

Abstract
Objective:
Lipodystrophy represents a group of rare diseases characterized by loss of body fat. While patients with generalized lipodystrophy exhibit near-total lack of fat, partial lipodystrophy is associated with selective fat loss affecting certain parts of the body. Although classical familial partial lipodystrophy (FPLD) is a well-described entity, recent reports indicate phenotypic heterogeneity among carriers of LMNA pathogenic variants.
Methods:
We have encountered 2 unique cases with complex phenotypes, generalized fat loss, and very low leptin levels that made the distinction between generalized versus partial lipodystrophy quite challenging.
Results:
We present a 61-year-old female with generalized fat loss, harboring the heterozygous pathogenic variant p.R541P (c.1622G>C) on the LMNA gene. The discovery of the pathogenic variant led to correct clinical diagnosis of her muscle disease, identification of significant heart disease, and a recommendation for the implantation of a defibrillator. She was able to start metreleptin based on her generalized fat loss pattern and demonstration of the genetic variant. Secondly, we report a 40-year-old Turkish female with generalized fat loss associated with a novel heterozygous LMNA pathogenic variant p.K486E (c.1456A>G), who developed systemic B cell follicular lymphoma.
Conclusion:
Clinicians need to recognize that the presence of an LMNA variant does not universally lead to FPLD type 2, but may lead to a phenotype that is more complex and may resemble more closely generalized lipo-dystrophy. Additionally, providers should recognize the multisystem features of laminopathies and should screen for these features in affected patients, especially if the variant is not at the known hotspot for FPLD type 2.
INTRODUCTION
Lipodystrophy is a heterogeneous group of congenital or acquired disorders characterized by partial to near total absence of subcutaneous adipose tissue. Among the known genetic causes of partial lipodystrophy, typically called familial partial lipodystrophy (FPLD), the most common form is type 2 which has been linked to heterozygous pathogenic variants of the LMNA gene which encodes lamin A/C nuclear envelope proteins (1).
Patients who have pathogenic variants of the LMNA gene have been mostly placed in the FPLD type 2 category due to the current clinical classification system which is based on clinical findings and presentation (2). Making this clinical distinction has gained even more importance in the U.S. after approval for metreleptin for only generalized lipodystrophy in February 2014. Although some patients who were classified as partial lipodystrophy had clear evidence of benefit (3), one of the reasons why the U.S. Food and Drug Admistration did not approve the treatment for partial lipodystrophy has been the lack of precise diagnostic criteria (4,5). Therefore, it is important to try to understand the caveats in the proper classification and diagnosis of lipodystrophy.
Here, we report 2 patients with pathogenic variants of the LMNA gene presenting with generalized fat loss. One is a patient with a heterozygous pathogenic variant p.R541P (c.1622G>C) the other patient has a novel heterozygous pathogenic variant p.K486E (c.1456A>G). Both cases also displayed unusual clinical features that expand the previously reported phenotypes.
CASE REPORT
Patient 1
A 61-year-old, Lebanese female presented at the age of 19 with secondary amenorrhea caused by central hypogonadism. Shortly after initiating hormone replacement with Premarin and Provera, she developed hypertriglyceridemia, xanthomas, and fatty liver disease. She reported that she had very little subcutaneous fat in her limbs, abdomen, and prominent muscles, especially in her extremities, since early childhood (Fig. 1 A).
Fig. 1.
