

Summary:
The kidney is a highly metabolic organ that requires substantial adenosine triphosphate for the active transport required to maintain water and solute reabsorption. Aberrations in energy availability and energy utilization can lead to cellular dysfunction and death. Mitochondria are essential for efficient energy production. The pathogenesis of acute kidney injury is complex and varies with different types of injury. However, multiple distinct acute kidney injury syndromes share a common dysregulation of energy metabolism. Pathways of energy metabolism and mitochondrial dysfunction are emerging as critical drivers of acute kidney injury and represent new potential targets for treatment. This review shows the basic metabolic pathways that all cells depend on for life; describes how the kidney optimizes those pathways to meet its anatomic, physiologic, and metabolic needs; summarizes the importance of metabolic and mitochondrial dysfunction in acute kidney injury; and analyzes the mitochondrial processes that become dysregulated in acute kidney injury including mitochondrial dynamics, mitophagy, mitochondrial biogenesis, and changes in mitochondrial energy metabolism.
Keywords: Acute kidney injury, metabolism, mitochondria
There is currently no targeted treatment for acute kidney injury (AKI), however, the incidence and associated costs of AKI are increasing.1,2 Energy metabolism pathways are emerging as critical drivers of AKI and represent new potential therapeutic targets for AKI prevention and treatment. Every living cell needs energy to function. Aberrations in energy availability and energy utilization can lead to cellular dysfunction and, ultimately, cell death. Mitochondria serve a vital role in energy creation from nutrient substrates. Mounting evidence has implicated mitochondrial dysfunction as a major determinant of renal tubular injury.
Purification of blood by the metanephric kidney requires two distinct sources of energy: the cardiac pump to generate the hydraulic force necessary for glomerular filtration and the electrochemical gradients established by the renal tubule to enable more than 99% reabsorption of filtered water and selective solute secretion. The latter may be pathologically jeopardized during AKI.
The tubular cells most responsible for creating electrochemical gradients reside in the renal cortex. These cells rely on oxidative metabolism, as opposed to glycolysis, to generate adenosine triphosphate (ATP). They are the most abundant in mitochondria, and they are the most severely injured in AKI. A growing body of literature has shown that during AKI, the utilization of fuel substrate is altered, mitochondrial oxidative function is attenuated markedly, and transcriptional regulation of key energy metabolism pathways is suppressed. Each of these abnormalities also highlights opportunities for novel therapeutic interventions.
OVERVIEW OF ENERGY METABOLISM
Metabolism is the set of chemical reactions that take place to maintain life. This includes the subset of reactions that harvest energy from nutrients. ATP is the most prominent intracellular energy carrier. It is generated in the cytoplasm from the oxidation of simple sugars to pyruvate via glycolysis, and within mitochondria from the oxidation of pyruvate, amino acids, and fatty acids (Fig. 1).
Figure 1.
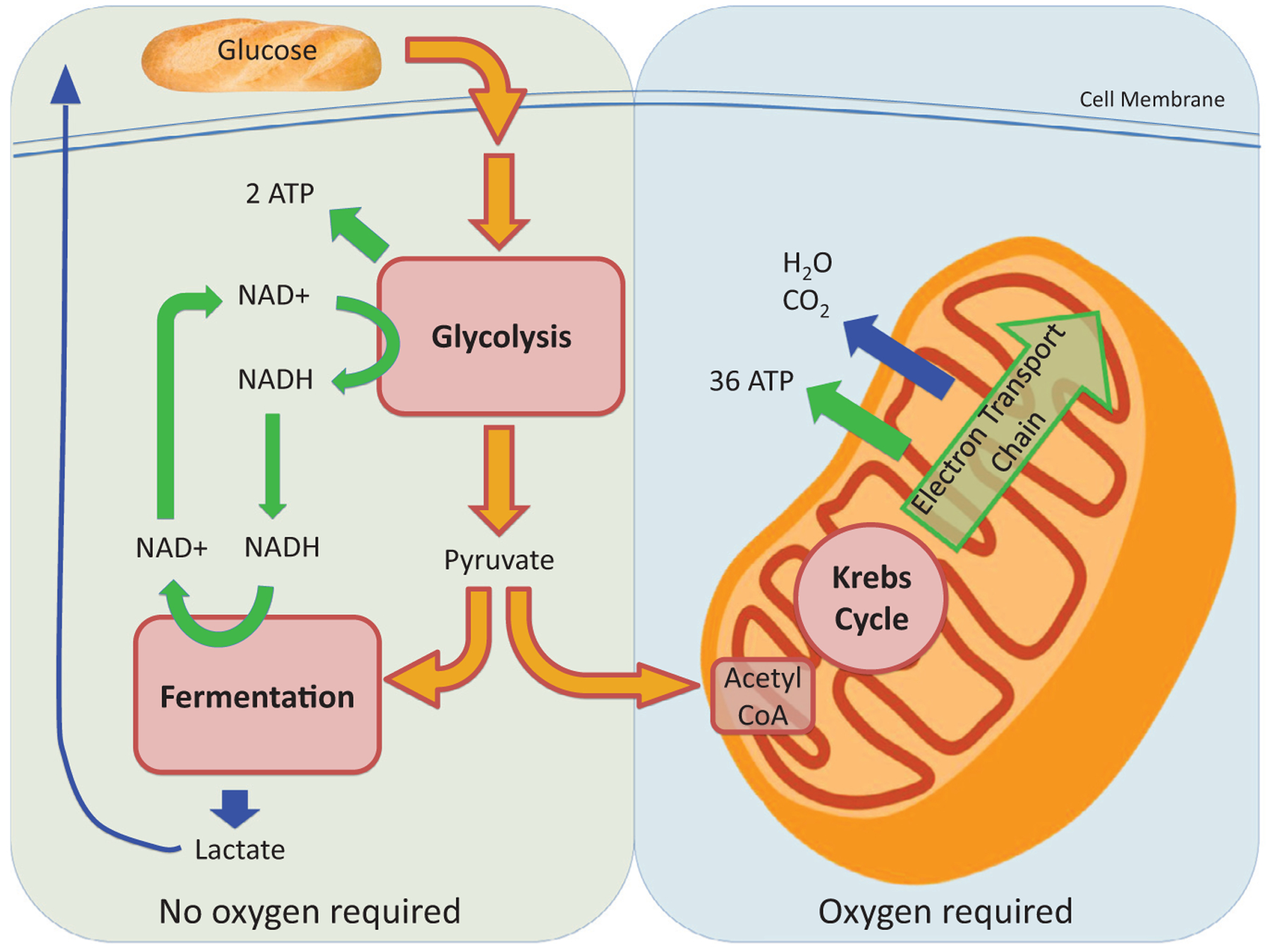
Overview of cellular energy metabolism including glycolysis, fermentation, Krebs cycle, and ETC. These combined reactions produce ATP, which is critical for cellular function. Glycolysis and fermentation take place in the cytoplasm and do not require oxygen. Oxidative phosphorylation via the electron transport chain takes place in mitochondria and requires oxygen. Abbreviation: NADH, nicotinamide adenine dinucleotide.
Glycolysis breaks down glucose into pyruvate, which subsequently is converted into acetyl-Coenzyme A (CoA). The process does not require oxygen and can take place in anaerobic environments. Pyruvate can either be converted to acetyl-CoA to participate in the Krebs cycle, or in the absence of oxygen it can be fermented to lactate.
Fatty acids are catabolized through β-oxidation to produce acetyl-CoA. When oxygen is present, acetyl-CoA derived from pyruvate or β-oxidation proceeds through the Krebs cycle. The Krebs cycle produces nicotinamide adenine dinucleotide and flavin adenine dinucleotide, which shuttle high-energy electrons to the mitochondrial electron transport chain (ETC). The series of reactions catalyzed by the ETC generate a strong proton gradient. In the final step of energy metabolism, the energy of this proton gradient is harvested by ATP synthase to phosphorylate adenosine diphosphate (ADP) to ATP.
To coordinate so many high-energy reactions without endangering cellular health, the mitochondrion is a highly specialized organelle. It has not one, but two membranes. The space between the outer and inner membranes is the intermembrane space. Nutrient substrates destined for the innermost space—the mitochondrial matrix—are transported from the cytoplasm through the intermembrane space. The intermembrane space retains the protons pumped out of the matrix during ETC reactions. The enzymes for the ETC reside within the inner membrane. That membrane is convoluted into layers called cristae that maximize surface area for ATP production. Cells with high-energy demands, such as cardiac muscle cells, have many more cristae per organelle and thus a larger surface area for energy production.
