

Abstract
Objective:
Hypophosphatemic rickets with hypercalciuria (HHRH) is a rare, recessively-inherited form of rickets caused by homozygous or compound heterozygous mutations in the SLC34A3 gene that encodes the renal tubular phosphate transporter protein NaPi2c. The bone phenotype varies from severe rickets to no disease. Accurate diagnosis is important as the treatment differs from other forms of rickets.
Methods:
The patient was a 12-year-old boy from the Indian subcontinent with florid hypophosphatemic rickets. A targeted gene panel to search for mutations in genes associated with inherited forms of rickets was performed. We also completed a literature search of published cases of HHRH.
Results:
The targeted gene panel demonstrated a novel homozygous SLC34A3 mutation: c.1339 G>A (p.Ala447Thr). His parents were heterozygous for the mutation. In our literature review we found that people with homozygous SLC34A3 mutations were more likely to have rickets than those with compound heterozygous mutations (85% versus 45%, p<0.002) and that serum phosphate z scores were lower in those with rickets than those without (−3.3 with a standard deviation of 1.5 versus −2.1 with a standard deviation of 1.5, p<0.005).
Conclusion:
The bone phenotype of HHRH is related to the nature of the mutation and serum phosphate levels. Targeted gene panels can aid in the accurate diagnosis of inherited forms of rickets, and facilitate correct treatment.
INTRODUCTION
Hereditary hypophosphatemic rickets with hypercalciuria (HHRH; Online Mendelian Inheritance in Man disorder number 241530) is a rare, autosomal recessive disorder originally described in consanguineous kindred in 1985 (1,2). In 2006, mutations in the gene SLC34A3 that encodes the renal tubular sodium-phosphate co-transporter NaPi2c were identified as the cause of the disorder in the original cases and several other families (3,4). Individuals who carry compound heterozygous or homozygous loss-of-function mutations in SLC34A3 have urinary phosphate wasting and chronic hypophosphatemia that can lead to hypophosphatemic rickets. Plasma calcitriol levels are usually elevated and cause hypercalciuria, primarily through enhanced intestinal calcium absorption. Hypercalciuria leads in turn to the development of kidney stones and/or nephrocalcinosis. The clinical manifestations in bone are highly variable; some patients have no obvious bone abnormality, while others have rickets that can range from mild to severe. It is uncertain what underlies this variation in phenotype (5).
In this paper we describe a case of unusually severe hypophosphatemic rickets in a boy in whom we identified a novel homozygous mutation in SLC34A3 by massive parallel gene sequencing, using a panel directed to hereditary forms of rickets and osteomalacia. We have also reviewed the published literature to examine whether the bone phenotype relates to the nature of the mutations in SLC34A3 or the severity of hypophosphatemia.
CASE REPORT
A 12-year-old boy was referred with complaints of recurrent fractures, bone pain, and severe progressive limb deformities. He had a renal stone at the age of 2 and rickets had been diagnosed at the age of 3. At first it had been thought that the rickets was nutritional in origin, but his condition deteriorated despite vitamin D and calcium treatment. Bone pain and deformities caused severe restriction of activity. By the age of 3 he was non-ambulant and unable to attend school. His parents were first cousins, but there was no family history of bone disease.
Physical examination showed short stature, with a height of 110 cm, 4.5 standard deviations (SDs) below the mean of age-matched Indian boys. The sclerae were white. He had classical rachitic deformities including pectus carinatum, Harrison sulcus, and rickety rosary, with bowing deformities of the arms and legs (Fig. 1). The long bones were painful to the touch.
Fig. 1.
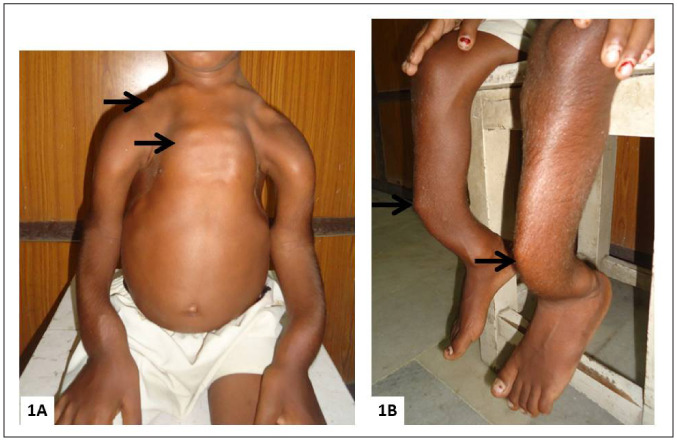
Clinical photographs showing severe rachitic deformities (A), pectus carinatum, rickety rosary (arrow), and deformities of the clavicles and upper limbs (arrows). (B) Anterior bowing deformity of the lower legs (arrow).