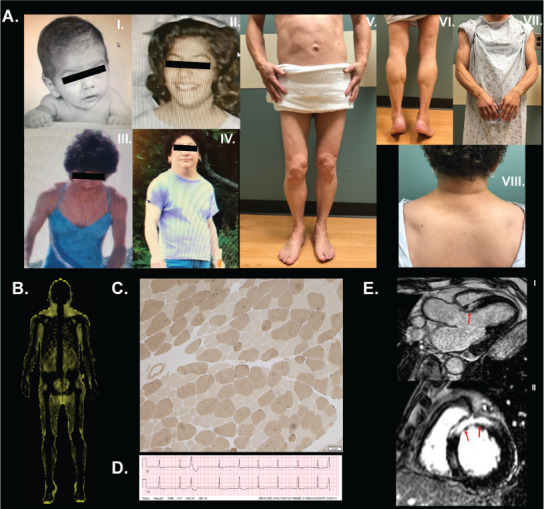
A, Photographs of patient 1 at various ages: (I) a few months old, (II) a teenager, (III) 25 years old, and (IV) 48 years old. We obtained the patient's pictures to demonstrate her current state in October of 2018 with her permission. Close-up pictures of (V) anterior view of the trunk and legs, (VI) back of the legs, (VII) arms, and (VIII) back of the neck. B, “Fat shadow” obtained from dual energy X-ray absorptiometry scan showing fat distribution consistent with generalized fat loss but with some preservation around the neck. Total fat percentage was reported as 22%, the trunk-to-total fat mass ratio was 0.61, and legs-to-total fat mass ratio was 0.24. C, Deltoid muscle biopsy studied with ATPase assay at pH 9.4 with remarkable fiber size disproportion where the type 1 fibers were 45% smaller than the type 2 fibers. Fiber size disproportion (FSD) is an important feature of the Emery-Dreifuss muscular dystrophy (EDMD) pathology and it was remarkably high in our first patient (Fig. 1). Fresh frozen tissue studied for abnormal lack of emerin protein expression by immunohistochemical staining would have been helpful to support the diagnosis of the x-linked form of EDMD caused by STA gene mutation on chromosome Xq28. Most LMNA gene mutations show no alteration in the lamin A/C protein and cannot be detected by immunohistochemical staining. FSD (formerly a subset of congenital fiber type disproportion) is a spectacular feature of a number of muscle abnormalities. The differential diagnosis of muscle diseases that cause FSD includes specific muscular diseases, polymyositis, mitochondrial myopathy, Pompe disease, mutations in the insulin receptor gene, congenital hypothyroidism, phosphofructokinase and carnitine palmitoyltransferase deficiency, and at least 5 congenital myopathies. In this case, the patient's age and presentation decrease this differential. Another important pathologic feature of this case is an ongoing non-inflammatory myopathy, revealed by regenerating muscle fibers. This and steadily elevated creatine kinase levels in this patient's blood suggest a muscular dystrophy. Specific diseases that cause FSD are Becker (more often than Duchenne) muscular dystrohy, myotonic dystrophy, rigid-spine syndrome, and EDMD. Other causes of FSD include certain central nervous system and peripheral nervous system diseases, skeletal disorders, and rare syndromes that do not pertain to this case. D, Electrocardiogram demonstrating first degree atrioventricular block, premature ventricular contractions, and nonspecific ST abnormalities. E, Cardiovascular magnetic resonance images demonstrating extensive late gadolinium enhancement involving the interventricular septum (arrows) consistent with myocardial fibrosis along the (I) apical long axis view and (II) short axis view.
After age 30, she developed progressive generalized and disabling muscle pain, numbness in the lower limbs and right arm, poor balance, and an unsteady walk. These complaints led to a muscle biopsy, which was interpreted as myositis, prompting steroid therapy and the use of other immunosuppressants intermittently, but never continuously. She was diagnosed with diabetes in her fifties. She developed cysts on both kidneys and mild proteinuria, diverticular bowel disease, gastroesophageal reflux disease and liver disease. A liver biopsy showed nonalcoholic steatohepatitis, and significant fibrosis (Table 1).
Table 1.
Patient Clinical and Metabolic Characteristics
| Patient 1 | Patient 2 | |
|---|---|---|
| Triangular face | Yes | Yes |
| Generalized fat loss | Yes | Yes |
| Hypertriglyceridemia | Yes | Yes |
| Pancreatitis | No | Yes |
| Clinical diabetes | Yes | Yes |
| Hepatomegaly or hepatic steatosis | Yes | Yes |
| Hypothyroidism | Yes | No |
| Irregular periods or infertility | Yes | Yes |
| Hypogonadism (with low follicle-stimulating hormone or luteinizing hormone) | Yes | No |
| Increased musculature | Yes | Yes |
| Muscle pain | Yes | Yes |
| Elevated creatine kinase | Yes | No |
| Raynaud phenomenon | Yes | No |
| Reduced left ventricle ejection fraction | Yes | No |
| Lymphoma | No | Yes |
When she was examined by us at age 58, her body mass index was 23.2 kg/m2. She had acromegaloid features, a triangular face, minimal fat palpable around the neck, and minimal fullness of the supraclavicular fossa. The rest of her body was virtually devoid of subcutaneous fat tissue, consistent with the phenotype of generalized lipodystrophy. Mid-tight skinfold was 4.5 mm. She displayed prominent and hypertrophic muscles with slightly elevated creatine kinase levels ranging from 179 to 299 IU/L (normal range is 26 to 180 IU/L), phlebomegaly on her arms, legs, and trunk, umbilical hernia, and acanthosis nigricans around the neck and armpits (Fig. 1 A). Her laboratory test results are shown in Table 2.
