

Abstract
G4C2 repeat expansions in an intron of C9ORF72 cause the most common familial amyotrophic lateral sclerosis and frontotemporal dementia (collectively, C9ALS/FTD). Mechanisms and mediators of C9ALS/FTD pathogenesis remain poorly understood. C9orf72 and Smcr8 form a protein complex. Here, we show that expression of Smcr8, like C9orf72, is reduced in C9ALS/FTD mouse models and patient tissues. Since Smcr8 is highly conserved between human and mouse, we evaluated the effects of Smcr8 downregulation in mice. Smcr8 knockout (KO) mice exhibited motor behavior deficits, which resemble those of C9ALS/FTD mouse models, and displayed axonal swellings in their spinal cords and neuromuscular junctions. These deficits are caused by impaired autophagy-lysosomal functions due to disrupted axonal transport in mutant motor neurons. Consistent with its interaction with C9orf72 and their downregulation in patient tissues, Smcr8 deficiency exacerbated autophagy-lysosomal impairment in C9orf72 KO mice. The disease relevance of Smcr8 downregulation was reflected by exacerbated axonal swellings and gain of toxicity pathology arising from Smcr8 haploinsufficiency in a mouse model of C9ALS/FTD. Thus, our in vivo studies suggested that Smcr8 deficiency impairs axonal transport dependent autophagy-lysosomal function and exacerbates axonal degeneration and gain of toxicity in C9ALS/FTD mouse models.
Introduction
Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are neurodegenerative disorders that occur simultaneously in certain populations of patients (1, 2). G4C2 hexanucleotide repeat expansion in a noncoding region of the gene chromosome 9 open reading frame 72 (C9ORF72) is the most common cause of familial ALS and FTD (C9ALS/FTD) (3–6). C9ORF72-associated ALS accounts for around 40% of familial ALS and about 5–10% of sporadic ALS cases (4, 5). Haploinsufficiency of C9ORF72 protein is a key proposed disease mechanism, which may act in parallel with gain of toxicity from toxic RNAs of repeat transcription and dipeptide repeat proteins (DPRs) from repeat-associated non-AUG (RAN) translation (1, 2). Consistent with this loss-of-function notion, C9ORF72 expression is reduced in C9ALS/FTD patient tissues (3, 5, 7, 8); haploinsufficiency of C9ORF72 is considered to contribute to the degeneration of motor neurons (MNs) derived from induced pluripotent stem cells of ALS patients (9); C9orf72 deficiency exacerbates motor behavior deficits of a C9ALS/FTD mouse model in vivo (10). Therefore, studying C9orf72 biology may provide new insights into pathogenesis of C9ALS/FTD from the perspective of loss-of-function.
C9orf72 forms a protein complex with Smcr8 (11–17). They are both DENN domain-containing proteins (18, 19) and regulate autophagy, a lysosome-dependent degradation process (20, 21). Enlarged lysosomal sizes and impaired lysosomal degradation have been observed in cells deficient in C9orf72 or Smcr8 (22, 23). These results suggest the possibility that autophagy-lysosomal function may be impaired in C9ALS/FTD. Interestingly, neither heterozygous nor homozygous knockout (KO) of C9orf72 in neurons leads to MN degeneration or motor deficits in mice at advanced ages (24). This raises the question whether and to what extent C9orf72’s functions are dysregulated in C9ALS/FTD in vivo. Consequently, the potential autophagy-lysosomal deficiency in C9ALS/FTD in vivo is unclear, and the mediators and underlying mechanisms remain poorly understood.
Using a C9ORF72 bacterial artificial chromosome (BAC) transgenic mouse model (C9-BAC) (25), we recently generated a genetic mouse model (C9orf72+/−;C9-BAC) of C9ALS/FTD with both loss- and gain-of-function features. We found that C9orf72 deficiency and haploinsufficiency exacerbate motor behavior deficits of C9-BAC mice in a dose-dependent manner, and this occurs early in the course of pathogenesis (10). To investigate the mechanisms underlying these motor deficits, we examined Smcr8 and found that it, like C9orf72, is reduced in C9ALS/FTD mouse models and patient tissues. Smcr8 mutant mice developed motor behavior deficits, suggesting that Smcr8 downregulation could be a mediator of motor deficits in C9orf72+/−;C9-BAC mice. Mechanistic studies revealed that Smcr8 deficiency disrupted axonal transport in MNs, impaired autophagy-lysosomal function, and led to abnormal axonal swellings in spinal cords and neuromuscular junctions (NMJs) in mice. These deficits were more severe in the background of C9orf72 KO. Smcr8 deficiency also exacerbated the accumulation of DPRs in C9-BAC mouse models, suggesting the interaction between loss- and gain-of-function in the pathogenesis of C9ALS/FTD.
Results
Smcr8 is downregulated in C9ALS/FTD mouse models and patient tissues
C9orf72 dose reduction exacerbates motor deficits of C9-BAC mice at an early stage (10), when C9-BAC mice display no behavior deficits (25). To investigate mechanisms underlying these motor deficits, we focused on Smcr8. Smcr8 and C9orf72 form a protein complex that regulates autophagy-lysosomal functions (11–17). They stabilize each other; Smcr8 protein level is reduced in C9orf72-deficient cells (13, 15, 26). However, it remains unknown whether the Smcr8 protein level changes under pathological conditions in which C9orf72 protein is partially lost. Therefore, we examined Smcr8 expression in spinal cords from C9-BAC mice with different C9orf72 dose reductions. Western blot analyses confirmed C9orf72 protein dose reduction with or without the presence of C9-BAC (Fig. 1A and B). Interestingly, Smcr8 protein levels were reduced accordingly in response to C9orf72 dose reduction. In contrast, the level of Atg101, another complex-associated protein, did not change (Fig. 1A and B) (12), suggesting the specificity of Smcr8 downregulation.
Figure 1.
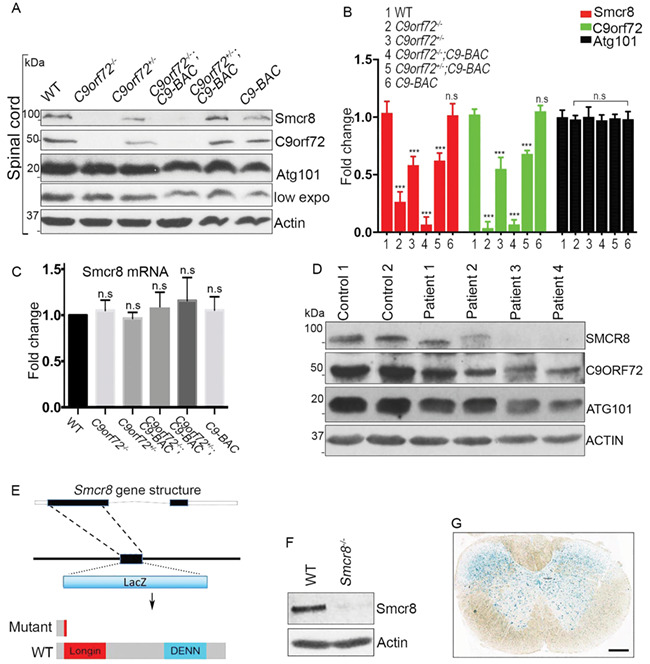
Smcr8 downregulation in C9ALS/FTD and mutant mouse generation. (A) Western blot analysis of protein expression in spinal cords. β-Actin serves as the loading control. (B) Quantification of protein levels of Smcr8 and other proteins in spinal cords. (C) RT-PCR analysis of Smcr8 mRNA expression. (D) Western blot analysis of the protein expression of Smcr8, C9orf72 and Atg101 in C9ALS/FTD patient brain tissues. β-Actin serves as the loading control. (E) Diagram of the generation of Smcr8−/− mutant mice. (F) Western blot analysis confirmed the absence of Smcr8 protein in mutant mice. β-actin serves as the loading control. (G) LacZ staining of coronal sections from 2-month-old heterozygous mice shows Smcr8 expression in spinal cord. Scale bars: 100 μm. Data are presented as mean ± SEM from three independent experiments containing one WT and one mutant mice in each experiment (n = 3). Statistical analyses were performed with one-way ANOVA with Bonferroni’s post hoc test (***P < 0.001, n.s. represents no significant difference detected).