Although most of the mitochondrial proteins are encoded in the nucleus, mitochondria also have their own circular genomes (mitochondrial DNA [mtDNA]) that are inherited maternally and distinct from nuclear DNA. The mtDNA resides in the mitochondrial matrix and encodes 37 gene products. In contrast, mammalian mitochondria may be comprised of more than 1,000 proteins, indicating the importance of nuclear gene transcription for mitochondrial health.3
METABOLIC NEEDS IN THE KIDNEY
The kidney is a highly metabolically active organ, containing more mitochondria per weight than any other organ, sparing the heart.3 Basolateral Na+-K+ adenosine triphosphatases throughout the nephron pump sodium out of tubular cells and potassium into tubular cells. The vectorial movement of sodium from filtrate through the tubular cell and back into blood is used to couple the transport of filtered sugars and amino acids back into the blood. As these solutes move back into the blood, water and urea follow passively, driven by the osmotic force. Second, these basolateral adenosine triphosphatases create a net-negative intracellular charge, three sodium cations out for each two potassium cations in, that further power the reabsorption of cations and cationic organic compounds. Nearly 100% of the ATP consumed by the renal tubules is used for active reabsorption, and the amount of ATP produced via oxidative phosphorylation varies to match tubular reabsorptive needs.4,5
Given this constant high demand for ATP, mitochondria are crucial for normal renal function. This is shown most vividly by patients who harbor rare mutations that disrupt mitochondrial enzymes, alter mitochondrial structural proteins, or impair oxidative phosphorylation. Among such individuals, the most severely affected organs are those requiring high energy consumption: skeletal muscle, central nervous system, heart, and the kidneys. In the kidneys, mitochondrial diseases present primarily as tubulopathies, although cystic and glomerular disease associations also have been reported.6
RELATIONSHIP OF RENAL VASCULAR ANATOMY TO ENERGY METABOLISM
As the final acceptor of electrons from the ETC, oxygen is necessary for cells to fully harness fuel substrates. Therefore, oxygen delivery to the nephron is critical for normal tubular function. The vascular anatomy of the kidney leads to heterogeneous organ perfusion. Blood enters the nephron through the afferent arteriole and then passes through the glomerulus. There, the hydraulic pressure generated by the heart coupled with afferent and efferent arteriolar tone enables bulk filtration of the blood.7 The remaining unfiltered blood continues with approximately 80% lower hydraulic pressure and a partial pressure of oxygen of approximately 40 to 42 mm Hg through the efferent arteriole to the peritubular capillary bed, where the proximal tubules extract and consume nearly 50% of the entire nephron’s oxygen consumption to generate the energy needed for reabsorption.8 From there, blood travels to the medulla. The blood flow that reaches the medulla has a partial pressure of oxygen near 25 to 30 mm Hg, however, the ascending loop of Henle still extracts sufficiently large amounts of oxygen to drive the active reabsorption of sodium to maintain the osmotic gradient that concentrates urine.9,10 The inner medulla is even more hypoxic, with an estimated partial pressure of oxygen near 10 mm Hg. This sharp corticomedullary gradient is likely the result of countercurrent exchange from descending to ascending vasa recta that effectively shunts oxygen in a fashion that bypasses the bottom of the vascular hairpin loop of the deep medulla.11
Energy metabolism throughout the kidney reflects the variation in oxygen supply. In the renal cortex, where there is abundant oxygen delivery and dense mitochondria, energy is created through aerobic metabolism with essentially no glycolysis. The renal cortex depends primarily on b-oxidation of fatty acids for Krebs cycle substrates.12,13 Compared with the renal cortex, the medulla has much less oxygen delivery and consumes roughly 5% of cortical oxygen consumption. Accordingly, medullary cells contain approximately 15 times fewer mitochondria than the cortex. The renal medulla relies primarily on anaerobic glycolysis for energy production, which is sufficient because the medulla has lower energy needs and less active transport.14 The corticomedullary junction exists between the high PO2 environment in the cortex and the hypoxic environment of the medulla, however, many metabolically active tubular cells are found there and depend on oxygen for normal function. For that reason, this area is particularly susceptible to hypoxic and ischemic injury.9,14
MITOCHONDRIA AND AKI
Mitochondria have been noted to be structurally abnormal in clinical AKI for decades. Abnormal-appearing mitochondria were described as early as the 1970s when transmission electron microscopy enabled direct visualization of mitochondria. Trump et al15 noted that mitochondria in the proximal tubule were abnormally swollen among patients who had died from shock. Since then, numerous other investigators have noted swollen mitochondria and disruption of the mitochondrial ultrastructure both in human beings and rodents with AKI, with proximal tubular cells being the most severely affected.16–26 Notably, mitochondrial structural changes have been visualized in the ischemic human kidney before clinical manifestations of AKI, implying that mitochondrial perturbation may not be an epiphenomenon arising after injury; rather, mitochondrial dysfunction may be a contributing factor to injury.27 Visible changes in the mitochondria typically are associated with decreased mitochondrial function because swelling classically represents a loss of inner-membrane permeability control, which is crucial for mitochondrial function (Fig. 2).
Figure 2.
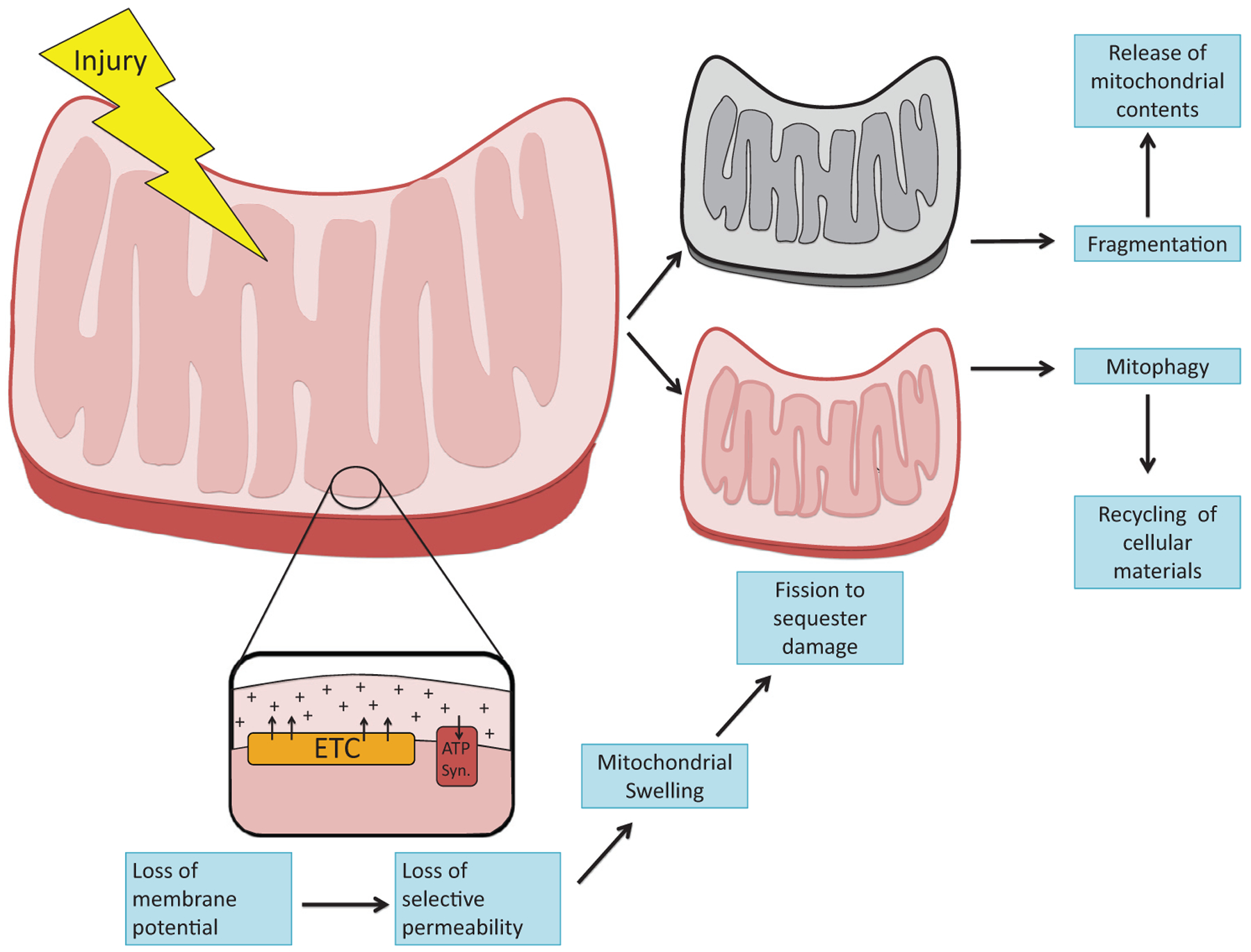
Mitochondrial structural changes after injury. Different noxious stimuli, including inflammatory cytokines, ischemia-reperfusion, and toxins, injure mitochondria. Injury disrupts the normal vectorial pumping of protons across the inner mitochondrial membrane by enzymatic complexes in the ETC. Subsequent loss of membrane potential impairs selective permeability. As a result, mitochondria swell. Fission is induced to sequester the damage and safely dispose of injured mitochondria through mitophagy. Excessive fission, also known as mitochondrial fragmentation, arises during severe injury and is associated with the release of mitochondrial contents that potentiate inflammation and cell death. Abbreviation: ATP syn, ATP synthase.
