

Graphical abstract
Keywords: COVID-19, SARS-CoV-2, Coronavirus, Receptor-binding domain, Receptor binding-motif
Highlights
-
•
SARS-CoV-2 S1-NTD presents different receptor binding motifs compared to the SARS-CoV.
-
•
Functional motifs similar to the S1-NTD GTNGTKR loop were identified in other proteins.
-
•
The GTNGTKR loop is very likely to allow the SARS-CoV-2 to bind other receptors.
-
•
The GTNGTKR motif is very likely an evolutionary acquisition under functional constraints.
Abstract
The 2019 novel coronavirus disease (COVID-19) that emerged in China has been declared as public health emergency of international concern by the World Health Organization and the causative pathogen was named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). In this report, we analyzed the structural characteristics of the N-terminal domain of the S1 subunit (S1-NTD) of the SARS-CoV-2 spike protein in comparison to the SARS-CoV in particular, and to other viruses presenting similar characteristic in general. Given the severity and the wide and rapid spread of the SARS-CoV-2 infection, it is very likely that the virus recognizes other receptors/co-receptors besides the ACE2. The NTD of the SARS-CoV-2 contains a receptor-binding motif different from that of SARS-CoV, with some insertions that could confer to the new coronavirus new receptor binding abilities. In particular, motifs similar to the insertion 72GTNGTKR78 have been found in structural proteins of other viruses; and these motifs were located in putative regions involved in recognizing protein and sugar receptors, suggesting therefore that similar binding abilities could be displayed by the SARS-CoV-2 S1-NTD. Moreover, concerning the origin of these NTD insertions, our findings point towards an evolutionary acquisition rather than the hypothesis of an engineered virus.
1. Introduction
A novel coronavirus has emerged in human population in the city of Wuhan (China) causing severe respiratory illness that the World Health Organization named 2019 novel coronavirus disease (COVID-19) and the pathogen named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Huang et al., 2020; Li et al., 2020). Since its emergence in December 2019, the viral infection has already spread in many Chinese cities and several countries, and the World Health Organization has declared it a public health emergency of international concern by the end of January 2020. The situation report 114 of the WHO (May 13th, 2020) indicated that more than 4 million confirmed cases had been reported globally with nearly 32 % of the cases in the USA alone; the total deaths caused by the disease reached 287 399 cases, mainly in the Americas and Europe (37 % and 55 %, respectively) (https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200513-covid-19-sitrep-114.pdf?sfvrsn=17ebbbe_4).
Coronaviruses belong to the family Coronaviridae in the order Nidovirales and can be classified into four genera: Alpha-coronavirus, Beta-coronavirus, Gamma-coronavirus, and Delta-coronavirus (Cui et al., 2019; Perlman and Netland, 2009). They are, enveloped, positive-stranded RNA viruses, containing the largest genome among all RNA viruses, ranging from 27 to 32 kb (Fehr and Perlman, 2015). After the release of the SARS-CoV-2 genome sequence, it has been classified as Beta-coronavirus, closely related to the severe acute respiratory syndrome coronavirus (SARS-CoV) that emerged in 2002–2003 (Ksiazek et al., 2003; Peiris et al., 2003).
The coronaviruses spike protein (S) forms large protrusions from the virus surface (spikes) giving the viral particles the appearance of having crowns (hence their name coronavirus) (Cui et al., 2019; Perlman and Netland, 2009; Zumla et al., 2016). These spikes represent the first contact with the host and mediate the virus entry into host cells; besides, the S protein has been linked to host and tissue tropism (Du et al., 2009; Li, 2016). Structurally, the coronavirus spikes are clove-shaped trimers of the S protein with the asymmetric unit containing a large ectodomain, a single-pass transmembrane anchor, and a short intracellular tail (Kirchdoerfer et al., 2016; Smith et al., 2016). The ectodomain consists of a receptor-binding subunit S1 and a membrane-fusion subunit S2, with the S1 subunit containing distinct N-terminal and C-terminal domains (S1-NTD and S1-CTD) (Beniac et al., 2006; Walls et al., 2016).
One of the major complexities of coronaviruses is their receptor recognition pattern. To date, several receptors have been found to be recognized by different coronaviruses (Li, 2015, 2016). Among these receptors: zinc peptidases such as angiotensin-converting enzyme 2 (ACE2) (Hofmann et al., 2005; Li et al., 2003) and aminopeptidase N (APN) (Delmas et al., 1993; Li et al., 2007); dipeptidyl peptidase 4 (DPP4) (Raj et al., 2013; Yang et al., 2014); carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) (Dveksler et al., 1991; Williams et al., 1991); and sialic acid-containing receptors (Schwegmann-Wessels and Herrler, 2006). In this report, we compared the composition and the structural features of the S protein receptor-binding domains between the SARS-CoV-2 and related viruses (especially the SARS-CoV) and tried to extrapolate the findings to shed some light on possible functioning of this S protein of the new coronavirus, especially the receptor-binding domains.
2. Results and discussion
2.1. Primary sequence alignment
A total of 1652 sequences of S proteins of SARS-CoV-2 isolates from 29 countries were retrieved. The majority of the sequences were for the USA (1451) followed by China (66) (Supplementary file 1). The sequence identity to the reference sequence (YP_009724390.1) varied from 99.7 to 100%, indicating thus a high degree of conservation. Nearly 40 % of these sequences were identical to the reference sequence, while the other 60 % presented mostly single mutations (one mutated position in each sequence). The mutated positions (77 in total) are reported in Supplementary file 1. However, of these mutated positions, three were shared by several sequences: position 791 where Thr was mutated to Ile in 5 of the Taiwanese sequences; position 829 where Ala was mutated to Thr in 9 sequences from Thailand; and more importantly the apparition of Gly instead of Asp at position 614 in 923 sequences. it is to note that all these three positions are located in the S2 subunit.