To investigate whether Smcr8 downregulation exists at the mRNA level, we performed RT-PCR analyses and found that there were no significant changes of Smcr8 mRNA levels (Fig. 1C). These results suggest that Smcr8 downregulation may occur post-transcriptionally. To determine whether SMCR8 downregulation occurs in patient tissues, we performed western blot analyses using postmortem patient brain tissues. As reported in published data (3, 4, 7), C9ORF72 protein levels were reduced in patient brain lysates (Fig. 1D). Consistent with Smcr8 downregulation from mouse models, SMCR8 protein expression was also decreased in patient brain tissues (Fig. 1D). Together, our studies revealed a Smcr8 protein reduction in C9ALS/FTD mouse models and patient brain tissues.
Smcr8 deficient mice exhibit motor behavior deficits
To investigate the importance of Smcr8 downregulation, we generated Smcr8 null mice in which the majority of Smcr8 exon 1 was replaced by a LacZ cassette (Fig. 1E). Western blot analyses confirmed the absence of Smcr8 proteins in the homozygous mutant mice (Fig. 1F); therefore, we refer to these mice as Smcr8−/−. To guide our functional studies, we examined Smcr8’s expression pattern by performing X-gal staining. We found that Smcr8 was ubiquitously expressed in the spinal cords and in different brain regions including the cerebral cortex, hippocampus and cerebellum (Fig. 1G, Fig. S1A). Immunohistochemical (IHC) staining revealed that Smcr8 was expressed in the cytoplasm of NeuN-positive neurons (Fig. S1B), including SMI32-labeled MNs (Fig. S1C).
Smcr8−/− mice were fertile, viable and displayed no visiable developmental defects. Offspring from heterozygous matings were born in the expected Mendelian ratios. To assess motor behaviors, we examined motor strength by measuring forelimb grip strength. Smcr8−/− mice exhibited a decrease in motor strength compared to WT controls, in both males and females, at 9 months of age (Fig. 2A). Grip strength deficits could be detected at 3, 9 and 12 months of age (Fig. 2B). After detecting grip strength deficits in both sexes, we focused on female mice because C9-BAC female mice exhibited more consistent behavior deficits than male mice (25). To measure motor coordination and motor learning, we performed an accelerating rotarod test. Smcr8+/− and Smcr8−/− mice had a significant reduction in latency to fall compared to age-matched WT controls (Fig. 2C). WT mice exhibited an increased trend of latency to fall in the rotarod test over a 4-day test period, indicating an active learning process during this period of time (Fig. 2C). In contrast, Smcr8−/− mice failed to display an increase in latency to fall, suggesting motor learning deficits. Open field analyses revealed that Smcr8−/− mice had a decrease in the total distance traveled and the percentage of time spent in the center (Fig. 2D–F). Together, these results suggest that Smcr8 deficiency developed motor behavior deficits in mice.
Figure 2.
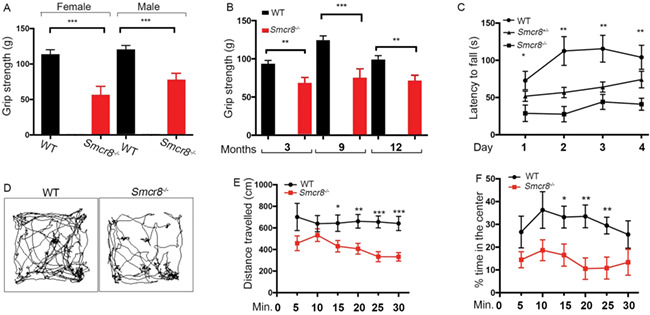
Smcr8 KO mice exhibit motor behavior deficits. (A) Forelimb grip strength assay on 9-month-old WT (n = 9 females, 13 males) and Smcr8−/− (n = 8 females, 12 males) mice. (B) Forelimb grip strength is reduced in Smcr8−/− (n = 11) female mice compared to WT Smcr8+/+ (n = 10) mice at 3, 9 and 12 months of age. (C) Rotarod assay reveals the decreased latency to fall in Smcr8−/− (n = 11) female mice compared to WT Smcr8+/+ (n = 10). Smcr8−/− mice exhibited a decreased rate of motor learning in the rotarod test over a 4-day test period compared to WT controls. (D) Representative traces showing the paths of mice in the open field chambers (snapshots taken at 5-minute intervals). (E) Smcr8−/− (n = 12) mutant mice traveled less distance (5-minute interval) compared to Smcr8+/+ (n = 10) control mice. (F) Smcr8−/− (n = 12) mutant mice spent less time in the center of the open field chambers in every 5-minute trial compared to Smcr8+/+ (n = 10) control mice. Statistical analysis was performed by one-way ANOVA analysis with Bonferroni correction; n.s. represents no significant difference detected (*P < 0.05, **P < 0.01 and ***P < 0.001).
Smcr8 deletion disrupts autophagy-lysosomal degradation and leads to axonal swellings
To identify the cellular basis of motor deficits in Smcr8−/− mutant mice, we first examined the central nervous system in 7-month-old mice. H&E staining did not reveal obvious loss or disorganization of major brain or spinal cord regions; NeuN-positive neurons were not lost in the cortex or spinal cord of Smcr8−/− mice. Next, we assessed nerve fibers in spinal cords using Bielschowsky silver impregnation analysis, which is used for the visualization of neuronal processes including axons. Although swollen neuronal processes were seldom detected in WT controls, there was a robust increase in swollen neuronal processes in mutant spinal cords (Fig. 3A and B). We used antibodies against neurofilament (NF) to label axons. Smcr8−/− spinal cords exhibited mainly NF-positive swellings (white arrowheads in Fig. 3C). These results suggest that these neuronal processes were of axonal origin; therefore, we refer to them as axonal swellings. Statistical analysis revealed that axonal swellings increased as age advanced in Smcr8−/− spinal cords (Fig. 3D).
Figure 3.
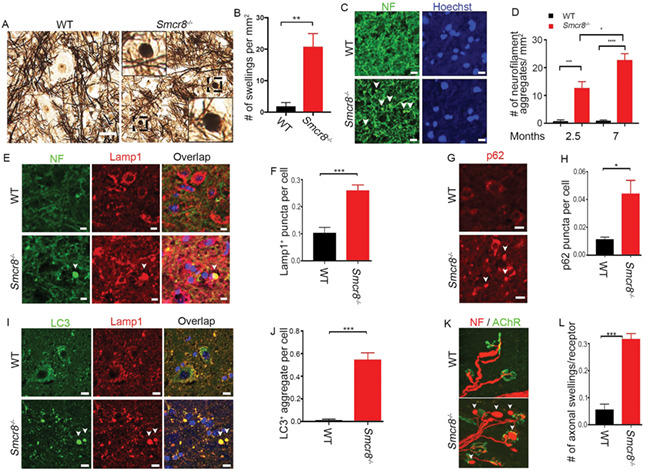
Impaired autophagy-lysosomal degradation leads to axonal swellings in Smcr8−/− mice. (A) Bielschowsky silver impregnation analyses revealed swelling neuronal processes in 7-month-old mutant spinal cords. Individual spheroid structures are enlargements of black boxed areas. Scale bars: 20 μm. (B) Quantification of numbers of swollen neuronal processes in spinal cord sections. (C) Representative imaging of spinal cords stained with antibodies against NF (NF, green). White arrowheads represent axonal swellings. Hoechst stains nuclei. Scale bars: 10 μm. (D) Quantification of NF-positive axonal swellings per mm2 in the spinal cords. Two-way ANOVA analysis with Bonferroni correction detected a significant difference between 2.5 and 7 months. (E, G, I) Confocal imaging of spinal cord sections stained with antibodies against NF (NF, green), LC3 (green), Lamp1 (red) and p62 (red). White arrowheads indicate axonal swellings, LC3-positive aggregates, or p62 puncta. Scale bars: 10 μm. (F, H, J) Quantification of Lamp1-positive puncta outside of perinuclear areas per cell (F), the number of p62 puncta per cell (H), and the number of LC3+ aggregate per cell (J). (K) Smcr8−/− mice display NMJ swellings stained with antibodies against NF (NF, red) and AChR (green). White arrowheads indicate swelling NMJs. (L) Quantification of numbers of NMJ swellings in Smcr8−/− mice. For all the experiments, error bars represent SEM using measurements averaged from ≥3 sections of each mutant mice (n = 3). Statistical analyses were performed with non-parametric Mann-Whitney test (*P < 0.05, **P < 0.01 and ***P < 0.001).
