

Abstract
Spatiotemporal protein reorganization at DNA damage sites induced by genotoxic chemotherapies is crucial for DNA damage response (DDR), which influences treatment response by directing cancer cell fate. This process is orchestrated by valosin-containing protein (VCP), an AAA+ ATPase that extracts polyubiquinated chromatin proteins and facilitates their turnover. However, because of the essential and pleiotropic effects of VCP in global proteostasis, it remains challenging practically to understand and target its DDR-specific functions. We describe a DNA-damage-induced phosphorylation event (Ser784), which selectively enhances chromatin-associated protein degradation mediated by VCP and is required for DNA repair, signaling, and cell survival. These functional effects of Ser784 phosphorylation on DDR correlate with a decrease in VCP association with chromatin, cofactors NPL4/UFD1, and polyubiquitinated substrates. Clinically, high phospho-Ser784-VCP levels are significantly associated with poor outcome among chemotherapy-treated breast cancer patients. Thus, Ser784 phosphorylation is a DDR-specific enhancer of VCP function and a potential predictive biomarker for chemotherapy treatments.
Keywords: VCP, phosphorylation, K48-linked polyubiquitin, chromatin-associated degradation, nucleus, DNA damage response, chemotherapy, cancer, biomarker, proteostasis
Graphical Abstract
Zhu et al. show that DNA-damage-induced, PIKK-mediated Ser784 phosphorylation is a specific enhancer of VCP function in chromatin-associated protein degradation. Phospho-Ser784-VCP is required for DNA repair, checkpoint signaling, and cell survival in response to a broad range of genotoxins and correlates with poor outcome among chemotherapy-treated breast cancer patients.
Introduction
Many anticancer chemotherapies are genotoxic and trigger DNA-damage-induced apoptosis. Unfortunately, their effects vary among patients, and our ability to both predict and improve therapeutic response remains limited. This is mainly due to the complex nature of the DNA damage response (DDR), an evolutionarily conserved mechanism involving extensive protein networks collectively serving to repair damaged DNA and to determine cell fate. Nevertheless, mounting evidence suggests that inherent DDR deficits and the resultant genome instability are an “Achilles’ heel” of cancer, which could be effectively targeted (Lord and Ashworth, 2012, O’Connor, 2015). The best modern example is the clinical success of treating cancers harboring defects in homologous recombination (the most faithful repair mechanism for DNA double-strand breaks [DSBs]) with inhibitors of PARP (a key repair enzyme for DNA single-strand breaks [SSBs]) (Bryant et al., 2005, Farmer et al., 2005). Thus, identifying cancer-specific DDR defects and understanding their mechanisms can guide effective therapeutic exploitation.
All DNA breaks trigger global changes in protein post-translational modifications (PTMs) at and near damage sites. These PTM events are tightly coordinated and together orchestrate a rapid and orderly recruitment of DNA repair factors and signaling molecules to DNA-damage sites to ensure the successful execution of all functional aspects of DDR, including DNA repair, cell cycle checkpoint activation, and transcriptional, translational, and metabolic reprogramming (Dantuma and van Attikum, 2016, Polo and Jackson, 2011). Among the various PTMs, K48-linked polyubiquitination of various chromatin-binding proteins occurs rapidly and transiently at DNA-damage sites and signals for their physical removal and subsequent recycling or degradation by proteasomes. Although our knowledge about the identity and functional significance of these K48-polyubiquinated proteins remains incomplete, it is evident that their timely removal governs proteostasis at DNA-damage sites by coordinating the protein flux between chromatin and the surrounding nuclear environment (Brinkmann et al., 2015, Brown and Jackson, 2015, Ghosh and Saha, 2012).
Certain polyubiquinated proteins, because of tight association with membranes, DNA, and protein partners, cannot dissociate spontaneously. In such cases, they are extracted in an energy-dependent manner by valosin-containing protein (VCP), a highly conserved, hexameric AAA+ ATPase essential for global cellular proteostasis. Dubbed a protein “segregase,” VCP is present throughout the cell and extracts “trapped” K48-polyubiquinated proteins from various organelles (endoplasmic reticulum, mitochondria, and endosomes), structures (chromatin), and macromolecular complexes (ribosomes and aggresomes) (Meyer et al., 2012, Meyer and Weihl, 2014). VCP function is facilitated by multiple cofactors (e.g., p47 and NPL4/UFD1); most of which associate with its N-terminal domain and directly bind polyubiquinated client proteins (Meyer et al., 2012, Meyer and Weihl, 2014, Ramadan et al., 2017, Vaz et al., 2013). The broad range of protein substrates functionally involved in nearly all cellular processes underlie the essentiality of VCP for multiple organisms (Fröhlich et al., 1991, Lamb et al., 2001, León and McKearin, 1999, Müller et al., 2007). In the context of DDR, a number of chromatin-associated VCP substrates have been identified. These include Ku70/80 and L3M6BTL1 for DSB repair (Acs et al., 2011, van den Boom et al., 2016), DDB2 and XPC for nucleotide excision repair (Puumalainen et al., 2014), RNA polymerase II during transcription-coupled DNA repair (Verma et al., 2011), MCM7 of the CMG replicative helicase complex during DNA replication termination (Maric et al., 2014, Moreno et al., 2014) and interstrand cross-link repair (Fullbright et al., 2016), and CDT1 during DNA replication under normal and DNA-damaging conditions (Franz et al., 2011, Raman et al., 2011).
The essential role of VCP in chromatin-associated protein clearance necessary for different DNA-repair mechanisms highlights its uniqueness as a general genome caretaker. However, given the pleiotropic effects of VCP, abolishing its total function pharmacologically, although showing promising effects in preclinical cancer models, triggers global protein stress (e.g., by inhibiting ER-associated degradation [ERAD]) and will likely have dose-limiting toxicity in patients (Anderson et al., 2015, Her et al., 2016, Magnaghi et al., 2013). Here, we have characterized a serendipitously discovered DNA-damage-specific phosphorylation event of VCP (Ser784) mediated by members of the DDR master kinase family, phosphatidylinositol 3-kinase-related kinases (PIKKs). Our data suggest that Ser784 phosphorylation is a specific enhancer of VCP nuclear functions in DDR and a predictor of breast cancer response to genotoxic chemotherapies.
Results
Nuclear Antigen Recognized by the pSer137-Pfn1 Antibody Associates with Poor Survival on Chemotherapy in Breast Cancer
In a prior work, we generated a polyclonal antibody targeting phospho-Ser137 of the actin-binding factor profilin-1 (Pfn1) (Shao et al., 2008). Using that antibody to stain a few breast cancer samples, we observed positive signals in a subset of cases. However, both the intensity and the subcellular location of the signals varied (Figures 1 A and S1A). Intrigued by the nuclear staining because of our interest in nuclear Pfn1 functions (Diamond et al., 2015), we stained a clinically annotated invasive breast cancer tissue microarray (TMA) (Strategic Partnering to Evaluate Cancer Signatures [SPECS]) (Cheang et al., 2009, Graveel et al., 2009) and scored the nuclear signals using the Allred method (intensity + proportion) (Harvey et al., 1999). Univariate survival analysis revealed an inverse and statistically significant correlation between the individual nuclear Allred scores and clinical outcomes of overall survival (OS) (n = 97, p = 0.0025) and relapse-free survival (RFS) (n = 88, p = 0.0034) by the log-rank test (Figure 1B). To confirm that result, we next stained a larger and independent breast cancer TMA (UBC series) (Bortnik et al., 2016). We observed similar results that high individual nuclear Allred scores were significantly associated with a lower RFS (n = 295, p = 0.0034) and trended toward a lower OS (n = 295, p = 0.071) (Table S1; Figure S1B). To simplify the analysis, we binarized the nuclear Allred scores into low (0–3) (n = 191, 65%) versus high (4–8) (n = 104, 35%) subgroups (Table S2). No significant association was observed between nuclear Allred scores and patient age at diagnosis, tumor size, nodal status, or ER, PR, HER2, or Ki67 status in this dataset. However, there was a significant association between nuclear Allred scores and histologic grade (Table S2). Univariate survival analyses revealed that cases with high Allred scores displayed significantly lower OS (n = 295, p = 0.0032), RFS (n = 295, p = 0.0003), and breast-cancer-specific survival (BCSS) (n = 293, p = 0.016) (Figures 1C and S1C). Multivariate analyses, adjusted for the clinicopathological variables of age, tumor grade and size, lymphovascular invasion, nodal status, and ER, PR, HER2, and Ki67 confirmed these results and revealed that the prognostic value of the nuclear staining is independent of those variables (Tables S3, S4, and S5). Interestingly, the association of nuclear scores with patient survival was mainly observed in the ER-negative subgroup (Figure S1D). There was no detectable association between the cytoplasmic staining and survival outcomes (Figure S1E).
Figure 1.

Nuclear Antigen Recognized by the pSer137-Pfn1 Antibody Associates with Poor Survival Among Chemotherapy-Treated Breast Cancer Patients
(A) Representative images of the nuclear staining of human breast tumors by the pSer137-Pfn1 antibody. 20X magnification; scale bars, 100 μm.
(B–D) Univariate Kaplan-Meier analyses showing an inverse correlation between nuclear staining by the pSer137-Pfn1 antibody and patient survival in the SPECS series (B), UBC series (C), and the TNBC subset of the UBC series (D). OS, overall survival; RFS, relapse-free survival; BCSS, breast cancer-specific survival; TNBC, triple-negative breast cancer.
(E) Univariate Kaplan-Meier analysis of the BCCancer series showing the associations between nuclear staining by the pSer137-Pfn1 antibody and poor survival outcome (BCSS) of the chemotherapy-treated breast cancer patients. Log-rank and Wilcoxon tests were used. p < 0.05 was considered statistically significant. Unadjusted p values for the outcome in the chemotherapy-treated group of BCCancer series are displayed.
See also Figures S1–S3 and Tables S1–S9.
To further investigate the potential clinical relevance of nuclear staining by the pSer137-Pfn1 antibody, we evaluated its association with survival outcomes across the major immunohistochemical subtypes of breast cancer (Cheang et al., 2008, Goldhirsch et al., 2011). A significant association between nuclear scores and survival was only detected in those with triple-negative breast cancer (TNBC: n = 42; OS, p = 0.0007; BCSS, p = 0.0005; OS-adjusted p = 0.003; BCSS-adjusted p = 0.002) and basal cases (n = 28; OS, p = 0.02; BCSS, p = 0.007; OS-adjusted p = 0.10; BCSS-adjusted p = 0.03) (Figures 1D, S2A, and S2B), although a significant correlation with RFS was also detected in luminal A cases (n = 151; p = 0.0001; adjusted p = 0.0006) (Figure S2C). No significant correlation with survival was observed in luminal B and HER2+ tumors (Figures S2A–S2C).
To investigate these observations according to treatment, we used the pSer137-Pfn1 antibody to stain a third and substantially larger breast cancer TMA (BCCancer series) (Cheang et al., 2006, Liu et al., 2012). We split this dataset into training and validation sets, each constituting approximately 50% of the total cases. The distribution of nuclear Allred scores in the training set of the BCCancer series, compared with the UBC series, showed a shift to higher levels, possibly because they had been snap frozen before fixation (Table S6). Thus, we modified the binarization method to retain a comparable number of cases in the “low (0–4)” versus “high (5–8)” nuclear score groups and thereby maximize study power in the validation set. We detected a statistically significant association between high nuclear scores and multiple poor prognostic factors, including high grade, ER negativity, HER2 positivity, non-luminal subtype, and triple-negative phenotype (Tables S7, S8, and S9).
In the overall dataset, we did not detect significant association between nuclear scores and survival outcomes (Figure S3A). However, when dividing the cases into four sub-groups based on the types of adjuvant therapies given after surgery and performing univariate analysis using BCSS, we found that high nuclear scores were significantly associated with worse patient survival in the chemotherapy-treated group of the training set only (n = 236; BCSS, p = 0.012; adjusted p = 0.048) (Figure 1E) but not in the untreated group or in those receiving tamoxifen or tamoxifen plus chemotherapy (Figure S3B). To assess whether the failed validation is due to properties specific to the pre-specified validation set, we randomly repeated the training/validation set split 500 times. Nuclear scores were found to be significant (BCSS; within chemotherapy-treated subset; ‘significant’ defined by lower 95% confidence interval of the hazard ratio >1) on 306 training sets and 115 of the corresponding validation sets. Thus, the significant results in the training set can be validated by about 40% of the training/validation set splits.