Evidence from septic and ischemic rodent models has shown that mitochondrial dysfunction in AKI is not only a consequence of inadequate delivery of critical energy oxidation substrates. For example, renal oxygen extraction either stayed stable17 or increased during sepsis despite decreased17,28,29 or unchanged30,31 renal blood flow and decreased reabsorptive load secondary to decreased glomerular filtration rate (GFR).29,32,33 Because solute reabsorption is the major energy-consuming task, a decreased filtered load in septic AKI should reduce oxygen need. Therefore, the cellular injury observed in septic AKI may be unrelated to decreased metabolic substrate availability. Rather, the increased oxygen uptake in the setting of decreased solute load implicates an inefficient use of oxygen: evidence of suboptimal mitochondrial function. The same inefficient oxygen consumption has been observed in human beings after cardiac surgery: GFR and renal blood flow were reduced, but oxygen extraction was higher in patients with AKI than in patients without AKI.34 Many additional studies have assessed mechanisms whereby primary metabolic and mitochondrial dysfunction was associated with AKI in many disease models (Tables 1 and 2).
Table 1.
Metabolic Findings in Acute Kidney Injury Models
| Type of Injury | Accumulation of Intratubular Fat | Mitochondrial Swelling | Alterations in Dynamics and Mitophagy | Impaired Mitochondrial Biogenesis | Reduced NAD+. Energy Metabolism |
|---|---|---|---|---|---|
| Septic AKI | Increased cortical free cholesterol76 | Swollen, dysmorphic mitochondria16,17 | Increased LC3 accumulation and other markers of autophagy46 | Decreased expression of PGC1α17 | Reduced oxygen consumption17 |
| Increased cortical triglycerides76,77 | Increased apoptosis46 | Reduced expression and activity of mitochondrial oxidative phosphorylation complexes17 | |||
| Decreased ATP63 | |||||
| Toxic AKI | Increased cortical triglycerides76,77 | Swollen, dysmorphic mitochondria20,23,25,40 | Increased LC3 accumulation and other markers of autophagy22,23,44,45,51,23,36,43–45,51 | Increased expression of PGC1α, NRF-1, and Tfam36 | Increased urine glucose and decreased urine pyruvate25 |
| Increased expression of Drp-1 and increased mitochondrial fragmentation18,36,37 | Decreased expression of PGC1α37 | Reduced expression of glycolysis enzymes25 | |||
| Decreased expression of fusion protein Opa125 | Decreased mitochondrial density20,67 | Reduced expression and activity of mitochondrial oxidative phosphorylation complexes20,36,66,67,87 | |||
| Decreased markers of mitophagy22,23 | |||||
| Release of cytochrome C18,66 | |||||
| Increased ROS production20,22,23,25,37,51,66,91 | Depolarization of the mitochondrial membrane21,66 | ||||
| Increased apoptosis.18,23,25,36,44,51,91 | |||||
| Reduced mtDNA66 | Increased PARP activity91 | ||||
| Decreased SIRT3 levels37 | |||||
| Decreased expression of NAD+ biosynthesis enzymes37 | |||||
| Decreased NAD+87 | |||||
| Decreased NAM61 | |||||
| Decreased ATP25 | |||||
| Ischemia-reperfusion | Increased cortical triglycerides and fatty acids64,76 | Swollen, dysmorphic mitochondria18,19,21,24,25 | Increased expression of Drp-1 and increased mitochondrial fragmentation18,36 | Reduced expression and activity of mitochondrial oxidative phosphorylation complexes24,36,87 | |
| Increased LC3 accumulation and other markers of autophagy36,41,42 | Depolarization of the mitochondrial membrane21 | ||||
| Increased markers of mitophagy19,42 | |||||
| Increased ROS production24,65,87 | Reduced expression of ATP synthase36 | ||||
| Increased apoptosis24,36,42,65 | Decreased ATP24,64,65 | ||||
| Decreased expression of NAD+ biosynthesis enzymes86 | |||||
| Increased PARP activity90 | |||||
| Decreased NAD+86 | |||||
| Crystalline nephropathy | Swollen, dysmorphic mitochondria26 | Increased expression of fission protein, Drp-1, and increased mitochondrial fragmentation26 | Decreased PGC1α, NRF-1, and Tfam26 | Reduced expression and activity of mitochondrial oxidative phosphorylation complexes26 | |
| Decreased expression of fusion proteins Opa1 and Mfn126 | Depolarization of the mitochondrial membrane26 | ||||
| Increased markers of mitophagy26 | |||||
| Decreased LC3 accumulation and other markers of autophagy26 | |||||
| Increased ROS production26 | |||||
| Ureteral obstruction | Increased cortical triglycerides78 | ||||
| Acid load | Increased LC3 accumulation and other markers of autophagy58 |
Abbreviations: Drp-1, dynamin-related protein; Mfn, mitofusin; NRF-1, nuclear respiratory factor 1; Opa1, optic atrophy 1.
Table 2.
Metabolic Effects of Genetic Manipulations in Response to AKI
| Gene | Gene Role | Overexpression | Underexpression/Inhibition |
|---|---|---|---|
| PGC1α | Mitochondrial biogenesis | Increased mitochondrial density48 | Worse injury after cisplatin17,22 |
| Restoration of normal oxygen consumption17 | Decreased cellular respiration22 | ||
| Increased cellular respiration22,48 | Decreased ATP production22 | ||
| Increased ATP production22 | Decreased mitophagy22 | ||
| Resistance to cisplatin injury22 | Decreased TFEB and lysosomal abundance22 | ||
| Persistent mitophagy after cisplatin injury22 | |||
| Increased TFEB and lysosomal abundance22 | |||
| Bcl2 | Apoptosis regulation | Reduced autophagy43 | |
| Reduced cytochorome C release18 | |||
| Reduced apoptosis18 | |||
| Drp1 | Mitochondrial fission | Reduced mitochondrial fragmentation.18 | |
| Decreased cytochrome C release18 | |||
| Reduced apopotosis18 | |||
| OMA1 | Mitochondrial quality control | Decreased Opa1 proteolysis (decreased inactivation)39 | |
| Decreased mitochondrial fragmentation39 | |||
| Reduced cytochrome C release39 | |||
| Reduced apoptosis39 | |||
| Mfn1 and Mfn2 | Mitochondrial fusion | Prevention of mitochondrial fragmentation40 | Increased mitochondrial fragmentation40 |
| Reduced cytochrome C release40 | Increased cytochrome C release40 | ||
| Reduced apoptosis40 | Increased apoptosis40 | ||
| Beclin-1 | Autophagy | Decreased autophagy43–45 | |
| Increased apoptosis41,44,45 | |||
| Atg5 and Atg7 | Autophagy | Decreased autophagy44,46,49,50,58 | |
| Increased apoptosis41,44,46,49–51 | |||
| Increased mitochondrial fragmentation58 | |||
| More severe injury after cisplatin and IRI49–51 | |||
| Increased oxidative stress accumulation50,51 | |||
| Reduced cellular respiration58 | |||
| Reduced mitochondrial membrane potential58 | |||
| Sestrin-2 | Metabolic homeostasis | Increased autophagy47 | Reduced autophagy47 |
| Decreased apoptosis47 | |||
| BNIP3 | Mitophagy | Increased mitophagy47 | Decreased mitophagy47 |
| Increased caspase-3 but decreased apoptosis47 | |||
| PINK1, PRKN, and PINK2 | Mitophagy | Decreased mitophagy59 | |
| Increased apoptosis59 | |||
| Increased mitochondrial fragmentation59 | |||
| Increased oxidative stress accumulation59 | |||
| More severe injury after IRI59 | |||
| PARP1 | DNA repair/NAD+ consumption | Less severe injury after IRI or cisplatin65,91 | |
| Preservation of ATP level65 | |||
| Decreased apoptosis65,91 | |||
| Decreased accumulation of ROS91 | |||
| SIRT1 and SIRT3 | Energy homeostasis/NAD+ consumption | Resistance to cisplatin injury37 | Worse injury after cisplatin or glycerol37,88 |
| Prevention of Drp1 recruitment and PINK1 expression37 | Increased mitochondrial fragmentation88 | ||
| Increased expression of Opa137 | Increased apoptosis88 | ||
| Prevention of mitochondrial depolarization37 | |||
| PPARα | Energy homeostasis/fatty acid catabolism | Reduced inhibition of fatty acid oxidation81 | |
| Reduced inhibition of mitochondrial protein expression81 | |||
| Decreased accumulation of lipid peroxidation products81 | |||
| Less severe injury after cisplatin and IRI81 | |||
| QPRT | NAD+ biosynthesis | More severe injury after IRI86 |
Abbreviation: Opa1, optic atrophy 1.
Impaired Mitochondrial Dynamics in AKI
Changes in mitochondrial dynamics have been implicated in AKI. Mitochondria are not static organelles. Their life cycles include constant remodeling with fission, fusion, and mitophagy, the last being a process of intracellular disposal (Fig. 3). The cell uses fission and fusion to exchange substrates and metabolites and to create daughter organelles.35 There is increasing evidence that the balance between fission and fusion is tilted toward fission during AKI. Excessive fission is described as mitochondrial fragmentation.
Figure 3.