Smcr8 is a lysosome-associated protein and regulates autophagy-lysosomal functions (12, 13). Massive accumulation of Lamp1-positive axonal lysosomes has been reported in patients with neurodegenerative conditions (27). Therefore, we examined Lamp1 and found that it mainly co-localized with NF in the axonal swellings (Fig. 3E), and was barely detected in GFAP-labeled astrocytes and Iba1-labeled microglia (Fig. S2A and B). Lamp1-positive aggregate numbers were significantly increased in mutants (Fig. 3F). Lamp1-positive aggregates in mutant spinal cords were scattered in the cytoplasm without the perinuclear concentration seen in WT (Fig. 3E), suggestive of immature lysosome precursors with deficient degradation capacity. In support of this notion, Smcr8−/− spinal cords displayed abnormal accumulation of p62, a substrate and receptor of autophagy degradation (Fig. 3G and H). Mutant spinal cords also exhibited an increase in the numbers of LC3-positive aggregates, which co-localize with Lamp1 (Fig. 3I and J). These results suggested impaired autophagy-lysosomal degradation coupled with axonal swellings in Smcr8−/− spinal cords. To determine whether these deficts occur at the axonal terminals of MNs, we examined the NMJs. The presynaptic terminals of mutant MNs exhibited swellings with NF accumulation (white arrowheads, Fig. 3K). Quantification confirmed that the numbers of axonal swellings were significantly increased in mutant NMJs (Fig. 3L).
To further investigate autophagy-lysosomal deficits, we examined axons using transmission electron microscopy (TEM) for the presence of autophagic vacuoles (AVs). Multiple criteria have been used to define AVs, including size, the presence of a double membrane, and the existence of multiple membrane-derived structures within a single vacuole (28). WT spinal cords had axonal structures that appeared typical, with no abnormal accumulation of organelles or vesicles (Fig. 4A). In contrast, Smcr8−/− spinal cords displayed swollen axons accompanied by large numbers of accumulated organelles and vesicles, including mitochondria (black arrows in Fig. 4B), double-membrane AVs containing engulfed organelles in the early stages of autophagy (green arrowheads in Fig. 4B and C), and late-stage autolysosomes containing degraded organelles (red arrowheads in Fig. 4C). Together, these results suggest that Smcr8 deletion disrupted autophagy-lysosomal degradation and led to axonal degeneration characterized by dystrophic neurites.
Figure 4.
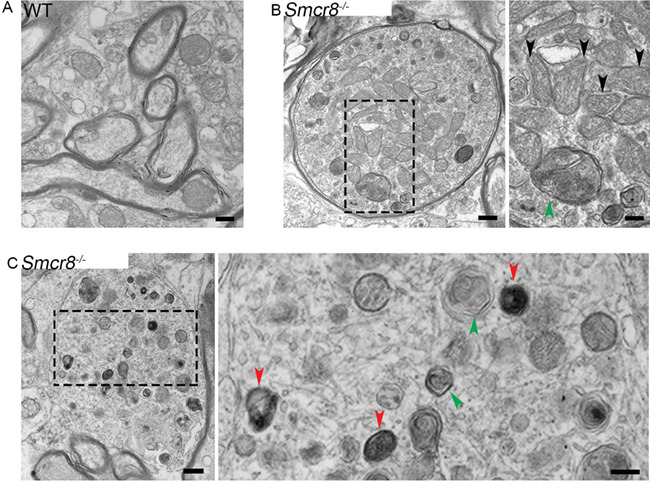
Transmission electronic microscopy (TEM) study reveals swollen axons with organelle accumulation in mutant spinal cords. (A) Representative TEM imaging of axons in WT spinal cords. Scale bar: 400 nM. (B) TEM images of mutant spinal cords showing an axonal swelling filled with double-membrane autophagosomes (green arrowhead) and multiple mitochondria (black arrowheads) in mutants. Scale bars: 400 (left panel), and 200 nM (right panel). (C) A typical axonal swelling filled with large numbers of lysosome-like organelles at different maturation stages. Red arrowheads represent late stage-autolysosomes containing degraded organelles; green arrowheads represent autophagosomes in the early stage of autophagy. Scale bars: 400 (left panel) and 200 nM (right panel).
Axonal transport is disrupted in Smcr8−/− MNs
Our previous mass spectrometry studies revealed that the C9orf72/Smcr8 complex associates with Dynein (12). Dynein is the major motor that drives retrograde transport of autophagosomes from the distal axons to somas of neurons (29, 30). Disruption of dynein-mediated axonal transport leads to autophagic stress and axonal terminal enlargement (31, 32), which were observed in Smcr8−/− mice. Therefore, we hypothesized that Smcr8 promotes axonal transport, disruption of which leads to impaired autophagy-lysosomal functions and axonal swellings. To test this hypothesis, we examined axonal transport of GFP-LC3-labeled autophagosomes in mouse spinal MNs using time-lapse imaging approaches. In WT MNs, autophagosomes appeared as smooth vesicular structures distributed evenly along neuronal processes. In contrast, they were clustered in Smcr8−/− MNs (Fig. 5A and B). Time-lapse imaging revealed that autophagosomes in WT MNs exhibited robust motility along the processes, with a predominant retrograde direction. However, the net run speed and run length of autophagosomes were significantly reduced in Smcr8−/− MNs (Fig. 5C and D). The stall numbers per trace were increased in mutant MNs (Fig. 5E). Smcr8−/− MNs had a significantly reduced percentage of retrograde transport accompanied by an increased percentage of stationary autophagosomes (Fig. 5F). Thus, Smcr8 deletion disrupted axonal transport in MNs, which could result in autophagy-lysosomal impairment and axonal swellings in mutant mice.
Figure 5.
