

Abstract
Background
Secondary hemophagocytic lymphohistiocytosis (HLH) is a rare hyperinflammatory syndrome that requires prompt diagnosis and appropriate treatment. A risk-stratification model that could be used to identify high-risk pediatric patients with HLH who should be considered for second-line therapies, including salvage regimens and allogeneic hematopoietic cell transplantation (HCT), was developed.
Methods
The medical records of 88 pediatric patients (median age 1.4 years, range 0.2–15 years) with non-malignancy associated secondary HLH were retrospectively reviewed. Treatment strategies included dexamethasone, etoposide, and cyclosporine.
Results
Survival analysis showed HLH patients with infections other than Epstein-Barr virus (EBV) and unknown causes experienced better 5-year overall survival (OS) than patients with HLH due to autoimmune disease, EBV or immunodeficiency (76% vs. 65, 33.3, 11%, p < 0.001). On multivariate analysis, among all patients, non-response at 8 weeks was the most powerful predictor of poor OS. When treatment response was excluded, hemoglobin < 60 g/L and albumin < 25 g/L at diagnosis were associated with poor OS. In patients with EBV-HLH, hemoglobin < 60 g/L at diagnosis was associated with poor OS. A prognostic risk score was established and weighted based on hazard ratios calculated for three parameters measured at diagnosis: hemoglobin < 60 g/L (2 points), platelets < 30 × 109/L (1 point), albumin < 25 g/L (2 points). Five-year OS of low-risk (score 0–1), intermediate-risk (score 2), and poor-risk (score ≥ 3) patients were 88, 38, and 22%, respectively (p < 0.001).
Conclusions
These findings indicate that clinicians should be aware of predictive factors at diagnosis and consider 8-week treatment response to identify patients with high-risk of disease progression and the need for second-line therapy and allogeneic HCT.
Keywords: Hemophagocytic lymphohistiocytosis; Prognostic factor, risk stratification
Background
Hemophagocytic lymphohistiocytosis (HLH) is a potentially fatal condition of genetic or functional hypercytokinemia that is characterized by uncontrolled proliferation of activated lymphocytes and macrophages. HLH is associated with a wide spectrum of signs and symptoms, including fever, hepatosplenomegaly, pancytopenias, hypertriglyceridemia, hypofibrinogenemia, neurological symptoms and pathological findings of hemophagocytosis in the bone marrow or other organs [1]. HLH is a life-threatening condition. Prompt diagnosis and initiation of treatment with dexamethasone and etoposide are essential [1]. Treatment of HLH can be associated with high morbidity and mortality, including severe sepsis and multi-organ failure in relapsed/refractory patients [1].
There are two major subtypes of HLH, which have been classified as “primary HLH” and “secondary HLH” based on family history, and the identification of a series of genetic variations that are responsible for the decreased functions of T/NK cells, such as PRF1/STXBP2/ITK [2]. Recently, Jordan et al. proposed two concepts to clarify how HLH is diagnosed and treated: within the broader syndrome of HLH, “HLH disease” should be distinguished from “HLH disease mimics” and HLH subtypes should be categorized by specific etiologic associations, not an ambiguous dichotomy of “primary” and “secondary” [3].
Traditionally, secondary HLH is considered a rare hyperinflammatory syndrome that occurs in individuals without a family history or underlying genetic defect. Secondary HLH may be triggered by infections, rheumatologic diseases, malignancy, acquired immune deficiency states, and drugs [4]. Diagnosis and initiation of treatment for patients with secondary HLH is difficult because signs and symptoms overlap with several chronic conditions, including sepsis, multi-organ dysfunction, and progression of malignancies or rheumatic disease [5, 6].
There remains an umet need to increase clinicians’ knowledge of HLH. In particular clinicians must be aware of factors that are predictive of treatment response and survival in patients with HLH. Previous studies showed that indicators such as age, associated infection, cerebrospinal fluid pleocytosis, and family history were not associated with treatment outcomes [7], while hyperbilirubinaemia, hyperferritinaemia, and cerebrospinal fluid pleocytosis (> 100 × 106/L) at diagnosis and thrombocytopenia and hyperferritinaemia 2 weeks after the initiation of therapy were significantly associated with adverse outcomes [8].
