

Abstract
The recent outbreak of coronavirus disease-19 (COVID-19) continues to drastically affect healthcare throughout the world. To date, no approved treatment regimen or vaccine is available to effectively attenuate or prevent the infection. Therefore, collective and multidisciplinary efforts are needed to identify new therapeutics or to explore effectiveness of existing drugs and drug-like small molecules against SARS-CoV-2 for lead identification and repurposing prospects. This study addresses the identification of small molecules that specifically bind to any of the three essential proteins (RdRp, 3CL-protease and helicase) of SARS-CoV-2. By applying computational approaches we screened a library of 4574 compounds also containing FDA-approved drugs against these viral proteins. Shortlisted hits from initial screening were subjected to iterative docking with the respective proteins. Ranking score on the basis of binding energy, clustering score, shape complementarity and functional significance of the binding pocket was applied to identify the binding compounds. Finally, to minimize chances of false positives, we performed docking of the identified molecules with 100 irrelevant proteins of diverse classes thereby ruling out the non-specific binding. Three FDA-approved drugs showed binding to 3CL-protease either at the catalytic pocket or at an allosteric site related to functionally important dimer formation. A drug-like molecule showed binding to RdRp in its catalytic pocket blocking the key catalytic residues. Two other drug-like molecules showed specific interactions with helicase at a key domain involved in catalysis. This study provides lead drugs or drug-like molecules for further in vitro and clinical investigation for drug repurposing and new drug development prospects.
Keywords: Drug repurposing, SARS-CoV-2, RdRp, Helicase, Rimantadine, Grazoprevir
Graphical abstract
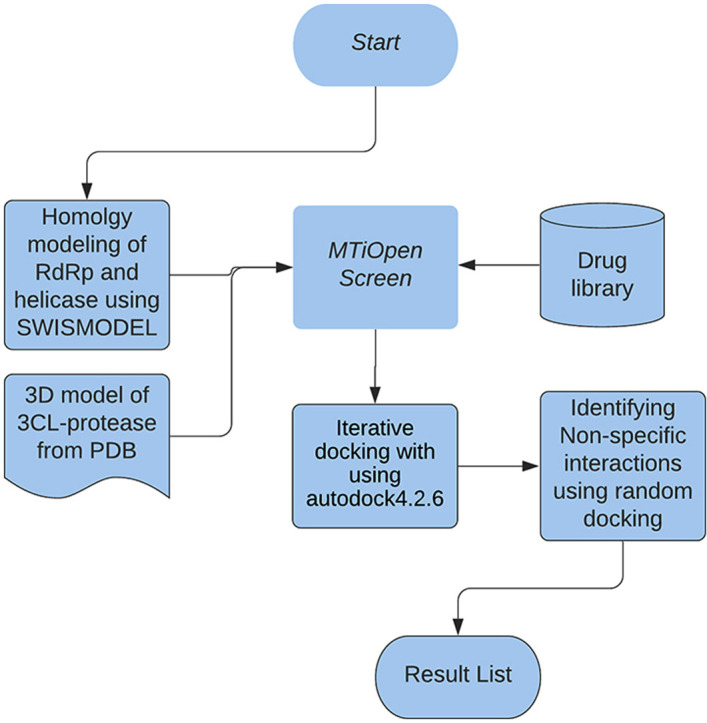
1. Introduction
COVID-19 after its emergence in China, continues to spread across the globe leading to global health emergencies and accounting for devastating economic and social impacts [1,2]. As of April 9, 2020, over 1.7 million people have been confirmed with COVID-19 leading to over 111,000 deaths since the emergence of the infection [3]. To date, no specific treatment regimen is available against COVID-19. However, previously approved antiviral drug Remdesivir and an antimalarial drug Chloroquine have shown promising effects, at least in reducing the duration of symptoms of COVID-19 in limited clinical studies [4,5]. SARS-CoV-2 that causes this disease belongs to the family Coronaviridae and genus Coronavirus [6]. Angiotensin-converting enzyme 2 (ACE2) that is expressed on the cellular surfaces of different organs including lungs has been identified as the primary receptor of the virus in addition to the role of another host protease enzyme, transmembrane serine protease 2 (TMPRSS2) in viral entry [[7], [8], [9]]. The viral genome consists of 30,000 base pairs that encode different structural and non-structural proteins. The structural proteins include the membrane protein, spike, envelope and nucleocapsid. Non-structural proteins include NSP1-NSP16. NSP3 (papain-like protease), NSP5 (3-chymotrypsin-like protease “3-CL-protease”), NSP12 (RNA-dependent RNA polymerase), NSP13 (helicase), NSP14 (N7-methyltransferase) and NSP16 (2′-O-methyl transferase) are important viral enzymes while NSP7-NSP10 are regulatory proteins. The SARS-CoV-2 orf1a and orf1ab genes encode polyprotein 1a (pp1a) and polyprotein 1 ab (pp1ab), respectively. These polyproteins are further processed by protease enzymes to produce different functional proteins. In this regard, 3CL-protease cleaves the polyprotein at 10 different specific positions thereby producing individual functional proteins [10].
