with short lead-in spikes on the right (Figure 2D).
Discussion
SLC13A5 citrate transporter disorder is rare, with the largest group collected by the TESS Research Foundation, 87 patients with 36 different mutations identified to date. The rarity of the diagnosis and having patients from multiple countries makes it challenging to collect prospective data or obtain a large retrospective cohort of patients. Here, we present data from a cohort of SLC13A5 disorder patients (23 patients from 17 families). Families were included based upon voluntary caregiver participation in an email questionnaire. Based on our current experience, this can be an effective way to amass phenotypic information on rare disorders that require international identification of patients to power studies. Our methodology does have multiple shortcomings, it is possible that patients may have been incorrectly diagnosed with SLC13A5 mutations as the cause of their epilepsy, though only one of the patients’ caregivers did not provide specific genetic testing results. Luckily, there are multiple shared similarities in the patients’ epilepsy phenotype to support a common etiology. We were provided with a mixture of clinical reports, EEG reports, and some EEGs for our review that also seemed to support a relatively consistent EEG and medical history but we were not able to review medication dosing and extensive seizure histories from the medical records to corroborate or further explore some findings. We only supported participation in English and via the internet further limiting participation from some countries and skewing toward a higher socioeconomic status of participants.
All the reported mutations to date are autosomal recessive with several families having more than one affected child. The disorder appears to be secondary to a loss of protein function with inability to transport citrate into homologous expression systems in vitro. The protein is widely expressed throughout the body with high levels in liver, testes, teeth, and brain. Mouse models did find mild decreased bone length and tooth enamel production.7,10 The overall mean growth parameters (weight and height) in our patients were below average, but within the normal range for the majority of patients. Multiple patients, however, were below 2 standard deviations for age. The questionnaire did not directly address the etiology of this growth failure, the patients are known to have tooth enamel issues and significant motor delays, that might impact normal food consumption. An elevation in citrate in the CSF and serum of patients with SLC13A5 has been reported, but how elevated citrate could contribute to growth or to seizures, cognitive, and/or motor delay is not understood.9,11
Epilepsy was present in all patients and 22 of 23 had onset in the first week of life. Later in life, the patients had a variety of seizure semiologies but 17 of them describe seizures with a prominent clonic, hemiclonic, or whole-body clonic movements though they do not all start in a similar fashion some have tonic onset and some with change in behavior suggesting a focal onset. Although most patients stay on seizure medications, it is possible for patients to have rare seizures and one patient was reported to be seizure-free off medication. The wide range of genetic mutations and seizure burden makes identification of a genotype phenotype correlation unlikely in this patient population. However, the presence of 5 pairs of siblings, and one family with 3 children in our cohort having markedly different seizure frequency suggests a contribution to their epilepsy beyond the SLC13A5 genotype.
The treatment regimens used in our cohort varied widely and with varied success in controlling seizures. There are no current recommendations for specific SLC13A5 epilepsy treatment; this study suggests several medications clinicians could consider when treating seizures. The 2 most commonly used medications phenobarbital and valproic acid were also associated with seizure freedom in the most patients and share a common GABA augmentation mechanism of action, though valproic acid has numerous targets.12 However, seizure frequency may relate to phenotypic or other variability irrespective of medication. Acetazolamide is an atypical medication that may also be considered. Although only 2 of the 8 patients taking it were having rare seizures (the rest more frequent seizures), one caregiver felt it was the most effective medication for their child’s seizures. The mechanism of acetazolamide’s central nervous system (CNS) effect is thought to be via decreasing CNS intracranial pressure and lowering Ph, though how this suppresses seizures is not well understood. It is possible that acetazolamide could alter excess citrate found in the CNS and support the need for further study of SLC13A5 mutations physiologic effects.
Although limited in number, the EEGs reviewed in this study demonstrated some consistent phenotypic patterns. The 3 EEGs in the neonatal period consistently demonstrated status epilepticus, and a discontinuous background, followed by normalization of the background with diminished seizures but multifocal sharps persisting. Electroencephalographies outside of the neonatal period suggested a well-organized, mostly normal for age frequencies, even in the face of frequent seizures. The interictal spikes in these EEGs were high voltage and multifocal or generalized in appearance. In contrast to other genetic developmental epilepsy disorders, the EEG findings do not support an ongoing epileptic encephalopathy.13 Future studies will need to focus on understanding this disparity between satisfactory seizure control and even normal EEG background with persistent neurocognitive dysfunction.