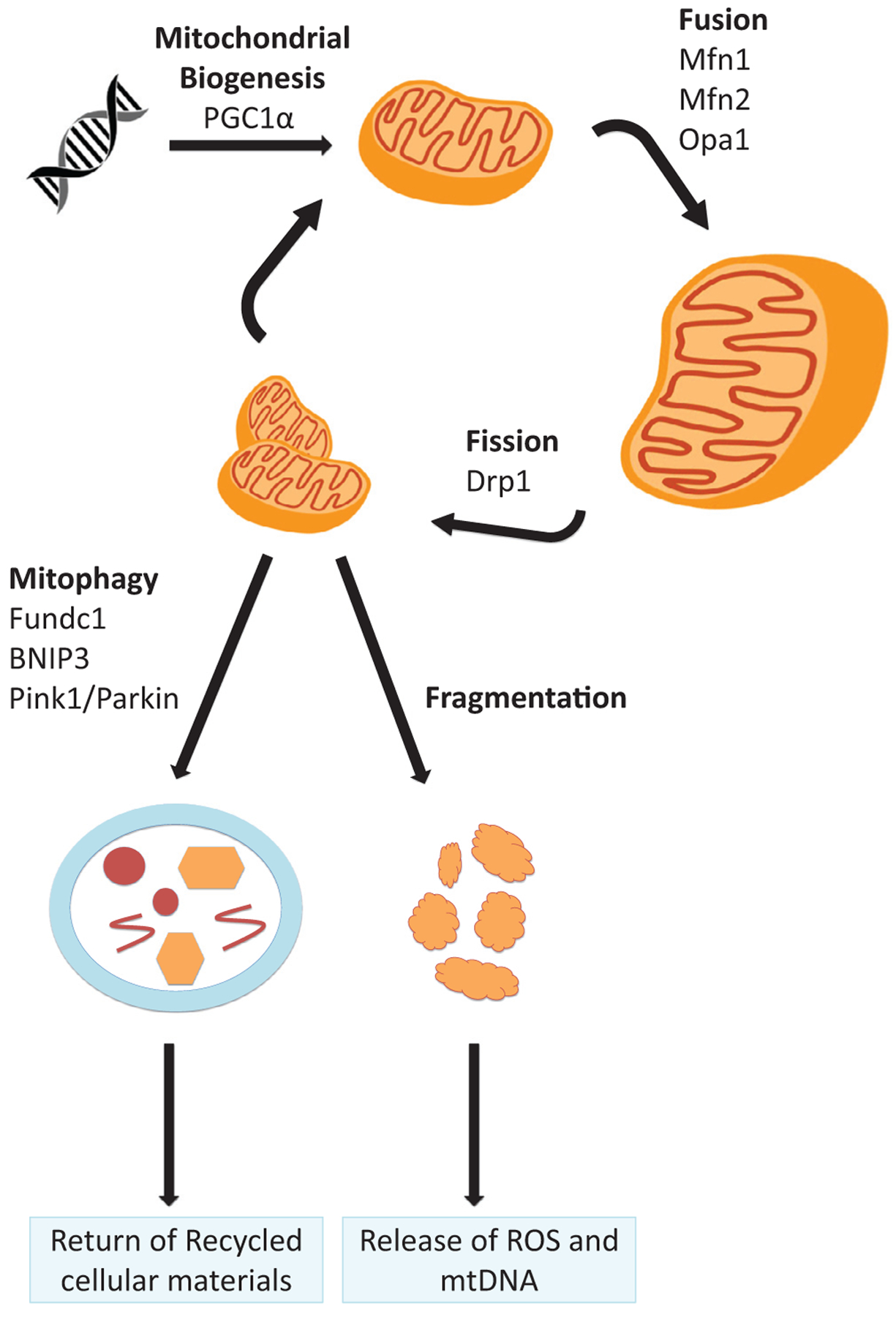
Mitochondrial life cycle. The life cycle of mitochondria includes biogenesis of mitochondrial structural proteins from nuclear DNA and dynamic remodeling of the mitochondrial network via fission and fusion to maintain an optimally functioning mass of mitochondria within the cell. Mitophagy enables intracellular disposal of mitochondria and recycling of their contents for cellular needs. Excessive fission, typically in the setting of injury, leads to fragmentation and release of cytotoxic mitochondrial contents including pro-apoptotic factors, ROS, and inflammatory mtDNA. PGC1α is a key regulator of mitochondrial biogenesis. Fusion proteins mitofusin 1 (Mfn1) and mitofusin 2 (Mfn 2) are necessary for outer-membrane fusion, while optic atrophy 1 (Opa1) is necessary for inner-membrane fusion. Dynamin-related protein 1 (Drp1) is critical for mitochondrial fission. Bcl-2/adenovirus E1B 19 KDa-interacting protein (BNIP)3, FUN14 containing 1 (FUNDC1), and PTEN-induced kinase-1 (PINK-1) with parkin are all proteins that can trigger mitophagy.
Mitochondrial fragmentation has been observed before tubular cell death in ischemia-reperfusion injury (IRI), folic acid, and cisplatin nephrotoxicity mouse models.18,26,36,37 Fragmentation is associated with the release of apoptotic factors such as cytochrome C, along with caspase activation and subsequent cellular apoptosis. Fragmentation may contribute to the pathogenesis of AKI.18 Dynamin-related protein 1, a protein that is critical for mitochondrial fission, was activated after AKI. Furthermore, inhibiting dynamin-related protein 1 prevented mitochondrial fission and protected against injury in cell models achieved by ATP depletion or cisplatin application, and in rodent models of IRI, cisplatin nephrotoxicity, and rhabdomyolysis.18,38
Fusion proteins mitofusin 1 and mitofusin 2 are necessary for outer-membrane fusion. Optic atrophy 1 is necessary for inner-membrane fusion. Mitofusin 2 and optic atrophy 1 were down-regulated and inactivated in rodent models of AKI.36,39 Inducing mitochondrial fusion by overexpressing mitofusins attenuated mitochondrial fragmentation, reduced cytochrome c release, and reduced apoptosis in cells treated with cisplatin and azides.40 Genetic manipulations to attenuate fission or to enhance fusion protect mice from different models of AKI.18 Given that a fused mitochondrial network is more efficient at ATP generation and less prone to the release of apoptotic mediators, these results imply that defense against excessive fission may be important to resist physiological impairment of kidney function.
Mitophagy and Lysosomal Biogenesis
Through autophagy, accumulated damage and debris within a cell are separated and then fused with an autophagosome for delivery to a lysosome to be safely degraded and recycled. In the uninjured state, autophagy is a method the cells use to facilitate macromolecule and organelle turnover. Autophagy is critical for cellular homeostasis because insufficient autophagy deprives the cell of nutrients and substrates and leads to accumulation of dysfunctional organelles that may be toxic.35 Autophagy also may play a critical role in AKI. Because fragmented mitochondria potentiate cell death, their safe disposal through autophagy, termed mitophagy, enables cells to achieve safe disposal of damaged organelles before those organelles inflict lethal damage.
Autophagic flux increases rapidly in proximal tubular cells after IRI, cisplatin injury, cyclosporine injury, and septic AKI models. Multiple studies have shown that this event precedes tissue damage or cellular apoptosis.41–48 Knocking out critical autophagy genes in the IRI and cisplatin models led to worsened AKI.49–51 Furthermore, using specific autophagy inhibitors in all models led to worsened AKI.41,43,44,46 Conversely, inducing autophagy protected against AKI in a septic mouse model.52 Together, these results suggest that the induction of autophagy early after a noxious stimulus may be an adaptive response to a lethal stressor. In turn, the data also imply that cellular injury may be related to a late failure of this adaptive response.
Mitophagy prevents the release of toxic intramitochondrial substances, such as mitochondrially derived reactive oxygen species (ROS), pro-apoptotic caspases, and proinflammatory mtDNA, into the cytoplasm.35,53 Mitophagy is coordinated closely with mitochondrial fission and fusion. Under physiological conditions, normal mitophagic flux functions as a quality control surveillance in which depolarized or dysfunctional mitochondria detach from the larger intracellular network and are targeted for removal.