SARS-CoV-2 is a positive sense RNA virus that requires RNA-templated RNA synthesis. RNA-dependent RNA polymerase (RdRp) catalyzes the synthesis of new viral RNA and plays a crucial role in the SARS-CoV-2 replication cycle [11]. Similarly, SARS-CoV-2 helicase (NSP13) is also crucial for the viral replication and proliferation as it catalyzes the unwinding of duplex RNA and DNA into single strand nucleic acid chain during replication [12]. Considering an urgent need of containing the pandemic, repurposing of previously approved drugs to use against SARS-CoV-2 is the first choice in the fight against this virus, as such drugs are not required to pass through most of the steps of an extensive drug testing process. In this regard, several recent studies have been conducted using computational methods to screen libraries of approved drugs or drug-like molecules to identify potential inhibitors of different viral proteins, particularly, RdRp and 3CL-protease [[13], [14], [15], [16], [17]]. Moreover, high throughput in vitro screening of FDA approved drug libraries followed by in vivo validation has also been applied on limited hits [18,19]. Many of the reported virtual screening based studies have targeted the FDA approved drugs and/or dug-like compound libraries. Since a vast range of compound libraries exist therefore, multiple efforts to screen diverse compound libraries are important to explore a large chemical landscape to identify potential inhibitors or the lead structures to design inhibitors of SARS-CoV-2. Here, we applied a computer aided drug discovery approach by targeting three important enzymes (RdRp, 3CL-protease and helicase) of SARS-CoV-2 and identified three FDA-approved drugs and three other drug-like molecules as potential therapeutics.
2. Material and methods
2.1. Homology modeling of RdRp and helicase
Structures of SARS-CoV-2 RdRp and helicase were modeled on the basis of amino acid sequence homology using SWISMODEL as crystal structure of these proteins are yet to be solved [20]. The genome sequence of SARS-CoV2 (NCBI Reference Sequence: NC_045512.2) was used to model the structures of RdRp and helicase [21]. The model accuracy and its stereochemical quality was assessed using the PROCHECK analysis tools, which predicts the quality of modeled structure on the basis of distribution of backbone phi/psi angles with reference to the Ramachandran plot [22]. Recently published crystal structure of SARS-CoV-2 3CL-protease (PDB ID, 6LU7) was used for screening and docking simulations [23].
2.2. Virtual screening of FDA approved antivirals and drug-like compounds
First, a library of 4512 compounds also containing several FDA approved drugs was screened through MTiOpen screen automated virtual screening platform using the integrated Vina AutoDock program against all three proteins [24]. Hits from virtual screening were shortlisted on the basis of binding energy based Vina empirical scoring function using −8 kcal mol−1 as the threshold [25]. In this regard, 46, [50] and 35 compounds were shortlisted against 3CL-protease, RdRp and helicase, respectively and were downloaded from the ZINC database [26]. In parallel, 62 FDA approved antiviral drugs with random targets were selected and their structure files were obtained from DrugBank in the SDF file format [27]. Format of these compounds were converted from SMILES into PDB using an online tool (Online SMILES Translator, NCI) [28].
The AutoDock tools 1.5.6 program was used to prepare ligand and protein structures for further docking [29]. In ligand preparation, Gasteiger charges were computed, nonpolar hydrogen atoms were merged, torsion angles for all rotatable bonds were set as flexible and the file was saved in the pdbq format. Similarly, protein files were prepared using automated functions of the AutoDock tools that added all hydrogen atoms, computed Gasteiger charges, merged non-polar hydrogen atoms and created pdbqt files.
