

Abstract
Lrp6 is generally described as a receptor required for signal transduction in the Wnt/β-catenin pathway. Wnt5a, however, is a Wnt ligand that usually does not activate Wnt/β-catenin but rather activates noncanonical Wnt signaling. We have previously shown that Lrp6 can inhibit noncanonical Wnt5a/Wnt11 signaling and that Lrp5/6 loss-of-function produces noncanonical gain-of function defects, which can be rescued by loss of Wnt5a. Here, we describe other phenotypes found in Wnt5a/Lrp6 compound mutant mice, including a worsening of individual Wnt5a or Lrp6 loss of function phenotypes. Lrp6 haploinsufficiency in a Wnt5a−/− background caused spina bifida and exacerbated posterior truncation. Wnt5a−/−Lrp6−/− embryos displayed presomitic mesoderm morphogenesis, somitogenesis, and neurogenesis defects, which are much more severe than in either of the single mutants. Interestingly these results reveal a further level of complexity in processes in which Wnt5a and LRP6 cooperate, or oppose each other, during mouse development.
Keywords: Wnt5a, Lrp6, somitogenesis, development
INTRODUCTION
Wnts are a family of secreted glycoproteins that regulate several processes during embryonic development including proliferation, migration, cell polarity, and differentiation (Logan and Nusse, 2004; Montcouquiol et al., 2006). Signaling components have been classically divided into canonical and noncanonical pathway components depending on their ability to transform C57MG mammary cells (Wong et al., 1994) and induce axis duplication in Xenopus (Sokol et al., 1991). By these standards, and based on their effect (or lack thereof) on β-catenin stabilization, Wnt5a has been classified as a noncanonical Wnt (Moon et al., 1993; Wong et al., 1994; Torres et al., 1996), while the low-density lipoprotein-related receptor (Lrp)6 has been classified as a canonical Wnt co-receptor (for review, see He et al., 2004). As such, Wnt5a and Lrp6 were not previously thought to interact. However, recent reports have shown that Lrp6 can affect typical noncanonical processes, and modulate Wnt5a-regulated processes in both Xenopus and mouse (Tahinci et al., 2007; Bryja et al., 2009).
We have previously reported the derivation and characterization of mice harboring simultaneous mutations in both Wnt5a (Yamaguchi et al., 1999a; Andersson et al., 2008) and Lrp6 (Pinson et al., 2000; Bryja et al., 2009). Analysis of these compound mutant mice showed that loss of Wnt5a rescues exencephaly, a typical planar cell polarity (PCP)/convergent extension (CE) defect, as well as heart defects, otherwise seen in Lrp6−/− embryos, while Wnt5a−/−Lrp6−/− mutants are severely developmentally delayed (Bryja et al., 2009). These previous results hinted at a dose-dependent interaction between Wnt5a and Lrp6, but also indicated that Wnt5a and Lrp6 may cooperate in some developmental processes/tissues, while acting in opposing fashions in others (such as PCP/CE).
Here, we describe additional phenotypes that demonstrate a genetic interaction between Wnt5a and Lrp6, including spina bifida, posterior truncation, presomitic mesoderm morpho-genesis, somitogenesis, and neurogenesis defects, all of which confirm the crucial importance of Lrp6 and Wnt5a in the development of these structures. Furthermore, these results reveal further processes in which Wnt5a and Lrp6 cooperate or oppose each other during mouse development.
RESULTS
General Phenotype of Wnt5a/Lrp6 Compound Mutants
We have previously shown that Wnt5a+/−Lrp6+/− animals are born with the expected frequency and do not show any obvious morphological, fertility, or behavioral defects in comparison with parental Wnt5a or Lrp6 heterozygotes. Mating of Wnt5a+/−Lrp6+/− mice generated Wnt5a−/−Lrp6−/− embryos with the expected frequency at E10.5 (Bryja et al., 2009). All Wnt5a−/−Lrp6−/− embryos were severely developmentally delayed at E10.5, and we did not obtain any Wnt5a−/−Lrp6−/− embryos at E12.5. A general morphological assessment of all studied genotypes is shown in Figure 1. Wnt5a−/−Lrp6+/+ and Wnt5a−/−Lrp6+/− embryos are indistinguishable at this stage and display a phenotype characterized by limb defects and a shorter anteroposterior (AP) body axis, which can be clearly diagnosed in the tail region (here referred to as “Wnt5a phenotype”). In contrast, Wnt5a+/+Lrp6−/− embryos are more variable in their phenotype. Typically they display a compound phenotype characterized at this stage by slightly delayed development (embryos are smaller), a less well-defined isthmus, and defects in tail development, often manifesting in the form of a kinked tail (here referred to as “Lrp6 phenotype”). Exencephaly is observed in approximately 30% of Lrp6−/−Wnt5a+/+ embryos (yellow arrow in Fig. 1A, and brain section in Fig. 1B). Wnt5a+/−Lrp6−/− embryos are usually similar in morphology to Wnt5a+/+Lrp6−/− embryos, but in approximately 40% of Wnt5a+/−Lrp6−/− embryos we observed a severe developmental delay and defects in heart morphogenesis (see Fig. 1A, embryo #2 for this phenotype). As we have shown previously (Bryja et al., 2009), the incidence of exencephaly is less than 10% in Wnt5a+/−Lrp6−/− embryos. Wnt5a−/−Lrp6−/− embryos show a severe, although to some extent variable, phenotype which is characterized by general developmental delay (with a size less than 40% of wild-type littermates) and a lack of embryonic turning (red arrowhead in Fig. 1A). Furthermore, heart morphogenesis is disrupted, and embryos manifest a grossly enlarged pericardium (white arrowhead in Fig. 1A), as well as a tubular, unlooped heart (yellow arrowhead in Fig. 1A). All these phenotypes are highly penetrant and appear in almost 100% of embryos (Fig. 1C).
Fig. 1.