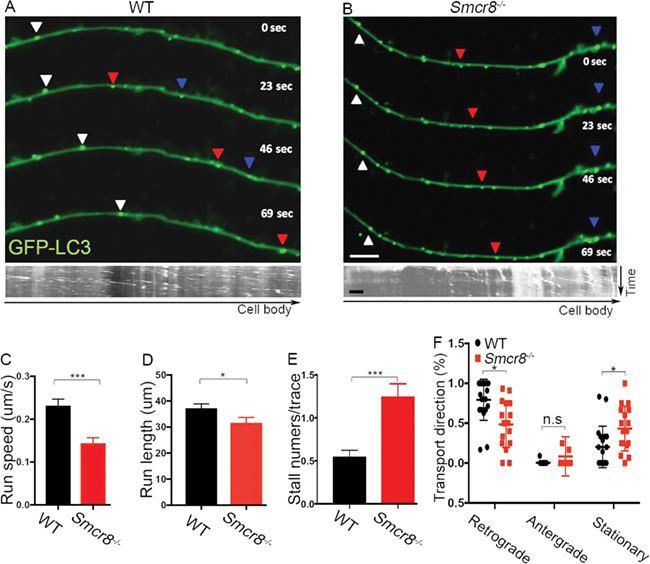
Axonal transport is disrupted in Smcr8−/− MNs. (A, B) Representative time-lapse images and corresponding kymographs of GFP-LC3-labeled autophagosome transport in spinal MNs isolated from E13.5 spinal cords. Individual autophagosomes marked by white triangles show robust retrograde transport in WT and diminished motility in mutant MNs. Scale bars: 2 μm. (C) Quantification of net run speed in WT (n = 126) and Smcr8−/− (n = 120) MNs. (D) Quantification of total run length in WT (n = 145) and Smcr8−/− (n = 87) MNs. (E) Quantification of stall numbers per trace in WT (n = 109) and Smcr8−/− (n = 125) MNs. (F) Quantification of the percentages of retrograde, anterograde, and stationary transport in WT (n = 20) and Smcr8−/− (n = 18) MNs. Error bars represent SEM of ≥5 independent experiments with more than five mice per group (n > 5). Statistical analyses were performed with non-parametric Mann-Whitney test (*P < 0.05, ***P < 0.001, n.s. represents no significant difference detected).
C9orf72 loss of function exacerbates autophagy-lysosomal deficits in Smcr8 mutant mice
C9orf72 depletion in neurons failed to generate neurodegeneration or motor deficits (24); C9orf72’s neuronal functions remain unknown. C9orf72 and Smcr8 form a protein complex and belong to the same DENN (differentially expressed in neoplastic versus normal cells) domain family (18, 33). Therefore, we hypothesized that Smcr8, which has robust neuronal function as described above, may compensate for the loss of C9orf72 in C9orf72−/− neurons. To test this hypothesis, we deleted C9orf72 in the background of Smcr8−/− mice. We examined axonal integrity and autophagy-lysosomal functions by performing side-by-side comparisons of spinal cords from WT, C9orf72−/−, Smcr8−/− and C9orf72−/−;Smcr8−/− double knockout (dKO) mice. In contrast to Smcr8−/− mice, C9orf72−/− mutant spinal cords displayed no significant changes in the numbers of Lamp1-positive organelles (Fig. 6A and B), NF aggregates (Fig. 6C and D), or p62 puncta (Fig. 6E and F). However, in the background of Smcr8−/−, C9orf72 deletion promoted deficits in the numbers of Lamp1- and p62-positive puncta as well as NF aggregates (Fig. 6B, D, and F). Together, these results suggest that C9orf72 deficiency exacerbates autophagy-lysosomal deficits and axonal swellings in Smcr8−/− spinal cords.
Figure 6.
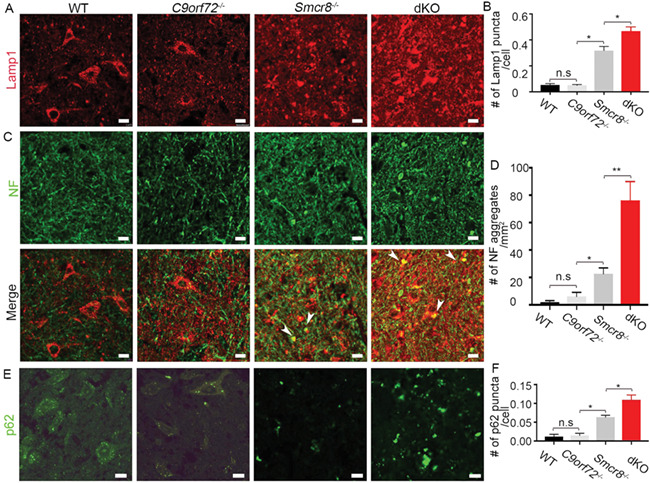
Smcr8 deficiency exacerbates autophagy-lysosomal impairment in C9orf72−/− spinal cords. (A, C, E) Representative confocal imaging of spinal cords from wild type (WT), C9orf72−/−, Smcr8−/− and C9orf72−/−; Smcr8−/− dKO mice at 7 months of age. Coronal sections were stained with antibodies against Lamp1 (red), NF (green), or p62 (green). Hoechst stains nuclei. White arrowheads represent axonal swellings with the co-localization of Lamp1 and NF. Scale bars: 20 μm. (B, D, F) Quantification of Lamp1-positive puncta per cell, numbers of NF aggregates per mm2 area, or numbers of p62-positive puncta per cell (F). All data are presented as mean ± SEM using measurements averaged from ≥3 sections of each mutant mice (n = 3). Statistical analyses were performed with one-way ANOVA with Bonferroni correction’s post hoc test (*P < 0.05, **P < 0.01); n.s. represents no significant difference detected.
Next, we extended our studies to the axonal terminals of MNs by focusing on diaphragm NMJs in 7-month-old mice. Similar to WT animals, C9orf72−/− diaphragms presented with normal NMJ morphology. NF-labeled presynaptic terminals were able to form synapses appropriately with AChR-positive postsynaptic terminals without Lamp1-positive organelle accumulation (Fig. 7A–C), while the size of NMJ axonal terminal was slightly increased in C9orf72−/− mice (Fig. 7E). Smcr8−/− mice exhibited multiple NMJ abnormalities, including an increase in the numbers and sizes of axonal terminals (Fig. 7D and E). In comparison to Smcr8−/− single mutants, dKO mice displayed a significant increase in the number of axonal swellings per NMJ receptor as well as an increase in the axonal terminal sizes (Fig. 7D and E). Together, these results suggest that C9orf72 has neuronal functions in promoting autophagy-lysosomal degradation, which are similar to and dominated by Smcr8 under physiological conditions.
Figure 7.
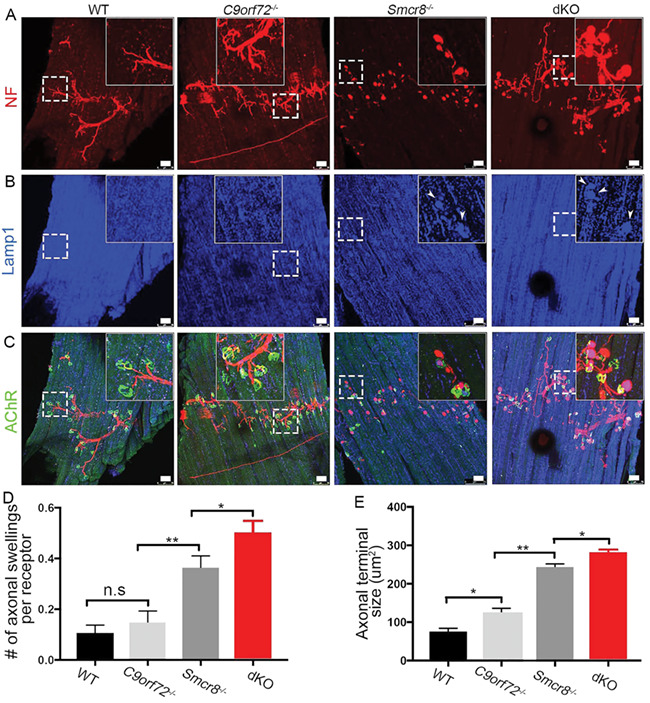
C9orf72 deficiency promotes terminal axon degeneration of Smcr8−/− NMJs. (A–C) Representative confocal imaging of diaphragms from wild type (WT), C9orf72−/−, Smcr8−/− and C9orf72−/−; Smcr8−/− dKO mice. Diaphragms from 7-month-old mice were stained with antibodies against NF (red), Lamp1 (blue), and acetylcholine receptor AChR (green). Hoechst stains nuclei. Inserts in top panels are enlargements of white boxed areas in individual panels. Scale bars: 50 μm. (D, E) Quantification of the numbers of axonal swellings associated with each AChR receptor, and sizes of axonal swellings. All data are presented as mean ± SEM using measurements averaged from ≥3 sections of each mutant mice (n = 3). Statistical analyses were performed with one-way ANOVA with Bonferroni’s post hoc test; n.s. represents no significant difference detected (*P < 0.05, **P < 0.01).