2.3. Refinement
Each of the shortlisted compounds was subjected to iterative docking with the respective protein using the AutoDock4.2.6 program [29]. Docking was performed by Lamarckian genetic algorithm (LGA) method using 270,000 generations and by applying at least 100 iterations. Pre-calculation of grid parameters for each protein was performed by using the Autogrid program. During docking, the size of grids was kept at maximum covering the whole surface of the protein to allow the ligand to bind in an unbiased binding pocket.
2.4. Analysis of the docking
The docking results were analyzed on the basis of a combination of binding energy, clustering score, shape complementarity, functional significance of the binding pocket and favorable interactions including H bonds and hydrophobic interactions. We used integrated clustering tool of the AutoDock4.2.6 program to cluster different docked structures of a ligand on the basis of root mean square deviation (RMSD) of 1 Å. The binding energy of every docked structure was predicted by the AutoDock program. Thirty percent population of the docked structures in a cluster with −7.5 kcal mol−1 binding energy was used as a cutoff for the first ranking of iterative docking using the AutoDock tools program. A cluster containing the highest number of docked structures was then selected. To further illuminate H bonds and hydrophobic interactions the LigPlot+ program was used [30]. The binding pockets with known functional significance on the basis of previously reported experimental data were considered. Shape complementarity was superficially accessed by visual analysis of the docked structures.
2.5. Identifying non-specific interactions
To further rule out non-specificity, 100 protein families were selected randomly from pfam to check for the specificity of the drug with a particular target. The details of the representative proteins from each family are given in Table S1.
Eight drugs or organic molecules (Table 2 ), which showed binding to any of the three target proteins of SARS-CoV-2, were subjected to docking simulation with all the 100 proteins by using the autodock4.2.6 program. Pre-calculation of grid parameters for each protein was performed by using the autogrid program. To rule out non-specific binding, docking was performed by Lamarckian genetic algorithm (LGA) method. The significance measure (probability of obtaining a better binding score by chance) for a drug was calculated by comparing the binding scores obtained for specific target (three SARS-CoV2 proteins) with the binding scores for 100 structures belonging to random protein families.
Table 2.
Identifying the specificity of the drug-target interactions using a significance score based on binding potential to random targets.
| Drug | Target | Binding score predicted by docking simulation | Probability to obtain better value by random selection of targets |
|---|---|---|---|
| Casopitant | RdRp | −8.2 | 0.22 |
| Atropineoxide | Proteinase | −6.5 | 0.63 |
| Bagrosin | Proteinase | −7.9 | 0.26 |
| Grazoprevir | Proteinase | −10.2 | 0.06 |
| Meclonazepam | Helicase | −10.5 | 0.01 |
| Oxiphenisatin | Helicase | −9.1 | 0.03 |
| Rimantadine | Proteinase | −7.6 | 0.26 |
| Stavudine | Proteinase | −4.2 | 0.78 |
3. Results
3.1. SARS-CoV-2 3CL-Protease
Recently published X-ray structure of 3CL-protease (PDB ID: 6LU7) was used for virtual screening [23]. A library consisting of 4574 compounds also including FDA approved drugs was screened and after applying first ranking with predicted binding energy cutoff of −8.0 kcal mol−1, 46 compounds were shortlisted. Detailed iterative docking and subsequent analysis resulted in the identification of three SARS-CoV-2 3CL-protease binding compounds as potential inhibitors. Rimantadine is an anti-flu FDA-approved drug with generic name as Flumadine. The known target of Rimantadine is the M-protein of influenza-A virus [31]. We observed that Rimantadine bound to SARS-CoV-2 3CL-protease at its substrate-binding site blocking about half of the binding pocket. Two highly conserved amino acid residues His-41 and Cys-145 are the main catalytic residues of 3CL-protease of corona viruses, however, many residues around these catalytic residues also play key role in the binding of peptide substrate and its subsequent catalysis. For example, His-163 and Phe-140 stabilize the position of glutamine present at the P1 position (Gln-P1) of the peptide substrate by developing two H bonds between the NΣ2 atom of His-163 and main carbonyl oxygen of Phe-140 with the OΣ1 and NΣ2 atoms of Gln-P1, respectively [32]. In our docked structure of Rimantadine, the adamantyl ring of the drug perfectly fits in the sub-pocket of the substrate binding site occupying the space between His-163 and Ph-140 of the protease and stabilized by hydrophobic and van der Waals interactions (Fig. 1 A). In the previously reported structure of His-41 mutant of SARS-CoV 3CL-protease in complex with its peptide substrate, carbonyl oxygen of Gln-P1 of the substrate occupies position in the oxyanion hole formed by the amide groups of Gly-143 and Cys-145 [32]. In the docked structure the nitrogen atom of amino group of Rimantadine occupies the position close to the oxyanion hole and is stabilized by four potential H bonds with backbone nitrogen of Cys-145, carbonyl oxygen of Leu-141 and backbone nitrogen and side chain oxygen of Ser-144 (Fig. 1B), thereby blocking the critical residues of the protease required for catalysis.