The objectives of this retrospective study were to evaluate treatment response and mortality and identify predictive factors for treatment response and survival in children with non-malignancy associated secondary HLH. A risk-stratification model that could be used to identify high-risk pediatric patients with HLH who should be considered for second-line therapies, including salvage regimens and allogeneic HCT, was developed.
Methods
Study subjects
Children diagnosed with HLH between 2005 and 2018 according to HLH-2004 criteria (fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis, ferritin ≥500 μg/L, low/absent NK-cell activity, and soluble CD25 [sIL-2r] [≥2400 U/ml], an affected sibling, and/or a molecular diagnosis based on familial hemophagocytic syndrome (FHL)-causative genes were eligible for his study [4]. The majority of patients satisfied at least 5/6 of these criteria as NK-cell activity and soluble CD25 were not evaluated. Exclusion criteria were 1) diagnostic criteria not met or 2) malignancy-associated HLH (MA-HLH). This research was conducted in accordance with the guidelines of the Ethics Committee of the Affiliated Hospital of Qingdao University. Consent was obtained from a parent or guardian on behalf of any participants under the age of 16 years.
Parameters associated with HLH
The medical records of the included patients were retrospectively reviewed. Pretreatment complete blood cell (CBC) count, levels of fibrinogen and albumin, and prothrombin time (PT) were extracted from serial laboratory analyses.
Clinical outcomes were assessed based on the highest ferritin, aspartate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin, lactate dehydrogenase (LDH), and triglyceride levels recorded during the 4 weeks after treatment initiation.
Epstein-Barr virus (EBV)-association was evaluated by viremia (including low level chronic viremia) and symptomatology of EBV infection, serologic tests (EBV early antigen [EA], EBV nuclear antigen [EBNA], EBV viral capsid antigen [VCA] IgG/IgM) and measuring EBV DNA level using real time polymerase chain reaction (RT-PCR).
Treatments
Treatment strategies were based on the HLH-94/04 protocol, and included dexamethasone, etoposide, and cyclosporine [9, 10]. If complete response was achieved at week 8, patients were provided maintenance therapy, while patients with known familial disease or persistent nonfamilial disease proceeded to continuation therapy.
Treatment response at 4 weeks and 8 weeks after treatment, and dynamic changes during the 8 weeks of treatment were evaluated. Complete response was defined as resolution of all clinical signs and symptoms, CBC recovery, and normalization of abnormal laboratory findings associated with HLH. Partial response was defined as either CBC recovery or normalization of laboratory findings. Progressive disease was defined as persistence of cytopenia and abnormal laboratory findings.
Early stable responders were defined as patients with a complete response at both 4 and 8 weeks after treatment. Late stable responders were defined as patients who failed to achieve complete response at 4 weeks after treatment but showed a continuous response until 8 weeks after treatment. Unstable responders were defined as patients who showed a transient response and eventually progressed to 8 weeks after treatment. Non-responders were defined as patients exhibiting no response (See Supplemental methods for Statistical analysis).
Statistical methods
The clinical characteristics and laboratory test results of children were described statistically. The normal distribution of measurement data is represented as mean ± standard deviation; the non normal distribution is represented by median (minimum maximum); the count data is represented by quantity and percentage. The total survival time was estimated using Kaplan Meier method, defined as the time from HLH diagnosis to death due to any cause. The total survival time of each group was compared by log rank test. Single factor and multivariate Cox analysis were used to evaluate the prognostic factors of HLH. In all the analyses, P < 0.05 was considered as statistically significant. SPSS software 22 was used for all statistical analysis.
Results
Baseline characteristics
The medical records of 88 patients (median age 1.4 years, range 0.2–15 years; mean age 3.5 years) with non-malignancy associated secondary HLH were retrospectively reviewed. All patients were negative on mutation analysis of all FHL-related genes. Causes of HLH were: EBV-associated (n = 24: recent primary infection n = 21, chronic active EBV infection n = 3), unknown cause (n = 25), infection other than EBV (n = 20:cytomegalovirus n = 10, brucella n = 3, Staphylococcus aureus n = 2, Streptococcus pneumoniae n = 2, Mycoplasm n = 1, parvovirus n = 1, herpes simplex n = 1), autoimmune disease (n = 13: systemic lupus erythematosus n = 4, juvenile rheumatoid arthritis n = 5, dermatomyositis n = 1, Kawasaki disease n = 2, anaphylactoid purpura n = 1), or immunodeficiency (n = 6: chronic granuloma n = 4, humoral immunodeficiency n = 1, combined immunodeficiency n = 1). Baseline characteristics and treatment response for the included patients are summarized in Table 1. Fever was observed in all patients and 49 (55.7%) patients had splenomegaly. Peak ferritin levels were reached at a median of 7 days after initiation.