Skeletal radiographs showed florid rickets with multiple fractures and Looser zones, gross widening of the metaphyses, thin cortices, and marked clavicular and long bone deformities (Fig. 2). A renal sonogram showed bilateral nephrocalcinosis. Laboratory studies showed normal blood cell count, renal function, and electrolytes. Biochemical measurements related to mineral metabolism are shown in Table 1. The plasma calcium was normal but the phosphate was low (4.7 SDs below the mean for his age). The alkaline phosphatase was elevated and the serum parathyroid hormone undetectable. Plasma concentrations of calcidiol, calcitriol, and fibroblast growth factor 23 (FGF23) were within the normal ranges. His 24-hour urinary calcium excretion was elevated. The ratio of tubular maximum reabsorption of phosphate to glomerular filtration rate was not formally assessed.
Fig. 2.
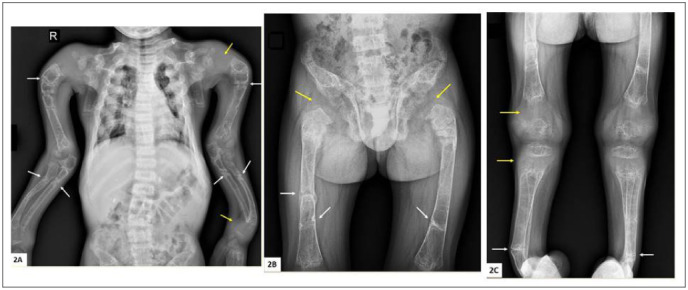
(A) Radiographs of spine and upper limb. There are multiple bilateral Looser zones of the humerus, radius, and ulna (white arrows), many of which have progressed to complete fracture. Gross epiphyseal widening can also be seen (yellow arrows), while the cortices are thin. There is a mild lumbar scoliosis. (B) Radiographs of pelvis and femora showing multiple Looser zones (white arrows), many of which have progressed to complete fracture, and gross epiphyseal widening (yellow arrows). The cortices are thin and the pubic rami are severely demineralized. (C) Radiographs of both lower limbs showing thin cortices, Looser zones or fractures (white arrows), and gross epiphyseal widening (yellow arrows).
Table 1.
Biochemical Profile of the Patient
| Result | Normal range | |
|---|---|---|
| Calcium (albumin-adjusted) | 2.25 mmol/L 9.0 mg/dL |
2.20–2.65 8.8–10.6 |
| Phosphate | 0.63 mmol/L 1.95 mg/dL |
1.07–1.74* 3.31–5.39* |
| Alkaline phosphatase | 1,182 U/L | 80–360* |
| Parathyroid hormone | <0.27 pmol/L <2.5 pg/mL |
0.53–6.4 5–60 |
| Calcidiol | 48 nmol/L 19 ng/mL |
25–100 10–40 |
| Calcitriol | 91 pmol/L 36 pg/mL |
48–138 19–55 |
| Fibroblast growth factor 23 | 69 RU/mL | <150 |
| 24-hour urine calcium | 3.8 mmol 152 mg |
0.3–1.5* 12–60* |
*Normal ranges for the patient's age.
METHODS
Genetic Analysis
The early presentation with rickets that did not respond to vitamin D therapy and parental consanguinity suggested a genetic cause. The patient's phenotype was thought to fit best with HHRH. We investigated this using a massive parallel gene sequencing panel directed toward hereditary rickets developed at the Department of Molecular Genetics, at the Children's Hospital at Westmead. Massive parallel gene sequencing was performed using the TruSight One panel (FC-141-1007, Illumina, Inc., San Diego, CA) on an Illumina NextSeq550.
