

Abstract
In this review, we focus on the metabolism of mammalian glycan-associated monosaccharides, where the vast majority of our current knowledge comes from research done during the 1960s and 1970s. Most monosaccharides enter the cell using distinct, often tissue specific transporters from the SLC2A family. If not catabolized, these monosaccharides can be activated to donor nucleotide sugars and used for glycan synthesis. Apart from exogenous and dietary sources, all monosaccharides and their associated nucleotide sugars can be synthesized de novo, using mostly glucose to produce all nine nucleotide sugars present in human cells. Today, monosaccharides are used as treatment options for a small number of rare genetic disorders and even some common conditions. Here, we cover therapeutic applications of these sugars and highlight biochemical gaps that must be revisited as we go forward.
Graphical Abstract
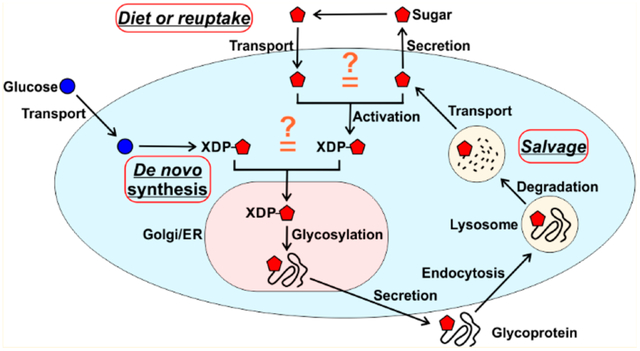
Far and away the most important role of monosaccharides is to provide energy for cells. Besides energy-generating catabolism, they can be utilized for the biosynthesis of multiple cellular components such as polysaccharides (e.g., glycogen and hyaluronan) and glycoproteins, proteoglycans, glycosphingolipids, and GPI anchors, all gathered under the biosynthetic umbrella of glycosylation. In mammalian cells, this enzymatic process involves at least nine monosaccharides (glucose, galactose, N-acetylglucosamine, N-acetylgalactosamine, xylose, glucuronic acid, sialic acid, mannose, and fucose) that are linked to proteins and lipids to generate hundreds of glycan structures (Figure 1).1 Glycosylation is important for the biosynthesis, folding, stability, trafficking, and turnover of many different macromolecules, all of which play an essential role in the interactions between the cells, tissue morpho-genesis, and the immune response.2
Figure 1.
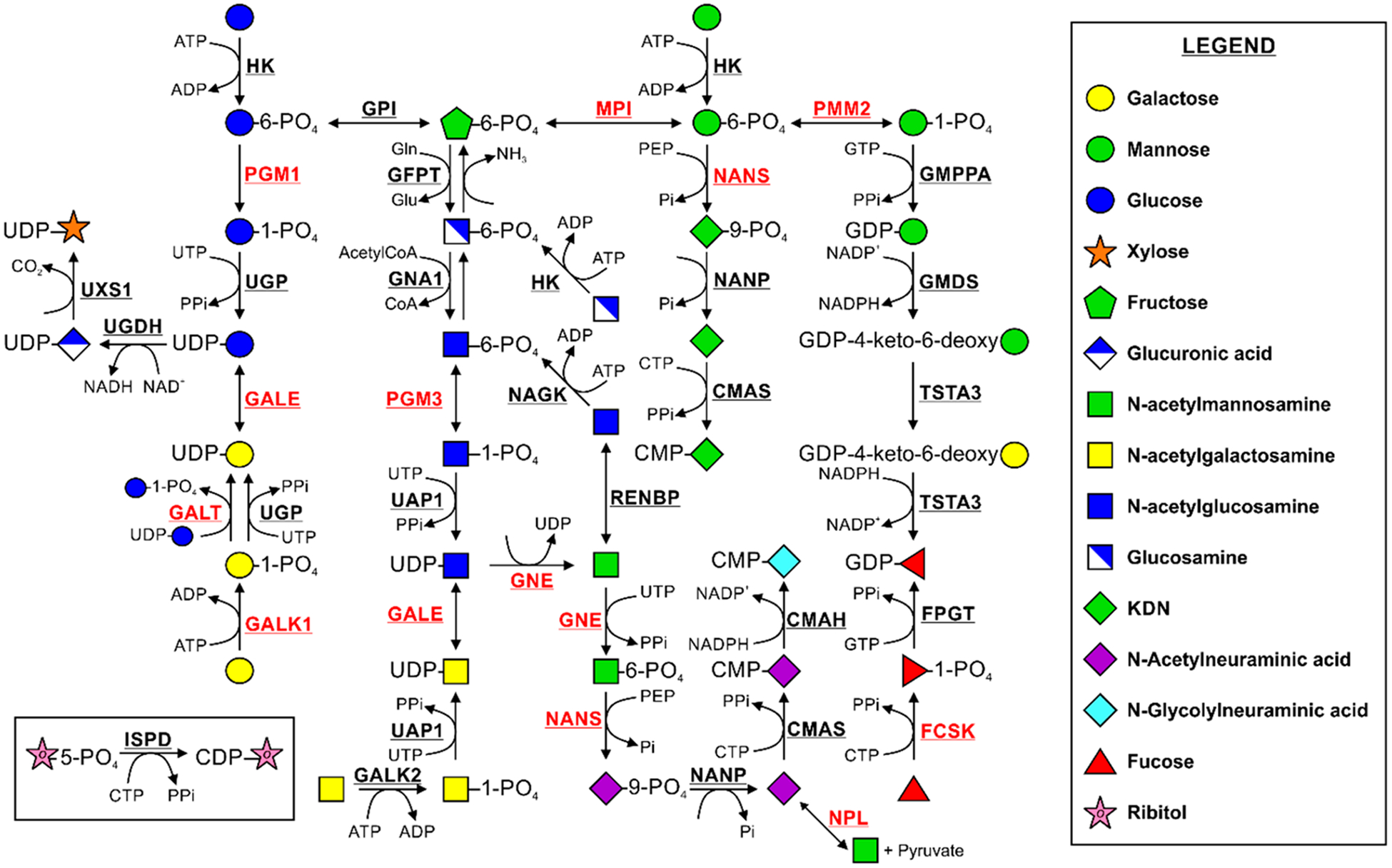
Biosynthesis and interconversion of monosaccharides. Names of enzymes catalyzing each reaction are shown in bold. Those with pathogenic variants discussed in this review are shown in bold red. Adapted from Essentials of Glycobiology.1
In the diet, glucose is the most important monosaccharide. The other sugars occur in various polysaccharides that are mostly indigestible and not bioavailable but are often degraded by gut bacteria, producing short chain fatty acids, some of which can be absorbed by the host.3 Besides their dietary origin, monosaccharides can be salvaged from glycans degraded in the lysosomes. They can be recycled within the cell or secreted.1 The bulk of glycan monosaccharides is likely synthesized de novo, ultimately from glucose (Figure 1). This review will cover time-honored, well-established tenants of monosaccharide use in glycan synthesis. We will also show where gaps in our understanding exist, especially in the relative contributions of multiple pathways. There is an urgency here because many simple sugars are serving as therapeutics for rare diseases and more common conditions.
BASICS OF GLYCAN-ASSOCIATED MONOSACCHARIDE BIOCHEMISTRY
Incorporation of monosaccharides into glycans first requires activation by the ATP-dependent specific kinases. These activated monosaccharide phosphates require further conversion to nucleotide sugars using UTP-, GTP-, or CTP-dependent reactions that generate free pyrophosphate and the activated pyrophosphate-containing sugar donors (Figure 1) Most nucleotide sugars are synthesized in the cytosol,4 except for CMP-sialic acid, which is made in the nucleus.5 Because glycosylation occurs predominantly in the Golgi apparatus and the endoplasmic reticulum (ER),1 the donors must be delivered there using the appropriate nucleotide sugar transporters (antiporters) (Figure 2).6 In the organellar lumen, the co-localized donors and respective glycosyltransferases add the monosaccharide to the acceptors, initiating glycan synthesis and providing glycans for further extension.1 An exception to this transport rule is UDP-N-acetylglucosamine (UDP-GlcNAc), which is utilized in the cytosol for protein modification by O-GlcNAc transferase or hyaluronan biosynthesis that requires cytoplasmic UDP-GlcNAc and UDP-glucuronic acid.7,8
Figure 2.

Nucleotide sugar transporters delivering activated forms of monosaccharides into the Golgi and ER lumen in humans. The red diamond denotes sialic acid. Adapted from Essentials of Glycobiology.1
DIETARY CARBOHYDRATES AFFECT THE HOST GUT MICROBIOME
Carbohydrates are important compounds in an infants’ diet. In addition to lactose, human milk contains more than 200 unique oligosaccharides.9 They occur both as free glycans and as glycoconjugates.10 The precise composition of milk glycans varies among women and during lactation.11 In contrast to human milk, infant formulas have a more limited amount and a more limited variety of free oligosaccharides.12 Interestingly, infants are already exposed to milk oligosaccharides in the uterus as they are found in amniotic fluid, highlighting their importance.13
Besides oligosaccharides, human milk contains different species of bacteria, including Bifidobacterium, Bacteroides, and Ruminococcus, which help in oligosaccharide digestion and assimilation qualifying milk as a probiotic food.14,15 Oligosaccharides present in human milk are an important factor that help to shape the gut microbiota and health of infants.16 Synergistic actions of sialylated milk oligosaccharides and infant intestinal microflora promote growth and have an impact on metabolism in different organs.17 Unlike breast-fed infants, newborn babies fed with formula develop different gut microbiota, with a lower content of beneficial bacteria.18
Modulating the host gut microbiome with dietary-provided mono- and oligosaccharides may improve the health and quality of life. However, at this point, we do not know the extent to which complex oligosaccharides directly contribute to host glycan synthesis versus serving as microbiome modulators.
CONGENITAL DISORDERS OF GLYCOSYLATION
Pathogenic mutations in glycosylation-related genes can perturb cellular glycosylation, resulting in developmental and physiological abnormalities in many organ systems.19 Congenital disorders of glycosylation (CDG) make up a rapidly expanding group of more than 135 rare metabolic diseases.20 They are rare because glycans are critical for development and many mutations in glycan pathways are probably lethal. More than half of the identified CDG types are due to deficiencies in the N-glycosylation pathway.20 Most of these disorders can be initially identified by carbohydrate-deficient transferrin (CDT) analysis, examining glycosylation of this abundant serum protein.21 However, in some CDGs, abnormal transferrin glycosylation is not seen or normalizes over time in authentic patients. New biomarkers are required to improve the diagnostic process, but ultimately, genetic analysis is necessary.22
Although the number of identified disorders is growing, there are few treatment options. Several therapeutic strategies rely on supplementing the individual’s diet with the limiting monosaccharides.20 Because there are few data on the relative contribution of each monosaccharide from diet/salvage to glycan production, it is impossible to predict which rate-limiting metabolic steps might respond to modest increases in circulating levels of monosaccharides. This is especially important for individuals with impaired glycosylation.