Table 1.
Baseline characteristics and treatment outcomes of included patients (n = 88)
| Characteristic | Value |
|---|---|
| Gender (male), % | 50 (56.8%) |
| Age, years old | 1.4 (0.2–15) |
| Fever | 88 (100%) |
| CMV % | 13 (14.8%) |
| Leukocyte | 3.22 (0.25–36.47) |
| Neutrophil | 1.195 (0.05–11.86) |
| Platelet | 67.00 (7–562) |
| Hemoglobin | 86.5 (31–131) |
| Fibrinogen | 1.29 (0.27–4.56) |
| Albumin | 29.27 ± 5.61 |
| Ferritin | 3564.5 (132.0–591,123.0) |
| AST | 249.15 (21.0–9420.0) |
| ALT | 207.50 (8.0–4953.0) |
| LDH | 944.5 (156.0–5311.0) |
| TG | 3.34 (0.39–13.56) |
| CRP | 15.05 (0.25–161.54) |
| GGT | 207.5 (7.0–1550.0) |
| D-dimer | 2489.0 (129.0–35,600.0) |
| DIC % | 28 (31.8%) |
| Splenomegaly % | 49 (55.7%) |
| Jaundice % | 34 (38.6%) |
| Bone marrow involvement % | 51 (58.0%) |
| Hepatomegaly % | 65 (73.9%) |
| Ascites % | 21 (23.9%) |
|
Lymphadenopathy % CR achievement after treatments |
28 (31.8%) 58 (65.9%) |
| Therapy | |
| HLH94/2004based therapy | 34 (38.6%) |
| Steroid + IVIG | 8 (9.1%) |
| Steroid | 22 (25%) |
| Only observation | 23 (26.1%) |
| Hematopoietic stem cell transplantation | 1 (1.1%) |
| Causes of HLH | 56 (76.7%) |
| EBV-associated | 30 (33.7%) |
| Immunodeficiency | 6 (6.7%) |
| Autoimmune disease (AID) | 13 (14.6%) |
| Unknown cause and infection other than EBV | 40 (44.9%) |
| Treatment response at 8 weeks | |
| Early stable responder | 29 (33.3%) |
| Late stable responder | 28 (32.2%) |
| Unstable responder | 17 (19.5%) |
| Non-responder | 13 (14.9%) |
Abbreviations: HLH hemophagocytic lymphohistiocytosis, AST aspartate aminotransferase, ALT alanine transferase, LDH lactate dehydrogenase, CRP C-reactive protein, DIC disseminated intravascular coagulation, EBV Epstein-Barr virus, CR complete remission, EBV Epstein-Barr virus, TG triglyceride, GGT gamma-glutamyl transpeptidase
Treatment outcomes
A total of 83/88 (94.3%) patients were treated. 65/88 (73.8%) (unknown cause and infection other than EBV n = 45, EBV infection n = 24, autoimmune disease n = 13, immunodeficiency n = 6) patients were administered therapy according to the HLH94 protocol. 5/88 (5.6%) patients were not treated. Of these, 3 patients had no signs of HLH activity and 2 patients died. 12/88 (13.6%) patients were administered dexamethasone alone. 5/88 (5.6%) patients were administered dexamethasone plus gamma globulin. 1/88 (1.1%) patient underwent HCT (Fig. 1). The frequency and dose of etoposide was not reduced for patients with substantially altered organ function or who were in a generally poor condition.
Fig. 1.