Analysis of 9 genes involved in hereditary rickets (PHEX, FGF23, ENPP1, DMP1, VDR, CYP27B1, SLC34A3, CLCN5, and ALPL) was performed on the proband, with an average coverage (>20×) of 98.9% across the 9 genes. Alignment of sequencing data was performed using NextGene v2.4.1 (SoftGenetics, State College, PA) to human genome assembly GRCh37/hg19 using software default settings. Only variants with >20% of read allele proportion were called. Variants were annotated using AlamutBatch (v.1.4.3, Interactive Biosoftware, Rouen, France), and only variants with a population allele frequency of <0.1% for dominant disorders, or <1% for recessive disorders, based on ExAC browser data, were considered of interest. Segregation testing of the variant of interest via Sanger sequencing was also performed on the parents.
Literature Review
We identified publications describing the phenotypes of 56 patients from 34 families who had either homozygous mutations or compound heterozygous mutations in SLC34A3 (1–4,6–18). We compared serum phosphate levels in those reported to have rickets with those reported to have only renal complications. As normal values for serum phosphate vary with age, we expressed them as the SD score (z score) for age-appropriate normal ranges (15). In some reports, the serum phosphate results were already reported as z scores. In those that were not, we estimated it using the following normal ranges, all in mmol/L: ages 1 to 5 years, mean 1.75 (SD 0.32); ages 5 to 15 years, mean 1.38 (SD 0.16); ages >15 years, mean 1.10 (SD 0.15). Proportions were compared using the χ2 test and mean values by unpaired t test.
Results
The proband's massive parallel gene sequencing data revealed a homozygous variant in the SLC34A3 gene, that results in the substitution of alanine to threonine at residue 446, p.(Ala447Thr). This gene was covered 100% in the proband and no other variant of interest was detected. Both parents were heterozygous for this SLC34A3 variant (Fig. 3). This variant, SLC34A3 (NM_080877.2): c. 1339G>A p. Ala447Thr, has been reported with a very low allele frequency (3 out of 246,256 alleles) in the Genome Aggregation Database (gnomAD) database. In silico analysis of this variant via PolyPhen2, MutationTaster, FATHMM, Provean, and SIFT all predicted it to be likely pathogenic. The alanine residue at position 447 lies between the fifth and sixth transmembrane domains of the NaPi2c transporter and is highly conserved across mammals with the exception of marsupials, in which it is replaced by a serine.
Fig. 3.
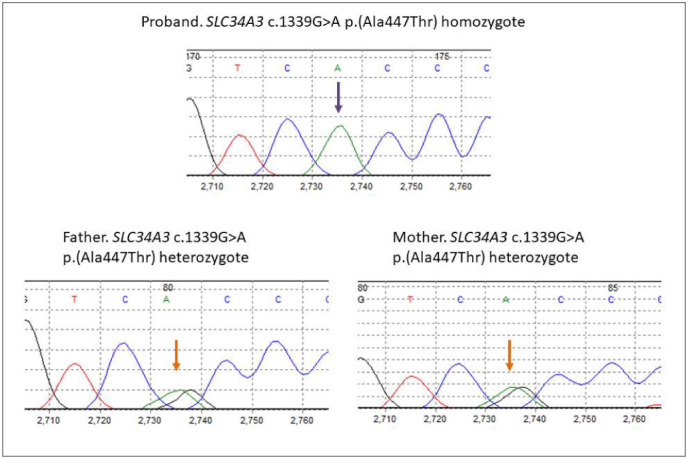
Electropherograms from the patient (top) and his parents (below). Both parents are heterozygous for the G>A missense mutation (orange arrows) and our patient is homozygous for the G>A mutation (purple arrow).
We identified from the literature 29 patients from 21 families who carried compound heterozygous SLC34A3 mutations, and 27 patients from 13 families who carried homozygous mutations including our patient (Table 2). Thirteen (45%) of those carrying compound heterozygous mutations were reported to have had rickets; the remainder presented with nephrocalcinosis and/or renal stones. In contrast, 23 of those carrying homozygous mutations were reported to have rickets (85%; p = 0.002 by χ2 test). The mean serum phosphate z score was significantly lower in patients reported to have rickets than those without (−3.3 with SD 1.5 versus −2.1 with SD 1.5, p<0.005). The mean serum phosphate z score was not significantly lower in people with homozygous mutations than those with compound heterozygous mutations (−3.1 with SD 1.5 versus −2.4 with SD 1.6, p = 0.12).
Table 2.