Smcr8 deficiency promotes axonal swellings of NMJs in C9-BAC mouse models
C9orf72 deficiency promotes motor behavior deficits in a gain-of-function mouse model of C9ALS/FTD (10), but the underlying mechanisms remain unknown. C9orf72 stabilizes its interaction protein Smcr8 (13, 15); Smcr8 expression is reduced in mouse models and patient tissues of C9ALS/FTD. Smcr8 deficiency is sufficient to disrupt autophagy-lysosomal functions and lead to axonal swellings and motor deficits, which resemble phenotypes in C9orf72+/−;C9-BAC mice. Therefore, we hypothesized that Smcr8 deficiency promotes autophagy-lysosomal impairment and leads to axonal swellings in C9-ABC mice. To test this hypothesis, we crossed Smcr8 deficient mice with C9-BAC mice, and focused our analyses on age-matched WT, Smcr8+/−, C9-BAC, and Smcr8+/−;C9-BAC mice. Impaired autophagy-lysosomal function led to axonal swellings. In comparison to WT, C9-BAC and Smcr8+/− mice exhibited an increase in the percentage of NMJs with axonal swellings (Fig. 8A–D), which was further exacerbated in Smcr8+/−;C9-BAC mice (Fig. 8E). Together, these results suggest that Smcr8 deficiency promotes axonal swellings of NMJs in C9-BAC mouse models.
Figure 8.
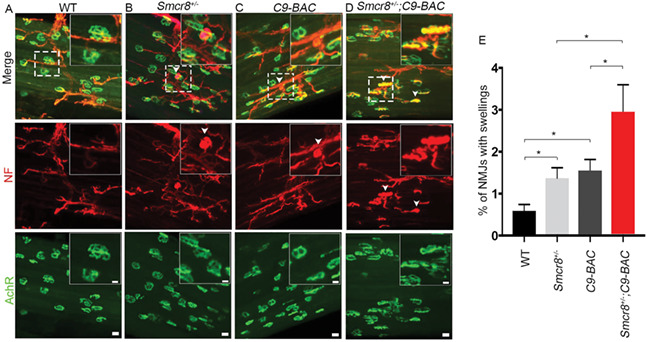
Smcr8 deficiency promotes axonal swellings in C9-BAC mice. (A–D) Representative confocal imaging of 9-month-old mouse diaphragms stained with antibodies against NF (NF, red), and acetylcholine receptor AChR (green). White arrowheads represent NF-positive axonal swellings or Lamp1-positive puncta, which are rarely detected in WT controls. Upper right panels are enlargements of white boxed areas in merged panels. Scale bars: 10 (upper panels) and 20 μm (bottom panels). (E) Quantification of NMJs with swellings. All data are presented as mean ± SEM using measurements averaged from ≥3 sections of each mutant mouse (n > 3). Statistical analysis was performed by one-way ANOVA analysis with Bonferroni correction (*P < 0.05).
Smcr8 deficiency exacerbates dipeptide repeat (DPR) abundance in C9-BAC mice
Gain of toxicity is attributed by DPRs from RAN translation in C9ALS/FTD (34, 35). Transgenic mice containing C9ORF72 BAC from patient DNA have been generated to model gain-of-toxicity (25, 36–38). In addition to axonal swellings, we attempted to test whether Smcr8 deficiency promotes the accumulation of DPRs, which are toxic to neurons. It has been documented that the amount of inclusions for poly (PR) and poly (PA) are relatively rare in postmortem C9ALS/FTD patient brains (39, 40). Whereas the poly (GP) is considered to be benign, both poly (GR) and poly (GA) have been shown to be particularly toxic in various cell types and animal models (41–43). We focused our studies on poly (GA) and poly (GP). We barely detected poly (GA) staining in 7-month-old WT mouse tissues. In contrast, age matched C9-BAC mouse spinal cords exhibited robust poly (GA) signals (Fig. 9A). The percentage of poly (GA)-positive cells was significantly increased in Smcr8+/−;C9-BAC mice compared to C9-BAC mice (Fig. 9B). Next, we examined poly (GP), and found poly (GP) staining in spinal cord tissues of 7-month-old WT mice (Fig. 9C). Age matched C9-BAC mice displayed an increase in the percentage of poly (GP)-positive cells, which was further increased by the Smcr8 heterozygosity (Fig. 9D). Together, these results suggest that Smcr8 deficiency exacerbates the DPR gain of toxicity in C9-BAC mice.
Figure 9.
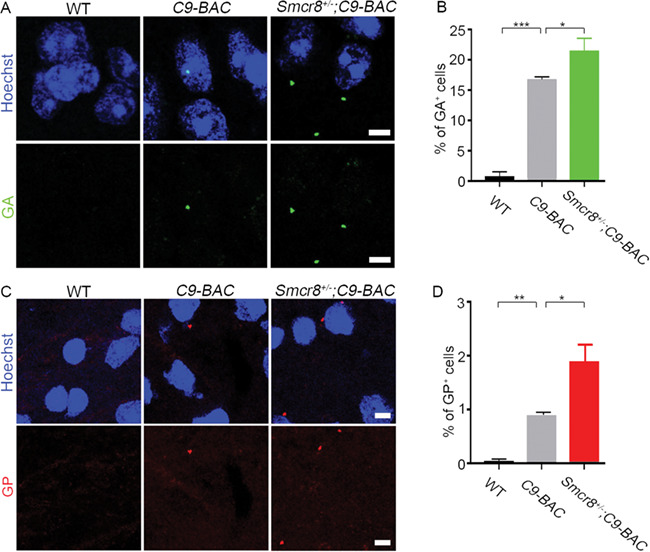
Smcr8 deficiency exacerbates gain of toxicity in C9-BAC mice. (A) Representative imaging of coronal sections stained with antibodies against poly (GA) (green). Age-matched 7-month-old mice were used to prepare sections in the area of spinal cords. Scale bars: 5 μm. (B) Quantification of the percentage of cells with poly (GA) puncta out of total cells from 3 WT, 4 C9-BAC and 4 Smcr8+/−;C9-BAC mice. (C) Representative imaging of coronal sections stained with antibodies against poly (GP) (red). Age-matched 7-month-old mice were used to prepare sections in the area of spinal cords. Scale bars: 5 μm. (D) Quantification of the percentage of cells with poly (GP) puncta out of total cells from 4 WT, 4 C9-BAC and 3 Smcr8+/−; C9-BAC mice. Error bars represent SEM of four independent experiments. Statistical analyses were performed with one-way ANOVA with Bonferroni’s post hoc test (*P < 0.05, **P < 0.01 and ***P < 0.001).
Discussion
Here, we discovered that Smcr8 deficiency disrupts MN axonal transport and lysosomal functions, leading to axonal swellings, which in turn contributes to motor behavior deficits in mice. The expression of Smcr8, like that of C9orf72, is reduced in mouse models and patient tissues. Smcr8 deficiency exacerbates axonal swellings and DPR gain of toxicity in C9-BAC mouse models, providing new insights into the pathogenesis of C9ALS/FTD.