The S protein of the SARS-CoV-2 shares 97.41 % amino acid similarity with the recently identified bat-CoV RatG13 isolate, 80.32 % with a bat SARS-like CoV and only 76.27 % identity with the SARS-CoV GZ02 isolate. Moreover, compared to the S1 subunit, the S2 subunit of the S proteins was found more conserved in the four strains (Fig. 1 and Table 1 ). Further, by comparing segments of 100aa of the SARS-CoV-2 S protein to the other three coronaviruses (Fig. 1a), the results indicated that the region spanning aa1-400 of the S protein was more similar to the new bat isolate (>90 %) than to the SARS- or the bat SARS-like strains. A more pronounced dissimilarity was noted at the regions spanning aa401-500 and aa601-700, which correspond to the C-terminal domain of the S1 subunit (S1-CTD).
Fig. 1.

a) The similarity of the S protein between the SARS-CoV-2 and SARS-CoV, bat SARS-like CoV and bat-nCoV as analyzed by 100aa segments alignment; b) The different regions identified in the similarity analysis were mapped on the SARS-CoV-2 S protein trimer predicted by the SWISS-model server: region aa1-400 is depicted in blue, region aa401-500 is depicted in red, region 501-600 is depicted in cyan, region aa601-700 is depicted in hot pink, and the C-terminal region starting from aa701 is depicted in yellow.
Table 1.
Amino acid sequence similarity of the SARS-CoV-2 S protein compared to SARS, bat SARS-like, and bat RatG13 coronaviruses.
| SARS-CoV (GZ02) | Bat SARS-like CoV | Bat-CoV (RatG13) | |
|---|---|---|---|
| Full length | 76.27 % | 80.32 % | 97.41 % |
| S1 subunit | 64.98 % | 68.74 % | 95.95 % |
| S1-CTD | 74.75 % | 64.14 % | 89.39 % |
| S1-NTD | 52.69 % | 66.77 % | 98.48 % |
| S2 subunit | 88.78 % | 92.90 % | 99.01 % |
2.2. Analysis of the S1-NTD receptor binding domain
The S1-NTD of the SARS-CoV-2 is highly similar to that of the newly isolated bat coronavirus RatG13 (>98 %) but shares roughly 53 % and 67 % with those of the SARS-CoV or bat SARS-like CoV, respectively (Table 1).
Structurally, we used the Dali server for aligning the S1-NTD of the SARS-CoV-2 with other coronaviruses from the different genera (Table 2 ). As expected the S1-NTD of the SARS-CoV-2 was more similar to those of the Beta-coronaviruses, especially the SARS-CoV (the highest Z-Score, highest sequence identity and the lowest RMSD). However, all the NTDs were aligned with Z-scores ranging from 6 to 22.1 and RMSDs ranging from 1.1 to 4.2. This similarity is due to the Galectin-like topology of the NTDs' core structures as previously documented (Li, 2012).
Table 2.
Structural alignment of the S1-NTDs of different coronaviruses.
| α genus | β genus |
δ genus | γ genus | |||||
|---|---|---|---|---|---|---|---|---|
| SARS-CoV-2 | NL63-CoV | MERS-CoV | SARS-CoV | MHV | Pd-CoV | IBV | hGALECTIN | |
| Z-score | ||||||||
| SARS-CoV-2 | 105.2 | 6.7 | 13.4 | 21.9 | 14.7 | 7.3 | 8.6 | 6 |
| NL63-CoV | 6.7 | 42.8 | 8.6 | 9.5 | 8.8 | 22.1 | 11.3 | 7 |
| MERS-CoV | 13.4 | 8.6 | 59.4 | 18.9 | 20.1 | 9 | 10.3 | 6.4 |
| SARS-CoV | 21.9 | 9.5 | 18.9 | 47.8 | 21 | 10.7 | 11.8 | 9 |
| MHV | 14.7 | 8.8 | 20.1 | 21 | 50.7 | 10 | 12.1 | 7 |
| Pd-CoV | 7.3 | 22.1 | 9 | 10.7 | 10 | 41.8 | 12.6 | 8.2 |
| IBV | 8.6 | 11.3 | 10.3 | 11.8 | 12.1 | 12.6 | 39.4 | 7.6 |
| hGALECTIN | 6 | 7 | 6.4 | 9 | 7 | 8.2 | 7.6 | 30.8 |
| RMSD (Angstrom, Å) | ||||||||
| SARS-CoV-2 | 0 | 4 | 2.7 | 1.1 | 2.2 | 3.4 | 3.1 | 2.6 |
| NL63-CoV | 4 | 0 | 4.2 | 3.8 | 4.2 | 2.1 | 3.4 | 3.3 |
| MERS-CoV | 2.7 | 4.2 | 0 | 3 | 3 | 4 | 3.5 | 3.1 |
| SARS-CoV | 1.1 | 3.8 | 3 | 0 | 2.6 | 3.4 | 3.3 | 2.4 |
| MHV | 2.2 | 4.2 | 3 | 2.6 | 0 | 3.7 | 3.4 | 2.8 |
| Pd-CoV | 3.4 | 2.1 | 4 | 3.4 | 3.7 | 0 | 3 | 2.7 |
| IBV | 3.1 | 3.4 | 3.5 | 3.3 | 3.4 | 3 | 0 | 3.3 |
| hGALECTIN | 2.6 | 3.3 | 3.1 | 2.4 | 2.8 | 2.7 | 3.3 | 0 |
| Sequence identity (%) | ||||||||
| SARS-CoV-2 | 100 | 13 | 23 | 63 | 23 | 11 | 11 | 12 |
| NL63-CoV | 13 | 100 | 9 | 14 | 12 | 28 | 11 | 9 |
| MERS-CoV | 23 | 9 | 100 | 21 | 18 | 10 | 11 | 7 |
| SARS-CoV | 63 | 14 | 21 | 100 | 20 | 12 | 13 | 10 |
| MHV | 23 | 12 | 18 | 20 | 100 | 11 | 14 | 8 |
| Pd-CoV | 11 | 28 | 10 | 12 | 11 | 100 | 16 | 9 |
| IBV | 11 | 11 | 11 | 13 | 14 | 16 | 100 | 3 |
| hGALECTIN | 12 | 9 | 7 | 10 | 8 | 9 | 3 | 100 |
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2 (PDB ID : 6vyb); NL63-CoV: NL63 respiratory coronavirus (PDB ID: 5szs); MERS-CoV: middle-east respiratory syndrome coronavirus (PDB ID: 5x5f); SARS-CoV: severe acute respiratory syndrome coronavirus (PDB ID: 5 × 58); MHV: mouse hepatitis coronavirus (PDB ID: 3jcl); Pd-CoV: porcine delta coronavirus (PDB ID: 6b7n); IBV: infectious bronchitis coronavirus (PDB ID: 6cv0); hGALECTIN: human galectin-3 (PDB ID: 1a3k).