Bone Phenotype and Plasma Phosphate Levels According to Mutation Status
| Case reports of homozygous mutations | |||
|---|---|---|---|
| Plasma phosphate z score | Rickets | Mutations | |
| Bergwitz et al (3) | −4.2 | Yes | c.228delC |
| −4.0 | Yes | ||
| −2.6 | Yes | ||
| −4.1 | Yes | ||
| −4.5 | Yes | ||
| −4.3 | Yes | ||
| Lorenz-Depiereux et al (4) | −6.3 | Yes | c.905delC |
| −4.0 | Yes | p.R353L | |
| Ichikawa et al (6) | −1.8 | Yes | g.2259_2359 |
| −2.3 | No | ||
| Kremke et al (10) | −3.6 | Yes | p.G196R |
| −1.4 | No | ||
| Yu et al (12) | −2.6 | Yes | p.Y588X |
| Braithwaite et al (21) | −3.6 | Yes | S168F |
| −3.9 | Yes | ||
| −5.5 | Yes | ||
| Areses-Trapote et al (22) | −3.3 | Yes | c.448+5G>A |
| Dasgupta et al (15) | −3.2 | Yes | p.G457S |
| −1.3 | No | ||
| −2.7 | Yes | ||
| −0.7 | Yes | ||
| −2.2 | Yes | p.R468W | |
| +0.1 | No | p.S192L | |
| Hasani-Ranjbar et al (18) | −2.0 | Yes | g.2259_2359 |
| −1.2 | Yes | ||
| NR | Yes | ||
| Bhadada et al (this paper) | −4.8 | Yes | p.A447T |
| Case reports of compound heterozygous mutations | |||
| Bergwitz et al (3) | −4.2 | Yes | p.G196R, p.R468W |
| −4.0 | Yes | p.G196R, g.2259_2359del | |
| −4.0 | Yes | p.S138F, p.S192L | |
| −4.1 | Yes | ||
| −3.8 | Yes | ||
| Lorenz-Depiereux et al (4) | −4.3 | Yes | c.846G>A, p.A413E |
| NR | Yes | ||
| −6.3 | Yes | c.304+2T>C. p.S192L | |
| Ichikawa et al (6) | −1.1 | Yes | g.1702G>A, g.2615-2699del |
| Jaureguiberry et al (7) | −3.1 | Yes | g.4225_50del, p.T137M |
| Page et al (8) | −4.0 | Yes | p.S192L, p.G49X |
| Tencza et al (9) | −1.2 | No | p.R182W, p.S192L |
Table 2.
Bone Phenotype and Plasma Phosphate Levels According to Mutation Status
| Case reports of homozygous mutations | |||
|---|---|---|---|
| Plasma phosphate z score | Rickets | Mutations | |
| Phulwani et al (11) | −2.6 | Yes | g.4225_50del, g.1226G>A |
| Yu et al (12) | −1.3 | Yes | c.560+27_561-38del, c.1046_47del |
| −1.4 | No | ||
| −1.7 | No | ||
| Ichikawa et al (13) | −1.2 | No | c.1304delG, g.1440_1469del |
| Chi et al (14) | −2.6 | Yes | p.G191R, p.R468W |
| Dasgupta et al (15) | −3.9 | No | g.1440_1469del, F453del |
| +0.2 | No | p.S138F, c.1304delG, p.L527del* | |
| +0.2 | No | ||
| +0.1 | No | ||
| −2.6 | No | S192L, g.2615_2699del | |
| −1.5 | No | S192L, c.367delC | |
| Abe et al (16) | −0.8 | No | c.175+1 G>A, p.R412W |
| Rafaelson et al (23) | −1.3 | No | c.757-1G>A, c.925+20_926-48del |
| Dhir et al (17) | −3.5 | No | p.R67G, p.G191S |
| Acar et al (24) | −2.9 | No | c.1335+2T>A, c.1639_1652del |
| −1.1 | No | ||
Abbreviation: NR = not reported.
*Cases had 3 mutations.
DISCUSSION
Our patient had severe, untreated rickets as a result of HHRH. He displayed the classical physical and physiological signs of rickets to a degree that is rarely seen nowadays (Fig. 1). The radiographic signs were similarly pronounced with Looser zones, fractures, cortical thinning, and metaphyseal widening. Looser zones are stress fractures that are a late manifestation of osteomalacia. They are usually multiple and often symmetric, and occur in both weight-bearing bones (pubic rami, medial aspects of the femur and tibia, and metatarsal bones) and non-weight-bearing bones (ribs and lateral border of the scapula).