d-MANNOSE
d-Mannose is required for N- and O-glycosylation, C-mannosylation, and GPI anchor synthesis. The mannose concentration in the human plasma is ~50 μM, which primarily comes from N-glycan processing.23 Members of the SLC2A family of hexose transporters commonly termed “GLUT” carry nearly all monosaccharides, including mannose, across the plasma membrane into the cytoplasm.23 Inside the cell, mannose is phosphorylated by hexokinase (HK) to produce mannose 6-phosphate (Man-6P),24 which can further serve as a substrate for three different pathways (Figure 1). First, it can be converted to mannose 1-phosphate (Man-1P) and further to GDP-mannose (Figure 1)25 and subsequently to dolichol-phosphate mannose, both of which can be directly utilized for N-glycan biosynthesis. Alternatively, Man-6P can serve as a substrate in 2-keto-3-deoxy-d-glycero-d-galactonononic acid (KDN) synthesis (Figure 1). The function of this pathway is unknown, but providing a high mannose concentration to cells increases the production of KDN.26 Man-6P can also be isomerized to fructose 6-phosphate (Fruc-6P), allowing it to be catabolized through glycolysis or to serve as a substrate for the production of different nucleotide sugars (Figure 1).27 While mannose is the most abundant monosaccharide in N-glycans, <2% of the mannose entering the cell is utilized for glycosylation. The great majority is converted to Fruc-6P and catabolized, presumably through glycolysis.23 Finally, GDP-mannose can be converted to GDP-fucose, resulting in its incorporation into glycans as fucose (Figure 1).28
Both mannose and glucose contribute to GDP-mannose synthesis through Man-6P (Figure 1);29 however, cells labeled with differentially tagged 13C mannose and glucose showed that under physiological conditions (5 mM glucose and 50 μM mannose), glucose is the major source (60–80%) of N-glycan mannose.30 Under this condition, 13C mannose cannot be detected in glycogen, galactose, or GlcNAc in N-glycans. However, when its concentration is increased to 1 mM and it is the sole monosaccharide source of mannose in N-glycans, mannose can be seen to contribute to glycolysis, galactose and GlcNAc, but it is never found in glycogen,30 implying some segregation of metabolic pathways. Mannose taken up by the cells is handled differently than after it is released within the cells due to N-glycan processing. Mannose released by intracellular processing is secreted from the cells as free mannose, suggesting that the monosaccharide origin is important for its future destination.23
Glycan-associated metabolism of mannose is significantly perturbed in at least two CDGs caused by mutations in phosphomannose isomerase (MPI) and phosphomannomutase 2 (PMM2) (MPI-CDG and PMM2-CDG, respectively). PMM2-CDG is the most prevalent CDG type, with more than 900 reported cases.31 It is caused by pathogenic variants in PMM2, whose encoded protein converts Man-6P to Man-1P, an intermediate in GDP-mannose synthesis (Figure 1). Reduced PMM2 enzymatic activity ultimately leads to depleted incorporation of mannose into glycoproteins and truncation of the lipid-linked oligosaccharides under low-glucose conditions.29 Some of these truncated glycans can still be transferred to proteins, as shown by recent analysis of patient sera.22 In MPI-CDG, the level of conversion of Fruc-6P to Man-6P is decreased because of reduced MPI activity (Figure 1), resulting in an abnormal serum glycosylation pattern. MPI-CDG was the first CDG type found to be effectively treated by dietary supplementation with a monosaccharide.32 Affected individuals responded quickly to mannose therapy, with correction of the glycosylation defect. However, liver abnormalities in some patients were irreversible and progressed to liver fibrosis that required a transplant.33
Mpi hypomorphic mice carrying a known pathogenic variant that generated a patient level of enzymatic activity showed very few abnormalities, unless mice were stressed.34 However, supplementing dams drinking water with 1–2% mannose reduced litter size and pup survival in a dose-dependent manner. Moreover, the increased level of mannose and reduced Mpi activity resulted in highly localized eye defects due to the failure of normal lens development along with the accumulation of Man-6P and related metabolites.34 Considering the data from Mpi hypomorphic mice, pregnant, at-risk women should be wary of using mannose as a preemptive therapy, because it could be harmful to a genetically compromised fetus. Besides being used to treat MPI-CDG patients, mannose has been extensively used as both a treatment and a prophylactic for urinary tract infections because its excretion in the urine competes with lectin-mediated binding of Escherichia coli to epithelial cells.35
In more recent studies, mannose showed therapeutic promise in other areas such as obesity and cancer. For example, when mannose was provided to mice early in life, it prevented high-fat-diet-induced obesity and promoted the lean and fit phenotype that correlated with changes in the gut microbiota.36 Furthermore, mannose was shown to cause the arresting of growth in several tumor types in mice as well as increased tumor cell death upon chemotherapy, suggesting mannose as a selective therapy for the treatment of cancer.37 While the mechanism has not been demonstrated, it is most likely due to the toxic accumulation of Man-6P that impairs glycolysis, starving the cells of metabolic fuel.
l-FUCOSE
l-Fucose is incorporated into N- and O-glycans, and glycolipids, using GDP-fucose as the donor substrate.38 GDP-fucose also can be synthesized de novo from either glucose or mannose. In this process, GDP-mannose is converted to GDP-fucose through a three-step reaction, catalyzed by two enzymes, GDP-mannose 4,6-dehydratase (GMDS) and GDP-fucose synthetase (TSTA3, also known as FX) (Figure 1).39 Alternatively, GDP-fucose can be synthesized via the diet/salvage pathway, directly from fucose.40–43 Here, fucose is first phosphorylated by fucokinase (FCSK) to fucose 1-phosphate (Fuc-1P), which is further converted to GDP-fucose by guanylyltransferase (FPGT) (Figure 1).44 In mid-1970s, isotope dilution studies showed that >90% of GDP-fucose in HeLa cells was supplied by the de novo pathway and <10% came from salvage.42,43 However, it should be pointed out that while the study was meticulously performed, only a single fucose concentration was used. Preliminary studies suggest that fucose may make a more substantial contribution than the previous results showed.45
Fucose occurs in glycans as α-fucose and is released by α-fucosidase.46 Fucose mutarotase converts it to β-fucose, and only the β-anomer is metabolized through the salvage pathway.44 The enzyme is highly expressed in the liver, suggesting that it may be important for the fucose salvage, but little is known about this protein.47
Not much is known about fucose transport by human cells. In 1994, it was shown that it is a Na+-independent mechanism, which occurs through an unidentified carrier system. Once transported into the cell, fucose is quickly incorporated into glycoproteins, and a large portion of these glycoproteins is rapidly secreted.48 Incorporation requires the delivery of GDP-fucose into the Golgi apparatus and ER lumen. In humans, this transport process is mediated by either SLC35C1 or SLC35C2 (Figure 2). While both are Golgi-localized, the former is thought to be the major provider of Golgi GDP-fucose,49 while the latter is used for Notch1 fucosylation, which occurs in the ER.50 In contrast to Drosophila, no ER-localized GDP-fucose transporter has been identified in humans.51
To date, several genetic disorders of fucosylation are known, including pathological variants in SLC35C1 that lead to leukocyte adhesion deficiency II (LAD II), also known as SLC35C1-CDG.52 In these individuals, fucosylation is perturbed due to the malfunction of SLC35C1 causing immunodeficiency and severe mental and growth retardation, in addition to its hallmark feature, an elevated level of circulating leukocytes.49 In a few cases, supplementing the diet with fucose can normalize elevated leukocytes, by promoting the synthesis of Sialyl-Lewis X on neutrophils and therefore allowing their rolling on the endothelium.53,54 How a small amount of fucose is such an effective treatment is poorly understood and requires further studies. One possibility is that responsive cells prefer the salvage pathway over the de novo pathway and are able to direct a larger amount of GDP-fucose to the Golgi. Presumably patients with some residual transporter activity might respond to a higher concentration of GDP-fucose, while others with more severe defects do not. No studies have been done to differentiate the responses of various mutations. Oral fucose therapy may also be helpful in FUK-CDG and FUT8-CDG. The former is caused by pathogenic variants in FCSK, which encodes fucose kinase,55 and the latter is a result of biallelic mutations in FUT8,56 which encodes the major fucosyltransferase required for adding core fucose to N-glycans.57 No trials on these very few individuals have been performed.
The benefits of dietary fucose supplementation could have broader implications. Feeding fucose to mice or increasing the level of expression of Fcsk attenuated primary melanoma growth, increased the number of tumor-associated natural killer cells, and decreased the extent of distal metastasis of the tumor.58 This appears to be due to the tumors’ downregulation of Fcsk. Like mannose, fucose supplementation can affect the host microbiome. Mouse models have shown that fucose decreased levels of endotoxin-producing bacteria from the Desulfovibrionaceae family. The same study showed that fucose supplementation reduced the incidence of both obesity and fatty liver induced by a high-fat diet.59 Interestingly, inhalation of a fucose and galactose solution was beneficial for individuals with cystic fibrosis because it reduced Pseudomonas aeruginosa levels in sputum and decreased the risk of bacterial infection.60
d-GALACTOSE
Galactose is a key source of energy for infants and a crucial component of glycoconjugates. It is also transferred across the plasma membrane using the GLUT transporters.61 Its concentration in blood does not exceed 10 μM.62 Most galactose is catabolized via the Leloir pathway (Figure 3).63 Galactose can also be utilized in glycosylation through multiple entry points. Before incorporation into the glycans, galactokinase (GALK1) phosphorylates galactose to generate galactose 1-phosphate (Gal-1P).64 The predominant pathway in liver converts Gal-1P to UDP-galactose using uridinyltransferase (GALT), which is an exchange reaction involving UDP-glucose, generating glucose 1-phosphate (Glc-1P).65 In addition, UDP-glucose pyrophosphorylase (UGP), the same enzyme that converts Glc-1P to UDP-glucose, exhibits residual activity toward Gal-1P (Figure 1).66,67 Alternatively, uridine diphosphate galactose pyrophosphorylase (EC 2.7.7.10) converts Gal-1P to UDP-galactose.68,69 Another pathway producing UDP-galactose from UDP-glucose involves an NAD-dependent reaction catalyzed by UDP-galactose epimerase (GALE) (Figure 1).70 The interplay and contribution of each pathway to glycan biosynthesis in different cell types have not been studied, and it may be difficult to extrapolate results from one type of cell to another.
Figure 3.
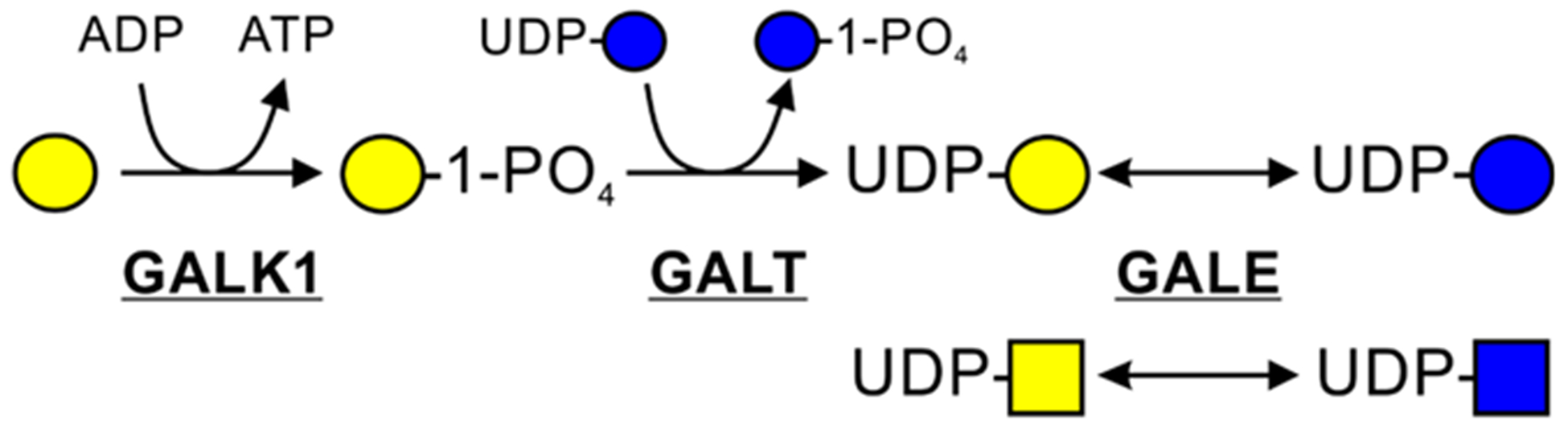
Leloir pathway of galactose catabolism. Adapted from Essentials of Glycobiology.1
UDP-galactose produced by the various pathways then serves as the substrate for galactosyltransferases located in the Golgi and ER.71 UDP-galactose is transported into these organelles’ lumen from the cytosol through the major UDP-galactose transporter (SLC35A2) (Figure 2).72 Galactose is released from glycans as the β-anomer using β-galactosidase, but only the α-anomer can be activated from salvage. This epimerization is catalyzed by galactose mutarotase (GALM).73 In 2019, studies showed that mutations in GALM are associated with galactosemia-like symptoms, suggesting the existence of a novel type (IV) galactosemia.74,75
Galactosemia is due to pathogenic variants in one of the genes involved in the Leloir pathway, namely, GALK1, GALT, or GALE (Figure 3).76 In this metabolic disorder, even small amounts of galactose are very harmful as this sugar and its metabolites accumulate without being further catabolized.77 As already mentioned, besides GALT and GALE, two other enzymes yield UDP-galactose. UGP (EC 2.7.7.9) exhibits very little activity toward Gal-1P. In the 1950s and 1960s, the existence of enzymatic activity catalyzing the reaction between UTP and Gal-1P was demonstrated in rat and human liver.68,69 UDP-galactose pyrophosphorylase (EC 2.7.7.10) is poorly described in the literature. It is unclear whether the UDP-galactose pyrophosphorylase enzymatic activity was catalyzed by UGP. Because some symptoms of galactosemia (GALT) remain even on an essentially galactose free diet, it is possible that some cells may prefer the UDP-galactose pyrophosphorylase pathway for glycosylation, and galactose limitation may lead to underglycosylation in those cells. Defective glycosylation is a typical feature of individuals with galactosemia, but it only partially improves with treatment.78–81 However, glycosylation defects and its improvement have not been the main focus of therapy.