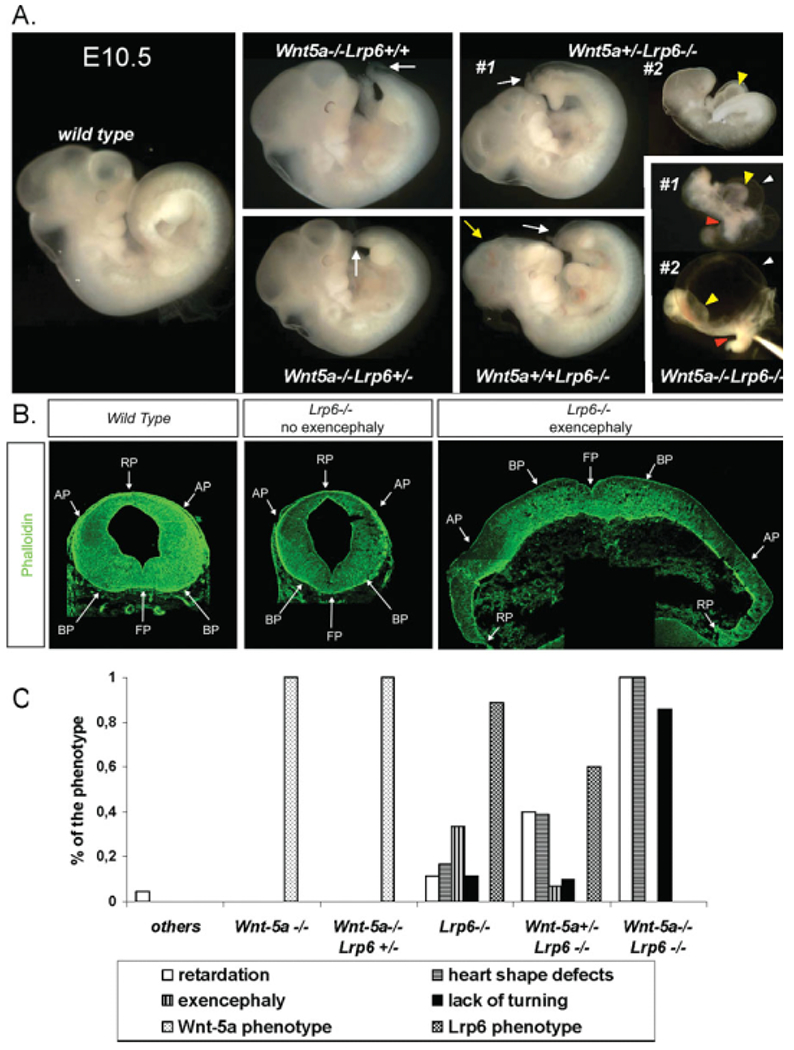
General developmental phenotypes of Wnt5a and Lrp6 single and double mutant mice at E10.5. A: General morphology of Wnt5a/Lrp6 compound mutants at E10.5. Arrows indicate the shortened tail in Wnt5a−/−Lrp6+/+ embryo and kinked tail and exencephaly in a Lrp6−/− embryo. Two specimens of Wnt5a+/−Lrp6−/− and Wnt5a−/−Lrp6−/− embryos are shown to demonstrate the variability of the phenotype. Yellow arrowheads indicate defective heart tube, white arrowheads indicate enlarged pericardium, and red arrowheads indicate defects in embryo turning. B: Normal and exencephalic midbrains of embryonic day (E) 12.5 wild-type (WT) and Lrp6−/− embryos were sectioned and stained with phalloidin to visualize morphogenetic changes, including exencephaly in the midbrain region. C: Quantification of the observed phenotype is based on 4 Wnt5a−/−, 12 Wnt5a−/−Lrp6+/−, 9 Lrp6−/−, 30 Wnt5a+/−Lrp6−/−, and 7 Wnt5a−/−Lrp6−/− embryos at E10.5. See text for details.
Later during development at E14.5, Wnt5a−/− mice show a complex phenotype characterized by craniofacial malformations, lack of digits in the limbs, shortening of the AP axis, and edema (Fig. 2A; Yamaguchi et al., 1999a; Yang et al., 2009). Lrp6−/− embryos at embryonic day (E) 14.5 display a complete penetrance in eye developmental defects, posterior truncation, spina bifida, and in defects in the limbs, which exhibit a reduced number of digits (Fig. 2A; Pinson et al., 2000; Adamska et al., 2005; Zhou et al., 2008) Wnt5a−/−Lrp6+/− embryos resemble Wnt5−/−Lrp6+/+ embryos.
Fig. 2.
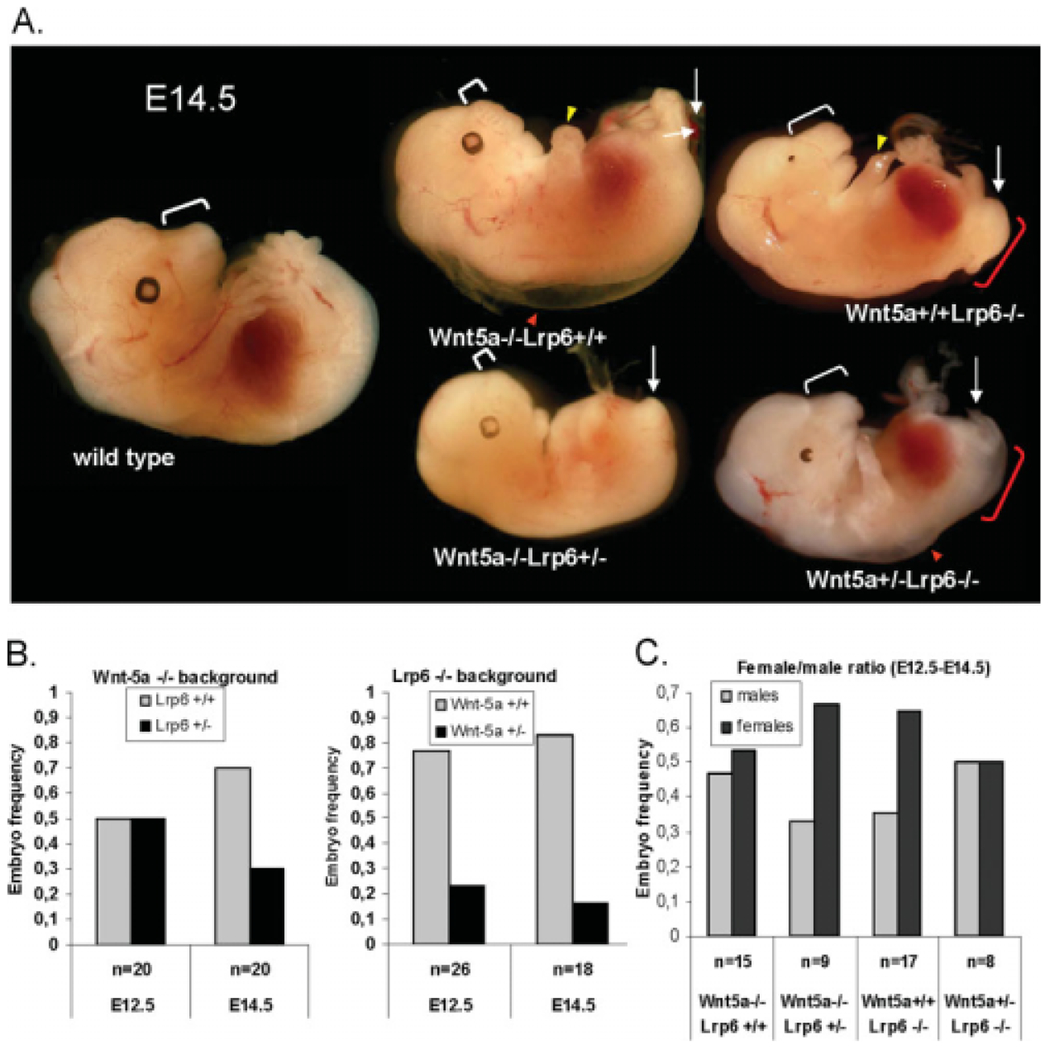
Impact of loss of Wnt5a or Lrp6 on phenotype at embryonic day (E) 14.5, survival and gender selection. A: General morphology of Wnt5a/Lrp6 compound mutants at E14.5. Yellow arrowheads indicate limb deformities, red arrowheads indicate edema, and white arrows indicate defects in the posterior development. White brackets indicate snout length and shortening in Wnt5a−/− embryos, and red bracket indicates spina bifida. B: Survival analysis of Wnt5a−/−Lrp6+/+ and Wnt5a−/−Lrp6+/− embryos (first graph), and Wnt5a+/+Lrp6−/−, and Wnt5a+/−Lrp6−/− embryos (second graph) at E12.5 and E14.5. Graphs indicate proportion of embryos with the indicated genotype within all Wnt5a−/− or Lrp6−/− embryos. n indicates number of embryos in the set. C: Analysis of female/male ratio as indicated by polymerase chain reaction (PCR) of embryos between E12.5 and E14.5. n indicates number of embryos in the set.