Using genetic KO mouse models, for the first time, our studies discovered Smcr8’s neuronal and behavioral functions in vivo. Specifically, Smcr8 promotes autophagy-lysosomal functions in MNs. It has been reported that local homeostasis of axon terminals, in comparison to dendrites or spines, is particularly vulnerable to autophagy impairment (45, 46). Indeed, progressive axonal dystrophic swellings were identified as early and pronounced deficits in Smcr8 mutant mice. Although Smcr8 is widely expressed, these axonal swellings were mostly detected in spinal cords and NMJs, not in different brain regions of mutant mice. These observations are consistent with the notion that MNs, with their unique long axons, are particularly vulnerable to the disruption of autophagy-lysosomal pathway (47). With progressive age, axonal swellings in Smcr8−/− mice become more severe, which correlates with motor behavior deficits in mutant mice. Multiple evidences suggest that NF-positive swellings are axon-derived, including TEM studies and NMJs. Recent studies suggest that Lamp1-labeled lysosomal precursors, including endosomes and autophagosomes, are initiated at the distal tips of axons and mature via axonal transport toward proximal regions where luminal proteases can be effectively obtained (27, 29–31). We found that Smcr8 mutant MNs exhibited disrupted axonal transport of autophagosomes, including reduced net run speed, run length and retrograde transport. Future studies should determine the mechanisms underlying the impaired axonal transport in Smcr8−/− MNs, such as potential deficits in motor activities, cargo loading, or cargo releasing. It is also important to investigate to what extent restoring axonal transport can rescue autophagy-lysosomal degradation and axonal swelling deficits in Smcr8−/− MNs. Overall, Smcr8 deletion impaired axonal transport and autophagy-lysosomal functions, which resulted in axonal swellings contributing to motor deficits in mutant mice.
Our Smcr8 studies provided new mechanistic insights into C9ALS/FTD pathogenesis from the loss-of-function perspective. We and others previously reported that C9orf72 and Smcr8 form a protein complex and regulate autophagy (11–17). Although these studies linked C9orf72 with autophagy, neuronal deletion of C9orf72 failed to produce neurodegeneration and motor deficits in mice (24). Whether and how autophagy-lysosomal functions are impaired in C9ALS/FTD in vivo remain unknown. We found that expression of Smcr8, like C9orf72, is reduced in C9ALS/FTD mouse models and patient tissues; Smcr8 heterozygous or homozygous mice displayed motor deficits; Smcr8 heterozygosity exacerbated axonal swellings in C9-BAC mouse models. Therefore, these results provide the in vivo evidences that Smcr8 downregulation impaires the autophagy-lysosomal function, which in turn promotes axonal degeneration in C9ALS/FTD. In addition to axonal swellings, Smcr8 deficiency also exacerbated poly (GA) and poly (GP) gain of toxicity, highlighting the cross talk between loss- and gain-of-function in the pathogenesis of C9ALS/FTD. It is possible that Smcr8 downregulation impaires the autophagy-lysosomal degradation of poly (GA)/(GP). Alternatively, Smcr8 deficiency may promote cellular stress and result in the increased DPR biogenesis. Future studies should identify mechanisms by which Smcr8 deficiency-mediated loss-of-function interacts with gain of toxicity in C9ALS/FTD.
We uncovered C9orf72’s neuronal functions in promoting autophagy-lysosomal degradation. Whereas C9orf72 deletion alone produced no neurodegeneration or MN deficits, its absence in the background of Smcr8 KO exacerbated Lamp1-positive organelle accumulation, p62 elevation and axonal swellings in spinal cords as well as NMJ swellings, all of which were more severe in dKO than single Smcr8−/− mutant mice. These results suggest that C9orf72, like Smcr8, promotes autophagy-lysosomal functions and prevents axonal degeneration. C9orf72’s neuronal functions were manifested in the absence of Smcr8, suggesting that these neuronal functions are dominated by the presence of Smcr8 under physiological conditions. We speculate that Smcr8 has similar but more potent neuronal functions than C9orf72 in regulating autophagy-lysosomal degradation; they act in parallel, not in a single linear pathway, such that Smcr8 can compensate for the loss of C9orf72 but not vice versa. Overall, our studies provided the first comprehensive functional studies of Smcr8 in neurons in vivo, and uncovered C9orf72’s neuronal functions in autophagy-lysosomal pathway. Furthermore, Smcr8 deficiency exacerbates axonal swellings and gain of toxicity in C9-BAC mice, suggesting of the interaction between loss- and gain-of-function in the pathogenesis of C9ALS/FTD.
Materials and Methods
Mice
To generate Smcr8βgeo/βgeo mutant mice, Smcr8tm1(KOMP)vlcg embryonic stem (ES) cells were obtained from the KO mouse project repository at the University of California, Davis. The majority of Smcr8 exon 1 was replaced by a cassette containing lacZ-polyA followed by a loxP-flanked hUbCpro-neo-polyA sequence. The Mouse Genetics Core Facility at National Jewish Health in Denver performed the ES cell injections into C57BK/6N blastocysts. The chimeric offsprings were mated to 129S1/SvImJ mice for germline transmission. Germline-transmitted heterozygous females were crossed with CMV-Cre males to remove the Neo cassette. Smcr8−/− mice were backcrossed into C57BL/6J for four generations. C9-BAC mice (Cat#: 029099) were ordered from Jackson laboratory, and were backcrossed into C57BL/6J for four generations. All animal studies were conducted under protocols approved by the Institutional Animal Care and Use Committee at the University of Georgia or University of Southern California.
Mutant characterization, immunofluorescence staining and western blot analysis
Histological processing and immunohistochemical labeling of sections were performed as described previously (48, 49). Mice were anesthetized with CO2 and perfusion-fixed with 4% paraformaldehyde (PFA). Spinal cords were harvested and post-fixed in 4% PFA, washed with PBS and immersed in 25% sucrose overnight at 4°C followed by standard OCT embedding and coronal sectioning. The following primary antibodies were used: anti-Lamp1 (1D4B, DSHB), anti-p62 (610 832, BD Biosciences), anti-160 kD NF Medium (ab65845, Abcam), anti-NF H, Nonphosphorylated (SMI 32, Biolegend), anti-beta Galactosidase (CGAL-45A-Z, Immune Systems), anti-GFAP (MAB3402, EMD Millipore), anti-Iba1 (019-19 741, Wako), anti-LC3 (5F10, NanoTools), Tuj1 (T3952, Sigma), ChAT (AB144P, EMP Millipore), GA (MABN889, Millipore) and poly GP (24494-1-AP, Proteintech). The secondary antibodies used were Alexa 488, Alexa 555 and Alexa 647 conjugated to specific IgG types (Invitrogen Molecular Probes). Axonal staining was performed by Bielschowsky staining with Hito Bielschowsky OptimStain™ Kit (Hito Biotec) according to the manufacturer’s protocol. X-gal activity was assessed by incubating sections with LacZ staining solution (1 mg/ml of X-gal, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide and 2 mM MgCl2 in PBS) overnight at 37°C.
TEM
Spinal cords were fixed in 2% PFA plus 2.5% glutaraldehyde in 0.1 M Sodium Cacodylate buffer (PH = 7.4) for 24 h at 4°C. Tissues were embedded and sectioned for TEM by the Department of Pathology, EM Division at the University of Georgia.
Mouse MN isolation, transfection and live imaging
Isolation and culture of spinal cord motoneurons from E 13.5 mice were performed as previously described (50). Spinal cords were isolated and dorsal root ganglia were removed carefully. MNs were enriched by lectin-based purification. Isolated cells were plated onto poly-DL-ornithine hydrobromide/laminin (Sigma) coated 35 mm glass bottom culture dishes (MatTek). Cells were cultured in Neurobasal medium (Invitrogen) with 1 × B27 supplement (Invitrogen), 5% inactivated horse serum (Invitrogen), 1% Glutamax (Invitrogen) and 10 ng/ml CNTF at 37°C in a 5% CO2 incubator. Half the volume of culture medium was exchanged every 2 days. At 4 days in vitro (DIV4), neurons were transfected using NeuroMag transfection reagent (Oz Biosciences) as previously described (51). Briefly, 1 h before magnetofection, complete medium was replaced with serum-free Neurobasal/B27/Glutamax medium. Plasmid DNA (1.5 μg) was incubated with 4 μl Neuromag in 300 μl OptiMEM for 15 minute, and then added drop-wise to the cultures. Cells were incubated on top of a magnetic plate (Oz Biosciences) for 15 minute and complete medium was restored after 1 h. For live cell imaging, 35 mm glass bottom dishes containing normal growth medium were mounted in a temperature-controlled stage and maintained at 37°C and 5% CO2 conditions. Cells were visualized at DIV6-9 on a Zeiss LSM 710 confocal microscope using a 100 × oil-immersion lens. Digital images were acquired with a camera using ZEN software. Laser lines at 488 nm were used. Time-lapse sequences of 1024 × 1024 pixels were collected at 7.8 second intervals for 15 minute.