A history of neonatal onset epilepsy, with no other known etiology, persistent motor and cognitive delay, warrants genetic testing for SLC13A5 even in the face of good seizure control and preserved EEG backgrounds later in life. Future prospective studies of children with SLC13A5 citrate transporter deficiency seem feasible due to the high level of family engagement. Focus on improving seizure control should be only one component of future therapeutic trials and research.
Supplemental Material
Supplementary_Table_1 for Epilepsy and EEG Phenotype of SLC13A5 Citrate Transporter Disorder by Qian-Zhou Yang, Emily M. Spelbrink, Kimberly L. Nye, Emily R. Hsu and Brenda E. Porter in Child Neurology Open
Acknowledgments
The authors thank the families who generously participated in this study.
Footnotes
Authors Contribution: Q-ZY and EMS are first authors.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval: The work described is consistent with the Journal’s guidelines for ethical publication.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this study was provided by the TESS Research Foundation. BEP has received funding from Jazz Pharmaceutical, Eisai, and NIH.
ORCID iD: Brenda E. Porter, MD, PhD  https://orcid.org/0000-0001-6346-7327
https://orcid.org/0000-0001-6346-7327
Supplemental Material: Supplemental material for this article is available online.
References
- 1. Thevenon J, Milh M, Feillet F, et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am J Hum Genet. 2014;95(1):113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hardies K, De Kovel CGF, Weckhuysen S, et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain. 2015;138(5):3238–3250. [DOI] [PubMed] [Google Scholar]
- 3. Klotz J, Porter BE, Colas C, et al. Mutations in the Na+/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol Med. 2016;22(2):310–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schossig A, Bloch-Zupan A, Lussi A, et al. SLC13A5 is the second gene associated with Kohlschütter-Tönz syndrome. J Med Genet. 2017;54(3):54–62. [DOI] [PubMed] [Google Scholar]
- 5. Kopel JJ, Bhutia YD, Ramachandran S, et al. Opinion tooth hypoplasia for differential diagnosis of childhood epilepsy associated with SLC13A5 mutations. Int J Neurol Disord [Internet]. 2017;1(3):033–037. [Google Scholar]
- 6. Selch S, Chafai A, Sticht H, et al. Analysis of naturally occurring mutations in the human uptake transporter NaCT important for bone and brain development and energy metabolism. Sci Rep. 2018;8:11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Birkenfeld AL, Lee HY, Guebre-Egziabher F, et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 2011;14(2):184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Willmes DM, Kurzbach A, Henke C, et al. The longevity gene INDY (I’m Not Dead Yet) in metabolic control: potential as pharmacological target. Pharmacol Ther. 2018;185(6):1–11. [DOI] [PubMed] [Google Scholar]
- 9. Bainbridge MN, Cooney E, Miller M, et al. Analyses of SLC13A5-epilepsy patients reveal perturbations of TCA cycle. Mol Genet Metab. 2017;121(4):314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Irizarry AR, Yan G, Zeng Q, et al. Defective enamel and bone development in sodium-dependent citrate transporter (NaCT) Slc13a5 deficient mice. PLoS One. 2017;12:e0175465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhutia YD, Kopel JJ, Lawrence JJ, Neugebauer V, Ganapathy V. Plasma membrane Na+-coupled citrate transporter (SLC13A5) and neonatal epileptic encephalopathy. Molecules. 2017;22(3):378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schachter SC. Review of the mechanisms of action of antiepileptic drugs. CNS Drugs. 1995;4(4):469–477. [Google Scholar]
- 13. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58(1):512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary_Table_1 for Epilepsy and EEG Phenotype of SLC13A5 Citrate Transporter Disorder by Qian-Zhou Yang, Emily M. Spelbrink, Kimberly L. Nye, Emily R. Hsu and Brenda E. Porter in Child Neurology Open