Three major mechanisms for mitophagy have been identified: receptor-mediated, ubiquitin-mediated, and cardiolipin-mediated mitophagy.54–57 All lead to a binding interaction with microtubule-associated protein 1 light chain (LC3). LC3 is an autophagy protein that binds mitochondria to autophagosomes to signal autophagosome formation and elongation.55–57 Receptor-mediated mitophagy is facilitated by transmembrane proteins expressed on the mitochondrial outer membrane that bind directly to LC3. Bcl-2/adenovirus E1B 19-kDa-interacting protein 3, Nix, and FUN14 containing 1 are such proteins. Nix expression is up-regulated during physiologic mitophagy (eg, reticulocyte maturation).55 FUN14 containing 1 is critical to mitochondrial regulation during cellular differentiation.58 All are up-regulated in response to hypoxia.54,55 Ubiquitin-mediated mitophagy typically is triggered by mitochondrial depolarization.57 After mitochondrial depolarization or other injuries that interfere with mitochondrial protein import, phosphatase and tensin homolog-induced kinase-1 accumulates on mitochondrial surfaces. That leads to recruitment of parkin and ultimate ubiquitination of outer-mitochondrial membranes. These proteins either undergo proteosomal degradation or bind LC3 to promote mitophagy.57 Finally, cardiolipin-mediated mitophagy involves translocation of cardiolipin from the inner-mitochondrial membrane to the outer surface where is interacts with LC3. It typically is triggered by mitochondrial injury.56
Even under stress, mitophagy is responsible for maintaining an optimally functioning pool of mitochondria. However, when mitophagy is unable to keep pace with mitochondrial fragmentation, affected cells are exposed to intramitochondrial contents leaked into the cytoplasm that further potentiate injury. For example, ROS from the ETC react with major macromolecules including proteins and lipids to alter their structure and impair their function.18,36,46 Second, the release of mtDNA into the cytoplasm activates inflammasomes, which in turn induce inflammatory cytokine cascades.59 Finally, mitochondrial disruption leads to the release of numerous pro-apoptotic mediators, such as cytochrome C, which trigger programmed cell death.60
Impaired mitophagy has been implicated in the pathogenesis of AKI. Renal IRI in mice has been shown to induce Bcl-2/adenovirus E1B 19-kDa-interacting protein in tubules.47 Metabolic acidosis induces mitophagy in proximal tubular cells. Knockout of a key mitophagy gene, Atg5, led to reduced respiratory chain activity, reduced mitochondrial membrane potential, increased mitochondrial fragmentation, and significant mitochondrial swelling.61 After ischemic injury, both cell and mouse models showed increased mitophagy. Deficiency in Pink1 and Park2 decreased mitophagy, worsened ischemic injury, and led to increased mitochondrial damage, ROS production, and inflammation.62 Although there is much more to be learned about the role of mitophagy in kidney disease, there is growing evidence that mitophagy offers a protective role against injury because it enables the safe elimination of cytotoxic and pro-apoptotic mitochondrial elements. Impaired or inhibited mitophagy likely contributes to the pathogenesis of kidney disease. Targeting this pathologic progress through (1) neutralizing mitochondrial ROS, (2) inhibiting downstream effectors of apoptosis, and (3) promoting more efficient mitophagy all hold promise for future acute and chronic kidney disease therapies.
Mitochondrial Biogenesis
The cell must replace the mitochondrial mass that is destroyed through mitophagy and also must generate new mitochondrial mass to respond to increased energy needs. This is accomplished through mitochondrial biogenesis. As mentioned earlier, the majority of mitochondrial proteins are transcribed from nuclear DNA even though each mitochondrion possesses multiple copies of mtDNA. Synthesis of mitochondrial proteins is regulated by an array of transcription factors including transcrip0tion factor A, mitochondrial (Tfam), mitochondrial transcription termination factor 3 and 4, and peroxisome proliferator activated receptors (PPARs) α, δ, and γ. PPARγ coactivator-1α (PGC1α) is a co-activator that binds noncovalently to these and other transcription factors to augment transcription of proteins that are critical for mitochondrial biogenesis. PGC1α is heavily expressed in highly metabolically active organs including the kidney, with the proximal tubule showing the most robust expression.17
In cellular and in vivo models of AKI, expression of PGC1α in cellular and animal models of AKI varies with time. Early measurements after noxious stimulus show suppression,17,26,37,63 and late measurements during the time window of functional recovery show increased expression of PGC1α and downstream transcription factors such as Tfam and nuclear respiratory factor 1.36,48 Tran et al17 found that proximal tubule-specific PGC1α knockout mice showed a normal renal phenotype at baseline, but were much more susceptible to septic AKI, indicating this protein’s significant role in renal recovery. They also showed that overexpression of PGC1α in cultured proximal tubule cells protected against decreased oxygen consumption induced by inflammatory factors. Similarly in human beings, PGC1α expression was strongly suppressed in AKI,64 and decreased expression in renal transplant patients was associated with prolonged and incomplete recovery from delayed graft function.65 These data propose that the renal tubular cell’s ability to engage in mitochondrial biogenesis is essential for standard recovery from transient inflammatory stress.
In the renal tubular epithelium, PGC1α appears to play a broader role in the maintenance of an optimal pool of mitochondria against diverse stressors. For example, although PGC1α knockout cells are more susceptible to death from cisplatin exposure, transgenic cells were found to be more resistant.17 Analogously, PGC1α knockout mice developed more severe AKI after cisplatin whereas tubule-specific transgenic animals were more resistant to nephrotoxicity. Transcriptomics proposed mitophagy and lysosomal biogenesis via transcription factor EB (TFEB) as a downstream mechanism of PGC1α renoprotection in these cisplatin studies. PGC1α defended mitophagy whereas cisplatin eventually suppressed mitophagy. Knockdown of TFEB abrogated the protective effect of PGC1α in cultured cells, and inhibition of lysosomes with chloroquine similarly nullified renoprotection of PGC1α -tubular transgenic mice to cisplatin. Indeed, in both instances, inhibition of this downstream mechanism from PGC1α unveiled a pro-oxidative, cytotoxic effect of excess PGC1α. Without mitophagy and lysosomal function intact, more PGC1α during a stress situation became a cellular liability. These results propose that mitochondrial biogenesis needs to be paired with mitophagy and lysosomal clearance, particularly under stress. Without effective clearance of injured mitochondria, having more mitochondrial mass exposed to noxious stimuli such as cisplatin actually potentiates injury.22
Thus, mitochondrial biogenesis via the master regulator, PGC1α, is not a simple on/off switch for mitochondrial production. Through traditional biogenesis transcription factors such as Tfam and more newly discovered partners such as TFEB, the PGC1α program enables dynamic coordination of production and safe disposal of mitochondria to maintain appropriate cellular mitochondrial abundance and function. As discussed later, PGC1α also coordinates fuel oxidation via nicotinamide adenine dinucleotide (NAD)+, which is closely regulated to meet a cell’s dynamic needs.
Mitochondrial Energy Metabolism in AKI
Multiple injury models have shown evidence of impaired mitochondrial energy metabolism after injury. A reduction in ATP production has been shown in sepsis models, crystalline AKI models, and IRI models.24,25,66–68 Decreases in the expression of mitochondrial genes and decreased mitochondrial DNA have been observed in cisplatin and glycerol-injured mice.36,69 Accompanying the decreased expression of key proteins, essential respiratory chain complexes also have shown decreased function in nearly every AKI model.17,20,26,36,69,70 Without a properly functioning ETC, there is loss of mitochondrial membrane polarization, loss of selective permeability, and loss of the ion gradient that powers ADP phosphorylation. This leads to decreased ATP and increased leakage of inflammatory and pro-apoptotic mediators (Fig. 2).18,21,69
With abnormal function of the ETC, ROS production also increases. Oxidative phosphorylation takes place in the mitochondria by passing high-energy electrons from nicotinamide adenine dinucleotide or flavin adenine dinucleotide through the complexes of the electron transport chain. The transfer of these electrons creates the proton gradient, which ultimately drives ATP synthase to produce ATP. The electron trail ends with the reduction of oxygen to water. Addition of one rather than two electrons to oxygen yields the superoxide ion, a free radical. Unstable flow of electrons through the ETC is thought to contribute to the generation of superoxide rather than H2O from oxygen. In this way, mitochondrial dysfunction induces a significant release of reactive oxygen species, which themselves trigger apoptosis and proinflammatory pathways that worsen injury.26,71–74 Several studies have shown that mitochondrially targeted antioxidants reduce the severity of AKI in rodent models.26,69,75–78
With the impairment in fuel combustion, AKI also leads to intracellular accumulation of the principal fuel for the renal cortex: fatty acids.67,79–81 In addition to cellular energy deprivation, the accumulation of fatty acids may contribute to cellular dysfunction and death via lipotoxicity. Indeed, accumulation of lipids in the kidney has been shown to induce inflammatory pathways that ultimately contribute to fibrosis.82 Excessive intracellular fatty acid content also has been associated with accumulation of toxic metabolites such as acyl-CoAs, ceramides, and ROS.12 PPARα is a transcription factor that induces expression of genes that encode enzymes of fatty acid oxidation and stimulate cellular uptake of free fatty acids.82 In both cellular and mouse models, PPARα agonists and transgenic overexpression of PPARa have mitigated renal injury after cisplatin and IRI.83,84
Finally, AKI leads to decreased local NAD+. NAD+ is a critical cofactor involved in many cellular oxidative-reduction reactions. As an electron carrier from glycolysis, the Krebs cycle, and β-oxidation to the ETC, NAD+ is essential for the efficient generation of ATP.85 NAD+ also serves a second role in eukaryotic cells as a substrate for signaling enzymes including poly-ADP ribose polymerases (PARPs), sirtuins, and ectonucleotidases that regulate broad swaths of cellular behavior.86 Impaired NAD+ homeostasis has been implicated in diverse pathologies including dementia, glaucoma, immune deficiencies, insulin resistance, diabetes, infertility, inflammation, cancers, obesity, cardiovascular disease, and autism.87 Similarly, genetic mutations that lead to impaired NAD+ biosynthesis in utero have been linked recently to renal dysplasia, implying a critical role for intact NAD+ homeostasis during renal development.88
Intracellular NAD+ and AKI
The concentration of NAD+ within a cell reflects the net actions of biosynthesis and consumption. Levels of renal NAD+ decrease precipitously in AKI as a result of both decreased biosynthesis and increased consumption.89,90 An experiment examining downstream mechanisms of PGC1α-dependent renoprotection showed de novo NAD+ biosynthesis as a novel downstream effector pathway. NAD+ levels were correlated with PGC1α expression both in genetic models and induced models of AKI.89 RNA sequencing showed that PGC1α coordinated the expression of an eight-enzyme cascade responsible for converting the essential amino acid tryptophan to NAD+. This cascade, also referred to as the de novo or kynurenine pathway, becomes suppressed in different models of AKI, but is induced by PGC1α.64 The importance of this effector arm for PGC1α-dependent renoprotection was shown by exogenous replenishment of NAD+ levels in PGC1α knockout mice. Although PGC1α knockout mice were highly sensitive to IRI, NAD+ augmentation via its precursor nicotinamide (NAM) normalized their response to the ischemic stress.64 An independent study showed that aged mice were protected from cisplatin-induced AKI with supplementation of nicotinamide mononucleotide, another nutritional NAD+ precursor.91 A third group showed that NAD+ augmentation also could be achieved by maintaining substrate flow through the de novo pathway to protect mice from IRI and cisplatin injury.90 Finally, in a pilot placebo-controlled randomized clinical trial assessing pharmacokinetics of NAM administration among patients undergoing cardiac surgery, participants who received 1 or 3 g of NAM orally per day perioperatively showed decreased rates of AKI.89 This preliminary clinical finding, buttressed by independent demonstrations of the salutary effects of augmenting NAD+ biosynthesis in preclinical models, has stoked significant enthusiasm for further clinical development of strategies to boost NAD+ for therapeutic benefit.92
Boosting NAD+ levels can be accomplished by supplementation of vitamin B3 precursors as described earlier or by reducing consumption. NAD+ consumption is accelerated in AKI.89 There are three major classes of NAD+ consuming enzymes: sirtuins, PARPs, and cyclic ADP-ribose synthases.86 Expression of each of these enzymes is altered in AKI (Fig. 4).