Table 2.
Laboratory Parameters
| Patient 1 | Patient 2a | Reference range | |
|---|---|---|---|
| Hemoglobin (g/dL) | 13 | 12 | 12-16 |
| White blood cells (103/μL) | 7.2 | 6.3 | 4.0–10.3 |
| Platelets (103/μL) | 194 | 372 | 156–373 |
| C-reactive protein (mg/L) | 0.9 | 1.9 | 0.2–5.0 |
| Insulin (μIU/mL) | 44 | 17 | 1–25 |
| Homeostatic model assessment of insulin resistance | 11.4 | 3.4 | <2.3 |
| Aspartate aminotransferase (IU/L) | 34 | 13 | 0–35 |
| Alanine aminotransferase (IU/L) | 26 | 13 | 0–35 |
| Total cholesterol (mg/dL) | 317 | 238 | <200 |
| Low-density lipoprotein (mg/dL) | 118 | 154 | <30 |
| High-density lipoprotein (mg/dL) | 34 | 31 | <50 |
| Triglyceride (mg/dL) | 1610 | 410 | <150 |
| Hemoglobin A1c (%, mmol/mmol) | 7.2 (55) | 5.8 (40) | 4.0–6.0 (20–42) |
| Leptin (ng/mL) | 0.6 | 0.2 | Adult females with body mass index of 22: 3.3–18.3 |
| Follicle-stimulating hormone (mIU/mL) | 1.2 | 3.6 | Follicular: 3.0–20.0 Midcycle: 1.0–12.0 Luteal: 18.0–153.0 Postmenopausal: 16.0–64.0 |
| Luteinizing hormone (mIU/mL) | <0.5 | 4.9 | Follicular: 2.0–15.0 Midcycle: 22.0–105.0 Luteal: 0.0–19.0 Postmenopausal: 16.0–64.0 |
| Estradiol (pg/mL) | 14 | 79 | Early follicular: 10–50 Late follicular: 60–200 Midcycle: 120–375 Luteal: 50–260 Postmenopausal: <20 |
aLabs were taken before starting systemic chemotherapy.
A “fat shadow” derived from the body composition scan using dual energy X-ray absorptiometry as described previously (6) highlighted the generalized pattern of fat loss (Fig. 1 B). A lipodystrophy genetic panel was performed at the University of Chicago, and it revealed a heterozygous pathogenic variant p.R541P (c.1622G>C) at exon 8 of the LMNA gene. She has no family members formally diagnosed with lipodystrophy. Her father died at the age of 51, reportedly of myocardial infarction her brother died suddenly at age 47 and no autopsy was performed. Upon identification of the variant, her previously obtained deltoid muscle biopsy was reanalyzed and was confirmed to represent muscular dystrophy as opposed to myositis (Fig. 1 C).
Transthoracic echocardiography demonstrated mild left ventricular hypertrophy with normal left ventricular systolic function Holter monitor showed premature ventricular contractions (Fig. 1 D) and a 5-beat run of non-sustained ventricular tachycardia. Given her conduction system disease, myocardial fibrosis (Fig. 1 E), and family history of sudden death, a dual-chamber implantable cardioverter-defibrillator was placed for primary prevention. She also underwent a sleep study demonstrating obstructive sleep apnea with total carbon dioxide values ranging from 42 to 44 mm Hg with a respiratory rate of 16 to 20 breaths per minute during supine quiet wakefulness during sleep her total carbon dioxide ranged from 45 to 52 mm Hg with a respiratory rate of 12 to 18 breaths per minute. Based on these results, she was prescribed bilevel positive airway pressure therapy.