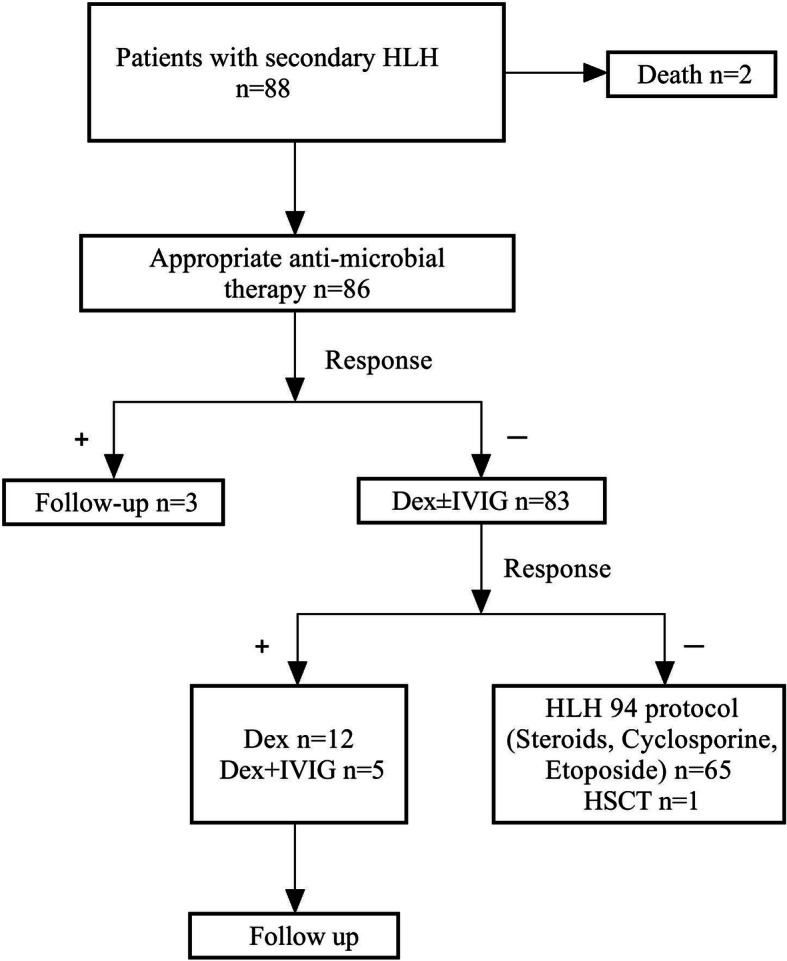
Flow-chart showing the treatment pathway
57/88 (64.7%) patients achieved a complete response; of these, 30 patients achieved complete response at 4 weeks after treatment and 27 achieved complete response at 8 weeks or later. Three patients relapsed; of these, 1 patient relapsed 8 weeks after treatment, 1 patient relapsed 4 months after treatment, and 1 patient relapsed 12 months after treatment. Relapsed patients had EBV-HLH, and were treated with rituximab (n = 1), alemtuzumab (n = 1), or allogeneic-HCT (n = 1) according to the HLH-94 protocol. Regarding dynamic response, 32/88 (36.4%) patients were early stable responders, 28/88 (31.8%) were late stable responders, 14/88 (15.9%) were unstable responders, and 13/88 (14.7%) were primary refractory non-responders.
Survival outcomes according to treatment response
The overall mortality for all patients was 35.2% (31/88). 5-year OS rates for patients with an unknown cause and infections other than EBV due to autoimmune disease were 76.0 and 65%, respectively, and higher than in patients with HLH from other causes (Fig. 2a). Among all patients, 5-year OS rates for patients who achieved complete response at 4 weeks (n = 30) and 8 weeks (n = 57) were each 100%. 5-year OS rates for patients who achieved partial response at 4 weeks (n = 38) and 8 weeks (n = 14) were 86 and 44.0%, respectively (Fig. 2b-c). 5-year OS rates for early stable responders and late stable responders were each 100% (Fig. 2d).
Fig. 2.

Survival outcomes according to the cause of secondary HLH and treatment response. a The cause of secondary HLH. b Treatment response at 4 weeks. c Treatment response at 8 weeks. d Dynamic treatment responses at 8 weeks
Prognostic factors and risk stratification
On univariate survival analysis, hemoglobin level, PLT, and albumin level at diagnosis, and dynamic treatment response were significantly correlated with survival outcomes. On multivariate analysis, non- response at 8 weeks was the most powerful predictor of poor OS. When treatment response was excluded, hemoglobin< 60 g/L and albumin< 25 g/L at diagnosis were associated with poor OS (Table 2). In patients with EBV-HLH, hemoglobin < 60 g/L at diagnosis was associated with poor OS.
Table 2.