Galactose supplementation successfully treated several different types of CDG. In a single individual with SLC35A2-CDG, galactose restored galactosylation and seemed to have a positive impact on the neurological symptoms,82 although it is unclear if this improvement was solely due to galactose supplementation, as some patients improve their abnormal glycosylation over time. Because the cohort of diagnosed SLC35A2-CDG individuals is growing rapidly, conclusions about the benefit of galactose therapy can be examined in more patients.83,84 In PGM1-CDG, pathogenic variants in PGM1 ultimately result in decreased enzyme activity, which interconverts glucose 6-phosphate (Glc-6P) to Glc-1P, resulting in slightly abnormal glycogen metabolism and dramatically abnormal glycosylation.85 The defect can be partially bypassed by galactose supplementation, because it contributes as an alternative source of Glc-1P and also increases the size of the cytosolic pools of UDP-glucose and UDP-galactose (Figure 1). This therapy is successfully applied in the growing number of PGM1-CDG individuals.86 However, it does not normalize glycosylation completely, and transferrin profiles may remain abnormal.62
SLC39A8-CDG results from mutations in the membrane-localized manganese transporter. Dysfunction in SLC39A8 transport activity affects the availability of cellular Mn2+ and thus the enzymatic activity of manganese-dependent glycosyltransferases, most notably β-1,4-galactosyltransferase, leading to impaired galactosylation. Oral galactose supplementation normalizes glycosylation in SLC39A8-CDG individuals, as it presumably increases the UDP-galactose concentration, which becomes more available for the β-1,4-galactosyltransferase.87 Glycosylation in individuals with TMEM165-CDG improves slightly with galactose therapy.88 TMEM165 most likely supplies Ca2+ and Mn2+ to the Golgi apparatus in exchange for protons, making it important in glycosyltransferase function and pH homeostasis.89 Probably the same mechanism underlies the successful galactose therapy of SLC39A8-CDG and TMEM165-CDG individuals. Studies of therapeutic applications of galactose will be expanded as part of a large multicenter U.S. consortium of physicians, scientists, and patients recently funded by the National Institutes of Health.
Besides congenital disorders of glycosylation, oral application of galactose is beneficial in individuals with idiopathic steroid-resistant nephrotic syndrome. This therapy decreases focal segmental glomerulosclerosis but fails to improve proteinuria.90 As mentioned previously, inhalation of galactose and fucose decreases the chances of P. aeruginosa infections in individuals suffering from cystic fibrosis.60 In addition, oral galactose treatment might be beneficial for people with sporadic Alzheimer’s disease being an alternative for incretin-based therapy. In a rat model of Alzheimer-like pathology induced with streptozotocin, exposure for 2 months to oral galactose improved the learning and memory function of rats. It also prevented the progress and improved already developed cognitive deficiency in the early stage of the disease.91,92
In the 1960s, HepG2 hepatocellular carcinoma cells were shown to increase respiration rates when grown in galactose-containing medium to maintain the ATP level.93 More recent studies showed that replacing glucose with galactose increases the sensitivity of HepG2 cells to mitochondrial toxicity,94 which most likely occurs due to the enhancement of mitochondrial oxidative phosphorylation by galactose.95 However, these studies use an unrealistic level of 10 mM galactose, which vastly exceeds the physiological plasma concentration (~10 μM) of this sugar.62 Therefore, its practical value is limited.
N-ACETYL-d-GLUCOSAMINE
GlcNAc is found in all types of glycans. How GlcNAc enters the cell is currently unknown, but to be incorporated into glycans, it must first be activated to UDP-GlcNAc. Different pathways contribute to the de novo biosynthesis of UDP-GlcNAc (Figure 1). One is the hexosamine biosynthetic pathway, which requires glucose, glucosamine, acetyl-coenzyme A, and UTP.96 Here, glucosamine:fructose-6-phosphate-amidotransferase (GFAT) catalyzes the rate-limiting step, resulting in the production of glucosamine 6-phosphate (GlcN-6P) from Fruc-6P.97 Alternatively, glucosamine can be directly phosphorylated by hexokinase (HK), allowing GlcN-6P to enter the pathway.98 Via the action of GNA1, an acetyl group is transferred from acetyl-coenzyme A to GlcN-6P to form GlcNAc 6-phosphate (GlcNAc-6P).99 Phosphoglucomutase 3 (PGM3) isomerizes GlcNAc-6P to GlcNAc 1-phosphate (GlcNAc-1P),100 and UDP-N-acetylhexosamine pyrophosphorylase (UAP1) catalyzes the production of UDP-GlcNAc.101 It is important to point out that human UDP-galactose 4-epimerase (GALE) can also interconvert UDP-GlcNAc and UDP-N-acetylgalactosamine, creating an alternative source of the de novo UDP-GlcNAc synthesis as well as its utilization.70 Its importance and contribution to various glycans are unknown.
Furthermore, UDP-GlcNAc can be provided by the salvage pathway (Figure 1).102 GlcNAc, which is recycled from lysosomal degradation of oligosaccharides or acquired from dietary sources, is phosphorylated by GlcNAc kinase (NAGK) to GlcNAc-6P and is processed further by PGM3.103 In addition to GlcNAc, NAGK is also able to phosphorylate N-acetylmannosamine (ManNAc).104 GlcNAc-2-epimerase (RENBP) catalyzes the interconversion of GlcNAc and ManNAc, preferably isomerizing the former monosaccharide to the latter.105 The relative contributions of all of these pathways in any cell type have not been studied.
Four UDP-GlcNAc transporters, SLC35A3,106 SLC35A5,107 SLC35B4,108 and SLC35D2,109 contribute to its transfer across the Golgi and ER membranes, where it is used by different glycosyltransferases in glycan synthesis (Figure 2). UDP-GlcNAc is the one of two nucleotide sugars utilized in the cytosol.7 Nucleocytoplasmic modification of serine and threonine residues by O-GlcNAc serves as an important metabolic sensor.110 In addition, the cytoplasmic pool of UDP-GlcNAc can be used for hyaluronan biosynthesis, which is the only glycosaminoglycan synthesized in the cytoplasm.8
PGM3-CDG is caused by pathogenic mutations in phosphoglucomutase 3, which interconverts GlcNAc-1P and GlcNAc-6P, disrupting the synthesis of UDP-GlcNAc and ultimately resulting in abnormal glycosylation.100 When PGM3-CDG fibroblasts were grown in medium supplemented with GlcNAc, abnormal glycosylation improved.111 This finding prompted a small trial in which PGM3-CDG individuals were supplemented with GlcNAc. Unfortunately, the treatment did not replicate the glycosylation improvements seen in the cell-based model, and the trial was stopped. In 2000, oral application of GlcNAc was adopted as a therapy in 12 children with chronic inflammatory bowel disease. This treatment resulted in histological improvement of rectal biopsy, significantly increasing epithelial and lamina propria glycosaminoglycans and intracellular GlcNAc.112 However, there is no information about further clinical trials for this therapy. Rabbits with experimental osteoarthritis showed improvement, when injected with GlcNAc, suggesting chondroprotective and anti-inflammatory effects of this monosaccharide.113 Glucosamine and GlcNAc treatment of mouse osteoblasts increased the rate of bone matrix deposition and decreased the rate of bone resorption modulating bone metabolism in osteoarthritis.114 In 2017, it was demonstrated that dietary supplementation with GlcNAc may improve the type II collagen metabolism of articular cartilage in healthy individuals, not exhibiting any arthritic syndromes, altogether suggesting GlcNAc as a potential therapeutic compound in osteoarthritis.115 In addition to GlcNAc, glucosamine was also in use for osteoarthritis, but the studies yielded conflicting results.116–118 In a mouse model for multiple sclerosis, an increased level of N-glycan branching was observed when GlcNAc was added to drinking water.119 This is an important observation because rare variants in MGAT5, which encodes GlcNAc transferase, were identified as being associated with the severity of multiple sclerosis.120 Disrupted N-glycan branching is thought to promote autoimmune demyelination and neurodegeneration in multiple sclerosis; therefore, correcting this phenotype by simply supplementing with GlcNAc is an attractive therapeutic strategy.119
Another rare disorder is caused by deficiency in NGLY1, which is responsible for deglycosylation of misfolded, retrotranslocated proteins prior their degradation in the cytosol.121 Affected individuals have developmental delay, liver disease, neurological symptoms, and alacrima.122 In dNGLY1-deficient fruit flies, GlcNAc supplementation partially rescued lethality and had a positive impact on the longevity of the flies, possibly by restoring UDP-GlcNAc levels, although this increase could not be demonstrated.123
SIALIC ACID
The term “sialic acids” refers to more than 50 unique variations of two parental compounds, N-acetylneuraminic acid (Neu5Ac) and N-glycolylneuraminic acid (Neu5Gc). These nine-carbon sugars are typically at the nonreducing ends of N- and O-glycans and glycolipids.124 Neu5Ac is the most abundant sialic acid in humans and can be synthesized de novo form UDP-GlcNAc in four steps (Figure 1). First, the bifunctional enzyme UDP-GlcNAc-2-epimerase/N-acetylmannosamine kinase (GNE) converts UDP-GlcNAc to ManNAc and subsequently to ManNAc 6-phosphate (ManNAc-6P).125 Next, Neu5Ac 9-phosphate (Neu5Ac-9P) is synthesized from ManNAc-6P by NANS. Finally, NANP removes the phosphate residue producing Neu5Ac.126 Recently, studies using NANP knockout cells showed that its activity is not essential for the de novo production of sialic acid, indicating an alternative phosphatase activity, bypassing NANP.127
Because of an inactivating mutation in CMAH, humans are genetically unable to synthesize Neu5Gc, which is the major sialic acid in most mammals (Figure 1).128 Surprisingly, small amounts of this sialic acid are still present in human cells, almost certainly of a dietary origin.129 While different pathways contribute to their uptake, including pinocytosis,130 both Neu5Ac and Neu5Gc can be taken up from plasma and incorporated directly into glycans.130,131 Neu5Ac can be salvaged from hydrolyzed glycans following its release by neuraminidases132,133 and transport from the lysosomes via Sialin, a transporter encoded by SLC17A5.134 Pathogenic mutations in SLC17A5 can cause either infantile sialic acid storage disorder (also known as sialuria) or an adult form of sialuria known as Salla disease.135
KDN is deaminated sialic acid, and as mentioned above, Man-6P serves as a substrate in its production (Figure 1).26 In contrast to Neu5Ac and Neu5Gc, KDN and KDN glycoconjugates are abundant in only lower vertebrates and pathogenic bacteria and undetectable in mammals.136 De novo synthesis of free KDN by human cells is enhanced in hypoxia.137
In contrast to the other nucleotide sugars, CMP-Neu5Ac is synthesized in the nucleus and is the only one activated with a monophosphonucleotide.5 Once synthesized, CMP-sialic acid is transported into the Golgi apparatus via SLC35A1 (Figure 2),138 where it serves as a substrate for different sialyltransferases. Only a few individuals with SLC35A1-CDG have been diagnosed, and it is unclear if either sialic acid or ManNAc could be used for monosaccharide therapy.139–142 One report showed that fibroblasts derived from a single individual do not improve glycosylation in media supplemented with sialic acid or ManNAc.141
Biallelic mutations in NANS encoding the Neu5Ac synthase led to elevated levels of ManNAc and resulted in hyposialylation of glycoproteins and glycolipids.143 Using nansa knockdown zebrafish embryos, it was demonstrated that addition of sialic acid to the water partially improved their phenotype by rescuing skeletal development. Interestingly, the effect of Neu5Ac supplementation was time-dependent, being successful immediately after its injection into embryos but not beyond 24 h after the fertilization.143 In 2018, an experimental theapy of orally supplemented Neu5Ac that aimed to treat NANS-deficient individuals was started.
N-ACETYL-d-MANNOSAMINE
As mentioned in the previous section, ManNAc is a hexosamine precursor on the biosynthetic pathway of sialic acids (Figure 1). It is synthesized by GNE from UDP-GlcNAc and converted further to ManNAc-6P.125 Then NANS and NANP use it as a substrate for Neu5Ac production (Figure 1).126 Sialic acids can be degraded to ManNAc and pyruvate in the reaction catalyzed by N-acetylneuraminate pyruvate lyase (NPL).144 This enzyme prevents Neu5Ac from being recycled.
GNE myopathy, also known as hereditary inclusion body myopathy, is a rare genetic muscle disorder caused by pathogenic variants in GNE.145 A GNE-deficient mouse model showed a 25% decrease in the level of membrane-bound sialic acids, mainly through the reduction of polysialic acids on NCAM and a shift of the transferrin isoelectric point toward basic pH.146 Different disease models showed that ManNAc or mannosamine can bypass the defect. For example, ManNAc added to the cell culture medium of GNE-CDG fibroblasts restored the observed glycosphingolipid defect.147 In a separate GNE-deficient mouse model, ManNAc, sialic acid, and mannosamine rescued both kidney and muscle hyposialylation but only ManNAc improved proteinuria.148 However, uptake of Neu5Ac administered orally to rats was insufficiently utilized and excreted mainly as free Neu5Ac via the kidneys. This could explain why the clinical trials of an extended release formulation of sialic acid (Ace-ER) were unsuccessful and discontinued in 2017.149 In contrast to Neu5Ac, ManNAc is still in clinical trials for GNE myopathy.