The primary sequence alignment also revealed some insertions shared by the SARS-CoV-2 and bat coronavirus RatG13 but not the SARS-CoV, located at positions aa72-82, aa144-147, aa244-246 and aa255-257 of the SARS-CoV-2 S protein (Fig. 2 ).
Fig. 2.

a) Alignment of the primary sequences the S1-NTDs of SARS-CoV-2, SARS-CoV and Bat-CoV RatG13 isolate. b) A cartoon presentation of 3D structure model of the SARS-CoV-2 S1-NTD with the four inserts identified in (a) shown as sticks: insert 1 colored in red, insert 2 colored in magenta, insert 3 colored in green and insert 4 colored in blue. c) A cartoon representation of the SARS-CoV-2 spike trimer showing the orientation of the S1-NTD towards the outside environment; the different inserts shown as sticks and colored as described in (b).
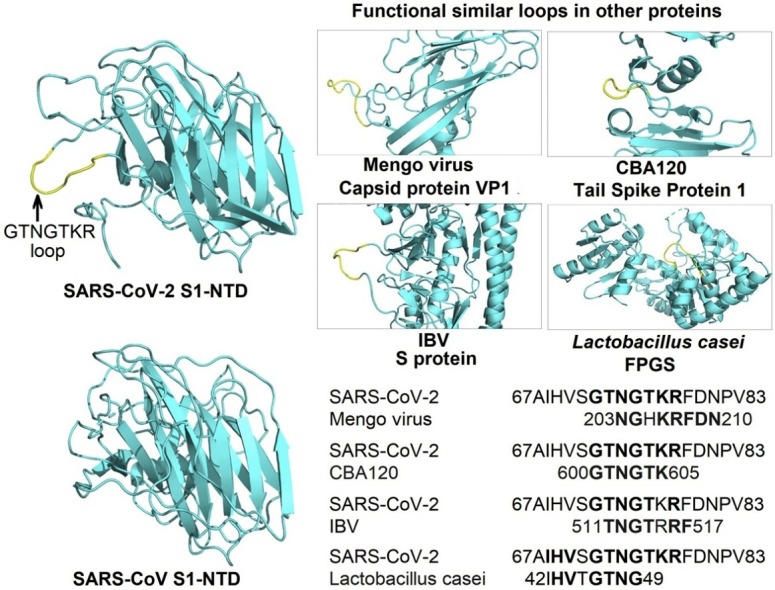
To further investigate the structural role of these inserts, we searched the Protein Data Bank using the largest insert 72GTNGTKR78 with 5 amino acids extensions on both N- and C-terminal sides, leading to 17aa long segment 67AIHVSGTNGTKRFDNPV83; then we analyzed the hits to see whether the aligned motifs were engaged in any identified structural function (Table 3 ).
Table 3.
Other proteins containing similar motifs than the 72GTNGTKR78 insert identified in the SARS-CoV-2 S1-NTD.
| Origin | Protein | Motif | PDB ID |
|---|---|---|---|
| SARS-CoV-2 | S protein | 67AIHVSGTNGTKRFDNPV83 | |
| Mengo virus | VP1 | 203NGHKRFDN210 | 2MEV |
| MHV | S protein | 168NTNGNK173 | 3R4D |
| CBA120 | Tail spike protein | 600GTNGTK605 | 4OJ6 |
| IBV | S protein | 511TNGTRRF517 | 6CV0 |
| Mimivirus | Cyclophilin | 221NGTKRF226 | 2OSE |
| Lactobacillus casei | Folylpolyglutamate synthetase | 42IHVTGTNG49 | 1FGS |
2.3. The GTNGTKR motif in binding protein receptors
When we searched the protein databank using the GTNGTKR motif, the first hit was the structure of the Mengo virus VP1 protein (Fig. 3 a). The aligned segment was located on the VP1 GH loop, which forms along with the VP3 C-terminal loop a depression on the capsid that has been associated with receptor recognition and binding (Kim et al., 1990; Krishnaswamy and Rossmann, 1990). Although the depression described in the Mengo virus capsid is absent in the S1-NTD of the SARS-CoV-2, the target motif 72GTNGTKRFDN81 forms a similarly exposed loop with two neighboring loops containing the 255SSG257 motif (the identified insert 4) and the N-terminal loop (18LTT20 motif) on both sides (Fig. 3b). Whether this formation could play the same role as the Mengo virus VP1 and VP3 loops, which would, in turn, allow the SARS-CoV-2 to interact with the same receptor, need further investigation. Moreover, the Mengo virus has been found to bind the murine cellular receptor vascular cell adhesion molecule 1 (VCAM-1) to enter and infect cells (Huber, 1994). This receptor molecule is restricted to endothelial cells and is subject to upregulation under cytokines stimulation (Hosokawa et al., 2006; Singh et al., 2005). Given the high cytokine amounts stimulated by the SARS-CoV-2 (Huang et al., 2020) and pre-existing heart disease (hypertension and coronary heart disease) being one of the major co-morbidities of the fatality cases (Deng and Peng, 2020), it is interesting to explore the possibility of the SARS-CoV-2 binding the VCAM-1 receptor via its S1-NTD.