Because the nuclear score appeared to be particularly prognostic for TNBC in the UBC dataset (Figure 1D), we performed an exploratory analysis to determine whether the combination of immunohistochemical (IHC) staining and TNBC status would provide additional information regarding survival in the cohort of patients treated with chemotherapy. We found that tumors classified as “TNBC and with high nuclear scores” had the worst BCSS compared with the other subgroups (p = 0.028, combined set; Figure S3C). Thus, our data suggest that the level of nuclear staining by the pSer137-Pfn1 antibody could potentially predict breast cancer response to chemotherapy treatments.
VCP Is the DNA-Damage-Induced Nuclear Antigen Detected by the pSer137-Pfn1 Antibody
Nearly all the chemotherapy drugs used to treat patients in the BCCancer series are genotoxins (cyclophosphamide, methotrexate, 5-fluorouracil, and anthracycline). To investigate whether there is a link between the nuclear antigen of the pSer137-Pfn1 antibody and the DNA-damage response, we treated HeLa and several breast cancer cell lines (BT549, MDA-MB-231, T47D, and HCC1806) with SN38 (DNA topoisomerase I inhibitor) and etoposide (DNA topoisomerase II inhibitor). Although the untreated cells mainly displayed cytoplasmic reactivity with the pSer137-Pfn1 antibody, as previously described (Shao and Diamond, 2012), both drugs induced nuclear signals to various degrees (Figures 2 A and S4A). Similar nuclear signals were induced by genotoxic agents with different mechanisms of action (Figure S4B) including hydroxyurea (HU), 5-fluorouracil (5FU), cisplatin, and gemcitabine. Unexpectedly, Pfn1 knockdown by two different short hairpin RNAs (shRNAs), although reducing Pfn1 to an undetectable level, showed no effect on etoposide-induced nuclear staining (Figures S4C and S4D), suggesting that the nuclear antigen is a different protein.
Figure 2.

pSer784-VCP Is the DNA-Damage-Induced Nuclear Antigen of the pSer137-Pfn1 Antibody
(A) HeLa cells were treated with DMSO, 200 nM of SN38, and 5 μM of etoposide for 6 h, followed by staining using the pSer137-Pfn1 antibody. DAPI was used to stain DNA.
(B) HeLa cells were treated with DMSO, 200 nM of SN38, and 1 μM of gemcitabine for 16 h, followed by western blot analysis using the pSer137-Pfn1 antibody. Red arrowhead indicates the DNA-damage-induced ~100-kDa protein.
(C) Nuclear extracts of DMSO- or SN38-treated (200 nM, 16 h) HeLa cells were immunoprecipitated by the pSer137-Pfn1 antibody, followed by SDS-PAGE analysis and silver staining. The drug-induced protein at ~100 kDa was excised and identified as VCP by mass spectrometry (Held et al., 2013).
(D) Pull-down samples from (C) were analyzed by western blot using a VCP-specific antibody.
(E) HeLa cells were infected with shLuc or two distinct shVCPs for 3 days, treated with 5 μM of etoposide for 6 h, and immunostained using the pSer137-Pfn1 antibody.
(F) HeLa cells were treated with 5 μM of etoposide for 6 h, extracted by the CSK buffer for 5 min, and stained by the pSer137-Pfn1 antibody.
(G) HEK293T cells were transfected with wild-type or mutant VCP-GFP, treated with 200 nM of SN38 for 16hr, and immunoprecipitated using the pSer137-Pfn1 antibody. Pull-down and input samples were analyzed by western blot using a GFP antibody.
(H) Input samples from (A) were analyzed by western blot using the pSer137-Pfn1 or VCP antibodies.
(I) HeLa cells were pre-treated for 30 min with DMSO, 10 mM of caffeine, or 10 μM of KU-55933, followed by 30 min of treatment with 50 μM of etoposide. They were lysed by RIPA (without SDS), and the soluble and insoluble fractions were analyzed by western blot. pSer784-VCP was detected by the pSer137-Pfn1 antibody.
All data, except (C), were independently confirmed two to three times. Scale bars, 20 μm (A) and (E) and 10 μm (F). See also Figures S4 and S5.
Because of its clinical importance (Figure 1), we sought to identify the unknown nuclear antigen. Treating multiple cell lines with different genotoxic agents reveals an ∼100-kDa protein recognized by the pSer137-Pfn1 antibody (Figures 2B, S5A, and S5B). To identify that protein, we subjected the nuclear extracts of untreated or SN38-treated HeLa cells to immunoprecipitation using the pSer137-Pfn1 antibody. Silver staining reveals an ∼100-kDa protein specifically present in the SN38-treated sample (Figure 2C), which was identified by mass spectrometry to be the AAA+ ATPase VCP. This was confirmed by western blot using a VCP antibody (Figure 2D). Incubating cell lysates with alkaline phosphatase before immunoprecipitation by the pSer137-Pfn1 antibody significantly reduced the binding of VCP, confirming that the reactivity is phosphorylation dependent (Figure S5C).
To confirm that VCP is the nuclear antigen of the pSer137-Pfn1 antibody by immunostaining, we silenced VCP in HeLa cells using two shRNAs, treated with SN38, and performed immunostaining with the pSer137-Pfn1 antibody. Although nuclear signals are observed in cells infected with a control shRNA, little to no signals were present in the VCP knockdown cells (Figure 2E). Notably, detergent extraction of etoposide-treated HeLa cells before fixation and staining revealed discrete nuclear structures characteristic of DNA-damage foci (Figure 2F). Thus, VCP is the DNA-damage-induced nuclear antigen of the pSer137-Pfn1 antibody.
Ser784 Is the Phosphorylation Site of VCP Recognized by the pSer137-Pfn1 Antibody
We next sought to identify the VCP epitope that reacts with the pSer137-Pfn1 antibody. Because there is no obvious similarity between the antigenic pSer137-Pfn1 peptide (MASHLRRpS137Q) and the VCP sequence, we took a candidate approach by individually mutating five serines to alanines in a VCP-GFP construct and determining the ability of the pSer137-Pfn1 antibody to recognize the VCP mutants upon DNA damage. The five serines are either preceded by 1–2 basic residues (R/KS) or followed by a glutamate (SQ) as Ser137 in the antigenic Pfn1 peptide. Upon transfection into HEK293T cells and SN38 treatment, we used the pSer137-Pfn1 antibody to immunoprecipitate wild-type versus mutant VCP-GFP, followed by western blot using a GFP antibody. One mutant, S784A, although abundantly expressed, was not pulled down by the pSer137-Pfn1 antibody (Figure 2G). It also does not react with the antibody directly on western blot (Figure 2H).
Phosphorylation of VCP at Ser784 has been reported to occur in response to different genotoxic treatments, but the functional significance remains completely unknown (Livingstone et al., 2005, Matsuoka et al., 2007, Stokes et al., 2007). Residing in a pSQ motif, it is believed to be directly phosphorylated by the PIKK family members ATM, ATR, or DNA-PKcs, known as master DDR regulators (Blackford and Jackson, 2017). Consistent with that, treating HeLa cells with caffeine (general ATM/ATR/DNA-PKcs inhibitor) (Sarkaria et al., 1999) and KU55933 (specific ATM inhibitor) (Hickson et al., 2004) significantly inhibits Ser784 phosphorylation caused by etoposide (predominantly inducing DSBs, which activate ATM) (Figure 2I). Interestingly, when cells are lysed with SDS-free RIPA buffer (radioimmunoprecipitation assay buffer; 1% NP40 and 1% sodium deoxycholate), pSer784-VCP is found exclusively in the soluble fraction, although a small fraction of total VCP is detected in the insoluble fraction (Figure 2I).
Generation and Characterization of the Monoclonal pSer784-VCP Antibody
To further confirm our results and better study pSer784-VCP, we generated monoclonal antibodies using a synthetic VCP-peptide-harboring pSer784 (GGAGPpS784QGSGGG). One clone (3E4) was evaluated in parallel to the pSer137-Pfn1 antibody. 3E4 shows superior specificity on western blot and detects the same 100-kDa protein recognized by the pSer137-Pfn1 antibody under DNA-damaging conditions (Figure 3 A) in an alkaline-phosphatase-sensitive manner (Figure 3B). This phospho-protein was confirmed to be VCP by anti-VCP pull-down under native and denaturing conditions followed by immunoblotting using the 3E4 or pSer137-Pfn1 antibodies (Figures 3C and S5D). In a reciprocal fashion, both antibodies pull down VCP specifically from genotoxin-treated cells under the native condition (Figure 3D), and the pSer137-Pfn1 antibody can also do this under the denaturing condition (Figure S5E). For immunostaining, 3E4 specifically detects nuclei in a DNA-damage-dependent fashion, which is abolished by VCP knockdown (Figure S5F), and shows no reactivity in untreated cells (Figure 3E).
Figure 3.

Monoclonal pSer784-VCP Antibody Confirms the Nuclear Antigen of the pSer137-Pfn1 Antibody
(A) HeLa cells treated with DMSO or SN38 (200 nM, 16 h) were analyzed by western blot using the pSer137-Pfn1 or pSer784-VCP antibodies.
(B) HeLa cells treated with etoposide (50 μM, 1 h) were lysed, incubated at 37°C for 1 h with or without calf intestinal alkaline phosphatase, and analyzed by western blot using the pSer137-Pfn1, pSer784-VCP, or pan-VCP antibodies.
(C) HeLa cells treated with DMSO or etoposide (50 μM, 6 h) were subjected to immunoprecipitation by control immunoglobulin G (IgG) or pan-VCP antibody, followed by western blot using the pSer137-Pfn1, pSer784-VCP, and pan-VCP antibodies.
(D) HeLa cells treated with DMSO or SN38, as in (A), were subjected to immunoprecipitation using the pSer137-Pfn1 or pSer784-VCP antibodies, followed by western blot using a pan-VCP antibody.
(E) HeLa cells were treated with 200 nM of SN38 for 16 h and immunostained with the pSer784-VCP antibody.
(F) HeLa cells were treated with 50 μM of etoposide for 1 h, followed by double immunostaining using the pSer137-Pfn1 and pSer784-VCP antibodies.
(G and H) U2OS cells were treated with 50 μM of etoposide for 1 h, recovered for 90 min, and detergent-extracted before fixation and double staining by the pSer137-Pfn1 and 53BP1 antibodies (G) or pSer784-VCP and γH2AX (H) antibodies.
(I) Representative images in the SPECS TMA immunostained in parallel by the pSer137-Pfn1 and pSer784-VCP antibodies.
(J) Univariate Kaplan-Meier analysis of the SPECS TMA stained in (G). Nuclear Allred scores were binarized into low (0–4) versus high (5–8) groups. Two extra interpretable cases stained by the pSer137-Pfn1 antibody (n = 46) (but not by pSer784-VCP, n = 44) were included in the analysis. p values were based on Log-rank test.
More than 100 cells per experiment were analyzed for (D)–(F). Data in (A)–(H) have been independently confirmed 2-3 times. Scale bars, 20 μm (E), 4 μm (F–H), and 10 μm (I). See also Figure S5.
Next, we tested whether the DNA-damage-induced nuclear foci detected by 3E4 and the pSer137-Pfn1 antibody (Figure 2F) are identical by double labeling of etoposide-treated, pre-extracted osteosarcoma U2OS cells. We observed complete co-localization of the nuclear foci recognized by the two antibodies in all focus-containing cells (Figure 3F). To confirm that they are DNA-damage foci, we co-stained the cells for well-known DSB markers (γH2AX) and repair factors (53BP1 and BRCA1). We detected co-localization of pSer784-VCP with nearly all the 53BP1-positive DNA-damage foci, consistent with the fact that DSB recruitment of 53BP1 requires VCP (Figure 3G) (Acs et al., 2011). We also detected significant co-localization between pSer784-VCP and γH2AX in more than 50% of the focus-containing cells (Figure 3H). Interestingly, we detected minimal co-localization between pSer784-VCP and BRCA1. Fewer cells contained BRCA1-positive nuclear foci than pSer784-VCP-positive foci, and of those positive for BRCA1, <10% showed only partial co-localization with pSer784-VCP (Figure S5G). Thus, pSer784-VCP is physically present at a subset of DNA-damage sites containing selective DNA repair factors.
To further confirm that the nuclear antigen of the polyclonal pSer137-Pfn1 antibody in human breast cancer tissues is pSer784-VCP (Figure 1), we performed parallel staining using the SPECS TMA. Comparison of nuclear Allred scores of the remaining tissues (n = 69, fewer cases than earlier staining because of the extensive cutting of the block) revealed highly concordant staining patterns of 3E4 and the pSer137-Pfn1 antibody (Pearson correlation coefficient, r2 = 0.9) (Figure 3I). Univariate Kaplan-Meier analysis using further filtered cases containing a sufficient number of tumor cells revealed that nuclear staining by the monoclonal 3E4 antibody was significantly correlated with worse RFS (n = 44, p = 0.0104, log-rank test), in the same trend as nuclear staining by the polyclonal pSer137-Pfn1 antibody (n = 46, p = 0.1232, log-rank test) (Figure 3J). Despite the limited statistical power because of the few cases, these data collectively support that the two antibodies detect a common nuclear antigen, pSer784-VCP.