In addition to GNE myopathy, pathogenic variants of GNE also lead to sialuria.150 Massive urinary excretion of unconjugated Neu5Ac151 is due to GNE mutations at an allosteric site that normally binds CMP-sialic acid and downregulates GNE activity.152,153 In this disorder, altered sialic acid metabolism also causes developmental delays and coarse facial features.151
Studies in 2018 showed that pathogenic variants of NPL affect heart and skeletal muscle function and development and a npl knockdown in zebrafish mimicked the human phenotype. Treatment of zebrafish embryos with ManNAc and GlcNAc efficiently improved somite morphology and muscle fiber arrangement. Neu5Ac could also rescue the knockdown zebrafish phenotype but to a lesser extent. Interestingly, ManNAc, but not GlcNAc or Neu5Ac, improved heart looping.154
RIBITOL
Ribitol is a sugar alcohol formed by the reduction of ribose and was initially identified as a component of the cell wall in Gram-positive bacteria. Most of their cell surface polysaccharides consist of teichoic acids, composed of ManNAc, GlcNAc, and ribitol phosphate.155 There was no precedent for ribitol in human glycans, but this dramatically changed in 2016 when two ribitol 5-phosphate (Rbo-5P) residues were found on α-dystroglycan.156 In humans, ISPD activates ribitol by CTP to CDP-ribitol (Figure 1).157 It serves as a substrate for FKRP and FKTN enzymes, critical for incorporation of Rbo-5P into the O-mannose-based glycan chains of α-dystrogycan.158 Prior to this discovery, pathogenic variants of SLC35A1, encoding the CMP-sialic acid transporter, produced abnormal α-dystroglycan O-mannosylation, independent of sialic acid,142 suggesting SLC35A1 as a strong candidate for a CDP-ribitol transporter (Figure 2).
Pathogenic variants of FKRP and FKTN result in multiple clinically defined muscular dystrophies, including Walker-Warburg syndrome, muscle-eye-brain disease, and a mild form of limb-girdle disease.159,160 Recently, mouse models demonstrated that oral supplementation with ribitol enhances the synthesis of CDP-ribitol and Rbo-5P. Importantly, this therapy significantly increases the level of functional glycosylation of α-dystroglycan in skeletal and cardiac muscles, thereby improving the condition of Fkrp mutant mice.161 This finding might be a breakthrough in the treatment of FKPR-related muscular dystrophy; however, studies have not yet shown whether ribitol can reverse muscle deterioration.
MODIFIED MONOSACCHARIDES AS A TOOL FOR STUDYING GLYCAN FUNCTION
Metabolic labeling of glycans with chemically modified monosaccharides ushers in a novel avenue for the study of glycosylation. When combined with click chemistry, modified monosaccharides are a powerful tool for improving our understanding of sugar utilization within the cell and organism. Azido sugars, such as ManNAz, GalNAz, GlcNAz, and 6-azidofucose, are broadly applied for the visualization of cellular glycans and for glycoproteomics.162 They can be utilized both in cell cultures and in model organisms, permitting the study of biochemical pathways involving different monosaccharides. Other analogues, including peracetyl, alkynyl, or alkenyl sugars, are also in common use. They are developed as not only glycan imaging probes but also glycosylation inhibitors and remodelers.163–166 Other applications of chemically modified monosaccharides include targeting toxins to tumor cells, inhibiting pathogen binding, or altering immune cell activity.167
Despite their broad application range, these monosaccharides must be cautiously utilized. As nonphysiological components, they may not fully mimic the behavior of naturally occurring sugars and might have secondary effects, which are hard to predict. For instance, 6-alkynyl fucose competitively inhibits de novo GDP-fucose synthesis via the FX enzyme.166 In contrast, 7-alkynyl fucose serves as a highly sensitive probe that labels fucose in glycans.165 Also, studies of zebrafish embryogenesis revealed that azide-modified fucose is inefficiently processed by the fucose salvage pathway enzymes.168
CONCLUDING REMARKS
Most of the studies of glycan-associated monosaccharides were performed in the 1960s and 1970s, using the best methods, reagents, and biological materials of that era. Today, researchers consider these studies as the dogma of bedrock biochemistry. For many monosaccharides, their mechanism of transport, contribution to nucleotide sugar synthesis, and turnover rates have not been studied in any detail. Results can vary according to cell lines as some may preferentially use one pathway over the others. Many studies were conducted using cancer cells or cell lines lacking entire pathways (e.g., HCT116, CHO-Lec13, or Neuro-2A deficient in the de novo GDP-fucose synthesis). In most cases, data from model organisms are lacking. A few studies demonstrate cross-talk between different pathways and the importance of using physiological concentrations of monosaccharides, which vastly differ between monosaccharides.23,30,45,153,169 Intermediate products of one pathway can produce allosteric inhibition of the enzymes in another pathway leading to the same product, as shown for GDP-fucose biosynthesis in the 1960s and 1970s when bacterial homologues of human enzymes were characterized.170 In addition, the origin of the activated monosaccharide may affect its utilization or access to seemingly continuous pathways, as shown by the lack of entry of mannose-derived glucose into glycogen or its exit.23 Most published studies do not differentiate between the salvage and dietary origin of monosaccharides (Table 1), and these two terms are often treated equivalently; here, we refer to only a few examples of this very common mistake.171,172 It is also very hard to assess if the sugar was salvaged or secreted and taken back into the same cell or a different cell before its reutilization. Incorporation of monosaccharides through the diet and salvage should be considered as distinct pathways as was shown for mannose, where dietary mannose is either used directly for glycan synthesis or used as fuel but released mannose is secreted into the medium.23 Salvage can be assessed by blocking lysosomal degradation of glycans or preventing the exit of the monosaccharide from the lysosome, as it was done for GlcNAc and sialic acid recycling.102,133 Another way is to add specific inhibitors to block activation and reincorporation of the released sugar into glycans.
Table 1.
Summary of the Most Important Information about the Monosaccharides Discussed Herein
| Monosaccharide | Major pathways | Therapeutic applications | Comments |
|---|---|---|---|
| Mannose | -Glycosylation (not glycolipids) *de novo synthesis from glucose29 *from diet, activated by GMPP23 *KDN synthesis26 -Catabolized23 |
-Supplement for MPI-CDG32 -Treatment and prophylactic for urinary tract infections35 |
-TOXIC:
Mpi hypomorphic mice34 -HARMFUL: may lead to diarrhea176 -Modulates host’s microbiome36 -Beneficial in obesity and cancer36, 37 -NOT converted to glucose in glycogen23 -NOT salvaged23 |
| Fucose | -Glycosylation *de novo synthesis from mannose and glucose39 *salvaged# 42, 43 or from diet#,40 activated by FPGT -Oxidized to lactate177 |
-Supplement for fucosylation abnormalities due to impaired Golgi transport of GDP - fucose (LADII)53, 54 |
-Modulates host microbiome59 -Beneficial in obesity and cancer58 -Decreases bacterial infection in cystic fibrosis60 |
| Galactose | -Glycosylation * synthesized de novo from glucose70 *salvaged# or form diet# activated by GALT65 -Catabolized through the Leloir pathway 63 -Oxidized to CO2178 |
Supplement for galactosylation abnormalities due to: -Decreased Golgi transport of UDP-galactose (SLC35A2-CDG)82 -Insufficient isomerization of Glu-6P to Glc-lP (PGM1-CDG)86 -Dysfunction in cellular transport of Mn2+ (SLC39A8-CDG)87 -Deficiency in the Golgi transport of Mn2+ and Ca2+ (TMEM165-CDG)88 |
-TOXIC: in galactosemia76 -HARMFUL: in lactose intolerance179 -Decreases the risk of bacterial infection in cystic fibrosis60 -Beneficial in idiopathic steroid-resistant nephrotic syndrome90 |
| GlcNAc | -Glycosylation *synthesized de novo in hexosamine biosynthetic pathway96 or from UDP-GalNAc70 * salvaged102 or from diet,112 activated by UAP1 |
-Supplement of glycosaminoglycans synthesis in chronic inflammatory bowel disease112 -Joint pain and damage in osteoarthritis113 -Impaired N-glycans branching in multiple sclerosis119 |
-GlcNAc as a therapy in GlcNAc-6P→ GlcNAc-1P isomerase activity deficiency (PGM3-CDG) failed clinical trials111 -Unclear if it has a positive impact in osteoarthritis -In dNGLY1 knock-down fruit flies GlcNAc increased survival and longevity123 |
| Sialic acid | -Glycosylation * synthesized de novo from GlcNAc or ManNAc125 *salvaged## 133 or from diet131, activated by CMAS -Catabolized to ManNAc and pyruvate144 |
-Defect in dephosphorylation of NeuNAc-9P mediated by NANS143 | -Ineffective in impaired CMP-sialic acid transport (SLC35A1-CDG) fibroblasts141 -Failed as a therapy for GNE myopathy149 |
| ManNAc | -Glycosylation *precursor for sialic acid and GlcNAc105, 125 -Product of sialic acid catabolism144 |
-GNE myopathy180 -NPL defects with perturbed sialic acid catabolism in zebrafish154 |
-ManNAc therapy of NPL deficiency used in humans -Not found in human glycans |
| Ribitol | -Glycosylation: *incorporated as Rbo-5P into α-dystroglycan156 |
Pathogenic variants of FKRP, adding Rbo-5P to α-dystroglycan, resulting in: -muscular dystrophy -Walker-Warburg syndrome -muscle-eye-brain disease -mild form of limb-girdle disease161 |
-Ribitol therapy untested in humans -Defect in α-dystroglycan O-mannosylation identified in SLC35A1-CDG individual142 -High ribitol concentrations found in ribose-5-phosphate isomerase deficiency (pentose-phosphate-pathway defect)181 |
The contribution and the difference between diet and salvage were not determined.
Salvage from glycoproteins was not studied.
As monosaccharides become a popular therapy for an ever-growing number of congenital disorders of glycosylation (Table 1), we need to revise some of the previous findings on the basis of new technologies not available to researchers 45–50 years ago. These new approaches may help overcome past limitations. It is also important to keep in mind that monosaccharides might be toxic and, under some conditions, are dangerous. Galactose is harmful for individuals with galactosemia;76 mannose exhibits toxicity in Mpi hypomorphic mice,34 and d-galactosamine leads to liver alterations in rats that resemble human viral hepatitis.173 Filling the gaps will allow us to better understand the mechanisms behind both the successes and failures of a therapy, potential toxicities, and therefore provide better help to the afflicted individuals treated with monosaccharides.
Finally, some companies market indigestible polysaccharides as sources of “essential glyconutrients” that have rightly been branded “shams”.174,175 It is important for the scientific community to guard against misrepresentation of the facts, and that is best done by filling in those blanks with data. If we do not, marketers of sham glyconutrients will be happy to present “alternative facts” on our behalf.