Fig. 3.

a) A cartoon representation of the Mengo virus capsid protein VP1, VP2 and VP3 with the adjacent active loops colored in red for the VP1 GH-loop harboring the 203NGHKRFDN210 motif, and in yellow for the VP3 loop (PDB ID: 2MEV). b) Cartoon representation of the SARS-CoV-2 S1-NTD with the 72GTNGTKRFDN82 loop colored in red showing a quiet similar orientation than the GH-loop of the Mengo virus VP1 (left); with two adjacent loops formed by the insert 4 (254SSSG257) and the first N-terminal amino acids (right). c) A surface representation of the MHV S1-NTD -mCEACAM1a complex (PDB ID: 3R4D) viewed from the top, with the receptor colored in yellow (70 % transparency) and the MHV S1-NTD colored in cyan; the residues in the MHV S1-NTD interacting with the mCEACAM1a are colored in blue and the 168NTNGNK173 is colored in red. d) A top view of the surface of the SARS-CoV-2 S1-NTD (cyan) with all the protruding residues colored in yellow. The N-terminal aa15-20 segment of both NTDs showing different conformations are circled (c and d).
The mouse hepatitis coronavirus (MHV) also binds another cell adhesion molecule, the murine carcinoembryonic antigen-related cell adhesion molecule 1a (mCEACAM1a), using its S1-NTD (Peng et al., 2011). Therefore, as a next step, we compared the MHV and SARS-CoV-2 S1-NTDs. We found that the receptor-binding motif of the MHV S1-NTD also presents a motif 168NTNGNK173 with some similarity to insert 1 (72GTNGTKR78) of the SARS-CoV-2 S1-NTD. However, when the S1-NTDs were compared in the quaternary structures of the S proteins, the above motifs seem to occupy opposite positions (Fig. 3c and d). Besides, Peng et al. identified four receptor binding motifs in the MHV S1-NTD (RBM1-4) (Peng et al., 2011). By comparing the MHV-receptor interaction interface and the exposed amino acids on the receptor-binding surface of the SARS-CoV-2 S1-NTD, we found that the N-terminal aa15-21 segments adopt different conformations (Fig. 3d), and this segment in the MHV (RBM1) contains three residues critical for receptor binding affinity (Peng et al., 2011). Therefore, it seems unlikely that the SARS-CoV-2 would bind the same receptor. However, this observation should be taken with care since it is based on the predicted model of the SARS-CoV-2 S1-NTD.
Taken all together, the presence of the GTNGTKR motif in the SARS-CoV-2 S1-NTD seems to be a potentially evolutionary feature that SARS-CoV-2 acquired to allow its S1-NTD to bind to protein receptors. We believe that the above observations are worth investigating.
2.4. The GTNGTKR motif in binding sugar receptors
Another structure containing the analyzed motif was the tail spike protein 1 of the bacteriophage CBA120 (Podoviridae) (Chen et al., 2014). The aligned motif GTNGTK was located within the receptor-binding domain, in the inverting region connecting the subdomain D3 and D4. Interestingly, unlike other tail spike proteins where the sugar-binding sites were located on the D3 subdomain (Barbirz et al., 2008; Muller et al., 2008; Steinbacher et al., 1996; Xiang et al., 2009), the D3-D4 inverting region of the CBA120 tail spike protein generates a hole that forms the sugar-binding site (Chen et al., 2014). Although the target motif was not directly involved in the sugar’s interactions, the binding site (hole) is formed in the opposite direction of the GTNGTK loop, and a quite similar orientation of the motif is observed in the SARS-CoV-2 S1-NTD (Fig. 4 ). Besides, what could be the counterparts of the sugar-binding pocket of the CBA120 tail spike protein is one of the two pockets formed in the SARS-CoV-2 S1-NTD: the first situated on the top part of the domain and the other located above the β-sandwich core in the opposite direction of the GTNGTKR loop (Fig. 4c). This latter pocket is also aligned with the sugar-binding site in the NTD of bovine coronavirus (BCoV) (Fig. 5 a and c). Peng et al. (2012) reported that the pocket above the β-sandwich core is the sugar-binding site in BCoV NTD and through mutagenesis studies, they identified 4 residues critical for the NTD-receptor interaction Y162, E182, W184, and H185 and the binding was stabilized by the loop 10–11 (146NDLNKL151) (Fig. 5c). Interestingly, the corresponding pocket in the SARS-CoV-2 NTD also contains three amino acids (E154, F157, and Y160) with the same orientation than the four key residues identified in the BCoV NTD. Moreover, the positions of E154 and Y160 are strikingly similar to that of Y162 and E182 in BCoV NTD (Fig. 5d). Besides, a counterpart of the stabilizing loop 10–11 is also present in the SARS-CoV-2 NTD, although shorter, but seems to share the NxxN motif.
Fig. 4.