Ser784 Phosphorylation Is a Late DNA-Damage-Induced Event
VCP is well-known for its rapid recruitment to DNA-damage sites to remove K48-linked polyubiquitinated protein substrates (Meyer et al., 2012, Vaz et al., 2013). Upon laser-induced DSB in MRC5 fibroblast cells, the biological half-life (t1/2) of VCP-GFP recruitment is reported to be 2 min (Acs et al., 2011). To determine the kinetics of Ser784 phosphorylation upon DNA damage, we performed laser micro-irradiation in U2OS and BT549 cells and compared the kinetics of damage site appearance of total VCP versus pSer784-VCP by immunostaining at different times. We detected VCP at DNA-damage sites (indicated by NBS1, part of the DSB sensor MRN complex) as early as 1 min after laser irradiation in the U2OS cells and 5 min in the BT549 cells. In contrast, pSer784-VCP was undetectable until 10–15 min in both cell lines. Interestingly, diffuse nucleoplasmic pSer784-VCP, although undetectable before laser irradiation, appeared upon DNA damage and intensified in parallel to pSer784-VCP at the laser-induced DNA wounds (Figures 4 A, S6A, and S6B). Thus, Ser784 phosphorylation occurs as a direct consequence of DNA damage and later than VCP recruitment to damage sites.
Figure 4.

Ser784 Phosphorylation Is a Late DNA-Damage-Induced Event
(A) BT549 cells were laser micro-irradiated (Chen et al., 2013) and double labeled at various time points by the pSer784-VCP/NBS1 or VCP/NBS1 antibody pairs (You et al., 2005). Experiment was independently performed twice with similar results.
(B) HeLa cells were treated continuously with 50 μM of etoposide, lysed by RIPA buffer (no SDS) at the indicated time points, and analyzed for soluble and insoluble fractions by western blot.
Band intensities were plotted over time for each indicated antibody. Data were independently confirmed three times. Scale bars, 10 μm. See also Figure S6.
We next chemically induced DSB in HeLa cells using etoposide and monitored pSer784-VCP by western blot at different times. We compared it to well-known DDR signaling events including auto-phosphorylation of ATM (pSer1981-ATM) and ATM-dependent phosphorylation of Chk2 (Thr68) and H2AX (Ser139) (Awasthi et al., 2016). Although these known DDR phosphorylation events occur quickly before (pThr68-Chk2 and pSer1981-ATM) or around 5 min (γH2AX) of treatment and reach maximal intensities around 30 min, pSer784-VCP is detectable after 20 min and gradually intensifies beyond 120 min. No changes in the total protein levels were observed (Figure 4B). Similarly delayed pSer784-VCP induction by hydroxyurea (inducing DNA replication stress and ATR activation) as compared with pSer345-Chk1 (known ATR substrate) was also observed (Figure S6C). Thus, Ser784 phosphorylation of VCP is a relatively late DDR event.
Ser784 Phosphorylation Increases VCP Activity on Chromatin
Given the essential function of VCP in the turnover of K48-polyubiquitinated proteins, we asked whether Ser784 phosphorylation has a role in these processes in response to DNA damage. We used the S784A and S784D mutations to mimic the unphosphorylated and phosphorylated forms of VCP, respectively. They were introduced into a human VCP-GFP construct containing silent mutations, which confer resistance to two different shRNAs (shVCP 1 and 2). Wild-type and mutant VCP-GFP were stably expressed in HeLa cells at similar levels to each other and relative to endogenous VCP (Figure 5 A). Upon VCP knockdown in the etoposide-treated GFP control cells, the level of K48-polyubiquitinated proteins increased significantly, both in the RIPA-soluble and insoluble fractions (Figures 5B and S7A). Interestingly, although the increase in the soluble fractions was similarly rescued by wild-type and mutant VCP-GFP, the increase in the insoluble fractions was rescued more effectively by VCP(S784D) and less effectively by VCP(S784A) compared with VCP(WT) (Figure 5B). To test whether the K48-polyubiquitin signals in the RIPA-insoluble fractions represent chromatin-associated proteins, we divided the cells into cytoplasm, nucleoplasm, and chromatin fractions. VCP knockdown increased K48-polyubiquitin in all three fractions, consistent with its importance in global protein clearance (Figures 5C and S7B). This was phenocopied by the specific VCP inhibitor NMS-873 (Magnaghi et al., 2013) (Figure S7C). Interestingly, the most notable differences between the rescuing activity of VCP(S784D) (more active) and VCP(S784A) (less active) relative to VCP(WT) were detected in the chromatin fractions, followed by subtle differences in the nucleoplasmic fractions. No difference in the activity of wild-type versus mutant VCP was detected in the cytoplasmic fraction (Figures 5C and S7B). Similar results were also observed in the U2OS cells (Figures S7D and S7E).
Figure 5.

Ser784 Phosphorylation Increases VCP Activity Specifically in the Nucleus
(A) HeLa cells stably expressing GFP or RNAi-resistant VCP-GFP (WT or mutants) were infected with shLuc or shVCP1 and 2 combined. Cells were analyzed 4 days later by western blot using antibodies against VCP (detecting both endogenous VCP and exogenous VCP-GFP) or actin.
(B) Cells in (A) were treated with 50 μM of etoposide for 30 min, recovered for 1 h, lysed by RIPA buffer (no SDS), and analyzed for soluble and insoluble fractions by western blot using a K48-linkage-specific polyubiquitin antibody controlled by histone H3 or GAPDH.
(C) Etoposide-treated HeLa cells, as in (B), were subjected to subcellular fractionation (Méndez and Stillman, 2000), followed by western blot analysis of the resulting cytoplasmic, nucleoplasmic, and chromatin fractions using the K48-ubiquitin antibody controlled by GAPDH, actin, and histone H3. Densitometry was performed to quantify K48-polyubiquitin levels in (B) and (C), which were subsequently normalized over the internal controls.
(D) Chromatin fractions from (C) were analyzed by western blot for Ku70.
(E) VCP knockdown and rescue HeLa cells were treated with 50 μM of etoposide for 30 min, recovered for 2 h in the presence of 20 μM of MG-132, and lysed with RIPA buffer. Soluble and insoluble fractions were analyzed by western blot for HIF1α and K48-ubiquitin, with tubulin and H3 as loading controls.
See also Figure S7.
To confirm that the observed changes in K48-polyubiquitin represented VCP substrates, we examined Ku70, the dimeric partner of Ku80, which is a known chromatin-associated VCP substrate essential for DSB repair (van den Boom et al., 2016). Chromatin-associated Ku70 level was increased by VCP knockdown in etoposide-treated HeLa cells, and that could be reduced by RNAi-resistant VCP(WT) and VCP(S784D) but not VCP(S784A) (Figure 5D). Next, we examined the level of HIF1α, a known soluble nuclear substrate of VCP (Alexandru et al., 2008). Because of the high turnover rate of HIF1α under normal conditions, cells were treated with MG-132 for 2 h to reduce their proteasomal degradation before harvest, as previously described (Alexandru et al., 2008). Interestingly, the level of RIPA-soluble HIF1α was significantly decreased by VCP(S784A) but increased by VCP(S784D) relative to VCP(WT), in clear contrast to the K48-ubiquitin levels in the insoluble fractions (Figures 5E and S7F). qRT-PCR showed no changes in the mRNA level of HIF1α (Figure S7G). These results suggest that Ser784 phosphorylation, induced by DNA damage, preferentially increases nuclear VCP ability to clear chromatin-associated but not soluble substrates.
Ser784 Phosphorylation Decreases VCP Association with NPL4/UFD1 and Polyubiquitinated Proteins
To study the regulatory mechanism of Ser784 phosphorylation, we first examined its effects on the VCP interaction with protein substrates. We immunoprecipitated total VCP (by a pan-VCP antibody) or pSer784-VCP (by 3E4) from etoposide or HU-treated HeLa cell lysates and immunoblotted for co-absorbed K48-polyubiquitinated proteins. Despite the similar amounts of precipitated VCP, 3E4 significantly enriched pSer784-VCP compared with the pan-VCP antibody. Interestingly, less K48-polyubiquitins were co-adsorbed by 3E4 than by the pan-VCP antibody, indicating that Ser784 phosphorylation may reduce VCP interaction with substrates (Figure S8A). To better separate pSer784-VCP from unphosphorylated VCP, we next performed sequential pull-downs from etoposide-treated HeLa cell lysates to first deplete pSer784-VCP using 3E4 and subsequently capture the remaining unphosphorylated VCP by the pan-antibody. We detected significantly less binding of K48-polyubiquitins to pSer784-VCP than of unphosphorylated VCP. Based on the levels of VCP in the input and supernatants after each pull-down, it is evident that Ser784 is phosphorylated at high stoichiometry in response to DNA damage (>50% of total VCP after overnight etoposide treatment) (Figure 6 A). The reduced binding of K48-polyubiquitins by pSer784-VCP was similarly observed in etoposide-treated MDA-MB-231 cells (Figure S8B) and was recapitulated by S784D versus S784A mutants of VCP-FLAG (Figure 6B). Interestingly, VCP interaction with NPL4 and UFD1, two core cofactors important for K48-polyubiquitin binding during chromatin-associated degradation, is similarly decreased by Ser784 phosphorylation (Figures 6B and S8B).
Figure 6.

Ser784 Phosphorylation Decreases VCP Association with Chromatin and Polyubiquitinated Proteins
(A) HeLa cells were treated with 5 μM of etoposide overnight, lysed with SDS-free RIPA, and immunoprecipitated first with the pSer784-VCP antibody; the supernatant of which was subsequently immunoprecipitated by the VCP pan-antibody.
(B) MDA-MB-231 cells stably expressing YFP-FLAG or VCP-FLAG (S784A versus S784D) were treated with 200 nM of SN38 overnight, and nucleoplasmic fractions were immunoprecipitated by the anti-FLAG antibody.
(C) HeLa cells expressing RNAi-resistant VCP-GFP were infected with combined shVCP1 and 2, treated with 50 μM of etoposide for 45 min, recovered for 1 h, lysed with SDS-free RIPA buffer, and analyzed by western blot.
(D) Similarly treated HeLa cells as in (C) were fractionated and analyzed by western blot using nucleoplasm and chromatin fractions. Data were independently confirmed two to three times.
See also Figures S7 and S8.
Next, we examined the effect of Ser784 phosphorylation on VCP association with chromatin. S784A mutation significantly increased VCP level in RIPA-insoluble, chromatin-enriched fractions of etoposide-treated VCP knockdown and rescue HeLa cells. Conversely, S784D caused a decrease of RIPA-insoluble VCP (Figures 6C and S7H). Consistent with that, subcellular fractionation showed more chromatin-associated VCP(S784A) than VCP(WT) and VCP(S784D) did, despite their similar nucleoplasmic levels (Figures 6D and S7I). Similar results were also observed in etoposide-treated MDA-MB-231 cells expressing wild-type and mutant VCP-FLAG (Figures S8C and S8D). Thus, the increased chromatin-selective activity of pSer784-VCP appears to correlate with decreased association with NPL4/UFD1 and polyubiquitinated substrates.
Ser784 Phosphorylation of VCP Is Important for DNA-Damage Response and Cell Survival upon Genotoxic Stress
To determine the functional significance of Ser784 phosphorylation for DNA-damage response, we performed VCP knockdown and rescue in etoposide-treated HeLa cells, as described in Figure 5. First, we examined DDR signaling by quantifying the phosphorylation events of the PIKK kinases. Using an antibody against the consensus pS/TQ motif and two well-known ATM substrates pSer343-NBS1 and pThr68-Chk2 functionally important for DSB repair and cell cycle checkpoint, we detected within RIPA-soluble fractions a significant decrease in PIKK signaling upon VCP knockdown (in GFP control cells), which was fully rescued by wild-type VCP (Figure 7 A). Chemical inhibition of VCP by NMS-873 similarly decreased PIKK signaling (Figure S9A). Interestingly, although VCP(S784D) showed similar rescuing activity as VCP(WT) did, VCP(S784A) was nearly inactive (Figure 7A). Similar results were observed in HU-treated HeLa cells (Figure S9B). In contrast, the increase in endoplasmic reticulum stress, indicated by Bip1 and CHOP induction, as a result of VCP knockdown can be fully rescued by both wild-type and mutant VCP (Figure 7A), consistent with their similar abilities to clear cytosolic polyubiquitinated proteins (Figure 5B).