Figure 4.
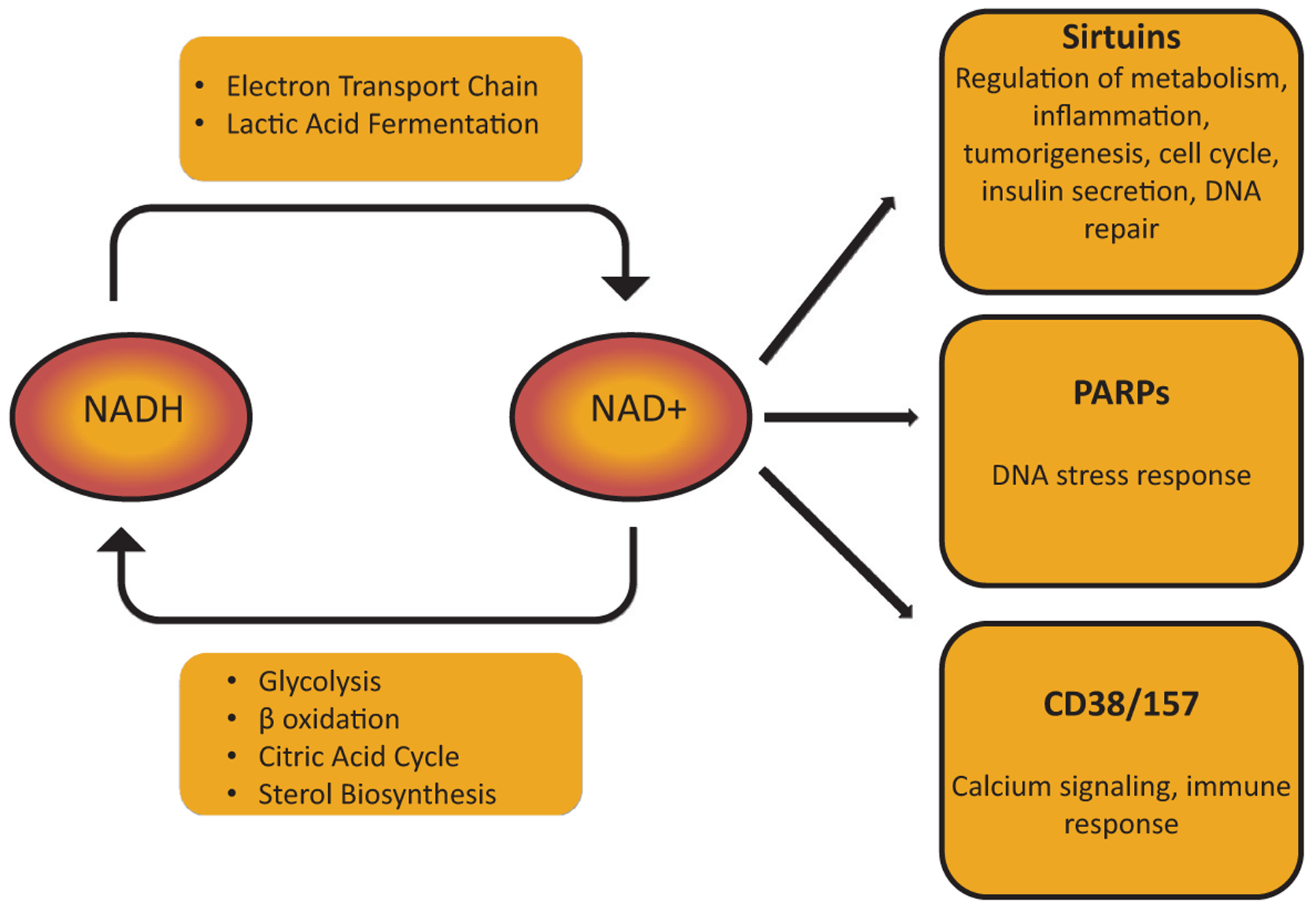
Overview of NAD+ metabolism. In highly metabolically active cells such as the renal tubular epithelium, NAD+ regulates broad aspects of cellular metabolism. Reduction of NAD+ to NADH is required for glycolysis, fatty acid oxidation, the tricarboxylic acid (Krebs) cycle, and sterol biosynthesis. Oxidation of NADH back to NAD+ provides high-energy electrons in the ETC and generates lactic acid from pyruvate. NAD+ also can be used by enzymes that cleave NAD+ to generate Nam. These include PARPs, sirtuins, and cyclic ADP-ribose (cADPR) synthetases (CD38/CD157), each of which play crucial roles in cellular function.
PARPs are activated after AKI as a repair mechanism in response to cellular stress and DNA damage. They use NAD+ as a substrate to attach ADP-riboses to target proteins. In doing so, they stabilize the conformation of damaged DNA and facilitate DNA access for repair enzymes.93 However, PARPs consume NAD+, and depletion of NAD+ leads to cellular ATP deprivation and cell death. Therefore, even though PARPs protect genome integrity after injury, excessive activation can be detrimental.68,93,94 Studies in rats with IRI showed that PARPs were overexpressed after injury, and pharmacologic inhibition of PARP led to more rapid improvement in blood urea nitrogen and creatinine after IRI, less histologic ischemic damage, more proximal tubule regeneration, and increased cellular ATP.93 Like-wise, mice with genetic deletion of parp1 developed a less severe decrease in GFR after IRI, less neutrophil infiltration, reduced expression of inflammatory mediators, and a less severe reduction in ATP compared with wild-type controls despite comparable levels of ROS production and DNA damage.68 Similar results were observed with pharmacologic inhibition or deletion of PARP1 in a mouse cisplatin AKI model and cisplatin-treated proximal tubular cells.94
The cyclic ADP-ribose synthases are much less studied in the context of AKI. These enzymes hydrolyze many nucleotide metabolites and closely modulate intracellular calcium levels. They typically are activated as a response to inflammation and lead to B-cell proliferation and differentiation and neutrophil trafficking.95 CD38 and CD157 are two of these enzymes that cleave NAD+. In septic mouse models, chemical blockade of CD38 led to improvement in blood urea nitrogen, less histologic evidence of tubular injury, decreased infiltration of macrophages and neutrophils, and decreased expression of inflammatory cytokines.95
Finally, the sirtuins are a class of NAD+ consumers that have been associated with longevity in many animal models; compared with the earlier-described classes of NAD+ consuming enzymes, sirtuins are considered to promote healthy metabolism.37 Sirtuins regulate metabolism reactions via NAD+-dependent deacetylation or deacylation of target proteins that includes histones, transcription factors, and coordinators of cell signaling.96 One widely cited early study showed that sirtuin 1 (SIRT1) deacetylated and thereby activated PGC1α.97 In the context of more recent results regarding PGC1α’s regulation of de novo NAD+ biosynthesis,64 the data collectively suggest a feed-forward loop in which PGC1α increases cellular NAD+ by up-regulating transcription of biosynthetic enzymes, which in turn provides more substrate for SIRT1 to activate PGC1α via deacetylation. The potential beneficial effects of sirtuins have been examined in AKI models. Renal SIRT3 was decreased after cisplatin injury in mice and cells, and overexpression of SIRT3 mitigated AKI.37 Renal SIRT1 and NAD+ were reduced in aged mice. Supplementing NAD+ in those mice via nicotinamide mononucleotide restored SIRT1 and protected against cisplatin-induced AKI.91 Together, these consumers highlight both the critical nature of NAD+ in cellular responses and also the various mechanisms of NAD+ depletion in AKI. PARP and CD38 may be pathologically activated in AKI settings, while sirtuins provide metabolic protection against stressors that trigger AKI. This creates a paradigm in which not only increasing NAD+ may be of utility, but targeting its application to ameliorate AKI could be a clinically advantageous possibility.