Funding
The Rocket Fund and National Institutes of Health Grant R01DK099551 supported this work.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, and Seeberger PH, Eds. (2015) Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, Plainview, NY. [PubMed] [Google Scholar]
- (2).Varki A, Freeze HH, and Manzi AE (2009) Overview of glycoconjugate analysis Current Protocols in Protein Science, Chapter 12, Unit 12.11, pp 12.11.11–12.11.18, Wiley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Flint HJ, Scott KP, Duncan SH, Louis P, and Forano E (2012) Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3, 289–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Coates SW, Gurney T Jr., Sommers LW, Yeh M, and Hirschberg CB (1980) Subcellular localization of sugar nucleotide synthetases. J. Biol. Chem 255, 9225–9229. [PubMed] [Google Scholar]
- (5).Munster AK, Eckhardt M, Potvin B, Muhlenhoff M, Stanley P, and Gerardy-Schahn R (1998) Mammalian cytidine 5′-monophosphate N-acetylneuraminic acid synthetase: a nuclear protein with evolutionarily conserved structural motifs. Proc. Natl. Acad. Sci. U. S. A 95, 9140–9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Caffaro CE, and Hirschberg CB (2006) Nucleotide sugar transporters of the Golgi apparatus: from basic science to diseases. Acc. Chem. Res 39, 805–812. [DOI] [PubMed] [Google Scholar]
- (7).Torres CR, and Hart GW (1984) Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem 259, 3308–3317. [PubMed] [Google Scholar]
- (8).Vigetti D, Karousou E, Viola M, Deleonibus S, De Luca G, and Passi A (2014) Hyaluronan: biosynthesis and signaling. Biochim. Biophys. Acta, Gen. Subj 1840, 2452–2459. [DOI] [PubMed] [Google Scholar]
- (9).Ruhaak LR, and Lebrilla CB (2012) Analysis and role of oligosaccharides in milk. BMB Rep 45, 442–451. [DOI] [PubMed] [Google Scholar]
- (10).Huang J, Kailemia MJ, Goonatilleke E, Parker EA, Hong Q, Sabia R, Smilowitz JT, German JB, and Lebrilla CB (2017) Quantitation of human milk proteins and their glycoforms using multiple reaction monitoring (MRM). Anal. Bioanal. Chem 409, 589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kunz C, Rudloff S, Baier W, Klein N, and Strobel S (2000) Oligosaccharides in human milk: structural, functional, and metabolic aspects. Annu. Rev. Nutr 20, 699–722. [DOI] [PubMed] [Google Scholar]
- (12).Bode L (2012) Human milk oligosaccharides: every baby needs a sugar mama. Glycobiology 22, 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wise A, Robertson B, Choudhury B, Rautava S, Isolauri E, Salminen S, and Bode L (2018) Infants Are Exposed to Human Milk Oligosaccharides Already in utero. Frontiers in Pediatrics 6, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Coppa GV, Gabrielli O, Zampini L, Galeazzi T, Ficcadenti A, Padella L, Santoro L, Soldi S, Carlucci A, Bertino E, and Morelli L (2011) Oligosaccharides in 4 different milk groups, Bifidobacteria, and Ruminococcus obeum. J. Pediatr. Gastroenterol. Nutr 53, 80–87. [DOI] [PubMed] [Google Scholar]
- (15).Bode L, McGuire M, Rodriguez JM, Geddes DT, Hassiotou F, Hartmann PE, and McGuire MK (2014) It’s alive: microbes and cells in human milk and their potential benefits to mother and infant. Adv. Nutr 5, 571–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Underwood MA, Davis JCC, Kalanetra KM, Gehlot S, Patole S, Tancredi DJ, Mills DA, Lebrilla CB, and Simmer K (2017) Digestion of Human Milk Oligosaccharides by Bifidobacterium breve in the Premature Infant. J. Pediatr. Gastroenterol. Nutr 65, 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Charbonneau MR, O’Donnell D, Blanton LV, Totten SM, Davis JC, Barratt MJ, Cheng J, Guruge J, Talcott M, Bain JR, Muehlbauer MJ, Ilkayeva O, Wu C, Struckmeyer T, Barile D, Mangani C, Jorgensen J, Fan YM, Maleta K, Dewey KG, Ashorn P, Newgard CB, Lebrilla C, Mills DA, and Gordon JI (2016) Sialylated Milk Oligosaccharides Promote Microbiota-Dependent Growth in Models of Infant Undernutrition. Cell 164, 859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Harmsen HJ, Wildeboer-Veloo AC, Raangs GC, Wagendorp AA, Klijn N, Bindels JG, and Welling GW (2000) Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J. Pediatr. Gastroenterol. Nutr 30, 61–67. [DOI] [PubMed] [Google Scholar]
- (19).Freeze HH, Eklund EA, Ng BG, and Patterson MC (2012) Neurology of inherited glycosylation disorders. Lancet Neurol 11, 453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ng BG, and Freeze HH (2018) Perspectives on Glycosylation and Its Congenital Disorders. Trends Genet 34, 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lacey JM, Bergen HR, Magera MJ, Naylor S, and O’Brien JF (2001) Rapid determination of transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry. Clin. Chem 47, 513–518. [PubMed] [Google Scholar]
- (22).Chen J, Li X, Edmondson A, Meyers GD, Izumi K, Ackermann AM, Morava E, Ficicioglu C, Bennett MJ, and He M (2019) Increased Clinical Sensitivity and Specificity of Plasma Protein N-Glycan Profiling for Diagnosing Congenital Disorders of Glycosylation by Use of Flow Injection-Electrospray Ionization-Quadrupole Time-of-Flight Mass Spectrometry. Clin. Chem 65, 653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sharma V, and Freeze HH (2011) Mannose efflux from the cells: a potential source of mannose in blood. J. Biol. Chem 286, 10193–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wood FC Jr., and Cahill GF Jr. (1963) Mannose utilization in Man. J. Clin. Invest 42, 1300–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Carminatti H, and Cabib E (1961) Phosphorolysis of the pyrophosphate bond of some nucleotides. Biochim. Biophys. Acta 53, 417–419. [DOI] [PubMed] [Google Scholar]
- (26).Go S, Sato C, Furuhata K, and Kitajima K (2006) Oral ingestion of mannose alters the expression level of deaminoneuraminic acid (KDN) in mouse organs. Glycoconjugate J 23, 411–421. [DOI] [PubMed] [Google Scholar]
- (27).Murrell MP, Yarema KJ, and Levchenko A (2004) The systems biology of glycosylation. ChemBioChem 5, 1334–1347. [DOI] [PubMed] [Google Scholar]
- (28).Becker DJ, and Lowe JB (2003) Fucose: biosynthesis and biological function in mammals. Glycobiology 13, 41R–53R. [DOI] [PubMed] [Google Scholar]
- (29).Panneerselvam K, and Freeze HH (1996) Mannose corrects altered N-glycosylation in carbohydrate-deficient glycoprotein syndrome fibroblasts. J. Clin. Invest 97, 1478–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ichikawa M, Scott DA, Losfeld ME, and Freeze HH (2014) The metabolic origins of mannose in glycoproteins. J. Biol. Chem 289, 6751–6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Peanne R, de Lonlay P, Foulquier F, Kornak U, Lefeber DJ, Morava E, Perez B, Seta N, Thiel C, Van Schaftingen E, Matthijs G, and Jaeken J (2018) Congenital disorders of glycosylation (CDG): Quo vadis? Eur. J. Med. Genet 61, 643–663. [DOI] [PubMed] [Google Scholar]
- (32).Niehues R, Hasilik M, Alton G, Korner C, Schiebe-Sukumar M, Koch HG, Zimmer KP, Wu R, Harms E, Reiter K, von Figura K, Freeze HH, Harms HK, and Marquardt T (1998) Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J. Clin. Invest 101, 1414–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Mention K, Lacaille F, Valayannopoulos V, Romano S, Kuster A, Cretz M, Zaidan H, Galmiche L, Jaubert F, de Keyzer Y, Seta N, and de Lonlay P (2008) Development of liver disease despite mannose treatment in two patients with CDG-Ib. Mol. Genet. Metab 93, 40–43. [DOI] [PubMed] [Google Scholar]
- (34).Sharma V, Nayak J, DeRossi C, Charbono A, Ichikawa M, Ng BG, Grajales-Esquivel E, Srivastava A, Wang L, He P, Scott DA, Russell J, Contreras E, Guess CM, Krajewski S, Del Rio-Tsonis K, and Freeze HH (2014) Mannose supplements induce embryonic lethality and blindness in phosphomannose isomerase hypomorphic mice. FASEB J 28, 1854–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kranjcec B, Papes D, and Altarac S (2014) D-mannose powder for prophylaxis of recurrent urinary tract infections in women: a randomized clinical trial. World J. Urol 32, 79–84. [DOI] [PubMed] [Google Scholar]
- (36).Sharma V, Smolin J, Nayak J, Ayala JE, Scott DA, Peterson SN, and Freeze HH (2018) Mannose Alters Gut Microbiome, Prevents Diet-Induced Obesity, and Improves Host Metabolism. Cell Rep 24, 3087–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Gonzalez PS, O’Prey J, Cardaci S, Barthet VJA, Sakamaki JI, Beaumatin F, Roseweir A, Gay DM, Mackay G, Malviya G, Kania E, Ritchie S, Baudot AD, Zunino B, Mrowinska A, Nixon C, Ennis D, Hoyle A, Millan D, McNeish IA, Sansom OJ, Edwards J, and Ryan KM (2018) Mannose impairs tumour growth and enhances chemotherapy. Nature 563, 719–723. [DOI] [PubMed] [Google Scholar]
- (38).Schneider M, Al-Shareffi E, and Haltiwanger RS (2017) Biological functions of fucose in mammals. Glycobiology 27, 601–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ginsburg V (1960) Formation of guanosine diphosphate L-fucose from guanosine diphosphate D-mannose. J. Biol. Chem 235, 2196–2201. [PubMed] [Google Scholar]
- (40).Coffey JW, Miller ON, and Sellinger OZ (1964) The metabolism of L-Fucose in the rat. J. Biol. Chem 239, 4011–4017. [PubMed] [Google Scholar]
- (41).Kaufman RL, and Ginsburg V (1968) The metabolism of L-fucose by HeLa cells. Exp. Cell Res 50, 127–132. [DOI] [PubMed] [Google Scholar]
- (42).Yurchenco PD, and Atkinson PH (1975) Fucosylglycoprotein and precursor polls in HeLa cells. Biochemistry 14, 3107–3114. [DOI] [PubMed] [Google Scholar]
- (43).Yurchenco PD, and Atkinson PH (1977) Equilibration of fucosyl glycoprotein pools in HeLa cells. Biochemistry 16, 944–953. [DOI] [PubMed] [Google Scholar]
- (44).Ishihara H, Massaro DJ, and Heath EC (1968) The metabolism of L-fucose. 3. The enzymatic synthesis of beta-L-fucose 1-phosphate. J. Biol. Chem 243, 1103–1109. [PubMed] [Google Scholar]
- (45).Ng BG, Xia Z-J, Scott DA, and Freeze HH (2018) Fucose Metabolism: Rethinking old concepts and identifying new mechanisms. Glycobiology 28, 1026. [Google Scholar]
- (46).Levvy GA, and McAllan A (1961) Mammalian fucosidases. 2. alpha-L-Fucosidase. Biochem. J 80, 435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Park D, Ryu KS, Choi D, Kwak J, and Park C (2007) Characterization and role of fucose mutarotase in mammalian cells. Glycobiology 17, 955–962. [DOI] [PubMed] [Google Scholar]
- (48).Wiese TJ, Dunlap JA, and Yorek MA (1994) L-fucose is accumulated via a specific transport system in eukaryotic cells. J. Biol. Chem 269, 22705–22711. [PubMed] [Google Scholar]
- (49).Luhn K, Wild MK, Eckhardt M, Gerardy-Schahn R, and Vestweber D (2001) The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nat. Genet 28, 69–72. [DOI] [PubMed] [Google Scholar]
- (50).Lu L, Hou X, Shi S, Korner C, and Stanley P (2010) Slc35c2 promotes Notch1 fucosylation and is required for optimal Notch signaling in mammalian cells. J. Biol. Chem 285, 36245–36254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ishikawa HO, Ayukawa T, Nakayama M, Higashi S, Kamiyama S, Nishihara S, Aoki K, Ishida N, Sanai Y, and Matsuno K (2010) Two pathways for importing GDP-fucose into the endoplasmic reticulum lumen function redundantly in the O-fucosylation of Notch in Drosophila. J. Biol. Chem 285, 4122–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Karsan A, Cornejo CJ, Winn RK, Schwartz BR, Way W, Lannir N, Gershoni-Baruch R, Etzioni A, Ochs HD, and Harlan JM (1998) Leukocyte Adhesion Deficiency Type II is a generalized defect of de novo GDP-fucose biosynthesis. Endothelial cell fucosylation is not required for neutrophil rolling on human nonlymphoid endothelium. J. Clin. Invest 101, 2438–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Marquardt T, Luhn K, Srikrishna G, Freeze HH, Harms E, and Vestweber D (1999) Correction of leukocyte adhesion deficiency type II with oral fucose. Blood 94, 3976–3985. [PubMed] [Google Scholar]