a) A cartoon representation of the tail spike protein 1 trimer of the bacteriophage CBA120 with chains A, B and C colored in cyan, blue and magenta, respectively (PDB ID: 4OJ6). b) a close-up on the D3-D4 inverting region of the receptor-binding domain of the tail spike protein 1 showing the 600GTNGTK605 loop in yellow and the sugar biding hole in the opposite side of the loop. c) A cartoon representation of the SARS-CoV-2 S1-NTD with the 72GTNGTKRFDN82 loop colored in red showing a similar orientation than the 600GTNGTK605 loop in (b) and the two possible sugar-binding pockets are shown in dotted circles.
Fig. 5.

a) A representation of the bovine coronavirus (BCoV) sugar-binding pocket with the sugar moiety shown as colored sticks and the critical loop 10-11 colored in red (PDB ID: 4H14). b) The SARS-CoV-2 S1-NTD in the same orientation as the BCoV S1-NTD in (a), with the 72GTNGTKR78 in the far side colored in red and the counterpart of the loop 10-11 with a conserved NxxN motif in the front (also colored in red). c and d) different orientations of the BCoV and SARS-CoV-2 S1-NTDs obtained by a 70° rotation of the x-axis of (a) and (b) respectively; the key residues for the interactions with sugar moieties in the BCoV S1-NTD and their possible counterparts in the SARS-CoV-2 S1-NTD are shown as red sticks in (c) and (d), respectively.
These observations suggest that SARS-CoV-2 NTD might recognize a sugar receptor as well, and it is likely to be the same Neu5,9Ac2 that BCoV NTD binds to (Peng et al., 2012).
2.5. The GTNGTKR motif in other structural functions
The TNGTRRF motif was also present in the infectious bronchitis coronavirus (IBV) spike protein. Despite the low primary sequence identity (11 %), the pairwise structural alignment revealed that the SARS-CoV-2 S1-NTD shared a relatively high structural similarity with the S1-NTD of the IBV with a Dali Z-score of 8.6 and RMSD of 3.1 over 159 aligned residues (Table 2). The aligned motif TNGTRRF was located within the S1-CTD of the IBV, in the subdomain connecting S1 and S2 (Shang et al., 2018). Although no functional features have been described for the subdomains of the S1-NTDs and S1-CTDs in the coronaviruses spikes, the target motif was found protruding from the surface of the trimer (Fig. 6 a), suggesting that such protrusion might interact with the surrounding environment.
Fig. 6.

a) Surface representation of the S protein trimers of IBV (PDB ID: 6CV0), MERS-CoV (PDB ID: 5 × 5F), SARS-CoV (PDB ID: 5 × 58), and MHV (PDB ID: 3JCL); chains A, B and C are colored in cyan, blue and magenta, respectively; the protruding 511TNGTRRF517 motif in the IBV trimer is colored in yellow in chains A and B. b) The folylpolyglutamate synthetase (FPGS) of Lactobacillus casei (PDB ID: 1FGS), with the putative nucleotide-binding P loop, harboring the 42IHVTGTNG49 motif, is shown in red, the Ω loop is shown in yellow, and the ligand is shown as magenta spheres. c) A top view of the SARS-CoV-2 S1-NTD showing the adjacent 72GTNGTKR78 (insert 1) and 254SSSG257 (insert 4) loops in red and yellow respectively. d) Structural alignment of the IHVxGTNG loops of SARS-CoV-2 S1-NTD (blue) and FPGS (yellow).
The aligned fragment was also located at the C-terminal of the Mimivirus cyclophilin but it was missing from the deposited structure (Thai et al., 2008). Further, the authors did not link any structural function of the segment of interest. However, it is to note that this is the first virus-encoded cyclophilin but it lacks peptidyl-prolyl isomerase, an activity that several viruses such human immunodeficiency virus type 1 and SARS-CoV exploit the host cyclophilin for (Chen et al., 2005; Sorin and Kalpana, 2006). Interestingly, the viral cyclophilin was located on the surface of mature Mimivirus virions, and given the absence of the catalytic activity, the authors suggested that the protein may play a structural role yet to be identified in the Mimivirus life cycle (Thai et al., 2008). Since the Mimivirus can cause pneumonia in humans (La Scola et al., 2005; Saadi et al., 2013), and the exact position of the cyclophilin is yet to be determined, a question could be asked whether the 221NGTKRF226 motif could play a role in the virus pathogenesis that could be shared by the SARS-CoV-2.
Besides viruses, the search for a functional GTNGTKR motif in the deposited protein structures revealed a similar motif in the folylpolyglutamate synthetase (FPGS) of Lactobacillus casei, an enzyme that catalyzes the MgATP-dependent glutamylation of folate coenzymes (Fig. 6b). The aligned motif 42IHVTGTNG49 was located on the putative nucleotide-binding P loop (GTNGKGS) that resembles the consensus P-loop sequence found in many other adenylate and uridylate kinase (Smith and Rayment, 1996; Sun et al., 1998). Moreover, a Ω loop near the P loop binding site was also suggested to play a role in the activity of the FPGS, especially the Serine residue. Interestingly, a similarly shaped loop is also found in the SARS-CoV-2 S1-NTD adjacent to the GTNGTKR loop, formed by the insert 4 (254SSSG257) also rich in serine residues (Fig. 6c and d).
3. Conclusions
Given the severity and the widespread nature of the infection, it is safe to assume that the SARS-CoV-2 has a more efficient way to penetrate and infect cells. Besides, based on the comparison of the SARS-CoV-2 and SARS-CoV receptor-binding domains, it seems that SARS-CoV-2 evolved in a way that allowed it to maintain and enhance the binding of the ACE2 receptor via its S1-CTD, but also acquired a different S1-NTD that according to our analysis might bind other receptors (protein or sugar receptor). More precisely, the acquisition of the GTNGTKR motif, found at the active sites of structural and nonstructural proteins of other viruses and organisms, might allow the SARS-CoV-2 to recognize other receptors/co-receptors besides the ACE2.