SUMMARY AND CONCLUSIONS
Mitochondria encompass a significant fraction of total cellular biochemistry, particularly for cells requiring high energy. The renal tubule requires a constant source of ATP for its core function of transporting water and solutes against gradients. Intact mitochondria furnish the energy for this process, but relying on mitochondria also exposes these cells to the risk of injury from different noxious stimuli that injure mitochondria.
Primary mitochondrial abnormalities lead to heritable clinical tubulopathies, and acquired mitochondrial abnormalities appear to be a pathogenic hallmark of different AKI syndromes. Mitochondria are not only a conserved target of unrelated stressors, such as inflammatory mediators, genotoxins, and ischemia-reperfusion, but mitochondrial impairment secondarily amplifies cellular injury through the production of mitochondrial ROS, the release of pro-apoptotic factors, and the release of factors such as mtDNA, which triggers inflammatory reactions. Virtually every aspect of mitochondrial biology, ranging from the dynamics of fission/fusion to biogenesis to the biochemistry of energy harvesting, is perturbed in AKI.
Although outside the scope of this review, many of the mitochondrial pathways involved in AKI also have been implicated in experimental and clinical CKD.98–100 Given that AKI itself can lead to CKD, and, conversely, that CKD increases the risk for AKI, intimate pathogenic connections between these two ends of the kidney disease spectrum are not surprising. The studies summarized earlier suggests a growing list of measurable and modifiable targets within metabolic and mitochondrial biology that could impact how AKI is diagnosed, monitored, prevented, and treated in the future.
Financial support:
National Institutes of Health grants K12-HD000850 (A.J.C.), and R35-HL139424, R01-DK095072, R01-AG027002, and R01-HL125275 (S.M.P.).
Footnotes
Conflict of interest statement:
Samir M. Parikh is listed as an inventor on patent filings from Beth Israel Deaconess Medical Center, holds equity in Raksana Therapeutics, and has received consulting fees from Astellas, Cytokinesis, Mission Therapeutics, and Aerpio, where he serves on the Scientific Advisory Board.
REFERENCES
- 1.Sawhney S, Fraser SD. Epidemiology of AKI: utilizing large databases to determine the burden of AKI. Adv Chronic Kidney Dis. 2017;24:194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pavkov ME, Harding JL, Burrows NR. Trends in hospitalizations for acute kidney injury - United States, 2000–2014. MMWR Morb Mortal Wkly Rep. 2018;67:289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pagliarini DJ, Calvo SE, Chang B, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008; 134:112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris SI, Balaban RS, Mandel LJ. Oxygen consumption and cellular ion transport: evidence for adenosine triphosphate to O2 ratio near 6 in intact cell. Science. 1980;208:1148–50. [DOI] [PubMed] [Google Scholar]
- 5.Kurnik BR, Weisberg LS, Kurnik PB. Renal and systemic oxygen consumption in patients with normal and abnormal renal function. J Am Soc Nephrol. 1992;2:1617–26. [DOI] [PubMed] [Google Scholar]
- 6.Emma F, Montini G, Parikh SM, Salviati L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol. 2016;12:267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalal R, Bruss ZS, Sehdev JS. Physiology, renal blood flow and filtration. Treasure Island, FL: StatPearls Publishing, LLC; 2019. [PubMed] [Google Scholar]
- 8.Layton AT, Laghmani K, Vallon V, Edwards A. Solute transport and oxygen consumption along the nephrons: effects of Na+ transport inhibitors. Am J Physiol Renal Physiol. 2016;311: F1217–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brezis M, Rosen S. Hypoxia of the renal medulla-its implications for disease. N Engl J Med. 1995;332:647–55. [DOI] [PubMed] [Google Scholar]
- 10.Wilcox CS, Palm F, Welch WJ. Renal oxygenation and function of the rat kidney: effects of inspired oxygen and preglomerular oxygen shunting. Adv Exp Med Biol. 2013;765:329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Epstein FH. Oxygen and renal metabolism. Kidney Int. 1997;51: 381–5. [DOI] [PubMed] [Google Scholar]
- 12.Bobulescu IA. Renal lipid metabolism and lipotoxicity. Curr Opin Nephrol Hypertens. 2010;19:393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balaban RS, Mandel LJ. Metabolic substrate utilization by rabbit proximal tubule. An NADH fluorescence study. Am J Physiol. 1988;254:F407–16. [DOI] [PubMed] [Google Scholar]
- 14.Bernanke D, Epstein FH. Metabolism of the renal medulla. Am J Physiol. 1965;208:541–5. [DOI] [PubMed] [Google Scholar]
- 15.Trump BF, Valigorsky JM, Jones RT, Mergner WJ, Garcia JH, Cowley RA. The application of electron microscopy and cellular biochemistry to the autopsy. Observations on cellular changes in human shock. Hum Pathol. 1975;6:499–516. [DOI] [PubMed] [Google Scholar]
- 16.Takasu O, Gaut JP, Watanabe E, et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. 2013;187:509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran M, Tam D, Bardia A, et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 2011;121:4003–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szeto HH, Liu S, Soong Y, et al. Mitochondria protection after acute ischemia prevents prolonged upregulation of IL-1 β and IL-18 and arrests CKD. J Am Soc Nephrol. 2017;28:1437–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zsengeller ZK, Ellezian L, Brown D, et al. Cisplatin nephrotoxicity involves mitochondrial injury with impaired tubular mitochondrial enzyme activity. J Histochem Cytochem. 2012;60: 521–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall AM, Rhodes GJ, Sandoval RM, Corridon PR, Molitoris BA. In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int. 2013;83:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lynch MR, Tran MT, Ralto KM, et al. TFEB-driven lysosomal biogenesis is pivotal for PGC1alpha-dependent renal stress resistance. JCI Insight. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui J, Bai XY, Sun X, et al. Rapamycin protects against gentamicin-induced acute kidney injury via autophagy in mini-pig models. Sci Rep. 2015;5:11256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szeto HH, Liu S, Soong Y, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. 2011;22:1041–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi YM, Kim HK, Shim W, et al. Mechanism of cisplatin-induced cytotoxicity is correlated to impaired metabolism due to mitochondrial ROS generation. PLoS One. 2015;10:e0135083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aparicio-Trejo OE, Reyes-Fermin LM, Briones-Herrera A, et al. Protective effects of N-acetyl-cysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free Radic Biol Med. 2019;130:379–96. [DOI] [PubMed] [Google Scholar]
- 27.Parekh DJ, Weinberg JM, Ercole B, et al. Tolerance of the human kidney to isolated controlled ischemia. J Am Soc Nephrol. 2013;24:506–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benes J, Chvojka J, Sykora R, et al. Searching for mechanisms that matter in early septic acute kidney injury: an experimental study. Crit Care. 2011;15:R256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z, Holthoff JH, Seely KA, et al. Development of oxidative stress in the peritubular capillary microenvironment mediates sepsis-induced renal microcirculatory failure and acute kidney injury. Am J Pathol. 2012;180:505–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langenberg C, Bellomo R, May C, Wan L, Egi M, Morgera S. Renal blood flow in sepsis. Crit Care. 2005;9:R363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langenberg C, Wan L, Egi M, May CN, Bellomo R. Renal blood flow and function during recovery from experimental septic acute kidney injury. Intensive Care Med. 2007;33:1614–8. [DOI] [PubMed] [Google Scholar]
- 32.Heemskerk AE, Huisman E, van Lambalgen AA, et al. Renal function and oxygen consumption during bacteraemia and endo-toxaemia in rats. Nephrol Dial Transplant. 1997;12:1586–94. [DOI] [PubMed] [Google Scholar]
- 33.Porta F, Takala J, Weikert C, et al. Effects of prolonged endotoxemia on liver, skeletal muscle and kidney mitochondrial function. Crit Care. 2006;10:R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sward K, Valsson F, Sellgren J, Ricksten SE. Differential effects of human atrial natriuretic peptide and furosemide on glomerular filtration rate and renal oxygen consumption in humans. Intensive Care Med. 2005;31:79–85. [DOI] [PubMed] [Google Scholar]
- 35.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Funk JA, Schnellmann RG. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Renal Physiol. 2012;302:F853–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morigi M, Perico L, Rota C, et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest. 2015;125:715–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang WX, Wu WH, Qiu HY, Bo H, Huang SM. Amelioration of rhabdomyolysis-induced renal mitochondrial injury and apoptosis through suppression of Drp-1 translocation. J Nephrol. 2013;26: 1073–82. [DOI] [PubMed] [Google Scholar]
- 39.Xiao X, Hu Y, Quiros PM, Wei Q, Lopez-Otin C, Dong Z. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am J Physiol Renal Physiol. 2014;306:F1318–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brooks C, Cho SG, Wang CY, Yang T, Dong Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am J Physiol Cell Physiol. 2011;300:C447–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol. 2010;176:1181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki C, Isaka Y, Takabatake Y, et al. Participation of autophagy in renal ischemia/reperfusion injury. Biochem Biophys Res Commun. 2008;368:100–6. [DOI] [PubMed] [Google Scholar]