Figure 7.

Ser784 Phosphorylation of VCP Is Important for DNA-Damage Response and Cell Survival upon Genotoxic Stress
(A) shLuc- or shVCP-infected stable HeLa cells were treated with 50 μM of etoposide for 30 min, recovered for 1 h, lysed by RIPA, and analyzed by western blot.
(B) shLuc- or shVCP-infected stable U2OS cells were treated with vehicle or different drugs for 16 h, and subjected to colony formation assays for 10–14 days (Guzmán et al., 2014). Relative effects represent normalized drug/vehicle percentages. Shown are means ± SEM of three technical replicates of single biological experiments for each drug. p Values were based on unpaired t tests (S784A versus WT or S784D).
(C) shVCP2-infected U2OS stable cells were treated with 25 μM of etoposide for 30 min followed by 1 h of recovery or 1 mM HU for 16 h. Cells were subjected to the comet assay under the alkaline condition, and tail DNA percentages were calculated (Gyori et al., 2014). Shown are single biological experiments with 150–300 cells analyzed per condition. Error bars represent SEM. p Values were based on unpaired t tests. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Results in (B) and (C) were confirmed by three biological replicates.
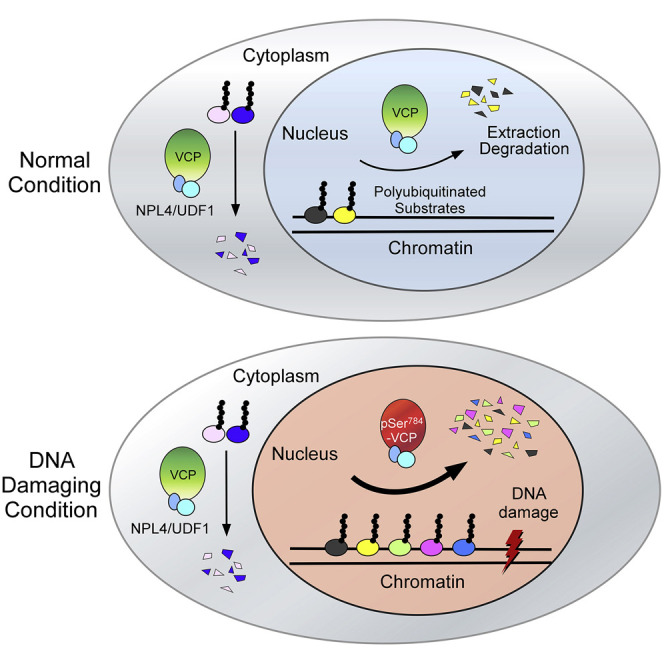
(D) Working model of enhanced substrate extraction from chromatin by VCP upon DNA-damage-induced Ser784 phosphorylation. In this model, Ser784 phosphorylation is a relatively late DDR event that occurs either in the nucleoplasm or on chromatin after VCP binding to polyubiquitinated substrates (both scenarios are depicted). Ser784 phosphorylation does not abolish chromatin recruitment of VCP but promotes substrate extraction and subsequent degradation at least partially because of its weakened interaction with cofactors NPL4 and UFD1, which directly bind substrates. Dissociated pSer784-VCP can regain access to chromatin and extract more substrates.
Scale bars, 40 μm. See also Figures S9 and S10.
We next tested whether Ser784 phosphorylation of VCP regulates PIKK signaling by affecting its chromatin binding upon DNA damage. Unexpectedly, in the etoposide or HU-treated VCP knockdown HeLa cells rescued by VCP(S784A), we detected more chromatin-associated ATM and ATR, respectively, than in the cells expressing VCP(WT) and VCP(S784D) (Figures S9C and S9D). Higher levels of phosphorylated ATM substrates, pSer343-NBS1 and γH2AX, are found in chromatin fractions of the VCP(S784A) cells (Figure S9C). This is in contrast to the reduced pSer1981-ATM and pSer343-NBS1 in the soluble nucleoplasm of VCP(S784A) cells compared with those expressing VCP(WT) and VCP(S784D) (Figure S9C). We could not detect pSer1981-ATM in the chromatin fractions, possibly because only a small fraction of total ATM is retained on chromatin after fractionation (Figure S9E). Nonetheless, immunostaining revealed more VCP(S784A)-rescued cells containing pSer1981-ATM-positive DNA-damage foci (Figure S9F). Thus, Ser784 phosphorylation of VCP appears to differentially affect PIKK signaling within DNA-damage sites on chromatin versus in the soluble nucleoplasm.
Next, we investigated whether Ser784 phosphorylation of VCP affects cell survival after DNA damage. Consistent with its essentiality, VCP knockdown caused cell death within 5–7 days without genotoxic stress, and that could be rescued by VCP, regardless of Ser784 mutations (Figures S10A and S10B). This enabled us to use the knockdown and rescue strategy (as in Figure 5) to “replace” endogenous VCP with RNAi-resistant wild-type and mutant VCP and specifically to study the effect of Ser784 phosphorylation on DNA-damage-induced cell death. Treating the resultant U2OS cells with a panel of genotoxic agents (5FU, HU, cisplatin, SN38, and PARP inhibitors olaparib and niraparib) revealed that cells expressing VCP(S784A) survive significantly less than those expressing VCP(WT) and VCP(S784D) (Figures 7B and S10C). Similar effects were also observed in HeLa cells (Figure S10D).
Last, we tested whether the pro-survival effect of pSer784-VCP upon genotoxic stress correlates with increased DNA-damage repair. We treated the VCP knockdown and rescue U2OS cells with etoposide and HU and used the comet assay to quantify single- and double-stranded DNA breaks under alkaline conditions. In response to both drugs, significantly fewer DNA breaks were detected in VCP(S784D) than detected in VCP(WT) and VCP(S784A) cells (Figure 7C). Collectively, these results demonstrate the functional importance of VCP phosphorylation at Ser784 for DNA-damage repair, global PIKK-dependent DDR signaling, and cell survival, further supporting the idea that pSer784-VCP may be used to predict the outcome of chemotherapy-treated patients with breast cancer.
Discussion
In this study, we discovered that Ser784 phosphorylation is an important regulatory event of VCP, specifically for its nuclear DDR functions. This work was motivated by the serendipitous finding that an unknown nuclear phospho-protein, cross-reacting with the pSer137-Pfn1 antibody, significantly correlated with poor survival for breast cancer patients receiving genotoxic chemotherapies. This unexpected, yet clinically important, finding led us to the identification of pSer784-VCP as the underlying nuclear antigen and our subsequent investigation that revealed its functional importance for DDR.
Although several phosphorylation events of VCP have been reported, Ser784 is the only DNA-damage-induced phospho-site unbiasedly and independently detected by different studies. It was first described by Livingstone et al. (2005) in a similarly serendipitous fashion, using a cross-reacting phospho-Chk2 antibody, which detected pSer784-VCP at DNA-damage foci in response to various genotoxic treatments. Subsequently, two unbiased proteomic studies independently confirmed Ser784 phosphorylation (the only detectable phospho-site on VCP) in response to IR and UV (Matsuoka et al., 2007, Stokes et al., 2007). However, its physiological significance remained unknown before our study.
Here, more than a decade later, we provide clinical and mechanistic evidence for the functional importance of Ser784 phosphorylation in DDR. Clinically, our data suggest that the level of nuclear pSer784-VCP, recognized by the cross-reacting pSer137-Pfn1 antibody, is associated with poor outcome among patients with breast cancer who received genotoxic therapies (cyclophosphamide, methotrexate, 5FU, and anthracyclines) but not among those receiving the ER-antagonist tamoxifen or no adjuvant systemic therapy at all. Notably, pSer784-VCP level used for our analysis is in the surgical tumor samples taken before the adjuvant treatments. In other words, it is the baseline pSer784-VCP level induced by endogenous DNA damage that predicts the survival for patients subsequently treated with adjuvant genotoxic chemotherapy. However, discrepancy is conceivable between the “baseline” and the “drug-induced” pSer784-VCP levels. For instance, certain tumors containing low levels of endogenous DNA damage would present low baseline pSer784-VCP levels, but may phosphorylate Ser784 to high levels upon exogenous drug treatments. Thus, although valid, our data likely underestimates the true chemo-predictive ability of pSer784-VCP. Future studies can better assess this by treating patients neoadjuvantly (before surgery) with genotoxic agents and staining biopsy samples for “induced” pSer784-VCP to correlate with short-term neoadjuvant tumor response and long-term survival during or after adjuvant treatment. Moreover, because Ser784 phosphorylation occurs in cell lines derived from various tissues (Figures S4A and S4B) (Livingstone et al., 2005, Matsuoka et al., 2007, Stokes et al., 2007) and protects them from a wide range of DNA-damaging agents, including PARP inhibitors (Figure 7B), it is likely a general predictor of genotoxic chemotherapy susceptibility for a broad range of cancer types.
Although our data suggest that tumors containing low levels of “induced” pSer784-VCP can be treated effectively by chemotherapy, they also implicate those containing high levels of pSer784-VCP as perhaps being sensitized by PIKK inhibitors, such as those against ATM and ATR. Multiple ATM and ATR inhibitors have entered clinical trials for different cancer types as monotherapies or radio- and chemo-sensitizers, supported by strong preclinical data (Brandsma et al., 2017, Durant et al., 2018, Weber and Ryan, 2015). However, because both kinases phosphorylate hundreds of proteins upon DNA damage, identifying the “driver” substrates with the functional capability of determining cell fate is important. VCP may be one such candidate because it is phosphorylated by both kinases and protects genome stability against a broad range of insults (Meyer et al., 2012, Meyer and Weihl, 2014, Ramadan et al., 2017, Vaz et al., 2013) (Figures 7B, S10C, and S10D). Nevertheless, inhibiting VCP function pharmacologically triggers global proteostatic stress throughout the cell and will likely cause dose-limiting toxicity when used in patients (Anderson et al., 2015, Her et al., 2016, Magnaghi et al., 2013). Instead, our work shows that nuclear, but not cytoplasmic, VCP can be selectively activated by ATM- and ATR-mediated Ser784 phosphorylation. Thus, pSer784-VCP levels may be an indicator of cellular reliance on ATM and ATR activities, and inhibiting ATM and ATR in pSer784-VCP-high tumors may be an effective chemo-sensitizing approach worth testing in the future.
As a substrate of PIKKs, VCP is reciprocally important for PIKK activation upon DNA damage (Figures 7A and S9B). Interestingly, this requires Ser784 phosphorylation in a context-dependent manner. Preventing Ser784 phosphorylation, although decreasing phosphorylation of soluble substrates of ATM/ATR, actually increases ATM/ATR presence and activity on chromatin (Figures S9C–S9F). Although somewhat surprising, this finding highlights the complexity of the regulatory mechanisms for PIKK signaling and suggests that the chromatin-associated and soluble signaling events can be differentially regulated. Ser784 phosphorylation may increase the ability of VCP to mobilize chromatin-activated PIKKs or increase their dynamic exchange between chromatin and nucleoplasm to more efficiently phosphorylate soluble substrates. Additionally, by increasing the ability of VCP to extract chromatin-associated substrates, such as KRAB-associated protein 1 (KAP1), Ser784 phosphorylation may promote the global chromatin relaxation necessary for fully activating ATM beyond the DNA-damage sites (Kuo et al., 2016, Wu et al., 2011, Ziv et al., 2006). As for ATR, because it is frequently activated at single and double DNA junctions during ATM-dependent DSB resections (Shiotani and Zou, 2009, You et al., 2009), similar regulatory mechanisms by pSer784-VCP may exist as well.
Our finding that Ser784 phosphorylation specifically increases VCP activity on chromatin is somewhat surprising, given that pSer784-VCP interacts with NPL4/UFD1 with lower affinity (Figures 6B and S8B). NPL4 and UFD1 are the core cofactors of VCP that directly bind polyubiquitins on many substrates and are important for chromatin-associated degradation (Meyer et al., 2012, Meyer and Weihl, 2014, Vaz et al., 2013). Thus, a reduced interaction with NPL4/UFD1 would be expected to inhibit VCP access to its substrates and can logically explain the higher level of HIF1α in cells expressing the phosphomimetic VCP(S784D) (Figure 5E). However, the increased activity of pSer784-VCP toward its chromatin-associated substrates seems counter-intuitive and mechanistically puzzling. In addition to the reduced interaction with NPL4/UFD1 and polyubiquitinated substrates, pSer784-VCP also seems to associate with chromatin with less affinity, biochemically speaking (Figures 6C and 6D), but is readily detected at DNA-damage sites by immunostaining (Figures 3G, 3H, and 4A). Based on all these data, we propose a working model that Ser784 phosphorylation may promote chromatin protein extraction by accelerating VCP dissociation from chromatin and NPL4/UFD1 to facilitate substrate release during the late stage of extraction (Figure 7D). Indeed, phosphorylation triggers chromatin release of several known DDR factors, including KAP1 (Goodarzi et al., 2008), Rad9 (Furuya et al., 2010), and Chk1 (Smits et al., 2006), each for a distinct purpose. However, the opposing effects of Ser784 phosphorylation on soluble (e.g., HIF1α) versus chromatin substrates of VCP and the clear presence of pSer784-VCP at DNA-damage sites suggest the involvement of additional cofactors that enable chromatin access of pSer784-VCP independently of NPL4/UFD1. An example of such cofactors during DNA-replication stress is DVC1, which delivers VCP to stalled replication forks to extract trans-lesion DNA polymerase (Davis et al., 2012, Mosbech et al., 2012).