Univariate and multivariate analysis of parameters affecting OS in non-malignancy associated HLH
| Univariate analysis | Multivariate analysis | |||||
|---|---|---|---|---|---|---|
| Treatment response included | Treatment response excluded | |||||
| 5-year OS | P | HR(95%CI) | P | HR(95%CI) | P | |
| Age < 1 years old | 80.0% VS (69.0%) | 0.425 | ||||
| EBV-associated | 79.0% VS (67.0%) | 0.262 | ||||
| Hemoglobin < 60 g/L | 25.0% VS (77.0%) | < 0.001 | 3.22 (2.01–19.28) | 0.002 | ||
| Platelet < 30 × 10^9/L | 36.0% VS (79.0%) | < 0.001 | 2.15 (0.88–5.24) | 0.093 | ||
| ALT > 200 U/L | 73.0% VS (71.0%) | 0.935 | ||||
| TG > 2.5 mg/dl | 76.0% VS (62.0%) | 0.173 | ||||
| Fibrinogen < 1.5 | 66.0% VS (80.0%) | 0.170 | ||||
| Ferritin > 10,000 | 75.0% VS (72.0%) | 0.744 | ||||
| Albumin < 25 g/L | 33.0% VS (82.0%) | < 0.001 | 7.71 (3.10–19.19) | < 0.001 | ||
| Dynamic treatment response | 0.010 | 0.010 | ||||
| Early stable responder | 100% | 0.00 | ||||
| Late stable responder | 100% | 0.00 | ||||
| Unstable responder | 36% | 0.21 (0.08–0.52) | ||||
| Non-responder | 0% | 1.0 | ||||
Abbreviations: OS overall survival, HLH hemophagocytic lymphohistiocytosis, HR hazard ratio, EBV Epstein-Barr virus, ALT alanine transferase, TG triglyceride
A prognostic risk score was established and weighted based on hazard ratios calculated for three parameters measured at diagnosis: hemoglobin < 60 g/L (2 points), platelets < 30 × 109/L (1 point), albumin < 25 g/L (2 points). Five-year OS of low-risk (score 0–1), intermediate-risk (score 2), and poor-risk (score ≥ 3) patients were 88, 38, and 22%, respectively (Fig. 3).
Fig. 3.
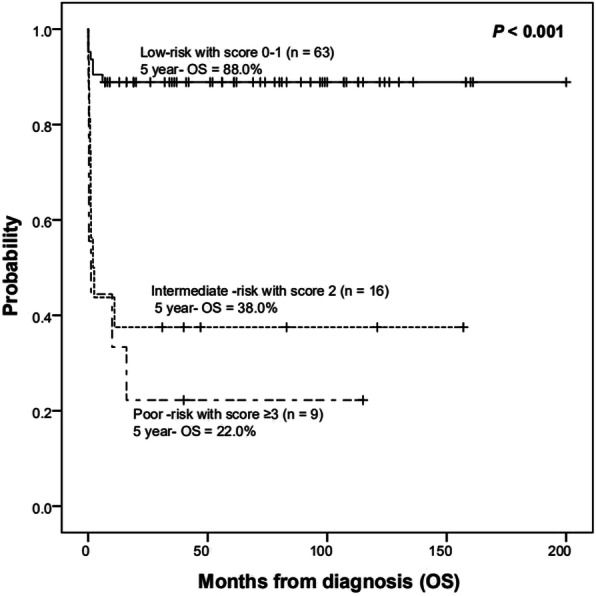
Survival outcomes according to HLH risk-score (low-risk [score 0–1], intermediate-risk [score 2], and high-risk [score ≥ 3])
Discussion
This study analyzed clinical symptoms, treatment response and prognostic factors in pediatric patients with secondary HLH after excluding MA-HLH. Consistent with a previous report [11], hepatomegaly (73.9%) was more prevalent than splenomegaly (55.7%) in our patient population, although hepatomegaly was not part of the diagnostic criteria included in the HLH-04 clinical trial.