- (54).van de Vijver E, Maddalena A, Sanal O, Holland SM, Uzel G, Madkaikar M, de Boer M, van Leeuwen K, Koker MY, Parvaneh N, Fischer A, Law SK, Klein N, Tezcan FI, Unal E, Patiroglu T, Belohradsky BH, Schwartz K, Somech R, Kuijpers TW, and Roos D (2012) Hematologically important mutations: leukocyte adhesion deficiency (first update). Blood Cells, Mol., Dis 48, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ng BG, Rosenfeld JA, Emrick L, Jain M, Burrage LC, Lee B, Craigen WJ, Bearden DR, Graham BH, and Freeze HH (2018) Pathogenic Variants in Fucokinase Cause a Congenital Disorder of Glycosylation. Am. J. Hum. Genet 103, 1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Ng BG, Xu G, Chandy N, Steyermark J, Shinde DN, Radtke K, Raymond K, Lebrilla CB, AlAsmari A, Suchy SF, Powis Z, Faqeih EA, Berry SA, Kronn DF, and Freeze HH (2018) Biallelic Mutations in FUT8 Cause a Congenital Disorder of Glycosylation with Defective Fucosylation. Am. J. Hum. Genet 102, 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Uozumi N, Yanagidani S, Miyoshi E, Ihara Y, Sakuma T, Gao CX, Teshima T, Fujii S, Shiba T, and Taniguchi N (1996) Purification and cDNA cloning of porcine brain GDP-L-Fuc:N-acetyl-beta-D-glucosaminide alpha1–>6fucosyltransferase. J. Biol. Chem 271, 27810–27817. [DOI] [PubMed] [Google Scholar]
- (58).Lau E, Feng Y, Claps G, Fukuda MN, Perlina A, Donn D, Jilaveanu L, Kluger H, Freeze HH, and Ronai ZA (2015) The transcription factor ATF2 promotes melanoma metastasis by suppressing protein fucosylation. Sci. Signaling 8, ra124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Wu G, Niu M, Tang W, Hu J, Wei G, He Z, Chen Y, Jiang Y, and Chen P (2018) L-Fucose ameliorates high-fat diet-induced obesity and hepatic steatosis in mice. J. Transl. Med 16, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Hauber HP, Schulz M, Pforte A, Mack D, Zabel P, and Schumacher U (2008) Inhalation with fucose and galactose for treatment of Pseudomonas aeruginosa in cystic fibrosis patients. Int. J. Med. Sci 5, 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Mueckler M, and Thorens B (2013) The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med 34, 121–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Wong SY, Gadomski T, van Scherpenzeel M, Honzik T, Hansikova H, Holmefjord KSB, Mork M, Bowling F, Sykut-Cegielska J, Koch D, Hertecant J, Preston G, Jaeken J, Peeters N, Perez S, Nguyen DD, Crivelly K, Emmerzaal T, Gibson KM, Raymond K, Abu Bakar N, Foulquier F, Poschet G, Ackermann AM, He M, Lefeber DJ, Thiel C, Kozicz T, and Morava E (2017) Oral D-galactose supplementation in PGM1-CDG. Genet. Med 19, 1226–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Frey PA (1996) The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J 10, 461–470. [PubMed] [Google Scholar]
- (64).Thoden JB, and Holden HM (2003) Molecular structure of galactokinase. J. Biol. Chem 278, 33305–33311. [DOI] [PubMed] [Google Scholar]
- (65).Leslie ND, Immerman EB, Flach JE, Florez M, Fridovich-Keil JL, and Elsas LJ (1992) The human galactose-1-phosphate uridyltransferase gene. Genomics 14, 474–480. [DOI] [PubMed] [Google Scholar]
- (66).Knop JK, and Hansen RG (1970) Uridine diphosphate glucose pyrophosphorylase. IV. Crystallization and properties of the enzyme from human liver. J. Biol. Chem 245, 2499–2504. [PubMed] [Google Scholar]
- (67).Turnquist RL, Gillett TA, and Hansen RG (1974) Uridine diphosphate glucose pyrophosphorylase. Crystallization and properties of the enzyme from rabbit liver and species comparisons. J. Biol. Chem 249, 7695–7700. [PubMed] [Google Scholar]
- (68).Isselbacher KJ (1958) A mammalian uridinediphosphate galactose pyrophosphorylase. J. Biol. Chem 232, 429–444. [PubMed] [Google Scholar]
- (69).Abraham HD, and Howell RR (1969) Human hepatic uridine diphosphate galactose pyrophosphorylase. Its characterization and activity during development. J. Biol. Chem 244, 545–550. [PubMed] [Google Scholar]
- (70).Thoden JB, Wohlers TM, Fridovich-Keil JL, and Holden HM (2001) Human UDP-galactose 4-epimerase. Accommodation of UDP-N-acetylglucosamine within the active site. J. Biol. Chem 276, 15131–15136. [DOI] [PubMed] [Google Scholar]
- (71).Zhang C, Griffith BR, Fu Q, Albermann C, Fu X, Lee IK, Li L, and Thorson JS (2006) Exploiting the reversibility of natural product glycosyltransferase-catalyzed reactions. Science 313, 1291–1294. [DOI] [PubMed] [Google Scholar]
- (72).Miura N, Ishida N, Hoshino M, Yamauchi M, Hara T, Ayusawa D, and Kawakita M (1996) Human UDP-galactose translocator: molecular cloning of a complementary DNA that complements the genetic defect of a mutant cell line deficient in UDP-galactose translocator. J. Biochem 120, 236–241. [DOI] [PubMed] [Google Scholar]
- (73).Thoden JB, Kim J, Raushel FM, and Holden HM (2003) The catalytic mechanism of galactose mutarotase. Protein Sci 12, 1051–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Wada Y, Kikuchi A, Arai-Ichinoi N, Sakamoto O, Takezawa Y, Iwasawa S, Niihori T, Nyuzuki H, Nakajima Y, Ogawa E, Ishige M, Hirai H, Sasai H, Fujiki R, Shirota M, Funayama R, Yamamoto M, Ito T, Ohara O, Nakayama K, Aoki Y, Koshiba S, Fukao T, and Kure S (2019) Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet. Med 21, 1286–1294. [DOI] [PubMed] [Google Scholar]
- (75).Timson DJ (2019) Type IV galactosemia. Genet. Med 21, 1283–1285. [DOI] [PubMed] [Google Scholar]
- (76).Berry GT (1993) Classic Galactosemia and Clinical Variant Galactosemia In GeneReviews (Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, and Amemiya A, Eds.) University of Washington, Seattle. [PubMed] [Google Scholar]
- (77).Coman DJ, Murray DW, Byrne JC, Rudd PM, Bagaglia PM, Doran PD, and Treacy EP (2010) Galactosemia, a single gene disorder with epigenetic consequences. Pediatr. Res 67, 286–292. [DOI] [PubMed] [Google Scholar]
- (78).Dobbie JA, Holton JB, and Clamp JR (1990) Defective galactosylation of proteins in cultured skin fibroblasts from galactosaemic patients. Ann. Clin. Biochem 27 (Part 3), 274–275. [DOI] [PubMed] [Google Scholar]
- (79).Charlwood J, Clayton P, Keir G, Mian N, and Winchester B (1998) Defective galactosylation of serum transferrin in galactosemia. Glycobiology 8, 351–357. [DOI] [PubMed] [Google Scholar]
- (80).Sturiale L, Barone R, Fiumara A, Perez M, Zaffanello M, Sorge G, Pavone L, Tortorelli S, O’Brien JF, Jaeken J, and Garozzo D (2005) Hypoglycosylation with increased fucosylation and branching of serum transferrin N-glycans in untreated galactosemia. Glycobiology 15, 1268–1276. [DOI] [PubMed] [Google Scholar]
- (81).Coss KP, Hawkes CP, Adamczyk B, Stockmann H, Crushell E, Saldova R, Knerr I, Rubio-Gozalbo ME, Monavari AA, Rudd PM, and Treacy EP (2014) N-glycan abnormalities in children with galactosemia. J. Proteome Res 13, 385–394. [DOI] [PubMed] [Google Scholar]
- (82).Dorre K, Olczak M, Wada Y, Sosicka P, Gruneberg M, Reunert J, Kurlemann G, Fiedler B, Biskup S, Hortnagel K, Rust S, and Marquardt T (2015) A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): molecular basis, clinical phenotype, and therapeutic approach. J. Inherited Metab. Dis 38, 931–940. [DOI] [PubMed] [Google Scholar]
- (83).Vals MA, Ashikov A, Ilves P, Loorits D, Zeng Q, Barone R, Huijben K, Sykut-Cegielska J, Diogo L, Elias AF, Greenwood RS, Grunewald S, van Hasselt PM, van de Kamp JM, Mancini G, Okninska A, Pajusalu S, Rudd PM, Rustad CF, Salvarinova R, de Vries BBA, Wolf NI, Ng BG, Freeze HH, Lefeber DJ, and Ounap K (2019) Clinical, neuroradiological, and biochemical features of SLC35A2-CDG patients. J. Inherited Metab. Dis 42, 553–564. [DOI] [PubMed] [Google Scholar]
- (84).Ng BG, Sosicka P, Agadi S, Almannai M, Bacino CA, Barone R, Botto LD, Burton JE, Carlston C, Chung BH, Cohen JS, Coman D, Dipple KM, Dorrani N, Dobyns WB, Elias AF, Epstein L, Gahl WA, Garozzo D, Hammer TB, Haven J, Heron D, Herzog M, Hoganson GE, Hunter JM, Jain M, Juusola J, Lakhani S, Lee H, Lee J, Lewis K, Longo N, Lourenco CM, Mak CCY, McKnight D, Mendelsohn BA, Mignot C, Mirzaa G, Mitchell W, Muhle H, Nelson SF, Olczak M, Palmer CGS, Partikian A, Patterson MC, Pierson TM, Quinonez SC, Regan BM, Ross ME, Guillen Sacoto MJ, Scaglia F, Scheffer IE, Segal D, Singhal NS, Striano P, Sturiale L, Symonds JD, Tang S, Vilain E, Willis M, Wolfe LA, Yang H, Yano S, Powis Z, Suchy SF, Rosenfeld JA, Edmondson AC, Grunewald S, and Freeze HH (2019) SLC35A2-CDG: Functional characterization, expanded molecular, clinical, and biochemical phenotypes of 30 unreported Individuals. Hum. Mutat 40, 908–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Tegtmeyer LC, Rust S, van Scherpenzeel M, Ng BG, Losfeld ME, Timal S, Raymond K, He P, Ichikawa M, Veltman J, Huijben K, Shin YS, Sharma V, Adamowicz M, Lammens M, Reunert J, Witten A, Schrapers E, Matthijs G, Jaeken J, Rymen D, Stojkovic T, Laforet P, Petit F, Aumaitre O, Czarnowska E, Piraud M, Podskarbi T, Stanley CA, Matalon R, Burda P, Seyyedi S, Debus V, Socha P, Sykut-Cegielska J, van Spronsen F, de Meirleir L, Vajro P, DeClue T, Ficicioglu C, Wada Y, Wevers RA, Vanderschaeghe D, Callewaert N, Fingerhut R, van Schaftingen E, Freeze HH, Morava E, Lefeber DJ, and Marquardt T (2014) Multiple phenotypes in phosphoglucomutase 1 deficiency. N. Engl. J. Med 370, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Radenkovic S, Bird MJ, Emmerzaal TL, Wong SY, Felgueira C, Stiers KM, Sabbagh L, Himmelreich N, Poschet G, Windmolders P, Verheijen J, Witters P, Altassan R, Honzik T, Eminoglu TF, James PM, Edmondson AC, Hertecant J, Kozicz T, Thiel C, Vermeersch P, Cassiman D, Beamer L,Morava E, and Ghesquiere B (2019) The Metabolic Map into the Pathomechanism and Treatment of PGM1-CDG. Am. J. Hum. Genet 104, 835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Park JH, Hogrebe M, Gruneberg M, DuChesne I, von der Heiden AL, Reunert J, Schlingmann KP, Boycott KM, Beaulieu CL, Mhanni AA, Innes AM, Hortnagel K, Biskup S, Gleixner EM, Kurlemann G, Fiedler B, Omran H, Rutsch F, Wada Y, Tsiakas K, Santer R, Nebert DW, Rust S, and Marquardt T (2015) SLC39A8 Deficiency: A Disorder of Manganese Transport and Glycosylation. Am. J. Hum. Genet 97, 894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Morelle W, Potelle S, Witters P, Wong S, Climer L, Lupashin V, Matthijs G, Gadomski T, Jaeken J, Cassiman D, Morava E, and Foulquier F (2017) Galactose Supplementation in Patients With TMEM165-CDG Rescues the Glycosylation Defects. J. Clin. Endocrinol. Metab 102, 1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Snyder NA, Palmer MV, Reinhardt TA, and Cunningham KW (2019) Milk biosynthesis requires the Golgi cation exchanger TMEM165. J. Biol. Chem 294, 3181–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Sgambat K, Banks M, and Moudgil A (2013) Effect of galactose on glomerular permeability and proteinuria in steroid-resistant nephrotic syndrome. Pediatr. Nephrol 28, 2131–2135. [DOI] [PubMed] [Google Scholar]
- (91).Salkovic-Petrisic M, Osmanovic-Barilar J, Knezovic A, Hoyer S, Mosetter K, and Reutter W (2014) Long-term oral galactose treatment prevents cognitive deficits in male Wistar rats treated intracerebroventricularly with streptozotocin. Neuropharmacology 77, 68–80. [DOI] [PubMed] [Google Scholar]
- (92).Knezovic A, Osmanovic Barilar J, Babic A, Bagaric R, Farkas V, Riederer P, and Salkovic-Petrisic M (2018) Glucagon-like peptide-1 mediates effects of oral galactose in streptozotocin-induced rat model of sporadic Alzheimer’s disease. Neuropharmacology 135, 48–62. [DOI] [PubMed] [Google Scholar]