The fact that Ser784 phosphorylation, within the C-terminal tail of VCP (763–806 amino acids [aa]), weakens the interaction with NPL4/UFD1 at the N-terminal domain suggests long-range conformational changes. Indeed, inter-domain communication is well-known for VCP and crucial for its ATP-dependent segregase activity (Tang and Xia, 2016). For example, nucleotide binding and hydrolysis in the central ATPase domains alter the relative position of the N-terminal domain, which presumably produces the mechanical force during protein extraction (Banerjee et al., 2016, Schuller et al., 2016, Tang et al., 2010). Conversely, conformation of the N-terminal domain and its occupancy by cofactors influence the enzymatic activity of the ATPase domains (Meyer et al., 1998, Niwa et al., 2012, Zhang et al., 2015). The C-terminal tail of VCP is poorly understood, both structurally and functionally. However, it can directly interact with several proteins, including PNGase, PLAA/UFD3, and UFD2, although none has been linked to DDR (Li et al., 2006, Qiu et al., 2010, Rumpf and Jentsch, 2006, Zhao et al., 2007). Interestingly, Tyr806 phosphorylation occurs in the C-terminal tail in response to T-cell receptor activation and abolishes the interaction with PNGase and PLAA (Egerton et al., 1992, Qiu et al., 2010, Zhao et al., 2007). These studies raise the possibility that Ser784 phosphorylation, in response to DNA damage, may alter the local interaction of VCP with certain C-terminally associated proteins and, in turn, cause long-range a conformational change to weaken the binding of NPL4/UFD1 at the N terminus.
In summary, we have provided clinical and experimental evidence that pSer784-VCP is a cancer prognostic biomarker that can potentially predict chemotherapy response because of its importance for DDR. However, regulatory mechanisms of Ser784 phosphorylation remain partially understood and will be further examined in the future. These include a validation and mechanistic investigation of the different effects of Ser784 phosphorylation on VCP substrates and PIKK signaling within soluble nucleoplasm versus on chromatin, as well as cell cycle analysis to confirm the role of Ser784 phosphorylation in PIKK-dependent checkpoint activation (which could not be performed because of the COVID-19 pandemic). Future studies should also investigate the changes in VCP conformation and its interactome upon Ser784 phosphorylation to mechanistically explain its functional importance for DDR. Last, proper validation of the prognostic value of pSer784-VCP using the monoclonal antibody will be crucial for translating it into a clinically useful biomarker.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-pSer137-Pfn1 | Previous Paper (Shao et al., 2008) | N/A |
| Mouse monoclonal anti-pSer784-VCP, clone (3E4) | This Paper | N/A |
| Rabbit monoclonal anti-GFP | Cell Signaling Technology | Cat#2956; RRID: AB_1196615 |
| Mouse monoclonal anti-VCP | Santa Cruz | Cat#sc-57492; RRID: AB_793927 |
| Cat#sc-136273; RRID: AB_2214651 | ||
| Mouse monoclonal anti-actin | Santa Cruz | Cat#sc-47778; RRID: AB_626632 |
| Mouse monoclonal anti-GAPDH | Santa Cruz | Cat#sc-365062; RRID: AB_10847862 |
| Rabbit monoclonal anti-Histone H3 | Cell Signaling Technology | Cat#4499; RRID: AB_10544537 |
| Rabbit monoclonal anti-γH2AX | Cell Signaling Technology | Cat#9718; RRID: AB_2118009 |
| Mouse monoclonal anti-Ku70 | Santa Cruz | Cat#sc-17789; RRID: AB_628454 |
| Mouse monoclonal anti-ATM | Santa Cruz | Cat#sc-377293 |
| Rabbit monoclonal anti-pSer1981-ATM | Cell Signaling Technology | Cat#5883; RRID: AB_10835213 |
| Rabbit polyclonal anti-pSer428-ATR | Cell Signaling Technology | Cat#2853; RRID: AB_2290281 |
| Mouse monoclonal anti-Chk2 | Cell Signaling Technology | Cat#3440; RRID: AB_2229490 |
| Rabbit monoclonal anti-pThr68-Chk2 | Cell Signaling Technology | Cat#2197; RRID: AB_2080501 |
| Rabbit monoclonal anti-pSer345-Chk1 | Cell Signaling Technology | Cat#2348; RRID: AB_331212 |
| Rabbit monoclonal anti-NBS1 | Cell Signaling Technology | Cat#14956; RRID: AB_2798660 |
| Rabbit polyclonal anti-pSer343-NBS1 | Cell Signaling Technology | Cat#3001; RRID: AB_10829154 |
| Rabbit polyclonal anti-K48-ubiquitin | Cell Signaling Technology | Cat#4289; RRID: AB_10557239 |
| Rat monoclonal anti-FLAG tag | BioLegend | Cat#637301; RRID: AB_1134266 |
| Rabbit polyclonal anti-NPL4 | Bethyl Laboratories | Cat#A304-102A; RRID: AB_2621351 |
| Rabbit polyclonal anti-UFD1 | Bethyl Laboratories | Cat#A301-875A; RRID: AB_1309801 |
| Mouse monoclonal anti-CHOP | Cell Signaling Technology | Cat#2895; RRID: AB_2089254 |
| Rabbit monoclonal anti-BiP | Cell Signaling Technology | Cat#3177; RRID: AB_2119845 |
| Rabbit monoclonal anti-phospho-ATM/ATR substrate motif | Cell Signaling Technology | Cat# 6966, RRID: AB_10949894 |
| Rabbit monoclonal anti-Pfn1 | Cell Signaling Technology | Cat#3246; RRID: AB_2163185 |
| Mouse monoclonal anti-BRCA1 | Santa Cruz | Cat#sc-6954; RRID: AB_626761 |
| Mouse monoclonal Anti-Human 53BP1(Clone19) | BD Biosciences | Cat#612522; RRID: AB_2206766 |
| Mouse anti-tubulin | Sigma | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-PELP1 | Bethyl Laboratories | Cat# A300-180A |
| Rabbit monoclonal anti-ATR | Cell Signaling Technology | Cat# 13934; RRID: AB_2798347 |
| Mouse monoclonal anti-HIF1 alpha | Cell Signaling Technology | Cat# 79233; RRID: AB_2799924 |
| Rabbit monoclonal anti-K48-linked polyubiquitin | Cell Signaling Technology | Cat# 8081; RRID: AB_10859893 |
| Mouse monoclonal anti-pSer1981-ATM | Rockland | Cat# 200-301-400; RRID: AB_217868 |
| Bacterial and Virus Strains | ||
| Stbl2 competent cells | Thermo Fisher | Cat#: 10268019 |
| Stbl3 competent cells | Thermo Fisher | Cat#: C737303 |
| DH5 alpha competent cells | Thermo Fisher | Cat#: 18258012 |
| XL10-Gold ultracompetent cells | Agilent | Cat#: 200314 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Etoposide | Selleckchem | Cat#S1225; CAS:33419-42-0 |
| KU55933 | Selleckchem | Cat#S1092; CAS:587871-26-9 |
| Niraparib | Selleckchem | Cat#S7625; CAS:038915-73-9 |
| Olaparib | Selleckchem | Cat#S1060; CAS:763113-22-0 |
| Cisplatin | Selleckchem | Cat#S1166; CAS:15663-27-1 |
| Gemcitabine | Selleckchem | Cat#S1714; CAS:95058-81-4 |
| Hydroxyurea | Sigma | Cat#H8627; CAS:27-07-1 |
| 5-Fluoracil | Sigma | Cat#F6627; CAS:51-21-8 |
| SN38 | Sigma | Cat#H1065; CAS:86639-52-3 |
| NMS-873 | Selleckchem | Cat#S7285; CAS:1418013-75-8 |
| MG-132 | Selleckchem | Cat# S2619; CAS:133407-82-6 |
| Critical Commercial Assays | ||
| ImmPACT DAB kit | Vectorlabs | Cat#SK-4105 |
| Single Cell Gel Electrophoresis Assay | Trevigen | Cat#4250-050-K |
| Experimental Models: Cell Lines | ||
| Human: T47D | ATCC | HTB-133 |
| Human: HCC1806 | ATCC | CRL-2335 |
| Human: HeLa | ATCC | CCL-2 |
| Human: HEK293T | ATCC | CRL-11268 |
| Human: U2OS | ATCC | HTB-96 |
| Human: MDA-MB-231 | ATCC | HTB-26 |
| Human: BT549 | ATCC | HTB-122 |
| Oligonucleotides | ||
| shLuc: ATATCCGAGTGTAGTAAACATT | This Paper | N/A |
| shPfn1#1: GTGGTTTGATCAACAAGAA | (Diamond et al., 2015) | N/A |
| shPfn1#2: TACGTGAATGGGCTGACACTT | (Diamond et al., 2015) | N/A |
| The primers for VCP S784A: sense TGGAGCTGGCCCCGCTCAGGGCAGTGGA | This Paper | N/A |
| The primer for VCP S784D: sense TGGAGCTGGCCCCGATCAGGGCAGTGGA | This Paper | N/A |
| The primers for VCP S784A: antisense TCCACTGCCCTGAGCGGGGCCAGCTCCA | This Paper | N/A |
| The primer for VCP S784D: antisense TCCACTGCCCTGATCGGGGCCAGCTCCA | This Paper | N/A |
| shVCP#1: CCTGATGTGAAGTACGGCAAA | This Paper | N/A |
| shVCP#2: AGGGAGGTAGATATTGGAATT | This Paper | N/A |
| Recombinant DNA | ||
| VCP(WT)-GFP (shVCP#1,Resistant) | This Paper | N/A |
| VCP(WT)-GFP (shVCP#2,Resistant) | This Paper | N/A |
| VCP(WT)-EGFP | Addgene | Cat#23971; RRID: Addgene_23971 |
| pEGFP-C1 | Clontech (discontinued) | https://www.addgene.org/vector-database/2487/ |
| pFLRu-NYFP-FH | Feng et al., 2010 | N/A |
| VCP(WT)-FLAG | This Paper | N/A |
| VCP(S784A)-EGFP | This Paper | N/A |
| VCP(S784D)-EGFP | This Paper | N/A |
| VCP(S326A)-EGFP | This paper | N/A |
| VCP(S652A)-EGFP | This paper | N/A |
| VCP(S746A,S748A)-EGFP | This paper | N/A |
| pFLRu-FH vector | (Feng et al., 2010) | N/A |
| VCP(S664A)-EGFP | This paper | N/A |
| VCP(S784A)-FLAG | This paper | N/A |
| VCP(S784D)-FLAG | This paper | N/A |
| VCP(S784A)-GFP (shVCP#1,Resistant) | This paper | N/A |
| VCP(S784A)-GFP (shVCP#2,Resistant) | This paper | N/A |
| VCP(S784D)-GFP (shVCP#1,Resistant) | This paper | N/A |
| VCP(S784D)-GFP (shVCP#2, Resistant) | This paper | N/A |
| Software and Algorithms | ||
| R software | The R foundation | N/A |
| SPSS version 23 | IBM corporation | N/A |
| ImageJ with the OpenComet plugin | (Gyori et al., 2014) | http://www.cometbio.org/index.html |
| GraphPad Prism 7 | GraphPad software | N/A |
| Cellsens software | Olympus Lifescience | N/A |
| Other | ||
| SignalStain Boost IHC Detection Reagent, mouse | Cell Signaling Technology | Cat#: 8125 |
| SignalStain Boost IHC Detection Reagent, rabbit | Cell Signaling Technology | Cat#: 8114 |
| RIPA lysis buffer | Cell Signaling Technology | Cat#: 9806 |
| HiTrap protein G column | GE healthcare | cat#: 17040403 |
| Mouse monoclonal anti-FLAG M2 affinity gel | Sigma | Cat#: A2220 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jieya Shao (shao.j@wustl.edu)
Materials Availability
Reagents generated in this study will be made available on request with a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate any unique datasets or code.