EBV infection is a common cause of HLH. Evidence suggests that the presence of VCA IgG and VCA IgM in the absence of EBNA IgG is indicative of an acute EBV infection and the presence of VCA IgG and EBNA IgG in the absence of VCA IgM shows a previous infection [12, 13]. In the present study, evaluation of EBV-specific antibodies (EBV VCA IgM, IgG and EBNA-1 IgG) and serous EB DNA virus load (> 105/copies) was used to confirm an EBV diagnosis. In our pediatric population, patients with EBV-HLH showed a worse treatment response and progressive OS (33.3%) than patients with secondary HLH due to other causes, except immunodeficiency, and patients with HLH caused by EBV infection had a very poor prognosis. Consistent with this, a study in adult patients with HLH reported that EBV was associated with poor survival outcomes, and the overall response to treatment at 4 weeks was similar in patients with HLH caused by EBV, autoimmune disease, other infections, or unknown causes, but decreased in the EBV group at 8 weeks [14]. Similarly, survival in pediatric patients with EBV-induced secondary HLH/macrophage activation syndrome (MAS) associated with rheumatic and nonrheumatic conditions was 50% [15].
Etoposide is very important for the treatment of HLH in patients who do not achieve remission with dexamethasone and gamma globulin. Etoposide, a chemotherapeutic agent, has high specificity against T-cell proliferation and cytokine secretion in mice [16]. Abnormal liver function in children with hemophagocytic syndrome is a cytokine storm syndrome. Therefore, the frequency and dose of etoposide was not reduced in patients with substantially altered organ function or who were in a generally poor condition, and liver function returned to normal with chemotherapy and remission.
Urgent preparation for allogeneic HCT should be considered for high-risk patients at HLH diagnosis [17]. In the present study, prognosis was very poor in patients with HLH caused by autoimmune diseases and immunodeficiency. HLH that results from rheumatic or other systemic diseases, including Still’s disease and sarcoidosis, is termed MAS [18–20]. In our cohort, 13 patients had HLH caused by autoimmune disease, including systemic lupus erythematosus, juvenile rheumatoid arthritis, dermatomyositis, Kawasaki disease, and anaphylactoid purpura, and the survival rate was 65%. In a previous study, 100% (n = 13) of pediatric patients with secondary HLH/MAS and underlying systemic juvenile idiopathic arthritis survived after treatment with a recombinant human interleukin-1 receptor antagonist [15].
There were 6 patients with HLH caused by immunodeficiency, including chronic granuloma, X-linked lymphocyte proliferative diseases (XLP), and humoral immune deficiency. In addition to abnormalities in cytotoxic granules and lysosomes, various primary immune deficiency disorders (PID) other than familial hemophagocytic lymphohistiocytosis (FHL) or XLP disorders have been identified among patients suffering from HLH [21]. In one report, patients with primary immunodeficiencies other than cytotoxicity defects or XLP disorders presenting with conditions fulfilling current criteria for HLH had chronic granulomatous disease with hemophagocytic episodes mainly associated with bacterial infections. The authors proposed that HLH syndrome in chronic granulomatous disease not only reflects an impaired response to infection, but also a genetic predisposition to the inflammatory reaction [22].
In the present study, 5-year OS rates for early stable responders and late stable responders were each 100%, indicating that patients can experience long-term survival if they fail to achieve a complete response at 4 weeks after treatment but exhibit a continuous and stable response until 8 weeks. The 5-year OS rate for patients who achieved partial response at 8 weeks was low at 44.0%. On multivariate analysis, non- response at 8 weeks was the most powerful predictor of poor OS. When treatment response was excluded, hemoglobin < 60 g/L and albumin < 25 g/L at diagnosis were associated with poor OS. In previous reports in adult patients with HLH, one study associated hypofibrinogenemia (≤150 mg/dl), fibrinogen ≤200 mg/dl (P = 0.04), and prothrombin time > 50% with increased mortality [23], while another showed that patients with disseminated intravascular coagulation, nosocomial infections and neurological symptoms had a statistically significant worse survival [23]. In pediatric patients with secondary HLH/MAS, thrombocytopenia was a predictor of mortality [15].
HLH is characterized by a hyperinflammatory phenotype [24, 25]. As clinical criteria for HLH include non-specific findings that overlap with other diseases, HLH may be an obsolete term that is better replace by hyperinflammatory syndrome and/or hypercytokinemia [26]. The HLH-94 treatment protocol includes 8 weeks of initial therapy that aims to achieve clinical remission, followed by continuation therapy that aims to keep patients alive and stable until an acceptable bone marrow transplantation donor becomes available [4]. In the present study, 12 (13.6%) patients received dexamethasone alone and 3 (3.4%) patients did not receive any treatment; these patients had a survival rate > 90%. These findings suggest treatment can be started before definite diagnosis in severe cases with high suspicion of HLH and progressive disease. Patients should be treated with dexamethasone. Clinical decision making relevant to etoposide administration should consider disease progression, treatment effect and diagnostic criteria, which will direct treatment duration and inform prognosis. In the present study, pediatric patients with HLH who did not achieve disease control by week 8 were considered at high risk of adverse outcomes.