Haploinsufficiency for Lrp6 did not affect the edema, craniofacial or limb defects seen in Wnt5a−/− mice (data not shown and Fig. 2A), but a slight reduction in size was observed in some Wnt5a−/−Lrp6+/− mice when compared with littermate Wnt5a−/−Lrp6+/+ embryos (Fig. 2A). Importantly, Wnt5a−/−Lrp6+/+ and Wnt5a−/−Lrp6+/− embryos were recovered at the expected equal frequencies at E12.5, whereas at E14.5, we obtained fewer Wnt5a−/−Lrp6+/− embryos than Wnt5a−/−Lrp6+/+ embryos, indicating increased mortality of Wnt5a−/−Lrp6+/− embryos between E12.5 and E14.5 (Fig. 2B). Haploinsufficiency for Wnt5a in the Lrp6−/ − background negatively affected embryo survival at both E12.5 and E14.5 (Fig. 2B). The decreased proportion of Wnt5a+/−Lrp6−/− embryos, observed as early as E12.5, may reflect the death of the group of Wnt5a+/−Lrp6−/− embryos that were severely developmentally delayed, with heart defects, seen at E10.5 (Fig. 1B). Wnt5a+/−Lrp6−/− embryos, which survived to E14.5, were often anemic and smaller than Wnt5a+/+Lrp6−/− embryos. However, approximately 60% of these embryos showed the same morphology as Wnt5a+/+Lrp6−/− embryos (Fig. 2A) and we did not observe any amelioration/aggravation of the eye phenotype, digit number, or spina bifida, which was present with almost 100% penetrance both in Wnt5a+/−Lrp6−/− and Wnt5a+/+Lrp6−/− embryos.
It has previously been shown that loss of certain Wnt signaling components, such as Dvl2, affects survival in a gender-specific manner, such that more females are born (Hamblet et al., 2002), whereas loss of Wnt4 revealed its role in gender development (Vainio et al., 1999). We have previously shown that Wnt5a treatment leads to the phosphorylation and mobility-shift of Dvl2 (Schulte et al., 2005; Bryja et al., 2007b), whereas other studies have shown that Dvl2 is involved in the phosphorylation of Lrp6 (Bilic et al., 2007). Because many of the phenotypes observed in the Wnt5a and Lrp6 single or compound mutant mice, such as heart outflow tract defects or exencephaly (Schleiffarth et al., 2007; Bryja et al., 2009), are reminiscent of Dvl2 mutant mouse phenotypes (Hamblet et al., 2002), we asked whether the proportion of male and female mice was affected by a genetic interaction between Wnt5a and Lrp6. Gender-specific polymerase chain reaction (PCR), using SMCX-1 and SMC4-1 primers, revealed that 53% of Wnt5a−/−Lrp6+/+ embryos recovered between E12.5 and E14.5 were female, whereas 63% of Wnt5a−/−Lrp6+/− embryos were female (Fig. 2C). Conversely, 67% of recovered Wnt5a+/+Lrp6−/− embryos at E12.5 to E14.5 were female, which was rescued to 50% in Wnt5a+/−Lrp6−/− mice. Although these differences were not statistically significant, the tendency for loss of Lrp6 to bias toward a female fate and the opposing effect of Wnt5a and Lrp6 on gender survival, indicate yet another level at which Wnt5a and Lrp6 may oppose one another.
Lrp6 Haploinsufficiency Aggravates Anteroposterior Defects Seen in Wnt5a−/− Mice
We next analyzed possible reasons for the decreased survival of Wnt5a−/−Lrp6+/− embryos between E12.5 and E14.5, and compared defects between Wnt5a−/−Lrp6+/− and Wnt5a−/−Lrp6+/+ embryos at E12.5. Wnt5a is required for the outgrowth of several structures including the snout, ears, genitals, and limbs, and loss of Wnt5a also causes AP elongation defects (Yamaguchi et al., 1999a; Andersson et al., 2008). At E12.5, 80% of Wnt5a−/− embryos display moderate AP defects (Fig. 3A, subpanel a, a’) with symmetrical hindlimbs (yellow arrowheads) and a rudimentary tail (black arrowhead). The remaining 20% display slightly more severe defects, with asymmetrical hindlimbs (Fig. 3A, subpanel b, b′). Interestingly ablation of a single allele of Lrp6 on a Wnt5a−/− background greatly affected the severity of these caudal defects, and thus no Wnt5a−/−Lrp6+/− embryos displayed mild defects. Instead, the majority of Wnt5a−/−Lrp6+/− embryos (60%) displayed intermediate defects (Fig. 3A, subpanel 2, 2′), and the remaining Wnt5a−/−Lrp6+/− embryos displayed a much more severely affected caudal region, with rudimentary asymmetrical hindlimbs (30%, Fig. 3A, subpanel c, c′) or a complete lack of caudal structures such as a tail rudiment or hindlimbs (10%, Fig. 3A, subpanel d, d′).
Fig. 3.
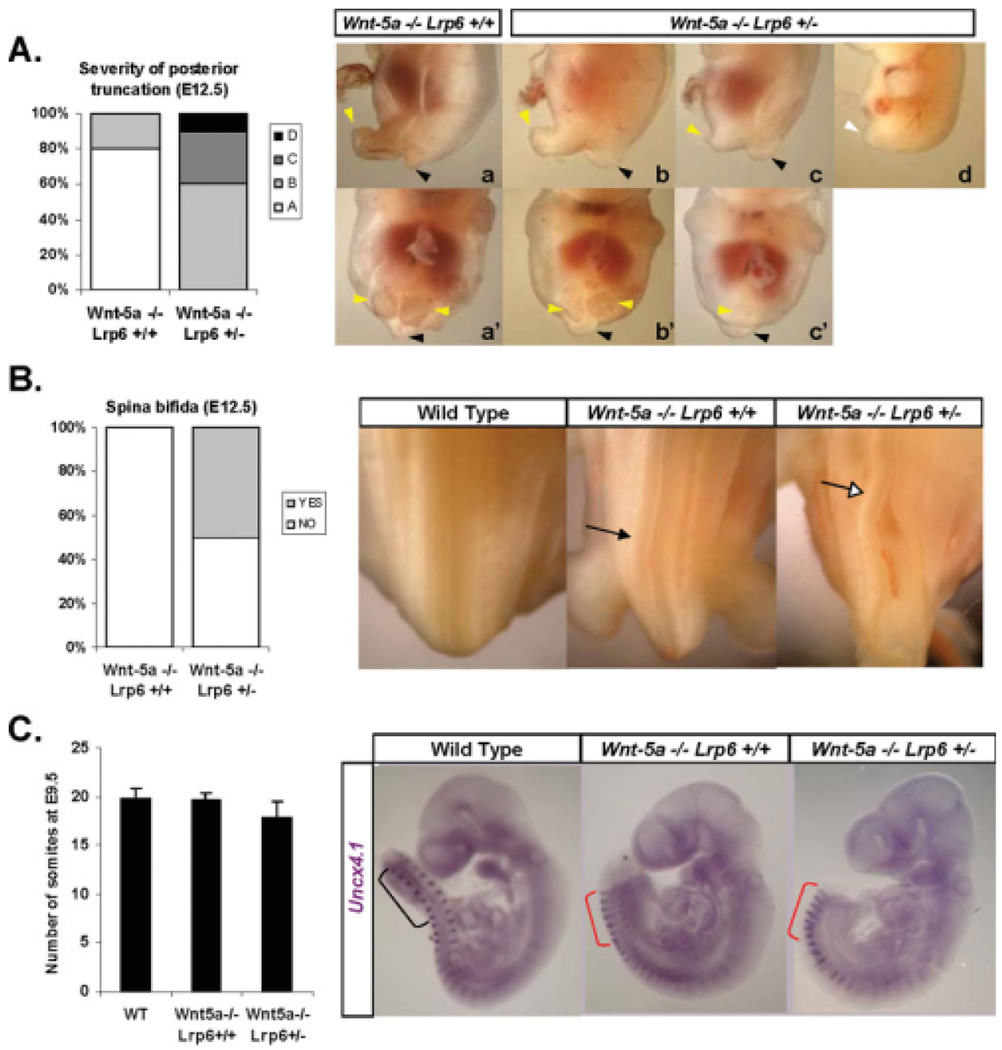
Ablation of Lrp6 on a Wnt5a−/− background aggravates caudal developmental defects. A: Wnt5a−/−Lrp6+/+ and Wnt5a−/−Lrp6+/− embryos were classified based on the severity of their posterior phenotype into four groups (A,B,C,D). Typical characteristics of these groups are depicted in subpanels a–d (lateral view) and a′–c′ (ventral view), and described in the text. Yellow arrowheads indicate residual hindlimbs, black arrowheads indicate tail rudiments. Analysis of 10 Wnt5a−/−Lrp6+/+ and 10 Wnt5a−/−Lrp6+/− demonstrates that the level of posterior truncation is more severe in Wnt5a−/−Lrp6+/−. B: The same set of embryos as in A was analyzed for presence of spina bifida (indicated by open arrow). Black arrow indicates nonstraight neural tube seen in Wnt5a−/− mice. Wnt5a−/−Lrp6+/− but not Wnt5a−/−Lrp6+/+ embryos frequently display spina bifida. C: Wild-type, Wnt5a−/−Lrp6+/+, and Wnt5a−/−Lrp6+/− embryos were collected at embryonic day (E) 9.5 and stained for Uncx4.1 to visualize somites. Somites were counted in each embryo, and the results (mean+SE) are shown in the graph. Embryos with Wnt5a−/− background show more compacted somites (red brackets).