Experimental Model And Subject Details
All cell lines were originally purchased from ATCC. HeLa, HEK293T, and U2OS cells were grown in high glucose DMEM supplemented with 5% or 10% fetal bovine serum and 50 μg/mL gentamicin. MDA-MB-231, BT549, T47D, and HCC1806 were grown in RPMI 1640 containing 5% or 10% fetal bovine serum (FBS) with gentamicin and supplements (50 μg/mL gentamycin, 1mM sodium pyruvate, 10 mM HEPES and glucose to 4.5 g/L). Transient transfection was performed using Fugene HD or Lipofectamine 2000. Lentiviruses were generated using HEK293T cells as previously described (Diamond et al., 2015). Stable HeLa and U2OS cells expressing GFP or VCP-GFP (WT and mutants) were generated by transfection, 2 weeks of G418 (0.8μg/ml) selection, and FACS. Stable MDA-MB-231 cells expressing YFP-FLAG and VCP-FLAG (WT and mutants) were generated by viral lentivirus and puromycin selection.
Method Details
Antibodies
Custom-made polyclonal rabbit pSer137-Pfn1 antibody was generated previously (Shao et al., 2008). The monoclonal pSer784-VCP antibody was custom generated by Genscript. Briefly, a peptide (GGAGPpSQGSGGGTGC, C-terminal cysteine was added) containing phospho-Ser784 of VCP was synthesized, KLH conjugated, used to immunize BALB/c mice. Two rounds of cell fusions were performed followed by screening of 20,000 clones by indirect ELISA (using the antigenic phospho-peptide and the control peptide without the phosphate on Ser784). Five positive clones were selected for subcloning and supernatants of each clone were tested for pSer784-VCP specificity by western blot. One clone (3E4) was selected, expanded, and grown in serum-free hydridoma-SFM media (GIBCO, cat # 12045-076) followed by purification using the HiTrap protein G column according to manufacturer’s protocol (GE healthcare, cat # 17040403). Commercial antibodies used for western blot include GFP (CST, #2956), VCP (Santa Cruz, sc-57492 and sc-136273), actin (Santa Cruz, sc-47778), GAPDH (Santa Cruz, sc-365062), histone H3 (CST, #4499), tubulin (Sigma, T9026), PELP1 (Bethyl, #A300-180A), γH2AX (CST, #9718), Ku70 (Santa Cruz, sc-17789), ATM (Santa Cruz, sc-377293), pSer1981-ATM (CST, #5883; Rockland, #200-301-400), pSer428-ATR (CST, #2853), ATR (CST, #13934), Chk2 (CST, #3440), pThr68-Chk2 (CST, #2197), pSer345-Chk1 (CST, #2348), NBS1 (CST, #14956), pSer343-NBS1 (CST, #3011), K48-ubiquitin (CST, #4289; CST, #8081), FLAG tag (BioLegend, #637301), NPL4 (Bethyl, #A304-102A), UFD1 (Bethyl, #A301-875A), CHOP (CST, #2895), BiP (CST, #3177), phospho-ATM/ATR substrate motif (CST, #6966), Pfn1 (CST, #3246), HIF1α (CST, #79233). Antibodies for immunofluorescence staining include BRCA1 (Santa Cruz, sc-6954), 53BP1 (BD Biosciences, #612522), γH2AX (CST, #9718), VCP (Santa Cruz, sc-57492), pSer1981-ATM (Rockland, #200-301-400)custom-made pSer137-Pfn1, pSer784-VCP, and NBS1 as previously described (You et al., 2005). Commercial antibodies for immunoprecipitation include VCP (sc-57492) and anti-FLAG M2 affinity gel (Sigma, A2220).
Drugs
Drugs were purchased from Selleckchem (NMS-873, S7285; Etoposide, S1225; KU55933, S1092; Niraparib, S7625; Olaparib, S1060; Cisplatin, S1166, Gemcitabine, S1714, MG-132, S2619), Sigma (Hydroxyurea, H8627; 5-Fluoracil, F6627; SN38, H1065).
DNA and shRNA constructs
Human VCP(wt)-GFP in the pEGFP-N1 vector was purchased from Addgene (#23971) (Tresse et al., 2010). VCP was digested out of pEGFP-N1 with BglII (5′) and Age I (3′) and cloned into the same sites in the lentiviral pFLRu-FH vector to generate VCP(wt)-FLAG-6His (Feng et al., 2010). S784A and S784D were introduced using QuickChange site-directed mutagenesis. S784A sense primer: TGGAGCTGGCCCCGCTCAGGGCAGTGGA; antisense primer: TCCACTGCCCTGAGCGGGGCCAGCTCCA. S784D sense primer: TGGAGCTGGCCCCGATCAGGGCAGTGGA; antisense primer: TCCACTGCCCTGATCGGGGCCAGCTCCA. Two human VCP-specific shRNAs in the pLKO.1 vector were purchased from the RNAi consortium at the Genome Institute at Washington University: CCTGATGTGAAGTACGGCAAA (#1) and AGGGAGGTAGATATTGGAATT (#2). To confer shRNA resistance, multiple synonymous mutations were introduced within the target sequences of VCP by Genewiz: CCAGACGTCAAATATGGTAAG (#1) and CGCGAAGTTGAGATAGGAATT (#2). Two human Pfn1-specific shRNAs were described previously (Diamond et al., 2015).
Immunohistochemistry staining and scoring
Formalin-fixed and paraffin-embedded human breast cancer TMAs were purchased from US Biomax or custom generated as described previously (Cheang et al., 2008, Cheang et al., 2009, Graveel et al., 2009, Liu et al., 2012). The UBC series refers to patients diagnosed with invasive breast cancer at the University of British Columbia hospitals between 1989 and 2002 as previously described (Bortnik et al., 2016). The BCCancer series was derived from 4543 samples from patients diagnosed with invasive breast cancer in the province of British Columbia in the period January 1986–September 1992. These cases are linked to well-annotated clinical data regarding age, staging, histology, pathological factors, long-term follow-up and information on adjuvant systemic therapy as previously published (Cheang et al., 2006, Liu et al., 2012). Tissues were subjected to standard rehydration and antigen retrieval by boiling in 10mM Tris-HCl, pH 9.0 for 10min. Following quenching with 3% hydrogen peroxide for 10min, they were washed with TBS/0.1% Tween 20 (TBST), and blocked for 1hr at room temperature with 5% normal goat serum and 4% BSA in TBST, and incubated with the primary antibodies (pSer137-Pfn1 at 1:1000; pSer784-VCP at 1:50) in blocking buffer at 4°C overnight. After TBST wash, they were incubated with HRP-conjugated secondary antibodies (SignalStain Boost IHC Detection Reagent Mouse, #8125 and rabbit, #8114) for 2hr at room temperature, and washed with TBST. They were developed using the ImmPACT DAB kit (Vectorlabs, SK-4105), counterstained with hematoxylin, dehydrated, and mounted. Bright field images were taken on an upright BX51 fluorescence microscope using the CellSens software. Nuclear staining was scored using the Allred scoring system, defined as the sum of the scores for average intensity (0: non, 1: weak, 2: moderate, 3: strong) and proportion of positive nuclei (0: none, 1: < 1%, 2: 1-10 %, 3: 10%–33%, 4: 33-66 %, 5: > 67%) scores (Harvey et al., 1999).
Immunofluorescence staining and image acquisition
Cultured cells grown on plastic or poly-D-lysine-coated glass coverslips were treated with vehicle (H2O or DMSO) or DNA damaging agents (in H2O or DMSO) for various times, rinsed with PBS, and fixed by 4% paraformaldehyde at room temperature for 20min. For detergent pre-extraction, cells were incubated with cytoskeletal buffer (25mM HEPES, pH 7.5, 50mM NaCl, 1mM EDTA, 3mM MgCl2, 300mM sucrose and 0.5% Triton X-100) for 1-5min, followed by PBS rinse and fixation with 4% paraformaldehyde. They were washed 4x with PBS/0.1% Triton X-100, blocked with 5% normal goat serum and 2% bovine serum albumin (BSA) in PBS/0.1% Triton X-100. Primary antibodies (pSer137-Pfn1 at 1:1000; pSer784-VCP at 1:500; 53BP1 at 1:1000; γH2AX at 1:1000; BRCA1 at 1:1000; NBS1 at 1:500; pSer1981-ATM at 1:1000 in the same blocking buffer were added and incubated at 4°C overnight. Cells were washed 4x with PBS/0.1% Triton X-100, and incubated with Alexa 488 or 594-conjugated secondary antibodies in the same blocking buffer for 2hr at room temperature. They were subsequently washed, counterstained with DAPI (1μg/ml), and mounted using the ProLong Gold antifade reagent (Invitrogen). Fluorescent images were acquired on an inverted Olympus IX70 and an upright BX51 fluorescence microscopes using the CellSens software.
Immunoprecipitation
Cells (5-10 × 106) were harvested by scraping in PBS, and pelleted at 500 g for 5min at 4°C. For native conditions, they were lysed by 200-500μl ice-cold 1x RIPA buffer (CST, #9806) with added protease (Pierce, A32955) and phosphatase inhibitors (Pierce, A32957) or fractionated to obtain the nucleoplasm (see below for details). For denaturing conditions, they were suspended in ∼50μl pre-heated lysis buffer (1% SDS, 20mM Tris-HCl, pH 7.4, 150mM NaCl), heated at 95°C for 10min, and diluted 20-fold with ice cold buffer containing 20mM Tris-HCl, pH 7.4, 150mM NaCl, protease and phosphatase inhibitors. Samples under native or denatured conditions were clarified at > 16,000 g at 4°C for 10min and supernatants were mixed with agarose pre-conjugated with various primary antibodies (10-20μl agarose with 1-10μg antibodies per sample) for 2-4hr at 4°C by gentle rotation. For sequential immunoprecipitations, supernatant from the first pulldown was collected by gently pelleting the beads (500 g, 5min), and added to the second antibody. Beads were washed and samples were denatured in SDS sample buffer.
Mass spectrometry
The unknown ∼100kDa protein band induced by DNA damage and immunoprecipitated by the pSer137-Pfn1 antibody was excised from silver-stained SDS gel and subjected to standard in-gel trypsin digest as previously described (Held et al., 2013). Tryptic peptides were analyzed on a TripleTOF 5600 nano LC-MS system (AB SCIEX). Mass spectrometry data were searched against the Uniprot Human Reference Proteome using Mascot to identify proteins. Search parameters were 25 ppm mass tolerance (MS1), 0.1 dalton (MS2), 2 missed cleavages and trypsin/P protease specificity, fixed carbamidomethylation (Cys) and variable methionine oxidation and protein N-terminal acetylation.
Subcellular fractionation
Cells were scraped from tissue culture dishes after rinsing with PBS and centrifuged for 5min at 500 g at 4°C, and subjected to fractionation using the protocol described by Méndez and Stillman, 2000. Briefly, cell pellets (2-5 × 106) were resuspended in 200μl buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M Sucrose, 10 % Glycerol, 1 mM DTT, Protease and Phosphatase inhibitor cocktails), and Triton X-100 was added to a final concentration of 0.1%. After 8min incubation on ice, samples were centrifuged at 1,300 g at 4°C for 5min, and supernatant was removed and saved as cytosolic fractions. Pellets were washed once with buffer A without Triton X-100 and centrifuged at 1,300 g again at 4°C for 5min. Supernatant was removed and discarded, and 100μl buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, Protease and Phosphatase inhibitor cocktails) was used to resuspend the pellets. Samples were incubated on ice for 30min with intermittent mixing, and centrifuged at 1,700 g at 4°C for 5min. Supernatant was removed and saved as the nucleoplasmic fraction. Pellets were washed once with buffer B and centrifuged again at 1,700 g at 4°C for 5min. Supernatant was discarded and pellets were solubilized in SDS sample buffer as the chromatin fraction.
Laser Microirradiation
A customized laser microirradiation system consisting of an inverted microscope (Nikon), a laser ablation unit (Photonic Instruments) and microscope automation and imaging software (Metamorph, Molecular Devices) was used. To introduce DNA damage to cells, a 551-nm dye laser was directed to irradiate cells cultured on 35-mm glass-bottomed dishes (MatTek Cultureware, P35G-15-14-C) in a line pattern. The laser energy delivered to each focused spot was set by an attenuator plate (50% transmission) and the number of pulses per spot. Cells were laser irradiated at the confluency of 50%, fixed, and immunofluorescence stained at different time points following irradiation as previously described (Chen et al., 2013, You et al., 2005). Total VCP and pSer784-VCP primary antibodies were used at 1:150, and NBS1 antibodies were used at 1:500. Cells were imaged using the Nikon microscope and MetaMorph software.