When her hemoglobin A1c rose to 7.2% on metformin monotherapy and her triglycerides remained above 250 mg/dL despite treatment with lipid-lowering agents, she was started on metreleptin therapy. The approval by her insurance company was enabled by review of her mutation and dual energy X-ray absorptiometry scan findings.
Patient 2
A 40-year-old, Turkish female first recognized increased musculature throughout her limbs and decreased fat in her face around puberty. She was diagnosed with polycystic ovarian syndrome in her high school years. At the age of 29, she experienced an episode of acute pancreatitis caused by severe hypertriglyceridemia. She was diagnosed with gestational diabetes mellitus and started on insulin treatment when she got pregnant at the age of 30. She had a history of local fat transplantation to her face from her mons pubis region at the age of 32.
Her family history was notable for lipodystrophic appearance in her sister who was deceased at age 49 due to myocardial infarction. Reportedly, her sister had a lean body shape with thin extremities and superficial vessels and no fat accumulation in the face and neck. She had high lipids. Per her death certificate, she had longstanding diabetes and preexisting ischemic heart disease. Her father also had metabolic abnormalities associated with insulin resistance and died after a cerebrovascular event.
Her body mass index was 20.7 kg/m2. There was decreased subcutaneous fat in her face, limbs, and trunk (Fig. 2 A), but fat tissue was preserved around her external genital region. Her physical examination revealed several palpable lymph nodes through the cervical and axillary regions that she had noticed approximately 3 months before presentation. The liver longitudinal diameter was 22.4 cm on magnetic resonance imaging. Laboratory test results are presented in Table 2.
Fig. 2.
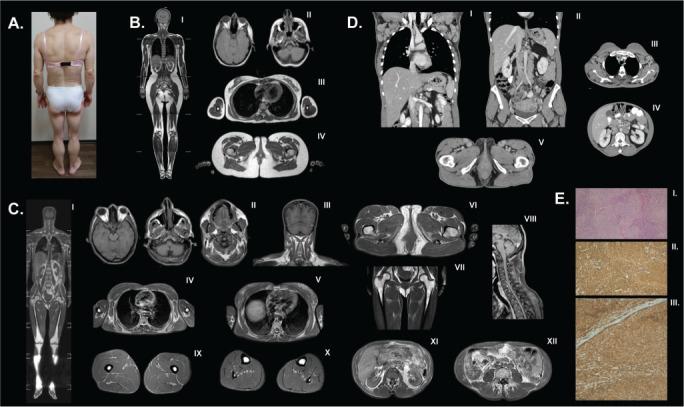
A, Generalized fat loss pattern in patient 2. B, Magnetic resonance images from a healthy control, who was a 28-year-old, healthy woman with a body mass index of 22.4 kg/m2 and normal fat distribution including (I) whole-body T1-weighted imaging, (II) face, head, and neck, axial T1, (III) trunk, axial T1, and (IV) pelvic region, axial T1. C, Whole-body magnetic resonance imaging showing adipose tissue is well preserved around the mons pubis and external genital region while fat tissue loss is noted in a generalized pattern in the scalp, mammary gland, visceral and subcutaneous abdomen, and extremities. A fluid-like signal is detected in the bone marrow. Retroorbital fat is protected. (I) Whole-body T1-weighted image, (II) head and neck, axial T1, (III) head and neck and shoulders, coronal T1, (IV) trunk and upper arms, axial T1, (V) trunk, axial T1, (VI) pelvic region, axial T1, (VII) pelvic region, coronal T1, (VII) back of the neck and upper trunk, sagittal T1, (IX) upper leg, axial T1, (X) lower leg, axial T1, (XI) retroperitoneal and perirenal fat, axial T1, and (XII) intraperitoneal and mesenteric fat residues, axial T1. D, Computed tomography scan with intravenous contrast showing multiple enlarged lymph nodes throughout the axillary, intraabdominal, and inguinal regions including the (I) thorax, coronal plane, (II) abdomen, coronal plane, (III) thorax, axial plane, (IV) abdomen, axial plane, and (V) inguinal region, axial plane. E, An excisional biopsy of the right cervical lymph node revealed neoplastic lymphoid infiltrate with a nodular pattern (more than 75%). These infiltrate cells were positively stained with CD20 and BCL-2. Additionally, many of these cells were BCL-6 and CD10 positive. There was no staining with CD5, CD38, or cyclin D1, while CD21 and CD23 were positive. Also, there were irregular dendritic cell networks in the areas showing a nodular pattern. The identified infiltrate was consistent with follicular lymphoma (low grade, or grade 1 to 2). (I) Neoplastic lymphoid infiltration showing nodular pattern. Hematoxylin and eosin stain, ×40. (II) CD20 positivity in the neoplastic cells, ×100. (III) BCL-2 positivity in the neoplastic cells ×100.