We have previously shown that 30% of Lrp6−/− mice exhibit exencephaly, a neural tube closure defect in the anterior neural tube, which is dose-dependently rescued by loss of Wnt5a (Fig. 1B) (Bryja et al., 2009; Castelo-Branco et al., 2009). Another neural tube closure defect often associated with abrogated convergent extension movements and/or defects in the caudal development is spina bifida, which is caused by a closure failure of the neural tube in caudal regions (for review, see Ybot-Gonzalez et al., 2007). Wnt5a−/− mice do not usually present spina bifida, although they frequently display a crooked or “wavy” neural tube (black arrow in Fig. 3B), which is associated with PCP defects. Intriguingly, haploinsufficiency for Lrp6 in a Wnt5a−/− background results in spina bifida (unfilled arrow in Fig. 3B) in 50% of Wnt5a−/−Lrp6+/− mice, revealing the crucial role of both in neural tube closure.
Anteroposterior defects such as those seen in Wnt5a−/−Lrp6+/− embryos could be the result of defective CE or additive defects in truncation of caudal regions. To assess these parameters, we examined the expression of Uncx4.1, a marker of the caudal portion of somites, in wild-type, Wnt5a−/−Lrp6+/+ and Wnt5a−/−Lrp6+/− embryos at E9.5. The numbers of somites in littermate mice of these genotypes were not significantly different, but a mild reduction could be observed in Wnt5a−/−Lrp6+/− embryos (Fig. 3C). The distance between somites was greatly decreased in the caudal portion of both Wnt5a−/−Lrp6+/+ and Wnt5a−/−Lrp6+/− embryos (Fig. 3C). These data indicate that the aggravated caudal phenotype in Wnt5a−/−Lrp6+/− mice could be a consequence of slight caudal truncation combined with deficient CE.
Wnt5a/Lrp6-Double Deficient Embryos Display Morphogenetic Defects in Presomitic Mesoderm
Wnt5a/Lrp6 mice exhibit a severe phenotype at E10.5 (see Fig. 1A), which is morphologically characterized by severe developmental delay, disrupted caudal development, and defects in heart morphogenesis. These widespread defects led us to speculate that the phenotype is a consequence of earlier defects occurring during gastrulation and early tissue specification. In the early embryo at E8.5, Wnt5a is expressed in the posterior part of embryo with high levels in the primitive streak (PS) and presomitic mesoderm (PSM; Yamaguchi et al., 1999a) To analyze the requirement of Wnt5a and Lrp6 in the development of these structures, we investigated the expression of somitogenesis markers and markers of streak-derived mesoderm. In the first set of experiments (Fig. 4A) we performed in situ hybridization for Hes7, a marker of the oscillating segmentation clock and Uncx4.1. In Lrp6−/− embryos, Hes7 was down-regulated and posterior Uncx4.1 was weaker or lost. In Wnt5a−/− embryos, Hes7 expression levels were normal, whereas the PS and PSM were shorter in length, and the somites were somewhat compressed but still expressed Uncx4.1. In Wnt5a−/−Lrp6−/− double mutants, expression of Hes7 was restored to wild-type levels but the PS instead displayed severe morphogenetic defects. Also Wnt5a−/−Lrp6−/− mutants expressed Uncx4.1 in a striped manner in anterior segmented somites, but appeared fused posteriorly. Finally, the somites of Wnt5a−/−Lrp6−/− double mutants were, in some embryos, very compressed compared with their non-Wnt5a−/−Lrp6−/− littermates.
Fig. 4.
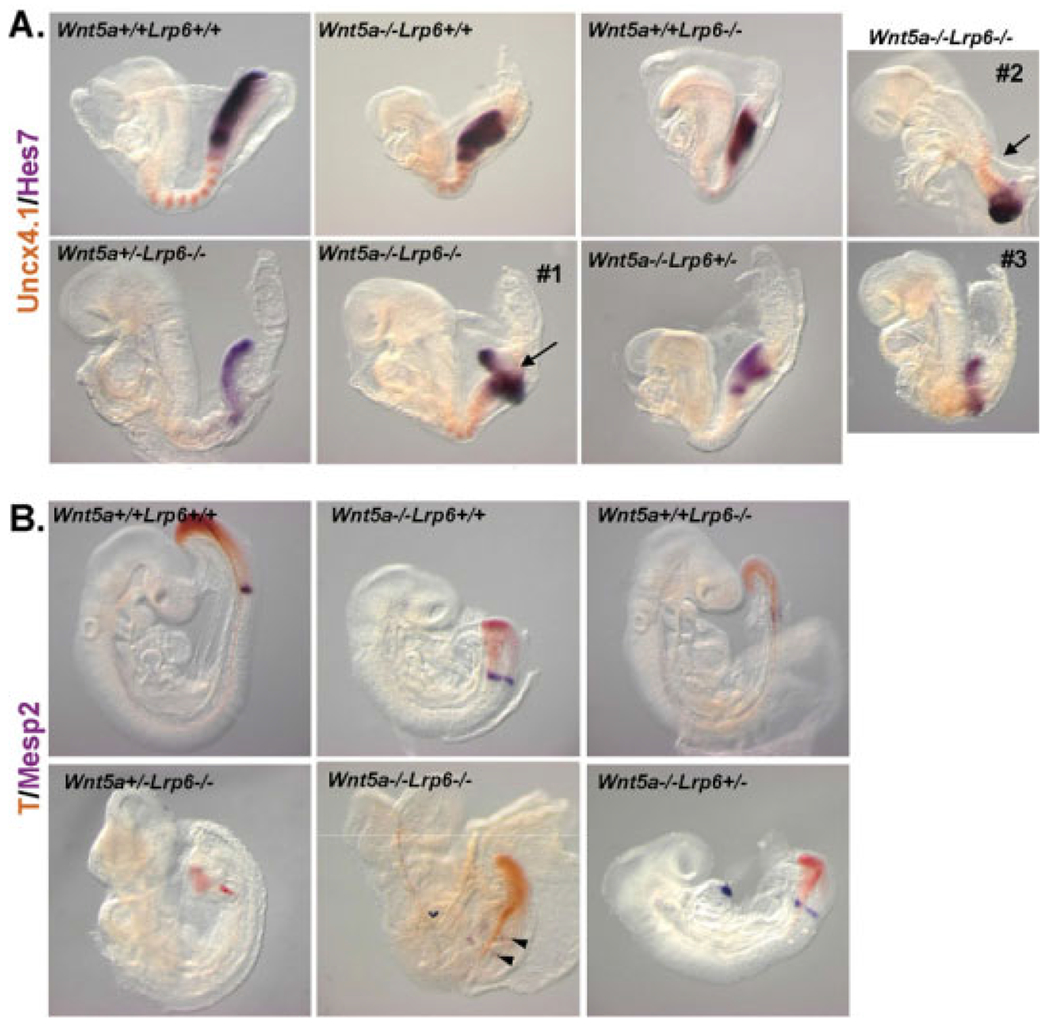
Defects in presomitic mesoderm in Wnt5a and Lrp6 compound mutants. A: Embryos with indicated genotypes were stained at embryonic day (E) 8.5 for Uncx4.1 (brown) and Hes7 (purple) using whole-mount in situ hybridization. Three different Wnt5a−/−Lrp6−/− embryos are shown. Defects in the morphogenesis of presomitic mesoderm are indicated by arrows. B: Embryos between embryonic day (E) 8.75–E9.0 were stained for T (brachyury; brown) and Mesp2 (purple). Abnormal expression of Mesp2 in Wnt5a−/−Lrp6−/− is indicated by arrowheads.