Comet assay
The experiment was performed using the kit for Single Cell Gel Electrophoresis Assay from Trevigen (cat # 4250-050-K). In brief, cells were diluted to 1 × 105/ml in PBS, combined with pre-melt LMAgarose (at 37°C) at a ratio of 1:10 (v/v) and immediately pipetted onto CometSlide (50 μl). Slides were placed flat at 4°C in the dark for 30min for sample adherence and subsequently immersed in 4°C lysis solution (cat # 4250-050-01) for 2h. Excess lysis buffer was drained, and slides were immersed in freshly prepared alkaline unwinding solution, pH > 13 (200mM NaOH, 1mM EDTA) for 20 min at RT. They were then subjected to gel electrophoresis using with the same alkaline electrophoresis solution at 21 V for 30 minutes. Slides were washed twice with dH2O and then 70% ethanol for 5 minutes. Samples were kept at 37°C until the gel circle was completely melted and stained with 100 μL 1xSYBR® Gold (diluted in TE Buffer, pH 7.5 containing 10 mM Tris-HCl pH 7.5 and 1 mM EDTA) for 30 min at RT. Slides were completely dried at 37°C. Fluorescent images were taken using the Cellsens software on a 10x objective of an Olympus IX70 microscope and analyzed by ImageJ under the OpenComet plugin (Gyori et al., 2014). Data were expressed by the tail DNA percentage (tail intensity out of total DNA intensity).
Colony formation assays
Cells were plated at low density (100-1000) in 24-well plates, allowed to adhere for several hours to overnight, and treated in triplicates with various drugs for overnight unless otherwise specified. They were washed twice with PBS and maintained in drug-free growth media for 10-14 days during which viable cells were periodically quantified using the Alamar blue assay. At the end of the experiments, cells were fixed by 4% paraformaldehyde and stained by 0.001% crystal violet followed by bright field image acquisition. Colonies were manually counted or semi-automatically quantified based on colony area (Guzmán et al., 2014).
Quantification and Statistical Analysis
Chi square or Fisher exact tests were used to examine associations between Allred scores of the nuclear staining with the pSer137-Pfn1 antibody and standard clinicopathological variables. For univariate survival analyses, RFS, OS and BCSS were quantified by Kaplan–Meier curves in the whole group and among different subgroups stratified according to estrogen receptor status, IHC subtype, or treatment. OS was defined as time from diagnosis to either death from any cause or to last follow-up. BCSS was defined as time from diagnosis to either death caused by breast cancer or to last follow-up. RFS was defined as time from diagnosis to any recurrence or to last follow-up. Significance of differences in survival was assessed by log-rank and Wilcoxon tests. For multivariate analyses, Cox proportional hazards models were used to assess the hazard ratio (HR) of different prognostic covariates with survival. These covariates included pathological age at diagnosis > 50 years versus ≤ 50 years, tumor grade (1 or 2) versus 3, nodal status negative versus positive, tumor size T1 versus (T2 or T3), lympho-vascular invasion (negative versus positive), ER status (negative versus positive), PR status (negative versus positive), HER2 status (negative versus positive). p < 0.05 was considered as statistically significant. Analyses including multiple comparisons were adjusted for multiple testing using the Benjamini-Hochberg FDR method. Numbers of the evaluable cases used in the final correlative analyses were depicted in the figures and tables. All statistical analyses were performed using R software (version 3.5.2) and SPSS version 23.
The analysis of the BC Cancer (BCCancer) series as described in the results section followed a training/validation approach used to cross-validate findings and to avoid overfitting that could result from data-driven cut-point determination. This series was originally divided into training (n = 2003) and validation (n = 1989) sets and each set was randomly selected to represent approximately 50% of the total study population with balanced clinicopathological characteristics. Optimal cut-points determination and biomarker analysis in association with clinical outcomes were first assessed on the training set to generate a specific and limited set of hypotheses for testing. After presentation at a minuted research group meeting, these were subsequently tested on the validation set by a research team member different from the one who did the training set analysis. Only results that were found to be significant in both training and validation sets were used for data interpretation and conclusions. For the robustness of our results and to ensure that they are not affected by properties specific to the pre-specified training/validation sets, the split was repeated 500 multiple times randomly.
Unpaired Student’s t test was used to compare the effects between wild-type and mutant VCP on cell survival, pSer1981-ATM foci formation, and levels of DNA break. P values < 0.05 were considered statistically significant. Linear regression was used to compare the staining patterns between pSer137-Pfn1 and pSer784-VCP antibodies, with the R2 value reflecting the goodness of the fit. Details can be found in figure legends.
Acknowledgments
We thank Dr. Matthew Ellis, Dr. Sherri Davies, and Jacqueline Snider for providing the SPECS TMA. We thank Dr. Craig Allred and Lin Li for help with the IHC. We thank Dr. Nima Mosammaparast and Andrea Byrum for help with DNA-damage foci analysis. J.S. was supported by the Longer Life Foundation Research Development Award (2014-006), Susan G. Komen Foundation Career Catalyst Research Grant (CCR14300139), Siteman Investment Program Pre-R01 Award (3948), and a National Cancer Institute R01 grant (5R01CA181671). Z.Y. is supported by a National Institute of General Medical Sciences (NIGMS) R01 grant (GM098535).
Author Contributions
J.S. conceived and designed the experiments. T.O.N. provided essential reagents and supported the design and execution of the patient outcome studies. C.Z., A.R., S.L., J.Z., J.M.H., and J.S. performed the experiments, including tissue and cell staining, image acquisition, cloning, cell line maintenance and generation, antibody characterization and purification, laser micro-irradiation, mass spectrometry, comet assays, colony formation, and western blotting. K.A., S.L., and J.W. performed the pathological and clinical data statistical analysis of the tissue microarrays. D.G. and K.R.V. performed the tissue scoring. C.Z., A.R., K.A., J.W., and J.S. analyzed the data. J.S. wrote the manuscript. Z.Y. and J.M.H. provided technical and intellectual input on the project. All authors read and edited the manuscript.
Declaration of Interests
A provisional patent has been filed by J.S. for the monoclonal pSer784-VCP antibody for its potential value as a prognostic and predictive cancer biomarker. T.O.N. had a role in the development of the PAM50 gene-expression classifier, which has been licensed to Veracyte Technologies.
Published: June 9, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107745.
Supplemental Information
References
- Acs K., Luijsterburg M.S., Ackermann L., Salomons F.A., Hoppe T., Dantuma N.P. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 2011;18:1345–1350. doi: 10.1038/nsmb.2188. [DOI] [PubMed] [Google Scholar]
- Alexandru G., Graumann J., Smith G.T., Kolawa N.J., Fang R., Deshaies R.J. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell. 2008;134:804–816. doi: 10.1016/j.cell.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D.J., Le Moigne R., Djakovic S., Kumar B., Rice J., Wong S., Wang J., Yao B., Valle E., Kiss von Soly S. Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell. 2015;28:653–665. doi: 10.1016/j.ccell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi P., Foiani M., Kumar A. ATM and ATR signaling at a glance. J. Cell Sci. 2016;129:1285. doi: 10.1242/jcs.188631. [DOI] [PubMed] [Google Scholar]
- Banerjee S., Bartesaghi A., Merk A., Rao P., Bulfer S.L., Yan Y., Green N., Mroczkowski B., Neitz R.J., Wipf P. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science. 2016;351:871–875. doi: 10.1126/science.aad7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford A.N., Jackson S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell. 2017;66:801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- Bortnik S., Choutka C., Horlings H.M., Leung S., Baker J.H., Lebovitz C., Dragowska W.H., Go N.E., Bally M.B., Minchinton A.I. Identification of breast cancer cell subtypes sensitive to ATG4B inhibition. Oncotarget. 2016;7:66970–66988. doi: 10.18632/oncotarget.11408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandsma I., Fleuren E.D.G., Williamson C.T., Lord C.J. Directing the use of DDR kinase inhibitors in cancer treatment. Expert Opin. Investig. Drugs. 2017;26:1341–1355. doi: 10.1080/13543784.2017.1389895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann K., Schell M., Hoppe T., Kashkar H. Regulation of the DNA damage response by ubiquitin conjugation. Front. Genet. 2015;6:98. doi: 10.3389/fgene.2015.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J.S., Jackson S.P. Ubiquitylation, neddylation and the DNA damage response. Open Biol. 2015;5:150018. doi: 10.1098/rsob.150018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N.J., Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Cheang M.C., Treaba D.O., Speers C.H., Olivotto I.A., Bajdik C.D., Chia S.K., Goldstein L.C., Gelmon K.A., Huntsman D., Gilks C.B. Immunohistochemical detection using the new rabbit monoclonal antibody SP1 of estrogen receptor in breast cancer is superior to mouse monoclonal antibody 1D5 in predicting survival. J. Clin. Oncol. 2006;24:5637–5644. doi: 10.1200/JCO.2005.05.4155. [DOI] [PubMed] [Google Scholar]
- Cheang M.C., Voduc D., Bajdik C., Leung S., McKinney S., Chia S.K., Perou C.M., Nielsen T.O. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res. 2008;14:1368–1376. doi: 10.1158/1078-0432.CCR-07-1658. [DOI] [PubMed] [Google Scholar]
- Cheang M.C., Chia S.K., Voduc D., Gao D., Leung S., Snider J., Watson M., Davies S., Bernard P.S., Parker J.S. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst. 2009;101:736–750. doi: 10.1093/jnci/djp082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Paudyal S.C., Chin R.I., You Z. PCNA promotes processive DNA end resection by Exo1. Nucleic Acids Res. 2013;41:9325–9338. doi: 10.1093/nar/gkt672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuma N.P., van Attikum H. Spatiotemporal regulation of posttranslational modifications in the DNA damage response. EMBO J. 2016;35:6–23. doi: 10.15252/embj.201592595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis E.J., Lachaud C., Appleton P., Macartney T.J., Näthke I., Rouse J. DVC1 (C1orf124) recruits the p97 protein segregase to sites of DNA damage. Nat. Struct. Mol. Biol. 2012;19:1093–1100. doi: 10.1038/nsmb.2394. [DOI] [PubMed] [Google Scholar]
- Diamond M.I., Cai S., Boudreau A., Carey C.J., Jr., Lyle N., Pappu R.V., Swamidass S.J., Bissell M., Piwnica-Worms H., Shao J. Subcellular localization and Ser-137 phosphorylation regulate tumor-suppressive activity of profilin-1. J. Biol. Chem. 2015;290:9075–9086. doi: 10.1074/jbc.M114.619874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant S.T., Zheng L., Wang Y., Chen K., Zhang L., Zhang T., Yang Z., Riches L., Trinidad A.G., Fok J.H.L. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv. 2018;4:eaat1719. doi: 10.1126/sciadv.aat1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerton M., Ashe O.R., Chen D., Druker B.J., Burgess W.H., Samelson L.E. VCP, the mammalian homolog of cdc48, is tyrosine phosphorylated in response to T cell antigen receptor activation. EMBO J. 1992;11:3533–3540. doi: 10.1002/j.1460-2075.1992.tb05436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H., McCabe N., Lord C.J., Tutt A.N., Johnson D.A., Richardson T.B., Santarosa M., Dillon K.J., Hickson I., Knights C. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Feng Y., Nie L., Thakur M.D., Su Q., Chi Z., Zhao Y., Longmore G.D. A multifunctional lentiviral-based gene knockdown with concurrent rescue that controls for off-target effects of RNAi. Genomics Proteomics Bioinformatics. 2010;8:238–245. doi: 10.1016/S1672-0229(10)60025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz A., Orth M., Pirson P.A., Sonneville R., Blow J.J., Gartner A., Stemmann O., Hoppe T. CDC-48/p97 coordinates CDT-1 degradation with GINS chromatin dissociation to ensure faithful DNA replication. Mol. Cell. 2011;44:85–96. doi: 10.1016/j.molcel.2011.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich K.U., Fries H.W., Rüdiger M., Erdmann R., Botstein D., Mecke D. Yeast cell cycle protein CDC48p shows full-length homology to the mammalian protein VCP and is a member of a protein family involved in secretion, peroxisome formation, and gene expression. J. Cell Biol. 1991;114:443–453. doi: 10.1083/jcb.114.3.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullbright G., Rycenga H.B., Gruber J.D., Long D.T. p97 Promotes a Conserved Mechanism of Helicase Unloading during DNA Cross-Link Repair. Mol. Cell. Biol. 2016;36:2983–2994. doi: 10.1128/MCB.00434-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya K., Miyabe I., Tsutsui Y., Paderi F., Kakusho N., Masai H., Niki H., Carr A.M. DDK phosphorylates checkpoint clamp component Rad9 and promotes its release from damaged chromatin. Mol. Cell. 2010;40:606–618. doi: 10.1016/j.molcel.2010.10.026. [DOI] [PubMed] [Google Scholar]