- (93).Warburg O, Geissler AW, and Lorenz S (1967) On growth of cancer cells in media in which glucose is replaced by galactose. Hoppe-Seyler’s Z. Physiol. Chem 348, 1686–1687. [PubMed] [Google Scholar]
- (94).Marroquin LD, Hynes J, Dykens JA, Jamieson JD, and Will Y (2007) Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci 97, 539–547. [DOI] [PubMed] [Google Scholar]
- (95).Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, and Capaldi RA (2004) Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res 64, 985–993. [DOI] [PubMed] [Google Scholar]
- (96).Schleicher ED, and Weigert C (2000) Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int. Suppl 58, S13–S18. [DOI] [PubMed] [Google Scholar]
- (97).Marshall S, Bacote V, and Traxinger RR (1991) Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem 266, 4706–4712. [PubMed] [Google Scholar]
- (98).Lange CF Jr., and Kohn P (1961) Substrate specificity of hexokinases. J. Biol. Chem 236, 1–5. [PubMed] [Google Scholar]
- (99).Wang J, Liu X, Liang YH, Li LF, and Su XD (2008) Acceptor substrate binding revealed by crystal structure of human glucosamine-6-phosphate N-acetyltransferase 1. FEBS Lett 582, 2973–2978. [DOI] [PubMed] [Google Scholar]
- (100).Stray-Pedersen A, Backe PH, Sorte HS, Morkrid L, Chokshi NY, Erichsen HC, Gambin T, Elgstøen KB, Bjoras M, Wlodarski MW, Kruger M, Jhangiani SN, Muzny DM, Patel A, Raymond KM, Sasa GS, Krance RA, Martinez CA, Abraham SM, Speckmann C, Ehl S, Hall P, Forbes LR, Merckoll E, Westvik J, Nishimura G, Rustad CF, Abrahamsen TG, Ronnestad A, Osnes LT, Egeland T, Rodningen OK, Beck CR, Boerwinkle EA, Gibbs RA, Lupski JR, Orange JS, Lausch E, and Hanson IC (2014) PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am. J. Hum. Genet 95, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Wang-Gillam A, Pastuszak I, and Elbein AD (1998) A 17-amino acid insert changes UDP-N-acetylhexosamine pyrophosphor ylase specificity from UDP-GalNAc to UDP-GlcNAc. J. Biol. Chem 273, 27055–27057. [DOI] [PubMed] [Google Scholar]
- (102).Rome LH, and Hill DF (1986) Lysosomal degradation of glycoproteins and glycosaminoglycans. Efflux and recycling of sulphate and N-acetylhexosamines. Biochem. J 235, 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Weihofen WA, Berger M, Chen H, Saenger W, and Hinderlich S (2006) Structures of human N-Acetylglucosamine kinase in two complexes with N-Acetylglucosamine and with ADP/glucose: insights into substrate specificity and regulation. J. Mol. Biol 364, 388–399. [DOI] [PubMed] [Google Scholar]
- (104).Allen MB, and Walker DG (1980) Kinetic characterization of N-acetyl-D-glucosamine kinase from rat liver and kidney. Biochem. J 185, 577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Takahashi S, Takahashi K, Kaneko T, Ogasawara H, Shindo S, and Kobayashi M (1999) Human renin-binding protein is the enzyme N-acetyl-D-glucosamine 2-epimerase. J. Biochem 125, 348–353. [DOI] [PubMed] [Google Scholar]
- (106).Maszczak-Seneczko D, Sosicka P, Olczak T, Jakimowicz P, Majkowski M, and Olczak M (2013) UDP-N-acetylglucosamine transporter (SLC35A3) regulates biosynthesis of highly branched N-glycans and keratan sulfate. J. Biol. Chem 288, 21850–21860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Sosicka P, Bazan B, Maszczak-Seneczko D, Shauchuk Y, Olczak T, and Olczak M (2019) SLC35A5 Protein-A Golgi Complex Member with Putative Nucleotide Sugar Transport Activity. Int. J. Mol. Sci 20, E276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Ashikov A, Routier F, Fuhlrott J, Helmus Y, Wild M, Gerardy-Schahn R, and Bakker H (2005) The human solute carrier gene SLC35B4 encodes a bifunctional nucleotide sugar transporter with specificity for UDP-xylose and UDP-N-acetylglucosamine. J. Biol. Chem 280, 27230–27235. [DOI] [PubMed] [Google Scholar]
- (109).Ishida N, Kuba T, Aoki K, Miyatake S, Kawakita M, and Sanai Y (2005) Identification and characterization of human Golgi nucleotide sugar transporter SLC35D2, a novel member of the SLC35 nucleotide sugar transporter family. Genomics 85, 106–116. [DOI] [PubMed] [Google Scholar]
- (110).Wells L, Vosseller K, and Hart GW (2003) A role for N-acetylglucosamine as a nutrient sensor and mediator of insulin resistance. Cell. Mol. Life Sci 60, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, Jing H, Kim ES, Biancalana M, Wolfe LA, DiMaggio T, Matthews HF, Kranick SM, Stone KD, Holland SM, Reich DS, Hughes JD, Mehmet H, McElwee J, Freeman AF, Freeze HH, Su HC, and Milner JD (2014) Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J. Allergy Clin. Immunol 133, 1400–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Salvatore S, Heuschkel R, Tomlin S, Davies SE, Edwards S, Walker-Smith JA, French I, and Murch SH (2000) A pilot study of N-acetyl glucosamine, a nutritional substrate for glycosaminoglycan synthesis, in paediatric chronic inflammatory bowel disease. Aliment. Pharmacol. Ther 14, 1567–1579. [DOI] [PubMed] [Google Scholar]
- (113).Shikhman AR, Amiel D, D’Lima D, Hwang SB, Hu C, Xu A, Hashimoto S, Kobayashi K, Sasho T, and Lotz MK (2005) Chondroprotective activity of N-acetylglucosamine in rabbits with experimental osteoarthritis. Ann. Rheum. Dis 64, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Igarashi M, Sakamoto K, and Nagaoka I (2011) Effect of glucosamine, a therapeutic agent for osteoarthritis, on osteoblastic cell differentiation. Int. J. Mol. Med 28, 373–379. [DOI] [PubMed] [Google Scholar]
- (115).Kubomura D, Ueno T, Yamada M, Tomonaga A, and Nagaoka I (2017) Effect of N-acetylglucosamine administration on cartilage metabolism and safety in healthy subjects without symptoms of arthritis: A case report. Exp. Ther. Med 13, 1614–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).McCarty MF (1997) Glucosamine may retard atherogenesis by promoting endothelial production of heparan sulfate proteogly-cans. Med. Hypotheses 48, 245–251. [DOI] [PubMed] [Google Scholar]
- (117).Cahlin BJ, and Dahlstrom L (2011) No effect of glucosamine sulfate on osteoarthritis in the temporomandibular joints–a randomized, controlled, short-term study. Oral Surg Oral Med. Oral Pathol Oral Radiol Endod 112, 760–766. [DOI] [PubMed] [Google Scholar]
- (118).Harrison-Munoz S, Rojas-Briones V, and Irarrazaval S (2017) Is glucosamine effective for osteoarthritis? Medwave 17, e6867. [DOI] [PubMed] [Google Scholar]
- (119).Grigorian A, Araujo L, Naidu NN, Place DJ, Choudhury B, and Demetriou M (2011) N-acetylglucosamine inhibits T-helper 1 (Th1)/T-helper 17 (Th17) cell responses and treats experimental autoimmune encephalomyelitis. J. Biol. Chem 286, 40133–40141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Brynedal B, Wojcik J, Esposito F, Debailleul V, Yaouanq J, Martinelli-Boneschi F, Edan G, Comi G, Hillert J, and Abderrahim H (2010) MGAT5 alters the severity of multiple sclerosis. J. Neuroimmunol 220, 120–124. [DOI] [PubMed] [Google Scholar]
- (121).Huang C, Harada Y, Hosomi A, Masahara-Negishi Y, Seino J, Fujihira H, Funakoshi Y, Suzuki T, Dohmae N, and Suzuki T (2015) Endo-beta-N-acetylglucosaminidase forms NGlcNAc protein aggregates during ER-associated degradation in Ngly1-defective cells. Proc. Natl. Acad. Sci. U. S. A 112, 1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Enns GM, Shashi V, Bainbridge M, Gambello MJ, Zahir FR, Bast T, Crimian R, Schoch K, Platt J, Cox R, Bernstein JA, Scavina M, Walter RS, Bibb A, Jones M, Hegde M, Graham BH, Need AC, Oviedo A, Schaaf CP, Boyle S, Butte AJ, Chen R, Clark MJ, Haraksingh R, Cowan TM, He P, Langlois S, Zoghbi HY, Snyder M, Gibbs RA, Freeze HH, and Goldstein DB (2014) Mutations in NGLY1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway. Genet. Med 16, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Owings KG, Lowry JB, Bi Y, Might M, and Chow CY (2018) Transcriptome and functional analysis in a Drosophila model of NGLY1 deficiency provides insight into therapeutic approaches. Hum. Mol. Genet 27, 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Angata T, and Varki A (2002) Chemical diversity in the sialic acids and related alpha-keto acids: an evolutionary perspective. Chem. Rev 102, 439–469. [DOI] [PubMed] [Google Scholar]
- (125).Reinke SO, Lehmer G, Hinderlich S, and Reutter W (2009) Regulation and pathophysiological implications of UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE) as the key enzyme of sialic acid biosynthesis. Biol. Chem 390, 591–599. [DOI] [PubMed] [Google Scholar]
- (126).Li Y, and Chen X (2012) Sialic acid metabolism and sialyltransferases: natural functions and applications. Appl. Microbiol. Biotechnol 94, 887–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Willems AP, Sun L, Schulz MA, Tian W, Ashikov A, van Scherpenzeel M, Hermans E, Clausen H, Yang Z, and Lefeber DJ (2019) Activity of N-acylneuraminate-9-phosphatase (NANP) is not essential for de novo sialic acid biosynthesis. Biochim. Biophys. Acta, Gen. Subj 1863, 1471–1479. [DOI] [PubMed] [Google Scholar]
- (128).Chou HH, Takematsu H, Diaz S, Iber J, Nickerson E, Wright KL, Muchmore EA, Nelson DL, Warren ST, and Varki A (1998) A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc. Natl. Acad. Sci. U. S. A 95, 11751–11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Tangvoranuntakul P, Gagneux P, Diaz S, Bardor M, Varki N, Varki A, and Muchmore E (2003) Human uptake and incorporation of an immunogenic nonhuman dietary sialic acid. Proc. Natl. Acad. Sci. U. S. A 100, 12045–12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Bardor M, Nguyen DH, Diaz S, and Varki A (2005) Mechanism of uptake and incorporation of the non-human sialic acid N-glycolylneuraminic acid into human cells. J. Biol. Chem 280, 4228–4237. [DOI] [PubMed] [Google Scholar]
- (131).Oetke C, Hinderlich S, Brossmer R, Reutter W, Pawlita M, and Keppler OT (2001) Evidence for efficient uptake and incorporation of sialic acid by eukaryotic cells. Eur. J. Biochem 268, 4553–4561. [DOI] [PubMed] [Google Scholar]
- (132).Pan X, De Aragao CBP, Velasco-Martin JP, Priestman DA, Wu HY, Takahashi K, Yamaguchi K, Sturiale L, Garozzo D, Platt FM, Lamarche-Vane N, Morales CR, Miyagi T, and Pshezhetsky AV (2017) Neuraminidases 3 and 4 regulate neuronal function by catabolizing brain gangliosides. FASEB J 31, 3467–3483. [DOI] [PubMed] [Google Scholar]
- (133).Chigorno V, Tettamanti G, and Sonnino S (1996) Metabolic processing of gangliosides by normal and Salla human fibroblasts in culture. A study performed by administering radioactive GM3 ganglioside. J. Biol. Chem 271, 21738–21744. [DOI] [PubMed] [Google Scholar]
- (134).Verheijen FW, Verbeek E, Aula N, Beerens CE, Havelaar AC, Joosse M, Peltonen L, Aula P, Galjaard H, van der Spek PJ, and Mancini GM (1999) A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat. Genet 23, 462–465. [DOI] [PubMed] [Google Scholar]
- (135).Aula N, Salomaki P, Timonen R, Verheijen F, Mancini G, Mansson JE, Aula P, and Peltonen L (2000) The spectrum of SLC17A5-gene mutations resulting in free sialic acid-storage diseases indicates some genotype-phenotype correlation. Am. J. Hum. Genet 67, 832–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Inoue S, Kitajima K, and Inoue Y (1996) Identification of 2-keto-3-deoxy-D-glycero–galactonononic acid (KDN, deaminoneuraminic acid) residues in mammalian tissues and human lung carcinoma cells. Chemical evidence of the occurrence of KDN glycoconjugates in mammals. J. Biol. Chem 271, 24341–24344. [DOI] [PubMed] [Google Scholar]
- (137).Go S, Sato C, Yin J, Kannagi R, and Kitajima K (2007) Hypoxia-enhanced expression of free deaminoneuraminic acid in human cancer cells. Biochem. Biophys. Res. Commun 357, 537–542. [DOI] [PubMed] [Google Scholar]