Moreover, our results suggest that the apparition of the GTNGTKR motif points more toward an evolutionary trait of the SARS-CoV-2 rather than the hypothesis of an engineered virus; Under functional constraints, proteins tend to evolve in a way that their tertiary structures could perform the needed functions regardless of the changes in their primary sequences (Goldstein, 2008; Siltberg-Liberles et al., 2011; Worth et al., 2009) and the two main factors driving the evolution of the S proteins are the need for better adaptation to the host receptors and the need to evade the immune system of the host to ensure better infectivity (Li, 2015, 2016). Therefore, it is more plausible to assume that the SARS-CoV-2 acquired the GTNGTKR motif during its evolutionary parkour under functional constraints. As for the exact mechanism of acquisition and the origin of this motif, we believe that further investigations are needed not only in the context of the SARS-CoV-2 infection but as a pertinent motif for viral proteins activity in general.
4. Material and methods
4.1. Sequences retrieval and alignment
A total of 1652 SARS-CoV-2 S protein complete sequences available at the NCBI Virus portal were retrieved. The sequences of SARS-CoV GC02 isolate (AY390556) and two bat isolates: a bat SARS-like coronavirus (MG772934) and the recently isolated RatG13 bat coronavirus (MN996532) were also retrieved, and their S glycoproteins were compared to that of the SARS-CoV-2 (RefSeq: YP_009724390.1).
First, we performed a multiple alignment of the S proteins of the 1652 SARS-CoV-2 strains to see if any dissimilarities were present and analyzed the occurrence of mutations in comparison to the reference sequence. Next, we compared the similarity of the S glycoprotein of the SARS-CoV-2 (RefSeq: YP_009724390.1) to that of the selected 4 related coronaviruses strains mentioned above: 1) aligning the full-length proteins of the 4 stains altogether; 2) aligning the full-length SARS-CoV-2 S protein to that of each of the related strains separately; 3) aligning portions (100aa windows) of the SARS-CoV-2 S protein by to that of each of the related strains separately.
All sequence alignments were performed using the Muscle algorithm implemented in the MEGA-X software or BLASTp suite of the U.S. National Library of Medicine.
For the search of motifs similar to the GTNGTKR motif in the Protein Data Bank deposited structures, the BLASTp suite of the U.S. National Library of Medicine was used by adjusting parameters to search for a short input sequence.
4.2. 3D structure models
Three crystal structures of the SARS-CoV-2 Spike protein (containing the S1-NTD) were retrieved from the Protein Data Bank (PDB ID: 6vyb, 6vxx, and 6vsb). Since all of these structures lacks some fragments of interest (especially the GTNGTKR motif), the sequence of S glycoprotein of the SARS-CoV-2 (Reference ID: YP_009724390.1) was submitted to I-Tasser (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) and Swiss-Model (https://swissmodel.expasy.org/) servers for the prediction of complete 3D structure models (Waterhouse et al., 2018; Yang and Zhang, 2015). The quality of the predicted 3D structures was evaluated using the MolProbity server (http://molprobity.biochem.duke.edu) (Williams et al., 2018) and the best models were selected for the analysis.
4.3. Structural alignment and analysis
All structural alignments were performed using the Dali server (http://ekhidna2.biocenter.helsinki.fi/dali/) (Holm, 2020). The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC was used for 3D structure visualization and analysis, and the preparation of all the figures containing 3D structures.
CRediT authorship contribution statement
Nouredine Behloul: Conceptualization, Methodology, Investigation, Formal analysis, Writing - original draft, Writing - review & editing. Sarra Baha: Methodology, Data curation, Formal analysis, Writing - review & editing. Ruihua Shi: Conceptualization, Supervision, Writing - review & editing. Jihong Meng: Conceptualization, Methodology, Resources, Supervision, Writing - review & editing.
Declaration of Competing Interest
The authors have no competing interests to declare.
Footnotes
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.virusres.2020.198058.
Contributor Information
Ruihua Shi, Email: ruihuashi@126.com.
Jihong Meng, Email: jihongmeng@163.com.