- 43.Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008;74:631–40. [DOI] [PubMed] [Google Scholar]
- 44.Yang C, Kaushal V, Shah SV, Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol. 2008;294:F777–87. [DOI] [PubMed] [Google Scholar]
- 45.Pallet N, Bouvier N, Legendre C, et al. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy. 2008;4: 783–91. [DOI] [PubMed] [Google Scholar]
- 46.Hsiao HW, Tsai KL, Wang LF, et al. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock. 2012;37:289–96. [DOI] [PubMed] [Google Scholar]
- 47.Ishihara M, Urushido M, Hamada K, et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol. 2013;305:F495–509. [DOI] [PubMed] [Google Scholar]
- 48.Rasbach KA, Schnellmann RG. Signaling of mitochondrial biogenesis following oxidant injury. J Biol Chem. 2007;282:2355–62. [DOI] [PubMed] [Google Scholar]
- 49.Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82:1271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu S, Hartleben B, Kretz O, et al. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy. 2012;8:826–37. [DOI] [PubMed] [Google Scholar]
- 51.Takahashi A, Kimura T, Takabatake Y, et al. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol. 2012; 180:517–25. [DOI] [PubMed] [Google Scholar]
- 52.Howell GM, Gomez H, Collage RD, et al. Augmenting autophagy to treat acute kidney injury during endotoxemia in mice. PLoS One. 2013;8:e69520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buelvas-Jimenez N, Suarez-Useche RJ, Vielma-Guevara JR. NLRP3 inflammasome: a therapeutic option for kidney disease? Rev Salud Publica (Bogota). 2017;19:118–22. [DOI] [PubMed] [Google Scholar]
- 54.Chen G, Ray R, Dubik D, et al. The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J Exp Med. 1997;186:1975–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Novak I, Kirkin V, McEwan DG, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11: 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawajiri S, Saiki S, Sato S, et al. PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett. 2010;584:1073–9. [DOI] [PubMed] [Google Scholar]
- 58.Lampert MA, Orogo AM, Najor RH, et al. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy. 2019;15:1182–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhong Z, Liang S, Sanchez-Lopez E, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int. 2011;80:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Namba T, Takabatake Y, Kimura T, et al. Autophagic clearance of mitochondria in the kidney copes with metabolic acidosis. J Am Soc Nephrol. 2014;25:2254–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang C, Han H, Yan M, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy. 2018;14:880–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Portilla D, Dai G, McClure T, et al. Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int. 2002;62:1208–18. [DOI] [PubMed] [Google Scholar]
- 64.Tran MT, Zsengeller ZK, Berg AH, et al. PGC1a drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. 2016;531:528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Drury ER, Zsengeller ZK, Stillman IE, Khankin EV, Pavlakis M, Parikh SM. Renal PGC1alpha may be associated with recovery after delayed graft function. Nephron. 2018;138:303–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Levy B, Mansart A, Bollaert PE, Franck P, Mallie JP. Effects of epinephrine and norepinephrine on hemodynamics, oxidative metabolism, and organ energetics in endotoxemic rats. Intensive Care Med. 2003;29:292–300. [DOI] [PubMed] [Google Scholar]
- 67.Feldkamp T, Kribben A, Roeser NF, Senter RA, Weinberg JM. Accumulation of nonesterified fatty acids causes the sustained energetic deficit in kidney proximal tubules after hypoxia-reoxygenation. Am J Physiol Renal Physiol. 2006;290: F465–77. [DOI] [PubMed] [Google Scholar]
- 68.Zheng J, Devalaraja-Narashimha K, Singaravelu K, Padanilam BJ. Poly(ADP-ribose) polymerase-1 gene ablation protects mice from ischemic renal injury. Am J Physiol Renal Physiol. 2005; 288:F387–98. [DOI] [PubMed] [Google Scholar]
- 69.Tanabe K, Tamura Y, Lanaspa MA, et al. Epicatechin limits renal injury by mitochondrial protection in cisplatin nephropathy. Am J Physiol Renal Physiol. 2012;303:F1264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kharbangar A, Khynriam D, Prasad SB. Effect of cisplatin on mitochondrial protein, glutathione, and succinate dehydrogenase in Dalton lymphoma-bearing mice. Cell Biol Toxicol. 2000;16:363–73. [DOI] [PubMed] [Google Scholar]
- 71.Andreyev AY, Kushnareva YE, Murphy AN, Starkov AA. Mitochondrial ROS metabolism: 10 years later. Biochemistry (Mosc). 2015;80:517–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Murphy Michael P How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fleury C, Mignotte B, Vayssière J-L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84:131–41. [DOI] [PubMed] [Google Scholar]
- 74.Nath KA, Norby SM. Reactive oxygen species and acute renal failure. Am J Med. 2000;109:665–78. [DOI] [PubMed] [Google Scholar]
- 75.Mitchell T, Rotaru D, Saba H, Smith RA, Murphy MP, MacMillan-Crow LA. The mitochondria-targeted antioxidant mitoquinone protects against cold storage injury of renal tubular cells and rat kidneys. J Pharmacol Exp Ther. 2011;336:682–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mukhopadhyay P, Horváth B, Zsengelér Z, et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic Biol Med. 2012;52:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Silachev DN, Plotnikov EY, Pevzner IB, et al. Neuroprotective effects of mitochondria-targeted plastoquinone in a rat model of neonatal hypoxic-ischemic brain injury. Molecules. 2018;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Szeto HH. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J. 2006;8:E521–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zager RA, Johnson AC, Hanson SY. Renal tubular triglyceride accumulation following endotoxic, toxic, and ischemic injury. Kidney Int. 2005;67:111–21. [DOI] [PubMed] [Google Scholar]
- 80.Johnson AC, Stahl A, Zager RA. Triglyceride accumulation in injured renal tubular cells: alterations in both synthetic and catabolic pathways. Kidney Int. 2005;67:2196–209. [DOI] [PubMed] [Google Scholar]
- 81.Tannenbaum J, Purkerson ML, Klahr S. Effect of unilateral ureteral obstruction on metabolism of renal lipids in the rat. Am J Physiol. 1983;245:F254–62. [DOI] [PubMed] [Google Scholar]
- 82.Simon N, Hertig A. Alteration of fatty acid oxidation in tubular epithelial cells: from acute kidney injury to renal fibrogenesis. Front Med (Lausanne). 2015;2:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nagothu KK, Bhatt R, Kaushal GP, Portilla D. Fibrate prevents cisplatin-induced proximal tubule cell death. Kidney Int. 2005;68:2680–93. [DOI] [PubMed] [Google Scholar]
- 84.Li S, Nagothu KK, Desai V, et al. Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-alpha in mice confers protection during acute kidney injury. Kidney Int. 2009;76:1049–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ralto K, Rhee EP, Parikh SM. NAD+ homeostasis in renal health and disease. Nat Rev Nephrol. 2020;16:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Verdin E NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–13. [DOI] [PubMed] [Google Scholar]
- 87.Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 2018;27:529–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shi H, Enriquez A, Rapadas M, et al. NAD deficiency, congenital malformations, and niacin supplementation. N Engl J Med. 2017; 377:544–52. [DOI] [PubMed] [Google Scholar]
- 89.Poyan Mehr A, Tran MT, Ralto KM, et al. De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat Med. 2018;24:1351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Katsyuba E, Mottis A, Zietak M, et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature. 2018;563:354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guan Y, Wang SR, Huang XZ, et al. Nicotinamide mononucleotide, an NAD(+) precursor, rescues age-associated susceptibility to AKI in a sirtuin 1-dependent manner. J Am Soc Nephrol. 2017;28:2337–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kellum JA, Prowle JR. Paradigms of acute kidney injury in the intensive care setting. Nat Rev Nephrol. 2018;14:217–30. [DOI] [PubMed] [Google Scholar]
- 93.Martin DR, Lewington AJ, Hammerman MR, Padanilam BJ. Inhibition of poly(ADP-ribose) polymerase attenuates ischemic renal injury in rats. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1834–40. [DOI] [PubMed] [Google Scholar]
- 94.Mukhopadhyay P, Horváth B, Kechrid M, et al. Poly(ADP-ribose) polymerase-1 is a key mediator of cisplatin-induced kidney inflammation and injury. Free Radic Biol Med. 2011;51:1774–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shu B, Feng Y, Gui Y, et al. Blockade of CD38 diminishes lipopolysaccharide-induced macrophage classical activation and acute kidney injury involving NF-kappaB signaling suppression. Cell Signal. 2018;42:249–58. [DOI] [PubMed] [Google Scholar]
- 96.Dominy JE, Puigserver P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb Perspect Biol. 2013;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–8. [DOI] [PubMed] [Google Scholar]
- 98.Sharma K, Karl B, Mathew AV, et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J Am Soc Nephrol. 2013;24:1901–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Galvan DL, Green NH, Danesh FR. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017;92: 1051–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Duann P, Lin PH. Mitochondria damage and kidney disease. AdvExp Med Biol. 2017;982:529–51. [DOI] [PMC free article] [PubMed] [Google Scholar]