Fig. 1.

Representative low energy conformation of Rimantadine docked with SARS-CoV-2 3CL-protease. (A) Molecular surface representation of 3CL-protease along with docked Rimantadine depicted in green sticks. The drug-interacting residues of 3CL-protease are shown in brown sticks. (B) Ligplot derived molecular interaction illustration of the complex where H bonds are represented by dotted green lines.
Another drug, Bagrosin was observed to bind at a potential allosteric site of the protease. Bagrosin is an anti-epileptic drug that belongs to a class of hydantoin derivatives (anticonvulsant compounds). It works by blocking the voltage-gated sodium channels of neurons and causes inhibition of calcium flux across neuronal membranes thereby stabilizing neurons. Compared to other hydantoin derivatives, Bagrosin has been reported to be safer in clinical trials [33,34]. SARS-CoV 3CL-protease has been reported to form a homodimer resulting in better catalytic efficiency as compared to its monomeric form [35,36]. The recently reported X-ray structure of SARS-CoV2 3CL-protease is very similar to that of SARS-CoV forming a dimer. The dimer formation is primarily mediated by several hydrophobic interactions between domain-3 of each monomer (Fig. 2 A) [37]. We observed that Bagrosin bound in a hydrophobic pocket in domain-3 of the protease and can potentially interfere with the dimer formation. In the docked structure, the phenanthryl ring of the compound perfectly fits in a hydrophobic pocket while the polar hydantoin ring is stabilized with two H bonds with backbone nitrogen and oxygen of Lys-5 (Fig. 2B and C). Another FDA approved drug, Grazoprevir was also found to bind at the allosteric site of SARS-CoV-2 protease and can potentially interfere with the dimer formation of the protease (Fig. 2D and C). Grazoprevir is known as hepatitis C protease (NS3/4A) inhibitor and is used to treat hepatitis C in combination therapy [38]. Several X-ray structures of the complexes of Grazoprevir and hepatitis C virus (HCV) NS3/4A have been reported [39,40]. In addition to performing docking of Grazoprevir with SARS-CoV2 3CL-protease, we also performed its docking with (HCV) NS3/4A and reproduced the binding interactions reported in different X-ray structures. Predicted binding score obtained through docking simulations of Grazoprevir with different NS3/4A variants is given in Table 1. The binding score of Grazoprevir with SARS-CoV2 3CL-protease was predicted to be −10.1, suggesting the potential stronger binding as compared to binding with HCV NS3/4A.
Fig. 2.

Representative low energy conformations of Bagrosin and Grazoprevir docked at an allosteric site of SARS-CoV-2 3CL-protease. (A) Cartoon presentation of SARS-CoV-2 3CL-protease monomer with individual domains labeled as I, II and III. The docked Bagrosin molecule is depicted in sticks with carbon, nitrogen and oxygen atoms colored yellow, blue and red, respectively. Two catalytic residues, His-41 and Cys-145 are also highlighted in purple sticks. The main region of domain III involved in forming the dimer interface is colored brown. (B) Molecular surface representation of 3CL-protease and bound Bagrosin depicted in sticks (C) Ligplot derived molecular interaction description of the docking complex, where H bonds are represented by dotted green lines. (D) and (E) Cartoon and surface representations of 3CL-protease, respectively including docked structure of Grazoprevir depicted in sticks.
Table 1.