To complement this analysis, we looked at the expression of T (brachyury), a target gene of Wnt/β-catenin pathway (Yamaguchi et al., 1999b) and a marker of PS/PSM, and Mesp2, another Wnt/β-catenin target gene, and marker of presumptive somites in the anterior PSM (Dunty et al., 2008) at E9.0 (Fig. 4B). In Lrp6−/− embryos, T was expressed in lower levels and Mesp2 was barely detectable, which confirms that Lrp6 deficiency causes a Wnt/β-catenin loss-of-function (LOF) in the PSM. In Wnt5a−/− embryos, no changes in the expression levels of T or Mesp2 were detected, although the Mesp2 expression domain had a clearly different shape, which reflects the broader and shorter somites in Wnt5a−/− embryos. In Wnt5a−/−Lrp6−/− embryos expression of T appeared normal and Mesp2 could be clearly detected. Interestingly, in some embryos, Mesp2 was present in two stripes, which is reminiscent of β-catenin gain-of-function (GOF) phenotype (Dunty et al., 2008).
In sum, Hes7 and T continued to be expressed strongly in the Wnt5a−/−Lrp6−/− double mutants, despite the PSM being shortened, the abnormal morphology of the PS, and the general AP axis shortening. Lrp6 deficiency resulted in a decrease in PSM markers (Hes, T), which was not further decreased by loss of Wnt5a. In fact, the reverse was seen, and expression of these markers was instead rescued in some double homozygous mice.
Wnt5a/Lrp6-deficient Embryos Exhibit Severe Defects in Neural Tube Development
At E10.5, embryos of all genotypes could still be obtained. While Wnt5a−/−Lrp6−/− mice were severely developmentally delayed (Fig. 1), they were still viable and exhibited, for example, a beating heart tube. Gross morphological examination of these embryos indicated that the neural tube was thinner and hollow, compared with littermate controls. We therefore collected sections of neural tube at the level of the forelimb, and stained for actin (with Phalloidin, green in Fig. 5A) and nuclei (with propidium iodide, red) to assess the general morphology of the neural tube.
Fig. 5.
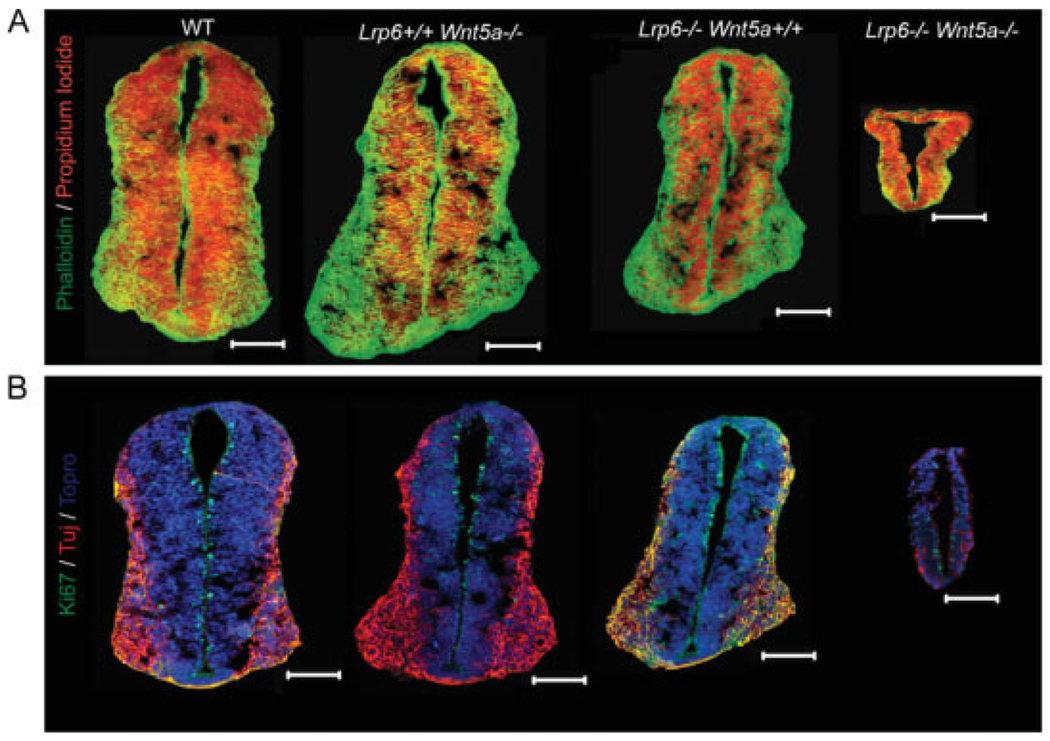
Decreased proliferation and neuron number in Wnt5a−/−Lrp6−/− mice. Neural tubes of mouse embryos with the indicated genotypes were sectioned at embryonic day (E) 10.5. A,B: Cross-sections of neural tube stained either for actin (A; phalloidin; green) and cell nuclei (propidium iodide, red), or for proliferation marker Ki67 (B; green), neuronal marker TuJ (red), and cell nuclei (Topro, blue). Scale bar = 100 μm.
While Wnt5a+/+Lrp6−/− neural tubes were slightly smaller than wild-type littermates, as a consequence of a mild developmental delay, Wnt5a−/−Lrp6−/− mice displayed a severely disrupted neural tube of only one- to two-cell layer thickness (Fig. 5A). It is important to note that the Wnt5a−/−Lrp6−/− embryo shown here is the most normal Wnt5a−/−Lrp6−/− embryo obtained and that others frequently displayed an open neural tube (data not shown) in the caudal region. We asked whether this great reduction in size was in part due to defects in proliferation or differentiation and therefore examined Ki67 (a marker of proliferating cells, green in Fig. 5B) and Tuj1 (a neuronal marker, red). We found that loss of Lrp6 or Wnt5a had negligible effects on the number of proliferating cells (Fig. 5B and data not shown), whereas loss of both Wnt5a and Lrp6 dramatically reduced the number of proliferating cells found in the neural tube at E10.5. Indeed, in some Wnt5a−/−Lrp6−/− embryos, no Ki67+ cells could be found (data not shown). The number of Tuj1+ cells was also greatly reduced in Wnt5a−/−Lrp6−/− mice (Fig. 5B). Thus, in the absence of Wnt5a and Lrp6, both proliferation and differentiation of neural tube stem cells is severely impaired.