The primary defect in HHRH is the loss of excessive amounts of phosphate in the urine because of hypofunction of the important renal tubular phosphate transporter, NaPi2c. Chronic hypophosphatemia is responsible for osteomalacia and rickets. Hypophosphatemia also increases the renal 1α-hydroxylation of calcidiol, so calcitriol levels are characteristically increased. This results in increased intestinal calcium absorption, that in turn that causes hypercalciuria, with its renal consequences of nephrocalcinosis and stones (5). This mechanism is in contrast to that seen in FGF23-mediated hypophosphatemic osteomalacia or rickets, in which calcitriol production is suppressed, so nephrocalcinosis and renal stones are uncommon (19).
HHRH is caused by homozygous or compound heterozygous mutations in the gene SLC34A3. People carrying a single disease-associated heterozygous mutation also have a phenotype of hypophosphatemia and hypercalciuria, often with renal stones and nephrocalcinosis, but not bone disease (2,15). The milder phenotypes of heterozygous mutation carriers is likely related to their lesser degree of urine phosphate loss and higher plasma phosphate levels (15). In people carrying biallelic mutations in SLC34A3 (homozygous or compound heterozygous), there are substantial variations in the skeletal phenotypes, with some patients having severe rickets, some mild symptoms, and some showing no skeletal abnormalities (1–18). Our analysis of the published data shows that patients with rickets have significantly lower plasma phosphate levels than those with only a renal phenotype, and that rickets is more prevalent in those with homozygous mutations than in those with compound heterozygous mutations. The latter effect may also be mediated at least in part by more marked hypophosphatemia in homozygous patients.
Nutritional rickets associated with vitamin D deficiency is common, particularly in the Indian subcontinent, whereas genetic causes are rare. Thus it is not surprising that, as in our case and others (7,8,11), inappropriate therapy with calciferol and calcium was prescribed. Failure to heal rickets with such treatment should prompt consideration of alternative diagnoses, as accurate diagnosis is important in selecting the correct therapy. In the case of HHRH, treatment with calcium and vitamin D may be harmful as it can worsen hypercalciuria. HHRH is best treated by oral phosphate salts alone at 1 to 2.5 g of elemental phosphorus per day over 4 to 5 doses (20 to 50 mg/kg of elemental phosphorus/day). When our patient was treated with phosphate, there was marked symptomatic, biochemical, and radiographic improvement, though he will need surgery to correct the severe deformities.
Accurate diagnosis is not only important for treatment, but also for genetic counseling. The number of genetic causes of osteomalacia and rickets that have been identified has increased substantially in recent years. The most frequently encountered are due to mutations in PHEX, DMP1, ENPP1, FGF23, or CLCN5, all of which were included in our gene panel. Rarer causes include somatic mutations in GNAS1, HRAS, NRAS, and, in 1 patient, a translocation close to the α-klotho gene.
Biochemical tests can be helpful, but some, such as plasma calcitriol, can be equivocal. The plasma concentration of calcitriol is typically elevated in HHRH, but was normal in our patient. In their review, Dasgupta et al (15) found that 23% of subjects with HHRH and biallelic SLC34A3 mutations had plasma calcitriol concentrations in the normal range. Explanations may include day-today variability in calcitriol concentrations, or assay difficulties. Plasma FGF23 levels are typically elevated in all the disorders mentioned above (20) with the exception of HHRH and Dent disease (CLCN5 mutation). In the latter 2 conditions, FGF23 levels may be low or in the normal range, depending on the type of assay used. Intact FGF23 hormone assays give low readings, but if assays that also detect C-terminal fragments of the hormone are used (as in our case) results in the normal range may be found (4). Nephrocalcinosis is characteristic of both HHRH and Dent disease, but not the FGF23-related conditions.
Targeted gene panels are increasingly used in circumstances where there are a number of genetic causes for a particular phenotype. In our case, a customized gene panel that covered genes involved in vitamin D metabolism, hypophosphatemia, and hypophosphatasia provided a rapid result at much reduced cost compared to that of sequencing all the genes of potential interest. The mutation identified is rare (approximately 1 in 105 in heterozygous form in the ExAc database) and no homozygous individuals have previously been reported.