Predicted binding scores of Grazoprevir with different HCV NS3/4A X-ray structures.
| PDB ID's | 3SUD | 3SUF | 5EPN | 5EPY | 53QQ |
|---|---|---|---|---|---|
| Predicted binding score with Grazoprevir | −7.3 | −7.26 | −8.05 | −6.54 | −7.48 |
3.2. SARS-CoV-2 RdRp
RdRp has been considered as an important target for the discovery of direct acting antiviral therapeutics against positive sense RNA viruses. In this study, we used homology modeling to model the structure of SARS-CoV-2 RdRp and performed virtual screening of over 4574 compounds including FDA-approved drugs. The SARS-CoV-2 RdRp sequence showed 96.35% identity to the RdRp sequence of SARS-CoV (PDB IDs 6NUR) that was used as a template for the model. RdRp homology modeled structure exhibited 92.6% and 100% of its residues in the most favorable and allowed regions of the Ramachandran plot, respectively. All of its side chain parameters were within the accepted limit of structure quality (Fig. S1 and Tables S2 and S3). Preliminary virtual screening and subsequent analysis resulted in the identification of 30 compounds potentially binding to the protein. These compounds were subjected to iteratively docking by selecting complete surface of the protein to map the binding. Final analysis resulted in the identification of one compound, Casopitant, tentative generic name Rezonic. Casopitant is a neurokinin-1 receptor antagonist and is under development to manage chemotherapy induced nausea [41].
Two aspartate residues, Asp-760, and Asp-761 are the main catalytic residues of RdRp. These two residues are highly conserved in all corona viruses [14]. Casopitant bound at the catalytic site of the RdRp enzyme (Fig. 3 A). The binding is stabilized by several favorable interactions including three H bonds between carbonyl oxygen of the acetyl piperazine ring and backbone nitrogen of Ala-762, and between the fluorine atoms of trifluoromethyl-phenyl moiety and the side chain nitrogen atoms of Arg-553 and Arg-836. The ligand perfectly fits in the catalytic cavity completely covering the two catalytic aspartate residues (Fig. 3A and B).
Fig. 3.

Docking of Casopitant with SARS-CoV-2 RdRp. (A) Surface representation of the RdRp protein with bound Casopitant depicted in sticks (The C, N, O and F atoms colored yellow, blue, red and cyan). The two catalytic aspartate residues of RdRp are colored cyan while two neighboring residues are colored purple. (B) Casopitant (yellow carbon) and surrounding amino acid residues of RdRp showing important intermolecular interactions.
3.3. SARS-CoV-2 helicase
SARS-CoV-2 helicase is considered an important target for therapeutic intervention due its sequence conservation among all corona viruses [42]. A typical CoV helicase consists of a zinc-binding domain, which is separated by stalk from the main catalytic domains 1A and 2A. It incorporates distinct DNA and ATP binding sites. In addition to these two important binding sites, the β19-β20 loop encompassing residues 331–357 plays a crucial role in nucleic acid unwinding [41]. We homology modeled the structure of SARS-CoV-2 helicase using the SWISS-MODEL online tools for virtual screening. The SARS-CoV-2 helicase sequence showed 99.8% sequence identity with helicase of SARS-CoV (PDB ID, 6JYT) that was used as a template in homology modeling. According to the Ramachandran plot analysis, 99% of the residues were in the allowed region with over 80% in the most favorable region, validating the quality of the modeled structure (Fig. S1 and Tables S4 and S5). We identified that Meclonazepam, a drug-like molecule with reported sedative, anxiolytic and anti-parasitic effects [43], bound to the viral helicase. According to the docking results, Meclonazepam binds to helicase at a pocket primarily formed by the β19-β20 loop and the binding is stabilized by several H bonds and hydrophobic interactions (Fig. 4 A and B).
Fig. 4.

Docking of Meclonazepam with modeled structure of SARS-CoV-2 helicase. (A) Cartoon presentation of SARS-CoV-2 helicase with docked ligand. Individual domains including the zinc-binding domain, the stalk, and domains 1A and 2A are colored differently and labeled accordingly. Residues involved in ATP and DNA binding are depicted in yellow and purple sticks, respectively. The ligand is depicted in brown sticks and highlighted in a circle. The β19-β20 loop is colored dark blue. (B) Ligplot derived details of interactions between Meclonazepam and helicase showing H bonds (dotted green lines) and hydrophobic interactions involving most of the β19-β20 loop residues.
Another drug-like molecule, Oxiphenisatin was found to bind SARS-CoV2 helicase also in a binding pocket primarily formed by the β19-β20 loop. The binding was stabilized by several favorable interactions including five H bonds and hydrophobic interactions involving primarily the residues from the β19-β20 loop (Fig. 5 A and B).
Fig. 5.

Docking of Oxiphenisatin with modeled structure of SARS-CoV-2 helicase. (A) Overall cartoon presentation of SARS-CoV-2 helicase with docked Oxiphenisatin depicted in purple sticks and highlighted in a circle. (B) Ligplot derived details of interactions between Oxiphenisatin and helicase showing several intermolecular H bonds (dotted green lines) in addition to hydrophobic interactions involving most of the β19-β20 loop residues.