DISCUSSION
Lrp6 is a co-receptor required for signal transduction in the Wnt/β-catenin pathway (He et al., 2004). After Wnt ligand binding, Lrp6 is phosphorylated, recruits axin, and in concert with Frizzled receptors and the cytoplasmic protein Dishevelled, blocks the function of the β-catenin destruction complex and promotes β-catenin-dependent transcription. It has been shown that, in most cell types, only some Wnts, for example, Wnt3a, are capable of promoting this signaling pathway, whereas some others, e.g., Wnt5a, fail to induce phosphorylation of intracellular parts of Lrp6, and instead activate other signaling pathways, collectively known as noncanonical Wnt signaling pathways (He et al., 2004; Tamai et al., 2004; Bryja et al., 2007a, 2009). This led to the hypothesis that the noncanonical and Wnt/β-catenin pathways are molecularly distinct. As a consequence, Wnt5a, a crucial component of the noncanonical pathways, and Lrp6, a crucial component of the Wnt/β-catenin pathway should not molecularly interact. This simple view is, however, currently being revised, and it has been suggested that Lrp6 and Wnt5a can functionally interact at several levels.
Evidence in the literature suggest that Lrp6 and Wnt5a could synergize/ antagonize in several ways: (1) Wnt5a could act as a β-catenin-activating ligand by means of Lrp6 (or Lrp5; He et al., 1997; Mikels and Nusse, 2006); (2) Wnt5a inhibits Lr_p6-mediated β-catenin activation by means of competition for Dvl, Frizzled, or Lrp6 itself or by means of other mechanisms (Topol et al., 2003; Westfall et al., 2003; Mikels and Nusse, 2006; Bryja et al., 2007b, 2009); (3) Lrp6 could inhibit Wnt5a-mediated noncanonical signaling (Tahinci et al., 2007; Bryja et al., 2009); and finally (4) Lrp6 and Wnt5a could control common developmental processes by means of distinct cellular or molecular mechanisms. Multiple modes of interaction between Lrp6 and Wnt5a in development could take place in different cell types, and/or at different time points in the embryo.
Our analyses of Wnt5a/Lrp6 compound mutants clearly show that Wnt5a and Lrp6 genetically interact. The analyses presented here, however, do not support the first mode of interaction, e.g., Wnt5a activates β-catenin signaling by means of Lrp6. Several overexpression experiments have shown that specific combinations of Fz receptors and Lrp5 on the cell surface can cause Wnt5a to activate β-catenin-dependent transcription both in Xenopus embryos and in cultured cells (He et al., 1997; Mikels and Nusse, 2006). We were not able, however, to observe any decrease in the expression of Wnt/β-catenin target genes in Wnt5a-deficient and/or Wnt5a/Lrp6-deficient embryos. Specifically, we looked at the posterior part of the developing embryo, where the biological interaction is the most obvious, but neither direct Wnt/β-catenin targets such as T (Yamaguchi et al., 1999a) and Mesp2 (Dunty et al., 2008), nor markers of the oscillating segmentation clock, such as Hes7, were negatively affected by loss of Wnt5a. These markers are down-regulated in Lrp6 mutants (this study) and in Wnt3a mutants (Dunty et al., 2008), and are completely absent from conditional β-catenin LOF mutants (Dunty et al., 2008) This suggests that Lrp6 acts as a receptor for canonical Wnts in PSM and that Wnt5a, which is also present in this region, does not contribute to the activation of β-catenin target genes in the PS-derived mesoderm. Alternatively, Wnt5a may primarily antagonize Wnt/b-catenin signaling in the PS/PSM, whereas Lrp6 functions to prevent Wnt5a from doing so by titrating Wnt5a away as it was shown to be the case in the noncanonical Wnt signaling (Bryja et al., 2009). In the absence of Lrp6, Wnt5a is no longer bound, and antagonizes Wnt/β-catenin signaling (as visualized by the expression of canonical targets such as T, or Mesp2) by means of other mechanisms (see below). However, this possibility does not exclude that Lrp6 acts as the direct positive regulator of Wnt/β-catenin signaling in the PS/PSM. The fact that somites form, visualized by Uncx4.1 expression, also suggests that the phenotypes observed in Wnt5a−/−Lrp6−/− mice are not the result of Wnt/β-catenin LOF.
Our analyses clearly suggest that Wnt5a may act, under some circumstances, as an inhibitor of Lrp6-mediated signaling. In most E8.5 embryos, loss of Wnt5a rescues down-regulation of Hes7 and T caused by Lrp6 deficiency. Moreover, in some Wnt5a/ Lrp6 double-deficient embryos Mesp2 is present in two stripes, which is a phenotype reminiscent of β-catenin GOF in the PS (Dunty et al., 2008). It is also worth noting that β-catenin GOF mice fail to turn, similar to Wnt5a−/−Lrp6−/− mice. On a molecular level, Wnt5a can inhibit Wnt/β-catenin signaling by means of several pathways, including competition for Lrp6 (Bryja et al., 2009), competition for Dvl (Bryja et al., 2007b), or activation of Ror2-driven signaling (Mikels and Nusse, 2006). Endogenous Wnt5a was shown previously to antagonize canonical signaling in vivo, and Wnt5a LOF results in Wnt/β-catenin GOF in several developmental processes (Topol et al., 2003; Westfall et al., 2003). We propose that the PSM may represent an additional structure in which endogenous Wnt5a reduces Wn^-catenm signaling. However, it remains to be clarified why Wnt5a LOF does not have any effect in Lrp6+/+ mice, and is only observed in an Lrp6−/− background when Wnt-β-catenin signaling is reduced.
We and others have recently provided evidence that inhibition of noncanonical Wnt5a- or Wnt11-driven signaling by Lrp6 (or by loss of Dkk1, an Lrp6 inhibitor) occurs during frog and mouse development (Caneparo et al., 2007; Tahinci et al., 2007; Bryja et al., 2009). We have shown previously that Lrp6 can bind Wnt5a and that Lrp6-deficiency results in noncanonical Wnt pathway GOF, which is rescued by depletion of noncanonical Wnt ligands such as Wnt5a. Importantly, this only takes place in the tissues where expression of Wnt5a is high. In mouse, this mode of interaction is observed in defects in the closure of the neural tube in the midbrain region and during heart outflow tract development (Bryja et al., 2009). Additional phenotypes described here, such as increased compaction of somites and the abnormal shape of PSM in some Wnt5a−/−Lrp6−/− embryos, suggest that this mode of interaction can act also in PSM morphogenesis. It is, however, difficult to distinguish this from the alternative possibility; that Lrp6 acts by means of the Wn^-catenm pathway in somite specification, segmentation clock, and definition of posterior structures, and Wnt5a affects convergent extension in this region by means of noncanonical Wnt signaling. Both processes take place in PSM: single Lrp6 mutants display severe defects in the development of posterior body parts, which is associated with decreased levels of Wn^-catenm target genes (Figs. 2, 4) and Wnt5a mutants show clear defects in convergent extension of PSM, which are visualized as higher compaction of somites (Uncx4.1 staining) and in the formation of somites, which are broader but shorter (Mesp2 staining). Similar CE-related phenotypes are observed in Wnt5a-deficient mice in the inner ear and neural tube (Qian et al., 2007; Andersson et al., 2008).