In people carrying biallelic mutations in SLC34A3, there is a substantial variation in the skeletal phenotype; some patients have severe rickets, some have mild symptoms, and some have no skeletal abnormalities (1–18). Our analysis of the published data shows that patients with rickets have significantly lower plasma phosphate levels than those with only a renal phenotype, and that rickets is more prevalent in those with homozygous mutations than in those with compound heterozygous mutations. The latter effect may be mediated at least in part by more marked hypophosphatemia in homozygous patients. Because of its rarity there is insufficient data to determine more detailed phenotype-genotype relationships in HHRH, in particular whether mutations affecting specific regions of the NaPi2c molecule are more damaging than others.
CONCLUSION
We have demonstrated the value of a targeted gene panel in identifying the cause of extremely severe rickets in a 12-year-old boy. The novel homozygous mutation we found in the SCL34A3 gene is predicted to disrupt the function of the NaPi2c transporter. Although we have not undertaken functional studies, it is highly conserved and predicted to be pathogenic. A review of the literature suggests that the severity of the bone disease in HHRH is worse with lower plasma phosphate levels and when there are homozygous mutations.
ACKNOWLEDGMENT
This study was funded in part by the Health Research Council of New Zealand.
Abbreviations
- FGF23
fibroblast growth factor 23
- HHRH
hypophosphatemic rickets with hypercalciuria
- SD
standard deviation
Footnotes
DISCLOSURE
The authors have no multiplicity of interest to disclose.
REFERENCES
- 1.Tieder M, Modai D, Samuel R et al. Hereditary hypophosphatemic rickets with hypercalciuria. N Eng J Med. 1985;312:611–617. doi: 10.1056/NEJM198503073121003. [DOI] [PubMed] [Google Scholar]
- 2.Tieder M, Modai D, Shaked U et al. “Idiopathic” hypercalciuria and hereditary hypophosphatemic rickets. Two pheno-typical expressions of a common genetic defect. N Engl J Med. 1987;316:125–129. doi: 10.1056/NEJM198701153160302. [DOI] [PubMed] [Google Scholar]
- 3.Bergwitz C, Roslin NM, Tieder M et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet. 2006;78:179–192. doi: 10.1086/499409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorenz-Depiereux B, Benet-Pages A, Eckstein G et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet. 2006;78:193–201. doi: 10.1086/499410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergwitz C, Miyamoto KI. Hereditary hypophosphatemic rickets with hypercalciuria: pathophysiology, clinical presentation, diagnosis and therapy. Pflugers Arch. 2019;471:149–163. doi: 10.1007/s00424-018-2184-2. [DOI] [PubMed] [Google Scholar]
- 6.Ichikawa S, Sorenson AH, Imel EA, Friedman NE, Gertner JM, Econs MJ. Intronic deletions in the SLC34A3 gene cause hereditary hypophosphatemic rickets with hypercalciuria. J Clin Endocrinol Metab. 2006;91:4022–4027. doi: 10.1210/jc.2005-2840. [DOI] [PubMed] [Google Scholar]
- 7.Jaureguiberry G, Carpenter TO, Forman S, Jüppner H, Bergwitz C. A novel missense mutation in SLC34A3 that causes hereditary hypophosphatemic rickets with hypercalciuria in humans identifies threonine 137 as an important determinant of sodium-phosphate co-transport in NaPi-IIc. Am J Physiol Renal Physiol. 2008;295:F371–F379. doi: 10.1152/ajprenal.00090.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Page K, Bergwitz C, Jaureguiberry G, Harinarayan CV, Insogna K. A patient with hypophosphatemia, a femoral fracture, and recurrent kidney stones: report of a novel mutation in SLC34A3. Endocr Pract. 2008;14:869–874. doi: 10.4158/EP.14.7.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tencza AL, Ichikawa S, Dang A et al. Hypophosphatemic rickets with hypercalciuria due to mutation in SLC34A3/type IIc sodium-phosphate cotransporter: presentation as hypercalciuria and nephrolithiasis. J Clin Endocrinol Metab. 