3.4. Minimizing the non-specific interactions
In order to minimize chances of false positive results in the docking simulations, we calculated a significance measure for the binding scores of prominent drug-protein pairs. The significance measure is calculated by comparing the binding scores obtained for potential targets of a drug with the binding scores of 100 structures belonging to random protein families with the respective drug and counting the number of times out of 100 that we see better docking value when docking the respective drug with the random protein families [44]. The significance measure enabled us to identify more promiscuous compounds where even the targets with most favorable docking score have an unfavorable significance measure (meaning that the drug is sticky and is interacting with many proteins with high probability). We set the cutoff value of the significance measure at 0.3 also to include slightly less specific compounds taking into account the identification of leads to develop more specific potential drugs. Drug-protein pairs with significance measure less than 0.3 were considered as specific (those with measure less than 0.1 are highly specific) as compared to the other pairs tested (Table 2). The two docked structures with binding score higher than −0.6 showed the significance measure higher than 0.3 indicating non-specific binding of these ligands.
4. Discussion
Over the last decade, computer aided drug discovery has emerged as a fundamental tool in drug discovery and design process [[45], [46], [47], [48]]. Identification of compounds that bind and inhibit normal function of proteins involved in viral replication has been the most successful strategy in direct acting antiviral drug discovery [[49], [51], [52]]. In this study, we used a virtual screening based strategy to identify already approved drugs or drug-like molecules that can bind to any of the three key viral enzymes, 3CL-protease, RdRp and helicase, and potentially inhibit the function of these enzymes. Virtual screening of over 4500 drugs and subsequent docking simulation resulted in the identification of three 3CL-protease binding drugs, one RdRp binding molecule and two helicase binding molecules. One of the identified 3CL-protease binding drugs, Rimantadine, showed binding at the catalytic site blocking the critical catalytic residues and half of the catalytic pocket. The two other compounds bound in a pocket present in domain III of the protease and can potentially interfere with the formation of dimer – it is well established that CoV 3CL-proteases are catalytically active primarily in dimeric form [53]. One RdRp binding compound, Casopitant was identified which perfectly fits in the catalytic cavity of the enzyme and completely blocks the two critical catalytic residues, Asp-260 and Asp-261. Casopitant is not an approved drug but it is in the process of development towards its approval. We identified two helicase-binding molecules, Meclonazepam and Oxiphenisatin both of these molecules bind at a pocket primarily formed by the β19-β20 loop of the enzyme. This loop has been reported to play a crucial role in the catalytic activity, nucleic acid unwinding, of the helicase enzyme [42]. Three of the identified compounds are already approved drugs that potentiate for their repurposing towards SARS-CoV-2 after in vitro and in vivo validation.
Moreover, all of these compounds represent leads for chemical modifications to develop new potential therapeutics. For instance, Rimantadine binds to 3CL-protease and fits in half of the substrate-binding pocket of the enzyme. This drug also shows non-specific binding to other proteins as determined from its specificity score (0.26) after performing its docking simulation with 100 irrelevant proteins. Its non-specific binding could be attributed to its smaller size as several side effects of this drug have also been reported [54]. Nevertheless, this docked structure provides necessary information for potential chemical modifications to incorporate chemical groups to fill the space of the binding pocket potentially leading to enhance the binding affinity and specificity. In specificity score measurement, we set the cutoff value of the significance measure at 0.3 considering two aspects. Firstly, to identify leads for chemical modification to synthesize more specific inhibitors and secondly, not to reject a ligand with good docking score even with moderate potential off target effects as in many cases a drug with several adverse effects is also acceptable when the question of life saving arises. For example, Rimantadine is an FDA approved drug to use against flu despite its several side effects.