A whole-body magnetic resonance image confirmed the near-total absence of fat with hepatic steatosis (Fig. 2 B and C). In contrast to classical FPLD type 2, she had loss of subcutaneous fat from the face, and magnetic resonance images revealed no significant fat tissue in the face except reduced buccal and parapharyngeal fat pads. Intraabdominal and mesenteric fat was also reduced. On the other hand, adipose tissue was well preserved in the mons pubis and external genital region. Orbital and retroperitoneal fat was also protected (Fig. 2 C). A computed tomography scan revealed multiple lymph nodes with lymphadenopathy throughout the axillary, intraabdominal, and inguinal regions (Fig. 2 D). An excisional biopsy of the right cervical lymph node revealed low-grade B cell follicular lymphoma (Fig. 2 E).
Genetic testing showed a novel heterozygous LMNA variant p.K486E (c.1456A>G) which was interpreted as pathogenic. Her 5-year-old son, who presented no distinct clinical attributes, was genetically tested with the consent of his mother, and the result for the genetic test is positive for the p.K486E LMNA variant. Because both her parents passed away, it is not possible to determine whether the pathogenic variant is inherited from the proband's parents.
DISCUSSION
Our unique patients point out that the distinction of generalized versus partial lipodystrophy may not be easy in some situations. De novo heterozygous LMNA p.T10I pathogenic variant has been associated with generalized lipodystrophy associated progeroid syndrome (7,8). Another report described 2 Caucasian brothers with more typical congenital generalized lipodystrophy due to a heterozygous missense LMNA pathogenic variant affecting lamin C p.(R571S) (9). Montenegro et al (10) reported 4 Brazilian patients from the same family with the p.R582C LMNA pathogenic variant. Interestingly, 1 of these family members with the homozygous p.R582C LMNA pathogenic variant had generalized fat loss unlike others, keeping in mind that the authors also questioned whether generalized fat loss could have been due to a situation of malnutrition. Therefore, at least a specific pathogenic variant in the heterozygous state, and other pathogenic variants in the homozygous state, can lead to more generalized fat loss in the clinical presentation.
The current classification system coined in the 80s and 90s and adopted into the 2016 Multi-Society Consensus Guidelines worked well before we started to understand the subtleties of molecular etiologies (2). Multi-Society Consensus Guidelines classify lipodystrophy based on the distribution of lost adipose tissue, affecting the entire body (generalized) or only several regions (partial). Principal features of FPLD type 2 are partial loss of subcutaneous fat from extremities and a variable reduction of subcutaneous adipose tissue from the trunk and abdomen. Our patients, however, had a remarkably distinct fat distribution characterized by fat loss primarily affecting the face, limbs, and the trunk, which was more similar to generalized lipodystrophy.
Loss of facial fat is an uncommon clinical presentation among carriers of pathogenic variants in the LMNA gene. In contrast, most affected individuals with FPLD type 2 have a rounded face, double chin, increased fat around the neck, and slender arms that can lead to misdiagnosis of Cushing syndrome. One exception to this is that loss of facial fat has been previously reported in carriers of the heterozygous missense LMNA pathogenic variant at p.R349W in exon 6 which probably is more progeroid in presentation than other variants (11). Despite the generalized fat loss, an accumulation of subcutaneous fat in the mons pubis region despite loss of fat tissue in the surrounding extremity regions may be a clue about the involvement of the LMNA gene in similar patients (6). This appearance on physical exam and radiological studies has been renamed the Dunnigan sign (6).
The LMNA gene is associated with a wide range of phenotypes, including neuromuscular and cardiac disorders in addition to lipodystrophy. Emery-Dreifuss muscular dystrophy (EDMD), limb-girdle muscular dystrophy (LGMD) type 1B, and LMNA-associated congenital muscular dystrophy are 3 major categories of muscle disease caused by laminopathies. Besides, various LMNA variants are associated with muscle abnormalities such as mild creatine phosphokinase elevations, muscle pain, stiffness, and discomfort.