2009;94:4433–4438. doi: 10.1210/jc.2009-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kremke B, Bergwitz C, Ahrens W et al. Hypophosphatemic rickets with hypercalciuria due to mutation in SLC34A3/NaPi-IIc can be masked by vitamin D deficiency and can be associated with renal calcifications. Exp Clin Endocrinol Diabetes. 2009;117:49–56. doi: 10.1055/s-2008-1076716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phulwani P, Bergwitz C, Jaureguiberry G, Rasoulpour M, Estrada E. Hereditary hypophosphatemic rickets with hypercalciuria and nephrolithiasis-identification of a novel SLC34A3/NaPi-IIc mutation. Am J Med Genet A. 2011;155A:626–633. doi: 10.1002/ajmg.a.33832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu Y, Sanderson SR, Reyes M et al. Novel NaPi-IIc mutations causing HHRH and idiopathic hypercalciuria in several unrelated families: long-term follow-up in one kindred. Bone. 2012;50:1100–1106. doi: 10.1016/j.bone.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ichikawa S, Tuchman S, Padgett LR, Gray AK, Baluarte HJ, Econs MJ. Intronic deletions in the SLC34A3 gene: a cautionary tale for mutation analysis of hereditary hypophosphatemic rickets with hypercalciuria. Bone. 2014;59:53–56. doi: 10.1016/j.bone.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chi Y, Zhao Z, He X et al. A compound heterozygous mutation in SLC34A3 causes hereditary hypophosphatemic rickets with hypercalciuria in a Chinese patient. Bone. 2014;59:114–121. doi: 10.1016/j.bone.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 15.Dasgupta D, Wee MJ, Reyes M et al. Mutations in SLC34A3/NPT2c are associated with kidney stones and nephrocalcinosis. J Am Soc Nephrol. 2014;25:2366–2375. doi: 10.1681/ASN.2013101085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abe Y, Nagasaki K, Watanabe T, Abe T, Fukami M. Association between compound heterozygous mutations of SLC34A3 and hypercalciuria. Horm Res Paediatr. 2014;82:65–71. doi: 10.1159/000360291. [DOI] [PubMed] [Google Scholar]
- 17.Dhir G, Li D, Hakonarson H, Levine MA. Late-onset hereditary hypophosphatemic rickets with hypercalciuria (HHRH) due to mutation of SLC34A3/NPT2c. Bone. 2017;97:15–19. doi: 10.1016/j.bone.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hasani-Ranjbar S, Ejtahed HS, Amoli MM et al. SLC34A3 intronic deletion in an Iranian kindred with hereditary hypophosphatemic rickets with hypercalciuria. J Clin Res Pediatr Endocrinol. 2018;10:343–349. doi: 10.4274/jcrpe.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carpenter TO. The expanding family of hypophosphatemic syndromes. J Bone Miner Metab. 2012;30:1–9. doi: 10.1007/s00774-011-0340-2. [DOI] [PubMed] [Google Scholar]
- 20.Endo I, Fukumoto S, Ozono K et al. Clinical usefulness of measurement of fibroblast growth factor 23 (FGF23) in hypophosphatemic patients: proposal of diagnostic criteria using FGF23 measurement. Bone. 2008;42:1235–1239. doi: 10.1016/j.bone.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 21.Braithwaite V, Pettifor JM, Prentice A. Novel SLC34A3 mutation causing hereditary hypophosphataemic rickets with hypercalciuria in a Gambian family. Bone. 2013;53:216–220. doi: 10.1016/j.bone.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Areses-Trapote R, López-García JA, Ubetagoyena-Arrieta M, Eizaguirre A, Sáez-Villaverde R. Hereditary hypophosphatemic rickets with hypercalciuria: case report. Nefrologia. 2012;32:529–534. doi: 10.3265/Nefrologia.pre2012.Apr.11321. [DOI] [PubMed] [Google Scholar]
- 23.Rafaelsen S, Johansson S, Ræder H, Bjerknes R. Hereditary hypophosphatemia in Norway: a retrospective population-based study of genotypes, phenotypes, and treatment complications. Eur J Endocrinol. 2016;174:125–136. doi: 10.1530/EJE-15-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Acar S, BinEssa HA, Demir K et al. Clinical and genetic characteristics of 15 families with hereditary hypophosphatemia: novel mutations in PHEX and SLC34A3. PLoS One. 2018;13:e0193388. doi: 10.1371/journal.pone.0193388. [DOI] [PMC free article] [PubMed] [Google Scholar]