- (138).Eckhardt M, Muhlenhoff M, Bethe A, and Gerardy-Schahn R (1996) Expression cloning of the Golgi CMP-sialic acid transporter. Proc. Natl. Acad. Sci. U. S. A 93, 7572–7576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Martinez-Duncker I, Dupre T, Piller V, Piller F, Candelier JJ, Trichet C, Tchernia G, Oriol R, and Mollicone R (2005) Genetic complementation reveals a novel human congenital disorder of glycosylation of type II, due to inactivation of the Golgi CMP-sialic acid transporter. Blood 105, 2671–2676. [DOI] [PubMed] [Google Scholar]
- (140).Mohamed M, Ashikov A, Guillard M, Robben JH, Schmidt S, van den Heuvel B, de Brouwer AP, Gerardy-Schahn R, Deen PM, Wevers RA, Lefeber DJ, and Morava E (2013) Intellectual disability and bleeding diathesis due to deficient CMP-sialic acid transport. Neurology 81, 681–687. [DOI] [PubMed] [Google Scholar]
- (141).Ng BG, Asteggiano CG, Kircher M, Buckingham KJ, Raymond K, Nickerson DA, Shendure J, Bamshad MJ, Ensslen M, and Freeze HH (2017) Encephalopathy caused by novel mutations in the CMP-sialic acid transporter, SLC35A1. Am. J. Med. Genet., Part A 173, 2906–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Riemersma M, Sandrock J, Boltje TJ, Bull C, Heise T, Ashikov A, Adema GJ, van Bokhoven H, and Lefeber DJ (2015) Disease mutations in CMP-sialic acid transporter SLC35A1 result in abnormal alpha-dystroglycan O-mannosylation, independent from sialic acid. Hum. Mol. Genet 24, 2241–2246. [DOI] [PubMed] [Google Scholar]
- (143).van Karnebeek CD, Bonafe L, Wen XY, Tarailo-Graovac M, Balzano S, Royer-Bertrand B, Ashikov A, Garavelli L, Mammi I, Turolla L, Breen C, Donnai D, Cormier-Daire V, Heron D, Nishimura G, Uchikawa S, Campos-Xavier B, Rossi A, Hennet T, Brand-Arzamendi K, Rozmus J, Harshman K, Stevenson BJ, Girardi E, Superti-Furga G, Dewan T, Collingridge A, Halparin J, Ross CJ, Van Allen MI, Rossi A, Engelke UF, Kluijtmans LA, van der Heeft E, Renkema H, de Brouwer A, Huijben K, Zijlstra F, Heise T, Boltje T, Wasserman WW, Rivolta C, Unger S, Lefeber DJ, Wevers RA, and Superti-Furga A (2016) NANS-mediated synthesis of sialic acid is required for brain and skeletal development. Nat. Genet 48, 777–784. [DOI] [PubMed] [Google Scholar]
- (144).Bergfeld AK, Pearce OM, Diaz SL, Pham T, and Varki A (2012) Metabolism of vertebrate amino sugars with N-glycolyl groups: elucidating the intracellular fate of the non-human sialic acid N-glycolylneuraminic acid. J. Biol. Chem 287, 28865–28881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Celeste FV, Vilboux T, Ciccone C, de Dios JK, Malicdan MC, Leoyklang P, McKew JC, Gahl WA, Carrillo-Carrasco N, and Huizing M (2014) Mutation update for GNE gene variants associated with GNE myopathy. Hum. Mutat 35, 915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Gagiannis D, Orthmann A, Danssmann I, Schwarzkopf M, Weidemann W, and Horstkorte R (2007) Reduced sialylation status in UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase (GNE)-deficient mice. Glycoconjugate J 24, 125–130. [DOI] [PubMed] [Google Scholar]
- (147).Patzel KA, Yardeni T, Le Poec-Celic E, Leoyklang P, Dorward H, Alonzi DS, Kukushkin NV, Xu B, Zhang Y,Sollogoub M, Bleriot Y, Gahl WA, Huizing M, and Butters TD (2014) Non-specific accumulation of glycosphingolipids in GNE myopathy. J. Inherited Metab. Dis 37, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Niethamer TK, Yardeni T, Leoyklang P, Ciccone C, Astiz-Martinez A, Jacobs K, Dorward HM, Zerfas PM, Gahl WA, and Huizing M (2012) Oral monosaccharide therapies to reverse renal and muscle hyposialylation in a mouse model of GNE myopathy. Mol. Genet. Metab 107, 748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Carrillo N, Malicdan MC, and Huizing M (2018) GNE Myopathy: Etiology, Diagnosis, and Therapeutic Challenges. Neurotherapeutics 15, 900–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Fontaine G, Biserte G, Montreuil J, Dupont A, and Farriaux JP (1968) Sialuria: an original metabolic disorder. Helv. Paediatr. Acta No. Suppl 17, 11–32. [PubMed] [Google Scholar]
- (151).Wilcken B, Don N, Greenaway R, Hammond J, and Sosula L (1987) Sialuria: a second case. J. Inherited Metab. Dis 10, 97–102. [DOI] [PubMed] [Google Scholar]
- (152).Weiss P, Tietze F, Gahl WA, Seppala R, and Ashwell G (1989) Identification of the metabolic defect in sialuria. J. Biol. Chem 264, 17635–17636. [PubMed] [Google Scholar]
- (153).Kornfeld S, Kornfeld R, Neufeld EF, and O’Brien PJ (1964) The feedback control of sugar nucleotide biosynthesis in liver. Proc. Natl. Acad. Sci. U. S. A 52, 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Wen XY, Tarailo-Graovac M, Brand-Arzamendi K, Willems A, Rakic B, Huijben K, Da Silva A, Pan X, El-Rass S, Ng R, Selby K, Philip AM, Yun J, Ye XC, Ross CJ, Lehman AM, Zijlstra F, Abu Bakar N, Drogemoller B, Moreland J, Wasserman WW, Vallance H, van Scherpenzeel M, Karbassi F, Hoskings M, Engelke U, de Brouwer A, Wevers RA, Pshezhetsky AV, van Karnebeek CD, and Lefeber DJ (2018) Sialic acid catabolism by N-acetylneuraminate pyruvate lyase is essential for muscle function. JCI Insight 3, 122373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Swoboda JG, Campbell J, Meredith TC, and Walker S (2010) Wall teichoic acid function, biosynthesis, and inhibition. ChemBioChem 11, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Kanagawa M, Kobayashi K, Tajiri M, Manya H, Kuga A, Yamaguchi Y, Akasaka-Manya K, Furukawa JI, Mizuno M, Kawakami H, Shinohara Y, Wada Y, Endo T, and Toda T (2016) Identification of a Post-translational Modification with Ribitol-Phosphate and Its Defect in Muscular Dystrophy. Cell Rep 14, 2209–2223. [DOI] [PubMed] [Google Scholar]
- (157).Riemersma M, Froese DS, van Tol W, Engelke UF, Kopec J, van Scherpenzeel M, Ashikov A, Krojer T, von Delft F, Tessari M, Buczkowska A, Swiezewska E, Jae LT, Brummelkamp TR, Manya H, Endo T, van Bokhoven H, Yue WW, and Lefeber DJ (2015) Human ISPD Is a Cytidyltransferase Required for Dystroglycan O-Mannosylation. Chem. Biol 22, 1643–1652. [DOI] [PubMed] [Google Scholar]
- (158).Gerin I, Ury B, Breloy I, Bouchet-Seraphin C, Bolsee J, Halbout M, Graff J, Vertommen D, Muccioli GG, Seta N, Cuisset JM, Dabaj I, Quijano-Roy S, Grahn A, Van Schaftingen E, and Bommer GT (2016) ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto alpha-dystroglycan. Nat. Commun 7, 11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, Benson MA, Ponting CP, Estournet B, Romero NB, Mercuri E, Voit T, Sewry CA, Guicheney P, and Muntoni F (2001) Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am. J. Hum. Genet 69, 1198–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Beltran-Valero de Bernabe D, Voit T, Longman C, Steinbrecher A, Straub V, Yuva Y, Herrmann R, Sperner J, Korenke C, Diesen C, Dobyns WB, Brunner HG, van Bokhoven H, Brockington M, and Muntoni F (2004) Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J. Med. Genet 41, e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Cataldi MP, Lu P, Blaeser A, and Lu QL (2018) Ribitol restores functionally glycosylated alpha-dystroglycan and improves muscle function in dystrophic FKRP-mutant mice. Nat. Commun 9, 3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Laughlin ST, and Bertozzi CR (2007) Metabolic labeling of glycans with azido sugars and subsequent glycan-profiling and visualization via Staudinger ligation. Nat. Protoc 2, 2930–2944. [DOI] [PubMed] [Google Scholar]
- (163).Rillahan CD, Antonopoulos A, Lefort CT, Sonon R, Azadi P, Ley K, Dell A, Haslam SM, and Paulson JC (2012) Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat. Chem. Biol 8, 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Schneider M, Kumar V, Nordstrom LU, Feng L, Takeuchi H, Hao H, Luca VC, Garcia KC, Stanley P, Wu P, and Haltiwanger RS (2018) Inhibition of Delta-induced Notch signaling using fucose analogs. Nat. Chem. Biol 14, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Kizuka Y, Funayama S, Shogomori H, Nakano M, Nakajima K, Oka R, Kitazume S, Yamaguchi Y, Sano M, Korekane H, Hsu TL, Lee HY, Wong CH, and Taniguchi N (2016) High-Sensitivity and Low-Toxicity Fucose Probe for Glycan Imaging and Biomarker Discovery. Cell Chem. Biol 23, 782–792. [DOI] [PubMed] [Google Scholar]
- (166).Kizuka Y, Nakano M, Yamaguchi Y, Nakajima K, Oka R, Sato K, Ren CT, Hsu TL, Wong CH, and Taniguchi N (2017) An Alkynyl-Fucose Halts Hepatoma Cell Migration and Invasion by Inhibiting GDP-Fucose-Synthesizing Enzyme FX, TSTA3. Cell Chem. Biol 24, 1467–1478. [DOI] [PubMed] [Google Scholar]
- (167).Moons SJ, Adema GJ, Derks MT, Boltje TJ, and Bull C (2019) Sialic acid glycoengineering using N-acetylmannosamine and sialic acid analogs. Glycobiology 29, 433–445. [DOI] [PubMed] [Google Scholar]
- (168).Dehnert KW, Beahm BJ, Huynh TT, Baskin JM, Laughlin ST, Wang W, Wu P, Amacher SL, and Bertozzi CR (2011) Metabolic labeling of fucosylated glycans in developing zebrafish. ACS Chem. Biol 6, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Barboza M, Wong M, Luke J, Cheng Z, Xu G, Gareau M, Raybould H, and Lebrilla CB (2018) Mapping the Incorporation of 13C-Labeled Monosaccharides into the Mouse Cell Surface Metaglycome by LC-MS/MS Analysis. J. Am. Soc. Mass Spectrom 29, n/a. [Google Scholar]
- (170).Kornfeld RH, and Ginsburg V (1966) Control of synthesis of guanosine 5′-diphosphate D-mannose and guanosine 5′-diphosphate L-fucose in bacteria. Biochim. Biophys. Acta, Gen. Subj 117, 79–87. [DOI] [PubMed] [Google Scholar]
- (171).Pouilly S, Piller V, and Piller F (2012) Metabolic glycoengineering through the mammalian GalNAc salvage pathway. FEBS J 279, 586–598. [DOI] [PubMed] [Google Scholar]
- (172).Lee SU, Li CF, Mortales CL, Pawling J, Dennis JW, Grigorian A, and Demetriou M (2019) Increasing cell permeability of N-acetylglucosamine via 6-acetylation enhances capacity to suppress T-helper 1 (TH1)/TH17 responses and autoimmunity. PLoS One 14, e0214253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Keppler D, Lesch R, Reutter W, and Decker K (1968) Experimental hepatitis induced by D-galactosamine. Exp. Mol. Pathol 9, 279–290. [DOI] [PubMed] [Google Scholar]
- (174).Schnaar RL, and Freeze HH (2008) A “glyconutrient sham”. Glycobiology 18, 652–657. discussion 658–663 [DOI] [PubMed] [Google Scholar]
- (175).Schnaar RL, and Freeze HH (2017) A “Glyconutrient Sham” and the Jenner Glycobiology and Medicine Symposium. Glycobiology 27, 383–384. [DOI] [PubMed] [Google Scholar]
- (176).Alton G, Hasilik M, Niehues R, Panneerselvam K, Etchison JR, Fana F, and Freeze HH (1998) Direct utilization of mannose for mammalian glycoprotein biosynthesis. Glycobiology 8, 285–295. [DOI] [PubMed] [Google Scholar]
- (177).Chan JY, Nwokoro NA, and Schachter H (1979) L-Fucose metabolism in mammals. The conversion of L-fucose to two moles of L-lactate, of L-galactose to L-lactate and glycerate, and of D-arabinose to L-lactate and glycollate. J. Biol. Chem 254, 7060–7068. [PubMed] [Google Scholar]
- (178).Segal S, Blair A, and Roth H (1965) The metabolism of galactose by patients with congenital galactosemia. Am. J. Med 38, 62–70. [DOI] [PubMed] [Google Scholar]
- (179).Deng Y, Misselwitz B, Dai N, and Fox M (2015) Lactose Intolerance in Adults: Biological Mechanism and Dietary Management. Nutrients 7, 8020–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Brasil S, Pascoal C, Francisco R, Marques-da-Silva D, Andreotti G, Videira PA, Morava E, Jaeken J, and Dos Reis Ferreira V (2018) CDG Therapies: From Bench to Bedside. Int. J. Mol. Sci 19, E1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Huck JH, Verhoeven NM, Struys EA, Salomons GS, Jakobs C, and van der Knaap MS (2004) Ribose-5-phosphate isomerase deficiency: new inborn error in the pentose phosphate pathway associated with a slowly progressive leukoencephalopathy. Am. J. Hum. Genet 74, 745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]