Changes in residues R541 and K486 have been previously reported. Scharner et al (12) reported 2 families with codon 541 pathogenic variants. The index patient with the p.R541P pathogenic variant was diagnosed with LGMD type 1B and also had atrial fibrillation. She had a child diagnosed with EDMD and another with LGMD type 1B. The index patient with the p.R541S pathogenic variant required a heart transplant because of severe dilated cardiomyopathy. Also, other members of the family were diagnosed with either dilated cardiomyopathy or LGMD type 1B.
Adipose tissue loss was reported in female mutation carriers. Van Tintelen et al (13) reported a 13-year-old boy with the p.R541K pathogenic variant who had dilated cardiomyopathy and muscular symptoms with a mildly elevated creatine phosphokinase level. Sylvius et al (14) studied heart tissue in end-stage dilated cardiomyopathy associated with the p.R541S pathogenic variant which revealed nonspecific myocyte damage and interstitial fibrosis and nonspecific nuclear membrane alterations.
In our patient 1, genetic testing uncovered the LMNA variant which ultimately led to the subsequent confirmation of muscular dystrophy and cardiomyopathy. Prior to the confirmation of the muscular dystrophy, our patient was followed as potentially having inflammatory myositis and was treated with steroids, worsening her metabolic complications. Interestingly, this patient also presented with hypogonadotropic hypogonadism which may be related to low leptin levels, but may also be the result of an additional genetic defect. We did not perform more extensive next-generation sequencing on this patient. It is possible that such a defect may have potentially modified the presentation and resulted in a blended phenotype.
Patient 2, on the other hand, represents the first report of lymphoma in lipodystrophy caused by a pathogenic variant of LMNA. Reduced or absent expression of nuclear lamins was reported in human malignancies (15), including leukemia and lymphomas (16). A variant similar to p.K486E, p.K486N, has previously been associated with partial lipodystrophy although no detailed description of the index patient was reported in the original paper (17). This novel heterozygous LMNA pathogenic variant p.K486E, however, may be associated with a distinct fat loss pattern. Another intriguing possibility is the potential modification of the fat depots with the presence of active lymphoma. Alternatively, the patient may have a currently undiscovered predisposition to more extensive fat loss. At this point, there is no evidence to suggest any causal relationship between the coexistence of B cell follicular lymphoma and lipodystrophy caused by the novel pathogenic variant of the LMNA gene.
CONCLUSION
We recommend screening for the LMNA gene in patients who present with the generalized lipodystrophy phenotype if their clinical presentation has complex features. Positive results in this genetic test change clinical practice and may bring the complex multisystem disease to light. These patients will require a multidisciplinary approach by specialists who are familiar with the potential comorbidities.
ACKNOWLEDGMENT
We thank our patients for their willingness to share their stories and pictures for publications. We also acknowledge the contributions of Adam H. Neidert, Mario Swaidan, and Rita Hench for their clinical coordination efforts.
Abbreviations
- EDMD
Emery-Dreifuss muscular dystrophy
- FPLD
familial partial lipodystrophy
- FSD
fiber size disproportion
- LGMD
limb-girdle muscular dystrophy
Footnotes
DISCLOSURE
E.A.O. and J.E.A.W. were partially supported by the Lipodystrophy Fund at the University of Michigan which is graciously funded by the Sopha Family and the White Point Foundation of Turkey. E.A.O. received grant support from and served as an advisor to Amylin Pharmaceuticals LLC, Bristol-Myers Squibb, and AstraZeneca in the past and is currently receiving grant support from Gemphire Therapeutics, Aegerion Pharmaceuticals, Ionis Pharmaceuticals, and Akcea Therapeutics and is serving as an advisor to Aegerion Pharmaceuticals, Akcea Therapeutics, and Regeneron Pharmaceuticals through funding paid to the University of Michigan. E.A.O. recently started grant support from GI Dynamics. B.A. has attended scientific advisory board meetings organized by Aegerion Pharmaceuticals and has received honoraria as a speaker from AstraZeneca, Lilly, MSD, Novartis, Novo Nordisk, Boehringer-Ingelheim, Servier, and Sanofi-Aventis. The other authors have no multiplicities of interest to disclose.