HLH is a rare condition, and rigorously conducted prospective studies are lacking. There remains an unmet need to identify prognostic factors for secondary HLH, as few data are available for risk-stratification. This study identified clinical symptoms, treatment response and prognostic factors in children with non-malignancy associated secondary HLH, but it was associated with several limitations. First, it was a retrospective study; despite this, medical records showed that patients were consistently treated and managed, and the sample size was adequate. Second, the risk-stratification model should be validated by larger studies. Third, there was significant heterogeneity in this cohort, in terms of attributed causes but also disease severity, since a number of patients were not treated. However, exclusion of cases of MA-HLH ensured that treatment response to the protocols and survival outcomes were applicable to risk- stratification.
Conclusions
Findings from this study suggest that clinicians should be aware of predictive factors at diagnosis, including hemoglobin < 60 g/L and albumin < 25 g/L, and consider treatment response at 8-weeks as a criterion to identify patients with non-malignancy associated secondary HLH who are at high-risk of disease progression and in need of second-line therapy and allogeneic HCT.
Acknowledgements
We thank Professor Yili WU for providing the medical statistics of the data.
Abbreviations
- HLH
Hemophagocytic lymphohistiocytosis
- ALL
Acute lymphoblastic leukemia
- AML
Acute myeloblastic leukemia
- IVIG
Intravenous Immunoglobulin
- Allo HSCT
Allogeneic Hematopoietic stem cell transplantation
- OS
Overall survival
- MA-HLH
Malignancy-associated HLH
- CBC
Complete blood cell
- PT
Prothrombin time
- AST
Aspartate aminotransferase
- ALT
Alanine aminotransferase
- LDH
Lactate dehydrogenase
- MAS
Macrophage activation syndrome
Authors’ contributions
HP conceptualized and designed the study, and reviewed and revised the manuscript. GYWand EBG drafted the initial manuscript. LS and AQS collected data. XDL, ZY and LRS was responsible for the follow-up. All authors have read and approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Availability of data and materials
Data are available from the authors upon reasonable request.
Ethics approval and consent to participate
The study was performed with the approval of the Ethics Committee of the Affiliated University of Qingdao University (no: 20190113). The protocol was designed and the study was conducted according to the principles of the Declaration of Helsinki (1964). Written informed consent was obtained from all participants. Consent was obtained from a parent or guardian on behalf of any participants under the age of 16 years.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Janka GE, Lehmberg K. Hemophagocytic syndromes. Blood Rev. 2014;28(4):135–142. doi: 10.1016/j.blre.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Janka GE. Familial and acquired Hemophagocytic Lymphohistiocytosis. Annu Rev Med. 2012;63:233–246. doi: 10.1146/annurev-med-041610-134208. [DOI] [PubMed] [Google Scholar]
- 3.Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the north American consortium for Histiocytosis (NACHO) Pediatr Blood Cancer. 2019;66(11):e27929. doi: 10.1002/pbc.27929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kleynberg RL, Schiller GJ. Secondary hemophagocytic lymphohistiocytosis in adults: an update on diagnosis and therapy. Clin Adv Hematol Oncol. 2012;10(11):726–732. [PubMed] [Google Scholar]
- 5.Apodaca E, Rodríguez-Rodríguez S, Tuna-Aguilar EJ, et al. Prognostic factors and outcomes in adults with secondary Hemophagocytic Lymphohistiocytosis: a single-center experience. Clin Lymphoma Myeloma Leuk. 2018;18(10):e373–e380. doi: 10.1016/j.clml.2018.06.014. [DOI] [PubMed] [Google Scholar]
- 6.Ho Yoon J, Soo Park S, Woo Jeon Y, et al. Treatment outcomes and prognostic factors of adult patients with non-malignancy associated secondary hemophagocytic lymphohistiocytosis. Haematologica. 2019;14(2):269–276. doi: 10.3324/haematol.2018.198655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aricò M, Janka G, Fischer A, et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996;10(2):197–203. [PubMed] [Google Scholar]