Interestingly loss of one allele of Lrp6 has no effect on an otherwise wild-type background (with the exception of low penetrance kinked tail phenotype in adult mice) but induces severe defects in the absence of Wnt5a. It is well established that Wn^-catenm signaling is crucial for development of posterior parts of the embryo and is critically required for proper somitogenesis (Yamaguchi, 2001; Dunty et al., 2008). Even partial loss of Wnt^-catenin activity (as demonstrated, for example, by Lrp6 hypomorph “ringelschwanz” mouse) can lead to defects in somitogenesis and posterior development, including spina bifida aperta (Kokubu et al., 2004). Malformations in the most posterior part of tail of Lrp6+/− adults, usually as a “kinked” tail, have been observed earlier (Pinson et al., 2000) and also in our experiments (not shown). One can speculate that the loss of Wnt5a, which results in a dramatically shortened tail, moves the domain of Lrp6-mediated activity more anteriorly to the level of hindlimbs (which is the most posterior part of Wnt5a-null embryos), where heterozygosity of Lrp6 function is sufficient to cause defects such as posterior truncation and associated spina bifida. This phenotype, which was found in Wnt5a−/−Lrp6+/− (compared with Wnt5a−/− mice), thus probably reflects the combination of highly compacted somites and slightly decreased somite number. Thus, our results indicate that the loss of one allele of Lrp6 affects posterior development, but only in the context of shorter body axis (Wnt5a-deficient bodies), when crucial temporal/spatial coordination of somitogenesis is disturbed because of CE defects.
In summary, we hereby provide evidence that Lrp6 and Wnt5a genetically interact in multiple tissues during mouse development. These interactions, while compatible with multiple mechanisms of interactions, do not support that Wnt5a acts as an Lrp6-activating ligand in vivo in the tissues examined. Moreover, in addition to the previously described phenotypes showing a crucial role of Lrp6 as the inhibitor of noncanonical Wnt signaling (Tahinci et al., 2007; Bryja et al., 2009), we hereby demonstrate that Lrp6 and Wnt5a interact to coordinate development of the neural tube, morphogenesis of the PSM, and somitogenesis in mouse.
EXPERIMENTAL PROCEDURES
Animal Maintenance and Embryo Collection
Wnt5a+/− (Yamaguchi et al., 1999a) and Lrp6+/− (Pinson et al., 2000) mice were kept on a C57bl6 background, and housed and bred in accordance with the approval of the local ethics committee (Stockholms Norra Djurforsoksetiskanamnd). Noon of day of plug was taken as embryonic day (E) 0.5. Embryos were dissected out in PBS, fixed in 4% paraformaldehyde overnight at 4°C, and then washed in phosphate buffered saline (PBS). Embryos were either dehydrated in a methanol gradient and stored in 100% methanol at −20°C until used in whole-mount in situ hybridization, or transferred to 30% sucrose, rocked overnight, embedded in OCT (Tissue-Tek), and frozen on dry ice for cryostat sectioning and immunohistochemistry. Serial transverse 14-μm sections were collected on SuperFrost glass slides on a cryostat.
Genotyping
DNA extraction and PCR for the Wnt5a or Lrp6 mutant alleles has been described previously (Yamaguchi et al., 1999a; Pinson et al., 2000; Bryja et al., 2009). SMCX-1 5′CCGCTGCCAAATTCTTTGG3′ and SMC4-1 5′TGAAGCTTTTGGCTTTGAG3′ primers and genotyping of male vs. female, which gives one band in females and two bands of different sizes in males, has been described previously (Agulnik et al., 1997).
In Situ Hybridization
Whole-mount in situ hybridization was performed as previously described (Wilkinson and Nieto, 1993). Embryos were photographed on a stereoscope (Leica, Germany) or and Axiophot compound microscope (Zeiss, Germany). At least three embryos were examined for each probe, and yielded similar results. The following probes were used: Uncx4.1, Hes7, T, and Mesp2 (Dunty et al., 2008).
Immunohistochemistry
Sections/slides were first immersed in PBS for 5 min, and then blocked with 5% goat serum (GS) in phosphate-buffered saline (PBS) with 0.1% Triton-X (PBST) for 1 hr. Primary antibodies were diluted in 5% GS/PBST overnight at 4°C. Slides were then washed in PBS, 3× 15 min, and secondary antibodies, diluted in 5%GS/PBST, were applied for 1 hr at room temperature. The slides were then washed again, 3× 15 min, counterstained with Topro-3 (Invitrogen) or propidium iodide (Invitrogen) and washed twice more. Slides were mounted in glycerol/PBS 9:1.
The following antibodies were used: rabbit anti-Ki67 (1:800, Abcam), mouse anti-Tuj1 (βIII tubulin, 1:1,000, BD Biosciences), Alexa488 anti-mouse and Alexa555 anti-rabbit (1:1,000, Molecular Probes). Phalloidin (Invitrogen) staining was carried out according to the manufacturer’s instructions.
All images were acquired on a Zeiss LSM 510 (Zeiss, Germany), with Zeiss software.
ACKNOWLEDGMENTS
We thank Johnny Söderlund and Alessandra Nanni for technical and secretarial assistance, respectively, as well as Susanne Ahnstrand and Cecilia Olsson of the MBB animal house facility, for excellent animal care. We also thank William Skarnes (Sanger Institute, Cambridge) for providing Lrp6-deficient mice. V.B. was funded by an EMBO Installation Grant and the Ministry of Education Youth and Sports of the Czech Republic. E.A. was funded by the Swedish Foundation for Strategic Research (INGVAR and CEDB), Swedish Research Council (VR2008:2811 and DBRM), Norwegian Research Council, Karolinska Institutet, Michael J. Fox Foundation, and European Commission (Eurostemcell). The authors declare no conflict of interest.
Grant sponsor: EMBO; Ministry of Education Youth and Sports of the Czech Republic; Grant number: MSM 0021622430; Grant sponsor: Swedish Foundation for Strategic Research (INGVAR and CEDB); Swedish Research Council; Grant number: VR2008:2811 and DBRM; Grant sponsor: Norwegian Research Council; Grant sponsor: Karolinska Institutet; Grant sponsor: Michael J. Fox Foundation; Grant sponsor: European Commission (Eurostemcell).