Transport analysis
Kymographs were generated using the Image J plugin Kymograph (https://www.embl.de//eamnet/html/kymograph.html). Net run speeds and lengths of vesicle movement were calculated by drawing a slope from the beginning to the end of the run on the kymograph. The transport direction was manually determined: vesicles were classified as retrograde (moved ≥ 1.5 μm in the retrograde direction), anterograde (moved ≥ 1.5 μm in the anterograde direction) or stationary (moved < 1.5 μm during the duration of the 15 minute video). The percentage motility of vesicles along the axon (retrograde, anterograde or stationary) was calculated as a percentage of the total number of imaged vesicles per neuron. The instantaneous velocities were quantified by Image J’s TrackMate v3.4.2 plugin (52), and stalls were defined as a vesicle with velocity < 0.1 μm/s for ≥8 s. The numbers of stalls per trace were calculated with a custom Python script.
NMJ analysis
Muscles were fixed for 10 minute in freshly prepared PBS containing 4% (w/v) PFA. After washing with PBS, further connective tissues were detached from the muscles, and individual fibers were carefully teased apart to facilitate antibody penetration. Muscles were permeabilized with 2% Triton X-100 in PBS for 30 minute and then blocked in 4% BSA and 1% Triton X-100 in PBS for 30 minute. Samples were incubated overnight at 4°C in blocking solution with α-Bungarotoxin, Alexa Fluor™ 488 conjugate (1:500 dilution), and primary antibodies against NF (2H3, 1: 70 dilution) and Lamp-1 (1D4B, 1:70 dilution) to visualize the AChRs, axons and lysosomes. The following day, muscles were washed three times for 10 minute each in PBS before incubation for 2 h with AlexaFluor 555 and 647 secondary antibodies and α-Bungarotoxin, Alexa Fluor™ 488 conjugate in PBS in the dark. Last, muscles were washed three times for 10 minute each in PBS and mounted on slides with coverslips. A Leica TCS SP8 microscope was used to image the NMJs with Z-stacks at 3 μm intervals.
Behavioral testing
The experimenter was blind to the animal’s genotype during all tests. Open filed test was performed using an overhead SMART video tracking system (Panlab), which measures distance traveled, time spent in each zone, and speed. The apparatus consisted of a gray open top plastic box (w × d × h: 45 × 45 × 40 cm) divided into four equal arenas. After a 2 h acclimatization to the behavioral testing room, each animal was placed in the center of the open field and left undisturbed for 30 minute. The apparatus was wiped between trials with a 70% ethanol solution. For rotarod test, an accelerating rotarod (Panlab) was used to analyze motor coordination and balance. Mice were trained three times on the rotarod with 4 rpm/minute speed 1 day before testing. During test conditions, we measured the latency (time) to the mouse falling from the rotating beam while ramping up the speed from 4 to 40 rpm over a 5-minute trial period. Mice were given four trials per day, with an intertrial interval of 20 minute. The average of the three trials was used to evaluate latency to fall. For grip strength, the grip strength of front paws was measured using a grip strength meter (Bioseb). Each mouse was held by the tail and lowered toward the apparatus. Front paws were allowed to grasp the assembly. The mouse was then pulled backward in the horizontal plane until the pull-bar was released. The trial was repeated four times, and the force generated by pulling the animal away from the wire mesh was recorded.
Author Contributions
C.L., Q.S., Q.C., M.Y., L.M., W.Z., J.-F.C conceived and performed all experiments. R.C. helped with manuscript writing. J.-F.C. designed and interpreted the experiments and wrote the manuscript.
Financial Statement
The authors declare that there are no competing financial interests that might be perceived as affecting the objectivity of these studies.
Data Availability
All relevant data are available from the corresponding authors upon reasonable request.
Supplementary Material
Acknowledgements
We thank Chen laboratory colleagues for stimulating discussions. We are grateful for Bridget Samuels’s critical reading of the manuscript. The authors wish to thank Dr Nikolay Filipov for help with behavioral experiments, and Ms Mary Ard for help with electron microscopy. Chen laboratory is supported by funds from the Associate Dean of Research Fund from the Center for Craniofacial Molecular Biology, Herman Ostrow School of Dentistry at the University of Southern California, and grants R01NS097231 (J.C.) and R01NS096176 (J.C.) from the National Institute of Health. Conflict of Interest statement. None declared.
References
- 1. Ling S.-C., Polymenidou M. and Cleveland D.W. (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron, 79, 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gao F.-B., Almeida S. and Lopez-Gonzalez R. (2017) Dysregulated molecular pathways in amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorder. EMBO J., 36, 2931–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gijselinck I., Van Langenhove T., van der Zee J., Sleegers K., Philtjens S., Kleinberger G., Janssens J., Bettens K., Van Cauwenberghe C., Pereson S. et al. (2012) A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol., 11, 54–65. [DOI] [PubMed] [Google Scholar]
- 4. Renton A.E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L. et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron, 72, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J. et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron, 72, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Majounie E., Renton A.E., Mok K., Dopper E.G.P., Waite A., Rollinson S., Chiò A., Restagno G., Nicolaou N., Simón-Sánchez J. et al. (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol., 11, 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Waite A.J., Bäumer D., East S., Neal J., Morris H.R., Ansorge O. and Blake D.J. (2014) Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging, 35, 1779.e5–e1779.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mizielinska S., Lashley T., Norona F.E., Clayton E.L., Ridler C.E., Fratta P. and Isaacs A.M. (2013) C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol., 126, 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shi Y., Lin S., Staats K.A., Li Y., Chang W.-H., Hung S.-T., Hendricks E., Linares G.R., Wang Y., Son E.Y. et al. (2018) Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med., 24, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shao Q., Liang C., Chang Q., Zhang W., Yang M. and Chen J.-F. (2019) C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose-dependent manner. Acta Neuropathol. Commun., 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jung J., Nayak A., Schaeffer V., Starzetz T., Kirsch A.K., Müller S., Dikic I., Mittelbronn M. and Behrends C. (2017) Multiplex image-based autophagy RNAi screening identifies SMCR8 as ULK1 kinase activity and gene expression regulator. elife, 6, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang M., Liang C., Swaminathan K., Herrlinger S., Lai F., Shiekhattar R. and Chen J.-F. (2016) A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv., 2, e1601167–e1601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Amick J., Roczniak-Ferguson A. and Ferguson S.M. (2016) C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol. Biol. Cell, 27, 3040–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sullivan P.M., Zhou X., Robins A.M., Paushter D.H., Kim D., Smolka M.B. and Hu F. (2016) The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun., 4, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ugolino J., Ji Y.J., Conchina K., Chu J., Nirujogi R.S., Pandey A., Brady N.R., Hamacher-Brady A. and Wang J. (2016) Loss of C9orf72 enhances autophagic activity via deregulated mTOR and TFEB signaling. PLoS Genet., 12, e1006443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sellier C., Campanari M.-L., Julie Corbier C., Gaucherot A., Kolb-Cheynel I., Oulad-Abdelghani M., Ruffenach F., Page A., Ciura S., Kabashi E. et al. (2016) Loss of C9ORF72 impairs autophagy and synergizes with poly Q Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J, 35, 1276–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Webster C.P., Smith E.F., Bauer C.S., Moller A., Hautbergue G.M., Ferraiuolo L., Myszczynska M.A., Higginbottom A., Walsh M.J., Whitworth A.J. et al. (2016) The C9orf72 protein interacts with Rab 1a and the ULK1 complex to regulate initiation of autophagy. EMBO J., 35, 1656–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang D., Iyer L.M., He F. and Aravind L. (2012) Discovery of novel DENN proteins: implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front. Genet., 3, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Levine T.P., Daniels R.D., Gatta A.T., Wong L.H. and Hayes M.J. (2013) The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics, 29, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mizushima N. and Komatsu M. (2011) Autophagy: renovation of cells and tissues. Cell, 147, 728–741. [DOI] [PubMed] [Google Scholar]