- 8.Trottestam H, Berglöf E, Horne A, et al. Risk factors for early death in children with haemophagocytic lymphohistiocytosis. Acta Paediatr. 2012;101(3):313–318. doi: 10.1111/j.1651-2227.2011.02501.x. [DOI] [PubMed] [Google Scholar]
- 9.Henter JI, Samuelsson-Horne A, Arico M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367–2373. doi: 10.1182/blood-2002-01-0172. [DOI] [PubMed] [Google Scholar]
- 10.Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018;6(5):1508–1517. doi: 10.1016/j.jaip.2018.05.031. [DOI] [PubMed] [Google Scholar]
- 11.Minoia F, Davì S, Horne AC, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol. 2014;66(11):3160–3169. doi: 10.1002/art.38802. [DOI] [PubMed] [Google Scholar]
- 12.Topp SK, Rosenfeldt V, Vestergaard H, et al. Clinical characteristics and laboratory findings in Danish children hospitalized with primary Epstein-Barr virus infection. Infect Dis (Lond) 2015;47(12):908–914. doi: 10.3109/23744235.2015.1082036. [DOI] [PubMed] [Google Scholar]
- 13.De Paschale M, Clerici P. Serological diagnosis of Epstein-Barr virus infection: problems and solutions. World J Virol. 2012;1(1):31–43. doi: 10.5501/wjv.v1.i1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoon JH, Park SS, Jeon YW, et al. Treatment outcomes and prognostic factors in adult patients with secondary hemophagocytic lymphohistiocytosis not associated with malignancy. Haematologica. 2019;104(2):269–276. doi: 10.3324/haematol.2018.198655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esraa M, Eloseily EM, Weiser P, et al. Benefit of Anakinra in treating pediatric secondary Hemophagocytic Lymphohistiocytosis. Arthritis Rheumatol. 2020;72(2):326–334. doi: 10.1002/art.41103. [DOI] [PubMed] [Google Scholar]
- 16.Johnson TS, Terrell CE, Millen SH, et al. Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. J Immunol. 2014;192(1):84–91. doi: 10.4049/jimmunol.1302282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Machaczka M, Nahi H, Karbach H, et al. Successful treatment of recurrent malignancy-associated hemophagocytic lymphohistiocytosis with a modified HLH-94 immunochemotherapy and allogeneic stem cell transplantation. Med Oncol. 2012;29(2):1231–1236. doi: 10.1007/s12032-011-9963-3. [DOI] [PubMed] [Google Scholar]
- 18.Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic,andneurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr. 1985;106(4):561–566. doi: 10.1016/s0022-3476(85)80072-x. [DOI] [PubMed] [Google Scholar]
- 19.Ravelli A, Davi S, Minoia F, et al. Macrophage activation syndrome. Hematol Oncol Clin North Am. 2015;29(5):927–941. doi: 10.1016/j.hoc.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 20.Abughanimeh O, Qasrawi A, Abu Ghanimeh M. Hemophagocytic Lymphohistiocytosis complicating systemic Sarcoidosis. Cureus. 2018;10(6):e2838. doi: 10.7759/cureus.2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faitelson Y, Grunebaum E. Hemophagocytic lymphohistiocytosis and primary immune deficiency disorders. Clin Immunol. 2014;155(1):118–125. doi: 10.1016/j.clim.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 22.Bode SF, Ammann S, Al-Herz W, et al. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies:implications for differential diagnosis and pathogenesis. Haematologica. 2015;100(7):978–988. doi: 10.3324/haematol.2014.121608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valade S, Azoulay E, Galicier L. Coagulation Disorders and Bleedings in Critically Ill Patients With Hemophagocytic Lymphohistiocytosis. Medicine (Baltimore) 2015;94(40):e1692. doi: 10.1097/MD.0000000000001692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otrock ZK, Eby CS. Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol. 2015;90(3):220–224. doi: 10.1002/ajh.23911. [DOI] [PubMed] [Google Scholar]
- 25.Teachey DT, Rheingold SR, Maude SL, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121(26):5154–5157. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otrock ZK, Daver N, Kantarjian HM, et al. Diagnostic challenges of Hemophagocytic Lymphohistiocytosis. Clin Lymphoma Myeloma Leuk. 2017;17S:S105–S110. doi: 10.1016/j.clml.2017.02.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available from the authors upon reasonable request.