- Ghosh S., Saha T. Central role of ubiquitination in genome maintenance: DNA replication and damage repair. ISRN Mol. Biol. 2012;2012:146748. doi: 10.5402/2012/146748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldhirsch A., Wood W.C., Coates A.S., Gelber R.D., Thurlimann B., Senn H.J., Panel M. Strategies for subtypes–dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. 2011;22:1736–1747. doi: 10.1093/annonc/mdr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi A.A., Noon A.T., Deckbar D., Ziv Y., Shiloh Y., Löbrich M., Jeggo P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- Graveel C.R., DeGroot J.D., Su Y., Koeman J., Dykema K., Leung S., Snider J., Davies S.R., Swiatek P.J., Cottingham S. Met induces diverse mammary carcinomas in mice and is associated with human basal breast cancer. Proc. Natl. Acad. Sci. USA. 2009;106:12909–12914. doi: 10.1073/pnas.0810403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán C., Bagga M., Kaur A., Westermarck J., Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE. 2014;9:e92444. doi: 10.1371/journal.pone.0092444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyori B.M., Venkatachalam G., Thiagarajan P.S., Hsu D., Clement M.V. OpenComet: an automated tool for comet assay image analysis. Redox Biol. 2014;2:457–465. doi: 10.1016/j.redox.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey J.M., Clark G.M., Osborne C.K., Allred D.C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 1999;17:1474–1481. doi: 10.1200/JCO.1999.17.5.1474. [DOI] [PubMed] [Google Scholar]
- Held J.M., Schilling B., D’Souza A.K., Srinivasan T., Behring J.B., Sorensen D.J., Benz C.C., Gibson B.W. Label-free quantitation and mapping of the ErbB2 tumor receptor by multiple protease digestion with data-dependent (MS1) and data-independent (MS2) acquisitions. Int. J. Proteomics. 2013;2013:791985. doi: 10.1155/2013/791985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Her N.G., Toth J.I., Ma C.T., Wei Y., Motamedchaboki K., Sergienko E., Petroski M.D. p97 Composition changes caused by allosteric inhibition are suppressed by an on-target mechanism that increases the enzyme’s ATPase activity. Cell Chem. Biol. 2016;23:517–528. doi: 10.1016/j.chembiol.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson I., Zhao Y., Richardson C.J., Green S.J., Martin N.M., Orr A.I., Reaper P.M., Jackson S.P., Curtin N.J., Smith G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- Kuo C.Y., Li X., Stark J.M., Shih H.M., Ann D.K. RNF4 regulates DNA double-strand break repair in a cell cycle-dependent manner. Cell Cycle. 2016;15:787–798. doi: 10.1080/15384101.2016.1138184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J.R., Fu V., Wirtz E., Bangs J.D. Functional analysis of the trypanosomal AAA protein TbVCP with trans-dominant ATP hydrolysis mutants. J. Biol. Chem. 2001;276:21512–21520. doi: 10.1074/jbc.M100235200. [DOI] [PubMed] [Google Scholar]
- León A., McKearin D. Identification of TER94, an AAA ATPase protein, as a Bam-dependent component of the Drosophila fusome. Mol. Biol. Cell. 1999;10:3825–3834. doi: 10.1091/mbc.10.11.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Zhao G., Zhou X., Schindelin H., Lennarz W.J. The AAA ATPase p97 links peptide N-glycanase to the endoplasmic reticulum-associated E3 ligase autocrine motility factor receptor. Proc. Natl. Acad. Sci. USA. 2006;103:8348–8353. doi: 10.1073/pnas.0602747103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Lachapelle J., Leung S., Gao D., Foulkes W.D., Nielsen T.O. CD8+ lymphocyte infiltration is an independent favorable prognostic indicator in basal-like breast cancer. Breast Cancer Res. 2012;14:R48. doi: 10.1186/bcr3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingstone M., Ruan H., Weiner J., Clauser K.R., Strack P., Jin S., Williams A., Greulich H., Gardner J., Venere M. Valosin-containing protein phosphorylation at Ser784 in response to DNA damage. Cancer Res. 2005;65:7533–7540. doi: 10.1158/0008-5472.CAN-04-3729. [DOI] [PubMed] [Google Scholar]
- Lord C.J., Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- Magnaghi P., D’Alessio R., Valsasina B., Avanzi N., Rizzi S., Asa D., Gasparri F., Cozzi L., Cucchi U., Orrenius C. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol. 2013;9:548–556. doi: 10.1038/nchembio.1313. [DOI] [PubMed] [Google Scholar]
- Maric M., Maculins T., De Piccoli G., Labib K. Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science. 2014;346:1253596. doi: 10.1126/science.1253596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S., Ballif B.A., Smogorzewska A., McDonald E.R., 3rd, Hurov K.E., Luo J., Bakalarski C.E., Zhao Z., Solimini N., Lerenthal Y. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- Méndez J., Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell. Biol. 2000;20:8602–8612. doi: 10.1128/mcb.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H., Weihl C.C. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J. Cell Sci. 2014;127:3877–3883. doi: 10.1242/jcs.093831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H.H., Kondo H., Warren G. The p47 co-factor regulates the ATPase activity of the membrane fusion protein, p97. FEBS Lett. 1998;437:255–257. doi: 10.1016/s0014-5793(98)01232-0. [DOI] [PubMed] [Google Scholar]
- Meyer H., Bug M., Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 2012;14:117–123. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- Moreno S.P., Bailey R., Campion N., Herron S., Gambus A. Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science. 2014;346:477–481. doi: 10.1126/science.1253585. [DOI] [PubMed] [Google Scholar]
- Mosbech A., Gibbs-Seymour I., Kagias K., Thorslund T., Beli P., Povlsen L., Nielsen S.V., Smedegaard S., Sedgwick G., Lukas C. DVC1 (C1orf124) is a DNA damage-targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks. Nat. Struct. Mol. Biol. 2012;19:1084–1092. doi: 10.1038/nsmb.2395. [DOI] [PubMed] [Google Scholar]
- Müller J.M., Deinhardt K., Rosewell I., Warren G., Shima D.T. Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem. Biophys. Res. Commun. 2007;354:459–465. doi: 10.1016/j.bbrc.2006.12.206. [DOI] [PubMed] [Google Scholar]
- Niwa H., Ewens C.A., Tsang C., Yeung H.O., Zhang X., Freemont P.S. The role of the N-domain in the ATPase activity of the mammalian AAA ATPase p97/VCP. J. Biol. Chem. 2012;287:8561–8570. doi: 10.1074/jbc.M111.302778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor M.J. Targeting the DNA damage response in cancer. Mol. Cell. 2015;60:547–560. doi: 10.1016/j.molcel.2015.10.040. [DOI] [PubMed] [Google Scholar]
- Polo S.E., Jackson S.P. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puumalainen M.R., Lessel D., Rüthemann P., Kaczmarek N., Bachmann K., Ramadan K., Naegeli H. Chromatin retention of DNA damage sensors DDB2 and XPC through loss of p97 segregase causes genotoxicity. Nat. Commun. 2014;5:3695. doi: 10.1038/ncomms4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu L., Pashkova N., Walker J.R., Winistorfer S., Allali-Hassani A., Akutsu M., Piper R., Dhe-Paganon S. Structure and function of the PLAA/Ufd3-p97/Cdc48 complex. J. Biol. Chem. 2010;285:365–372. doi: 10.1074/jbc.M109.044685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan K., Halder S., Wiseman K., Vaz B. Strategic role of the ubiquitin-dependent segregase p97 (VCP or Cdc48) in DNA replication. Chromosoma. 2017;126:17–32. doi: 10.1007/s00412-016-0587-4. [DOI] [PubMed] [Google Scholar]
- Raman M., Havens C.G., Walter J.C., Harper J.W. A genome-wide screen identifies p97 as an essential regulator of DNA damage-dependent CDT1 destruction. Mol. Cell. 2011;44:72–84. doi: 10.1016/j.molcel.2011.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpf S., Jentsch S. Functional division of substrate processing cofactors of the ubiquitin-selective Cdc48 chaperone. Mol. Cell. 2006;21:261–269. doi: 10.1016/j.molcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Sarkaria J.N., Busby E.C., Tibbetts R.S., Roos P., Taya Y., Karnitz L.M., Abraham R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- Schuller J.M., Beck F., Lössl P., Heck A.J., Förster F. Nucleotide-dependent conformational changes of the AAA+ ATPase p97 revisited. FEBS Lett. 2016;590:595–604. doi: 10.1002/1873-3468.12091. [DOI] [PubMed] [Google Scholar]
- Shao J., Diamond M.I. Protein phosphatase 1 dephosphorylates profilin-1 at Ser-137. PLoS ONE. 2012;7:e32802. doi: 10.1371/journal.pone.0032802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao J., Welch W.J., Diprospero N.A., Diamond M.I. Phosphorylation of profilin by ROCK1 regulates polyglutamine aggregation. Mol. Cell. Biol. 2008;28:5196–5208. doi: 10.1128/MCB.00079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiotani B., Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell. 2009;33:547–558. doi: 10.1016/j.molcel.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits V.A., Reaper P.M., Jackson S.P. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr. Biol. 2006;16:150–159. doi: 10.1016/j.cub.2005.11.066. [DOI] [PubMed] [Google Scholar]
- Stokes M.P., Rush J., Macneill J., Ren J.M., Sprott K., Nardone J., Yang V., Beausoleil S.A., Gygi S.P., Livingstone M. Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. USA. 2007;104:19855–19860. doi: 10.1073/pnas.0707579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.K., Xia D. Mutations in the Human AAA+ Chaperone p97 and Related Diseases. Front. Mol. Biosci. 2016;3:79. doi: 10.3389/fmolb.2016.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.K., Li D., Li C.C., Esser L., Dai R., Guo L., Xia D. A novel ATP-dependent conformation in p97 N-D1 fragment revealed by crystal structures of disease-related mutants. EMBO J. 2010;29:2217–2229. doi: 10.1038/emboj.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresse E., Salomons F.A., Vesa J., Bott L.C., Kimonis V., Yao T.P., Dantuma N.P., Taylor J.P. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 2010;6:217–227. doi: 10.4161/auto.6.2.11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom J., Wolf M., Weimann L., Schulze N., Li F., Kaschani F., Riemer A., Zierhut C., Kaiser M., Iliakis G. VCP/p97 Extracts sterically trapped Ku70/80 rings from DNA in double-strand break repair. Mol. Cell. 2016;64:189–198. doi: 10.1016/j.molcel.2016.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz B., Halder S., Ramadan K. Role of p97/VCP (Cdc48) in genome stability. Front. Genet. 2013;4:60. doi: 10.3389/fgene.2013.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R., Oania R., Fang R., Smith G.T., Deshaies R.J. Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol. Cell. 2011;41:82–92. doi: 10.1016/j.molcel.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A.M., Ryan A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015;149:124–138. doi: 10.1016/j.pharmthera.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Wu J., Chen Y., Lu L.Y., Wu Y., Paulsen M.T., Ljungman M., Ferguson D.O., Yu X. Chfr and RNF8 synergistically regulate ATM activation. Nat. Struct. Mol. Biol. 2011;18:761–768. doi: 10.1038/nsmb.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z., Chahwan C., Bailis J., Hunter T., Russell P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 2005;25:5363–5379. doi: 10.1128/MCB.25.13.5363-5379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z., Shi L.Z., Zhu Q., Wu P., Zhang Y.W., Basilio A., Tonnu N., Verma I.M., Berns M.W., Hunter T. CtIP links DNA double-strand break sensing to resection. Mol. Cell. 2009;36:954–969. doi: 10.1016/j.molcel.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Gui L., Zhang X., Bulfer S.L., Sanghez V., Wong D.E., Lee Y., Lehmann L., Lee J.S., Shih P.Y. Altered cofactor regulation with disease-associated p97/VCP mutations. Proc. Natl. Acad. Sci. USA. 2015;112:E1705–E1714. doi: 10.1073/pnas.1418820112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G., Zhou X., Wang L., Li G., Schindelin H., Lennarz W.J. Studies on peptide:N-glycanase-p97 interaction suggest that p97 phosphorylation modulates endoplasmic reticulum-associated degradation. Proc. Natl. Acad. Sci. USA. 2007;104:8785–8790. doi: 10.1073/pnas.0702966104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y., Bielopolski D., Galanty Y., Lukas C., Taya Y., Schultz D.C., Lukas J., Bekker-Jensen S., Bartek J., Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006;8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.