REFERENCES
- 1.Akinci B, Meral R, Oral EA. Phenotypic and genetic characteristics of lipodystrophy: pathophysiology, metabolic abnormalities, and comorbidities. Curr Diab Rep. 2018;18:143. doi: 10.1007/s11892-018-1099-9. [DOI] [PubMed] [Google Scholar]
- 2.Brown RJ, Araujo-Vilar D, Cheung PT et al. The diagnosis and management of lipodystrophy syndromes: a multi-society practice guideline. J Clin Endocrinol Metab. 2016;101:4500–4511. doi: 10.1210/jc.2016-2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oral EA, Gorden P, Cochran E et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with partial lipodystrophy. Endocrine. 2019;64:500–511. doi: 10.1007/s12020-019-01862-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Golden JK. Myalept (metreleptin for injection) Clinical Review. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125390Orig1s000MedR.pdf Accessed February 15, 2020.
- 5.Colman EC. Myalept/metreleptin. Summary Review. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125390Orig1s000SumR.pdf Accessed February 15, 2020.
- 6.Meral R, Ryan BJ, Malandrino N et al. “Fat Shadows” from DXA for the qualitative assessment of lipodystrophy: when a picture is worth a thousand numbers. Diabetes Care. 2018;41:2255–2258. doi: 10.2337/dc18-0978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hussain I, Patni N, Ueda M et al. A novel generalized lipodystrophy-associated progeroid syndrome due to recurrent heterozygous LMNA p.T10I mutation. J Clin Endocrinol Metab. 2018;103:1005–1014. doi: 10.1210/jc.2017-02078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Csoka AB, Cao H, Sammak PJ, Constantinescu D, Schatten GP, Hegele RA. Novel lamin A/C gene (LMNA) mutations in atypical progeroid syndromes. J Med Genet. 2004;41:304–308. doi: 10.1136/jmg.2003.015651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patni N, Xing C, Agarwal AK, Garg A. Juvenile-onset generalized lipodystrophy due to a novel heterozygous missense LMNA mutation affecting lamin C. Am J Med Genet A. 2017;173:2517–2521. doi: 10.1002/ajmg.a.38341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montenegro RM, Jr, Costa-Riquetto AD, Fernandes VO et al. Homozygous and heterozygous nuclear lamin A p.R582C mutation: different lipodystrophic phenotypes in the same kindred. Front Endocrinol (Lausanne) 2018;9:458. doi: 10.3389/fendo.2018.00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akinci B, Onay H, Demir T et al. Clinical presentations, metabolic abnormalities and end-organ complications in patients with familial partial lipodystrophy. Metabolism. 2017;72:109–119. doi: 10.1016/j.metabol.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 12.Scharner J, Brown CA, Bower M et al. Novel LMNA mutations in patients with Emery-Dreifuss muscular dystrophy and functional characterization of four LMNA mutations. Hum Mutat. 2011;32:152–167. doi: 10.1002/humu.21361. [DOI] [PubMed] [Google Scholar]
- 13.van Tintelen JP, Hofstra RM, Katerberg H et al. High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am Heart J. 2007;154:1130–1139. doi: 10.1016/j.ahj.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 14.Sylvius N, Bilinska ZT, Veinot JP et al. In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J Med Genet. 2005;42:639–647. doi: 10.1136/jmg.2004.023283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin F, Worman HJ. Expression of nuclear lamins in human tissues and cancer cell lines and transcription from the promoters of the lamin A/C and B1 genes. Exp Cell Res. 1997;236:378–384. doi: 10.1006/excr.1997.3735. [DOI] [PubMed] [Google Scholar]
- 16.Stadelmann B, Khandjian E, Hirt A, Lüthy A, Weil R, Wagner HP. Repression of nuclear lamin A and C gene expression in human acute lymphoblastic leukemia and non-Hodgkin's lymphoma cells. Leuk Res. 1990;14:815–821. doi: 10.1016/0145-2126(90)90076-l. [DOI] [PubMed] [Google Scholar]
- 17.Shackleton S, Lloyd DJ, Jackson SN et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24:153–156. doi: 10.1038/72807. [DOI] [PubMed] [Google Scholar]