Appendix A. Supplementary data
The following is Supplementary data to this article:
References
- Barbirz S., Muller J.J., Uetrecht C., Clark A.J., Heinemann U., Seckler R. Crystal structure of Escherichia coli phage HK620 tailspike: podoviral tailspike endoglycosidase modules are evolutionarily related. Mol. Microbiol. 2008;69(2):303–316. doi: 10.1111/j.1365-2958.2008.06311.x. [DOI] [PubMed] [Google Scholar]
- Beniac D.R., Andonov A., Grudeski E., Booth T.F. Architecture of the SARS coronavirus prefusion spike. Nat. Struct. Mol. Biol. 2006;13(8):751–752. doi: 10.1038/nsmb1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Mi L., Xu J., Yu J., Wang X., Jiang J., Xing J., Shang P., Qian A., Li Y., Shaw P.X., Wang J., Duan S., Ding J., Fan C., Zhang Y., Yang Y., Yu X., Feng Q., Li B., Yao X., Zhang Z., Li L., Xue X., Zhu P. Function of HAb18G/CD147 in invasion of host cells by severe acute respiratory syndrome coronavirus. J. Infect. Dis. 2005;191(5):755–760. doi: 10.1086/427811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Bales P., Greenfield J., Heselpoth R.D., Nelson D.C., Herzberg O. Crystal structure of ORF210 from E. coli O157:H1 phage CBA120 (TSP1), a putative tailspike protein. PLoS One. 2014;9(3) doi: 10.1371/journal.pone.0093156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J., Li F., Shi Z.L. Origin and evolution of pathogenic coronaviruses. Nature reviews. Microbiology. 2019;17(3):181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas B., Gelfi J., Sjostrom H., Noren O., Laude H. Further characterization of aminopeptidase-N as a receptor for coronaviruses. Adv. Exp. Med. Biol. 1993;342:293–298. doi: 10.1007/978-1-4615-2996-5_45. [DOI] [PubMed] [Google Scholar]
- Deng S.Q., Peng H.J. Characteristics of and public health responses to the coronavirus disease 2019 outbreak in China. J. Clin. Med. 2020;9(2) doi: 10.3390/jcm9020575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L., He Y., Zhou Y., Liu S., Zheng B.J., Jiang S. The spike protein of SARS-CoV--a target for vaccine and therapeutic development. Nature reviews. Microbiology. 2009;7(3):226–236. doi: 10.1038/nrmicro2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dveksler G.S., Pensiero M.N., Cardellichio C.B., Williams R.K., Jiang G.S., Holmes K.V., Dieffenbach C.W. Cloning of the mouse hepatitis virus (MHV) receptor: expression in human and hamster cell lines confers susceptibility to MHV. J. Virol. 1991;65(12):6881–6891. doi: 10.1128/jvi.65.12.6881-6891.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr A.R., Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol. 2015;1282:1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein R.A. The structure of protein evolution and the evolution of protein structure. Curr. Opin. Struct. Biol. 2008;18(2):170–177. doi: 10.1016/j.sbi.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Hofmann H., Pyrc K., van der Hoek L., Geier M., Berkhout B., Pohlmann S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. U. S. A. 2005;102(22):7988–7993. doi: 10.1073/pnas.0409465102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L. Using Dali for protein structure comparison. Methods Mol. Biol. 2020;2112:29–42. doi: 10.1007/978-1-0716-0270-6_3. [DOI] [PubMed] [Google Scholar]
- Hosokawa Y., Hosokawa I., Ozaki K., Nakae H., Matsuo T. Cytokines differentially regulate ICAM-1 and VCAM-1 expression on human gingival fibroblasts. Clin. Exp. Immunol. 2006;144(3):494–502. doi: 10.1111/j.1365-2249.2006.03064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber S.A. VCAM-1 is a receptor for encephalomyocarditis virus on murine vascular endothelial cells. J. Virol. 1994;68(6):3453–3458. doi: 10.1128/jvi.68.6.3453-3458.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Boege U., Krishnaswamy S., Minor I., Smith T.J., Luo M., Scraba D.G., Rossmann M.G. Conformational variability of a picornavirus capsid: pH-dependent structural changes of Mengo virus related to its host receptor attachment site and disassembly. Virology. 1990;175(1):176–190. doi: 10.1016/0042-6822(90)90198-z. [DOI] [PubMed] [Google Scholar]
- Kirchdoerfer R.N., Cottrell C.A., Wang N., Pallesen J., Yassine H.M., Turner H.L., Corbett K.S., Graham B.S., McLellan J.S., Ward A.B. Pre-fusion structure of a human coronavirus spike protein. Nature. 2016;531(7592):118–121. doi: 10.1038/nature17200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswamy S., Rossmann M.G. Structural refinement and analysis of Mengo virus. J. Mol. Biol. 1990;211(4):803–844. doi: 10.1016/0022-2836(90)90077-Y. [DOI] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J., Group S.W. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348(20):1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- La Scola B., Marrie T.J., Auffray J.P., Raoult D. Mimivirus in pneumonia patients. Emerging Infect. Dis. 2005;11(3):449–452. doi: 10.3201/eid1103.040538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Evidence for a common evolutionary origin of coronavirus spike protein receptor-binding subunits. J. Virol. 2012;86(5):2856–2858. doi: 10.1128/JVI.06882-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Receptor recognition mechanisms of coronaviruses: a decade of structural studies. J. Virol. 2015;89(4):1954–1964. doi: 10.1128/JVI.02615-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016;3(1):237–261. doi: 10.1146/annurev-virology-110615-042301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.X., Ge J.W., Li Y.J. Porcine aminopeptidase N is a functional receptor for the PEDV coronavirus. Virology. 2007;365(1):166–172. doi: 10.1016/j.virol.2007.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Guan X., Wu P., Wang X., Zhou L., Tong Y., Ren R., Leung K.S.M., Lau E.H.Y., Wong J.Y., Xing X., Xiang N., Wu Y., Li C., Chen Q., Li D., Liu T., Zhao J., Li M., Tu W., Chen C., Jin L., Yang R., Wang Q., Zhou S., Wang R., Liu H., Luo Y., Liu Y., Shao G., Li H., Tao Z., Yang Y., Deng Z., Liu B., Ma Z., Zhang Y., Shi G., Lam T.T.Y., Wu J.T.K., Gao G.F., Cowling B.J., Yang B., Leung G.M., Feng Z. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 2020;382(13):1199–1207. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller J.J., Barbirz S., Heinle K., Freiberg A., Seckler R., Heinemann U. An intersubunit active site between supercoiled parallel beta helices in the trimeric tailspike endorhamnosidase of Shigella flexneri Phage Sf6. Structure. 2008;16(5):766–775. doi: 10.1016/j.str.2008.01.019. [DOI] [PubMed] [Google Scholar]
- Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y., group, S.s Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361(9366):1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G., Sun D., Rajashankar K.R., Qian Z., Holmes K.V., Li F. Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc. Natl. Acad. Sci. U. S. A. 2011;108(26):10696–10701. doi: 10.1073/pnas.1104306108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G., Xu L., Lin Y.L., Chen L., Pasquarella J.R., Holmes K.V., Li F. Crystal structure of bovine coronavirus spike protein lectin domain. J. Biol. Chem. 2012;287(50):41931–41938. doi: 10.1074/jbc.M112.418210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S., Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nature reviews. Microbiology. 2009;7(6):439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj V.S., Mou H., Smits S.L., Dekkers D.H., Muller M.A., Dijkman R., Muth D., Demmers J.A., Zaki A., Fouchier R.A., Thiel V., Drosten C., Rottier P.J., Osterhaus A.D., Bosch B.J., Haagmans B.L. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495(7440):251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadi H., Pagnier I., Colson P., Cherif J.K., Beji M., Boughalmi M., Azza S., Armstrong N., Robert C., Fournous G., La Scola B., Raoult D. First isolation of Mimivirus in a patient with pneumonia. Clin. Infect. Dis. 2013;57(4):e127–134. doi: 10.1093/cid/cit354. [DOI] [PubMed] [Google Scholar]
- Schwegmann-Wessels C., Herrler G. Sialic acids as receptor determinants for coronaviruses. Glycoconj. J. 2006;23(1-2):51–58. doi: 10.1007/s10719-006-5437-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J., Zheng Y., Yang Y., Liu C., Geng Q., Luo C., Zhang W., Li F. Cryo-EM structure of infectious bronchitis coronavirus spike protein reveals structural and functional evolution of coronavirus spike proteins. PLoS Pathog. 2018;14(4) doi: 10.1371/journal.ppat.1007009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siltberg-Liberles J., Grahnen J.A., Liberles D.A. The evolution of protein structures and structural ensembles under functional constraint. Genes. 2011;2(4):748–762. doi: 10.3390/genes2040748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.J., Mason J.C., Lidington E.A., Edwards D.R., Nuttall R.K., Khokha R., Knauper V., Murphy G., Gavrilovic J. Cytokine stimulated vascular cell adhesion molecule-1 (VCAM-1) ectodomain release is regulated by TIMP-3. Cardiovasc. Res. 2005;67(1):39–49. doi: 10.1016/j.cardiores.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Smith C.A., Rayment I. Active site comparisons highlight structural similarities between myosin and other P-loop proteins. Biophys. J. 1996;70(4):1590–1602. doi: 10.1016/S0006-3495(96)79745-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.B., Simmonds P., Izopet J., Oliveira-Filho E.F., Ulrich R.G., Johne R., Koenig M., Jameel S., Harrison T.J., Meng X.J., Okamoto H., Van der Poel W.H.M., Purdy M.A. Proposed reference sequences for hepatitis E virus subtypes. J. Gen. Virol. 2016;97(3):537–542. doi: 10.1099/jgv.0.000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorin M., Kalpana G.V. Dynamics of virus-host interplay in HIV-1 replication. Curr. HIV Res. 2006;4(2):117–130. doi: 10.2174/157016206776055048. [DOI] [PubMed] [Google Scholar]
- Steinbacher S., Baxa U., Miller S., Weintraub A., Seckler R., Huber R. Crystal structure of phage P22 tailspike protein complexed with Salmonella sp. O-antigen receptors. Proc. Natl. Acad. Sci. U. S. A. 1996;93(20):10584–10588. doi: 10.1073/pnas.93.20.10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X., Bognar A.L., Baker E.N., Smith C.A. Structural homologies with ATP- and folate-binding enzymes in the crystal structure of folylpolyglutamate synthetase. Proc. Natl. Acad. Sci. U. S. A. 1998;95(12):6647–6652. doi: 10.1073/pnas.95.12.6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai V., Renesto P., Fowler C.A., Brown D.J., Davis T., Gu W., Pollock D.D., Kern D., Raoult D., Eisenmesser E.Z. Structural, biochemical, and in vivo characterization of the first virally encoded cyclophilin from the Mimivirus. J. Mol. Biol. 2008;378(1):71–86. doi: 10.1016/j.jmb.2007.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Tortorici M.A., Bosch B.J., Frenz B., Rottier P.J.M., DiMaio F., Rey F.A., Veesler D. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature. 2016;531(7592):114–117. doi: 10.1038/nature16988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L., Lepore R., Schwede T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.K., Jiang G.S., Holmes K.V. Receptor for mouse hepatitis virus is a member of the carcinoembryonic antigen family of glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 1991;88(13):5533–5536. doi: 10.1073/pnas.88.13.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B., Jain S., Lewis S.M., Arendall W.B., 3rd, Snoeyink J., Adams P.D., Lovell S.C., Richardson J.S., Richardson D.C. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018;27(1):293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worth C.L., Gong S., Blundell T.L. Structural and functional constraints in the evolution of protein families. Nature reviews. Mol. Cell Biol. 2009;10(10):709–720. doi: 10.1038/nrm2762. [DOI] [PubMed] [Google Scholar]
- Xiang Y., Leiman P.G., Li L., Grimes S., Anderson D.L., Rossmann M.G. Crystallographic insights into the autocatalytic assembly mechanism of a bacteriophage tail spike. Mol. Cell. 2009;34(3):375–386. doi: 10.1016/j.molcel.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Du L., Liu C., Wang L., Ma C., Tang J., Baric R.S., Jiang S., Li F. Receptor usage and cell entry of bat coronavirus HKU4 provide insight into bat-to-human transmission of MERS coronavirus. Proc. Natl. Acad. Sci. U. S. A. 2014;111(34):12516–12521. doi: 10.1073/pnas.1405889111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015;43(W1):W174–181. doi: 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A., Chan J.F., Azhar E.I., Hui D.S., Yuen K.Y. Coronaviruses - drug discovery and therapeutic options. Nature reviews. Drug Discov. 2016;15(5):327–347. doi: 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.