REFERENCES
- Adamska M, Billi AC, Cheek S, Meisler MH. 2005. Genetic interaction between Wnt7a and Lrp6 during patterning of dorsal and posterior structures of the mouse limb. Dev Dyn 233:368–372. [DOI] [PubMed] [Google Scholar]
- Agulnik AI, Bishop CE, Lerner JL, Agulnik SI, Solovyev VV. 1997. Analysis of mutation rates in the SMCY/SMCX genes shows that mammalian evolution is male driven. Mamm Genome 8:134–138. [DOI] [PubMed] [Google Scholar]
- Andersson ER, Prakash N, Cajanek L, Minina E, Bryja V, Bryjova L, Yamaguchi TP, Hall AC, Wurst W, Arenas E. 2008. Wnt5a regulates ventral midbrain morphogenesis and the development of A9-A10 dopaminergic cells in vivo. PLoS ONE 3:e3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M, Niehrs C. 2007. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 316:1619–1622. [DOI] [PubMed] [Google Scholar]
- Bryja V, Gradl D, Schambony A, Arenas E, Schulte G. 2007a. beta-arrestin is a necessary component of Wnt/beta-catenin signaling in vitro and in vivo. Proc Natl Acad Sci USA 104:6690–6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryja V, Schulte G, Rawal N, Grahn A, Arenas E. 2007b. Wnt-5a induces Dishevelled phosphorylation and dopaminergic differentiation via a CK1-dependent mechanism. J Cell Sci 120:586–595. [DOI] [PubMed] [Google Scholar]
- Bryja V, Andersson ER, Schambony A, Esner M, Bryjova L, Biris KK, Hall AC, Kraft B, Cajanek L, Yamaguchi TP, Buckingham M, Arenas E. 2009. The extracellular domain of Lrp5/6 inhibits noncanonical Wnt signaling in vivo. Mol Biol Cell 20:924–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caneparo L, Huang YL, Staudt N, Tada M, Ahrendt R, Kazanskaya O, Niehrs C, Houart C. 2007. Dickkopf-1 regulates gastrulation movements by coordinated modulation of Wnt/beta catenin and Wnt/PCP activities, through interaction with the Dally-like homolog Knypek. Genes Dev 21:465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelo-Branco G, Andersson ER, Minina E, Sousa KM, Ribeiro D, Kokubu C, Imai K, Prakash N, Wurst W, Arenas E. 2009. Delayed dopaminergic neuron differentiation in Lrp6 mutant mice. Dev Dyn 238:000–000. [DOI] [PubMed] [Google Scholar]
- Dunty WC Jr, Biris KK, Chalamalasetty RB, Taketo MM, Lewandoski M, Yamaguchi TP. 2008. Wnt3a/beta-catenin signaling controls posterior body development by coordinating mesoderm formation and segmentation. Development 135:85–94. [DOI] [PubMed] [Google Scholar]
- Hamblet NS, Lijam N, Ruiz-Lozano P, Wang J, Yang Y, Luo Z, Mei L, Chien KR, Sussman DJ, Wynshaw-Boris A. 2002. Dishevelled 2 is essential for cardiac outflow tract development, somite segmentation and neural tube closure. Development 129:5827–5838. [DOI] [PubMed] [Google Scholar]
- He X, Saint-Jeannet JP, Wang Y, Nathans J, Dawid I, Varmus H. 1997. A member of the Frizzled protein family mediating axis induction by Wnt-5A. Science 275: 1652–1654. [DOI] [PubMed] [Google Scholar]
- He X, Semenov M, Tamai K, Zeng X. 2004. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development 131:1663–1677. [DOI] [PubMed] [Google Scholar]
- Kokubu C, Heinzmann U, Kokubu T, Sakai N, Kubota T, Kawai M, Wahl MB, Galceran J, Grosschedl R, Ozono K, Imai K. 2004. Skeletal defects in ringelschwanz mutant mice reveal that Lrp6 is required for proper somitogenesis and osteogenesis. Development 131: 5469–5480. [DOI] [PubMed] [Google Scholar]
- Logan CY, Nusse R. 2004. The wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20:781–810. [DOI] [PubMed] [Google Scholar]
- Mikels AJ, Nusse R. 2006. Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol 4:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montcouquiol M, Crenshaw EB III, Kelley MW. 2006. Noncanonical Wnt signaling and neural polarity. Annu Rev Neurosci 29:363–386. [DOI] [PubMed] [Google Scholar]
- Moon RT, Campbell RM, Christian JL, McGrew LL, Shih J, Fraser S. 1993. Xwnt-5A: a maternal Wnt that affects morphogenetic movements after overexpression in embryos of Xenopus laevis. Development 119:97–111. [DOI] [PubMed] [Google Scholar]
- Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. 2000. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 407: 535–538. [DOI] [PubMed] [Google Scholar]
- Qian D, Jones C, Rzadzinska A, Mark S, Zhang X, Steel KP, Dai X, Chen P. 2007. Wnt5a functions in planar cell polarity regulation in mice. Dev Biol 306:121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleiffarth JR, Person AD, Martinsen BJ, Sukovich DJ, Neumann A, Baker CV, Lohr JL, Cornfield DN, Ekker SC, Petryk A. 2007. Wnt5a is required for cardiac outflow tract septation in mice. Pediatr Res 61:386–391. [DOI] [PubMed] [Google Scholar]
- Schulte G, Bryja V, Rawal N, Castelo-Branco G, Sousa KM, Arenas E. 2005. Purified Wnt-5a increases differentiation of midbrain dopaminergic cells and dishevelled phosphorylation. J Neurochem 92:1550–1553. [DOI] [PubMed] [Google Scholar]
- Sokol S, Christian JL, Moon RT, Melton DA. 1991. Injected Wnt RNA induces a complete body axis in Xenopus embryos. Cell 67:741–752. [DOI] [PubMed] [Google Scholar]
- Tahinci E, Thorne CA, Franklin JL, Salic A, Christian KM, Lee LA, Coffey RJ, Lee E. 2007. Lrp6 is required for convergent extension during Xenopus gastrulation. Development 134:4095–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai K, Zeng X, Liu C, Zhang X, Harada Y, Chang Z, He X. 2004. A mechanism for Wnt coreceptor activation. Mol Cell 13:149–156. [DOI] [PubMed] [Google Scholar]
- Topol L, Jiang X, Choi H, Garrett-Beal L, Carolan PJ, Yang Y. 2003. Wnt-5a inhibits the canonical Wnt pathway by promoting GSK-3-independent beta-catenin degradation. J Cell Biol 162:899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres MA, Yang-Snyder JA, Purcell SM, DeMarais AA, McGrew LL, Moon RT. 1996. Activities of the Wnt-1 class of secreted signaling factors are antagonized by the Wnt-5A class and by a dominant negative cadherin in early Xenopus development. J Cell Biol 133:1123–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainio S, Heikkila M, Kispert A, Chin N, McMahon AP. 1999. Female development in mammals is regulated by Wnt-4 signalling. Nature 397:405–409. [DOI] [PubMed] [Google Scholar]
- Westfall TA, Brimeyer R, Twedt J, Gladon J, Olberding A, Furutani-Seiki M, Slusarski DC. 2003. Wnt-5/pipetail functions in vertebrate axis formation as a negative regulator of Wnt/beta-catenin activity. J Cell Biol 162:889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DG, Nieto MA. 1993. Detection of messenger RNA by in situ hybridization to tissue sections and whole mounts. Methods Enzymol 225:361–373. [DOI] [PubMed] [Google Scholar]
- Wong GT, Gavin BJ, McMahon AP. 1994. Differential transformation of mammary epithelial cells by Wnt genes. Mol Cell Biol 14:6278–6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi TP. 2001. Heads or tails: Wnts and anterior-posterior patterning. Curr Biol 11:R713–724. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Bradley A, McMahon AP, Jones S. 1999a. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development 126:1211–1223. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Takada S, Yoshikawa Y, Wu N, McMahon AP. 1999b. T (Brachyury) is a direct target of Wnt3a during paraxial mesoderm specification. Genes Dev 13:3185–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DH, Yoon JY, Lee SH, Bryja V, Andersson ER, Arenas E, Kwon YG, Choi KY. 2009. Wnt5a is required for endothelial differentiation of embryonic stem cells and vascularization via pathways involving both Wnt/beta-catenin and protein kinase Calpha. Circ Res 104:372–379. [DOI] [PubMed] [Google Scholar]
- Ybot-Gonzalez P, Savery D, Gerrelli D, Signore M, Mitchell CE, Faux CH, Greene ND, Copp AJ. 2007. Convergent extension, planar-cell-polarity signalling and initiation of mouse neural tube closure. Development 134:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CJ, Molotkov A, Song L, Li Y, Pleasure DE, Pleasure SJ, Wang YZ. 2008. Ocular coloboma and dorsoventral neuroretinal patterning defects in Lrp6 mutant eyes. Dev Dyn 237:3681–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]