- 21. Mizushima N., Levine B., Cuervo A.M. and Klionsky D.J. (2008) Autophagy fights disease through cellular self-digestion. Nature, 451, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Rourke J.G., Bogdanik L., Yáñez A., Lall D., Wolf A.J., Muhammad A.K.M.G., Ho R., Carmona S., Vit J.P., Zarrow J. et al. (2016) C9orf72 is required for proper macrophage and microglial function in mice. Science, 351, 1324–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Corrionero A. and Horvitz H.R. (2018) A C9orf72 ALS/FTD ortholog acts in endolysosomal degradation and lysosomal homeostasis. Curr. Biol., 28, 1522–1535.e5. [DOI] [PubMed] [Google Scholar]
- 24. Koppers M., Blokhuis A.M., Westeneng H.-J., Terpstra M.L., Zundel C.A.C., Vieira de Sá R., Schellevis R.D., Waite A.J., Blake D.J., Veldink J.H. et al. (2015) C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol., 78, 426–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Y., Pattamatta A., Zu T., Reid T., Bardhi O., Borchelt D.R., Yachnis A.T. and Ranum L.P.W. (2016) C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron, 90, 521–534. [DOI] [PubMed] [Google Scholar]
- 26. Zhang Y., Burberry A., Wang J.-Y., Sandoe J., Ghosh S., Udeshi N.D., Svinkina T., Mordes D.A., Mok J., Charlton M. et al. (2018) The C9orf72-interacting protein Smcr 8 is a negative regulator of autoimmunity and lysosomal exocytosis. Genes Dev., 32, 929–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gowrishankar S., Yuan P., Wu Y., Schrag M., Paradise S., Grutzendler J., De Camilli P. and Ferguson S.M. (2015) Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer's disease amyloid plaques. Proc. Natl. Acad. Sci. U. S. A., 112, E3699–E3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Glaumann H., Ericsson J.L. and Marzella L. (1981) Mechanisms of intralysosomal degradation with special reference to autophagocytosis and heterophagocytosis of cell organelles. Int. Rev. Cytol., 73, 149–182. [DOI] [PubMed] [Google Scholar]
- 29. Maday S., Wallace K.E. and Holzbaur E.L.F. (2012) Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol., 196, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng X.-T., Zhou B., Lin M.-Y., Cai Q. and Sheng Z.-H. (2015) Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J. Cell Biol., 209, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tammineni P., Ye X., Feng T., Aikal D. and Cai Q. (2017) Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer's disease neurons. elife, 6, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xie Y., Zhou B., Lin M.-Y., Wang S., Foust K.D. and Sheng Z.-H. (2015) Endolysosomal deficits augment mitochondria pathology in spinal motor neurons of asymptomatic fALS mice. Neuron, 87, 355–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marat A.L., Dokainish H. and McPherson P.S. (2011) DENN domain proteins: regulators of Rab GTPases. J. Biol. Chem., 286, 13791–13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gendron T.F. and Petrucelli L. (2018) Disease mechanisms of C9ORF72 repeat expansions. Cold Spring Harbor Perspect. Med., 8, a024224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zu T., Pattamatta A. and Ranum L.P.W. (2018) Repeat-associated non-ATG translation in neurological diseases. Cold Spring Harbor Perspect. Biol., 10, a033019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. O'Rourke J.G., Bogdanik L., Muhammad A.K.M.G., Gendron T.F., Kim K.J., Austin A., Cady J., Liu E.Y., Zarrow J., Grant S. et al. (2015) C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron, 88, 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peters O.M., Cabrera G.T., Tran H., Gendron T.F., McKeon J.E., Metterville J., Weiss A., Wightman N., Salameh J., Kim J. et al. (2015) Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron, 88, 902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang J., Zhu Q., Gendron T.F., Saberi S., McAlonis-Downes M., Seelman A., Stauffer J.E., Jafar-Nejad P., Drenner K., Schulte D. et al. (2016) Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron, 90, 535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mackenzie I.R.A., Frick P., Grässer F.A., Gendron T.F., Petrucelli L., Cashman N.R., Edbauer D., Kremmer E., Prudlo J., Troost D. et al. (2015) Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol., 130, 845–861. [DOI] [PubMed] [Google Scholar]
- 40. Mackenzie I.R.A., Frick P. and Neumann M. (2014) The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol., 127, 347–357. [DOI] [PubMed] [Google Scholar]
- 41. May S., Hornburg D., Schludi M.H., Arzberger T., Rentzsch K., Schwenk B.M., Grässer F.A., Mori K., Kremmer E., Banzhaf-Strathmann J. et al. (2014) C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc 119 sequestration. Acta Neuropathol., 128, 485–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Y.-J., Gendron T.F., Grima J.C., Sasaguri H., Jansen-West K., Xu Y.-F., Katzman R.B., Gass J., Murray M.E., Shinohara M. et al. (2016) C9ORF72 poly (GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci., 19, 668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mizielinska S., Grönke S., Niccoli T., Ridler C.E., Clayton E.L., Devoy A., Moens T., Norona F.E., Woollacott I.O.C., Pietrzyk J. et al. (2014) C9orf72 repeat expansions cause neurodegeneration in drosophila through arginine-rich proteins. Science, 345, 1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Saberi S., Stauffer J.E., Jiang J., Garcia S.D., Taylor A.E., Schulte D., Ohkubo T., Schloffman C.L., Maldonado M., Baughn M. et al. (2018) Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol., 135, 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yue Z., Horton A., Bravin M., DeJager P.L., Selimi F. and Heintz N. (2002) A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron, 35, 921–933. [DOI] [PubMed] [Google Scholar]
- 46. Komatsu M., Wang Q.J., Holstein G.R., Friedrich V.L., Iwata J.-I., Kominami E., Chait B.T., Tanaka K. and Yue Z. (2007) Essential role for autophagy protein Atg 7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. U. S. A., 104, 14489–14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kulkarni V.V. and Maday S. (2018) Compartment-specific dynamics and functions of autophagy in neurons. Dev. Neurobiol., 78, 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang M., Yang S.-L., Herrlinger S., Liang C., Dzieciatkowska M., Hansen K.C., Desai R., Nagy A., Niswander L., Moss E.G. et al. (2015) Lin 28 promotes the proliferative capacity of neural progenitor cells in brain development. Development, 142, 1616–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shao Q., Herrlinger S., Yang S.-L., Lai F., Moore J.M., Brindley M.A. and Chen J.-F. (2016) Zika virus infection disrupts neurovascular development and results in postnatal microcephaly with brain damage. Development, 143, 4127–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Conrad R., Jablonka S., Sczepan T., Sendtner M., Wiese S. and Klausmeyer A. (2011) Lectin-based isolation and culture of mouse embryonic motoneurons. J. Vis. Exp. Sep 15;(55). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fallini C., Bassell G.J. and Rossoll W. (2010) High-efficiency transfection of cultured primary motor neurons to study protein localization, trafficking, and function. Mol. Neurodegener., 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tinevez J.-Y., Perry N., Schindelin J., Hoopes G.M., Reynolds G.D., Laplantine E., Bednarek S.Y., Shorte S.L. and Eliceiri K.W. (2017) TrackMate: an open and extensible platform for single-particle tracking. Methods, 115, 80–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available from the corresponding authors upon reasonable request.