Recently, several studies have been conducted using computational methods to identify already approved drugs as potential inhibitors of SARS-CoV-2. Elfiky et al. reported the binding of four approved anti-viral drugs to RdRp of SARS-CoV-2, including currently under clinical trial drug Remdesivir [14]. RdRp is a highly significant drug target due to its conservation across positive sense RNA viruses. However, this study was highly focused on nucleotide inhibitors and only eight already known drugs were screened against SARS-CoV-2 RdRp using a less vigorous docking program AutodockVina. Wang et al. recently conducted a study to screen a library consisting of diverse approved drugs against the SARS-CoV-2 3CL-protease and identified binding of several antiviral drugs, an anticancer drug, Carfilzomib and an antibiotic, Streptomycin [17]. This study is computationally vigorous as it also includes molecular dynamics simulation to approximate binding free energy. However, docking simulations using the autodock program provides quite a reliable approximation for further in vitro and in vivo testing as Elfiky et al. utilized only the less vigorous program of autodock (AutoDock Vina) to identify potential inhibitors and one of their identified drugs is currently in clinical trials, suggesting the reliability of docking. In our studies we performed computational screening by targeting three important enzymes of SARS-CoV-2 including RdRp, 3CL-protease and helicase, to identify not only the already approved drugs for repurposing but also the drug candidates or lead structures that can be chemically modified to develop potential drugs. Moreover, we incorporated an additional aspect of specificity score obtained by performing docking simulation of every identified compound with 100 irrelevant proteins.
A similar study was recently reported by Mirza et al. incorporating screening of drug candidates against three key enzymes of SARS-CoV-2 and reported several potential inhibitors of these enzymes [13]. However, they performed docking by selecting a very specific area around the predicted catalytic sites of the protease and RdRp, and the ATP binding site of helicase ignoring allosteric sites. These enzymes also incorporate allosteric sites that can be targeted for potential drug discovery. For example, 3CL-protease is known to be catalytically active in its dimeric form and any inhibitor of dimerization could be of high significance. Similarly, helicase incorporates several distinct sites including DNA and ATP binding sites, the zinc-binding domain and the β19-β20 loop involved in nucleic acid unwinding process. In our studies, during docking, the whole surface of the protein was selected to allow the ligand to bind in an unbiased binding pocket and results were analyzed considering the known functional significance of the binding pocket that resulted in the identification of potential allosteric inhibitors of these enzymes as well.
Moreover, several other studies have also been reported contributing to the efforts in identifying approved drugs for repurposing or drug candidates or leads to develop drugs against SARS-CoV-2. Overall, this computer-aided study represents a contribution to the efforts against SARS-CoV-2 and provides information about potentially repurposable drugs and drug-like molecules or lead structures that can be chemically modified to develop into potential drug candidates for further in vitro and clinical investigation.
Author contributions
H.I, H.N.A and S.F performed all analyses. S.S.H and H.N supervised and analyzed the data and wrote the manuscript.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Acknowledgments
This study was supported by an internal funding from Lahore University of Management Sciences. One of the authors, Hafsa Iftikhar was supported by the Higher Education Commission of Pakistan in the form of PhD stipend.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.compbiomed.2020.103848.
Contributor Information
Hammad Naveed, Email: hammad.naveed@nu.edu.pk.
Syed Shahzad-ul-Hussan, Email: shahzad.hussan@lums.edu.pk.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Burkle F.M. Declining public health protections within autocratic regimes: impact on global public health security, infectious disease outbreaks, epidemics, and pandemics. Prehospital Disaster Med. 2020:1–10. doi: 10.1017/S1049023X20000424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bayham J., Fenichel E.P. Impact of school closures for COVID-19 on the US health-care workforce and net mortality: a modelling study. Lancet Public Health. 2020;5:e271–e278. doi: 10.1016/S2468-2667(20)30082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization, Coronavirus Disease . April 14, 2020. COVID-19) Situation Report Number 84.https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200413-sitrep-84-covid-19.pdf?sfvrsn=44f511ab_2 (last time visited on April 14th, 2020) [Google Scholar]
- 4.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhong W., Xiao G., Gao J., Tian Z., Yang X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Cell Res. 2020;14:72–73. doi: 10.5582/bst.2020.01047. [DOI] [PubMed] [Google Scholar]
- 6.Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., Bi Y., Ma X., Zhan F., Wang L., Hu T., Zhou H., Hu Z., Zhou W., Zhao L., Chen J., Meng Y., Wang J., Lin Y., Yuan J., Xie Z., Ma J., Liu W.J., Wang D., Xu W., Holmes E.C., Gao G.F., Wu G., Chen W., Shi W., Tan W. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wan Y., Shang J., Graham R., Baric R.S., Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020;94 doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., Muller M.A., Drosten C., Pohlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. doi: 10.1016/j.cell.2020.02.052. e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wrapp D., Wang N. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shereen M.A., Khan S., Kazmi A., Bashir N., Siddique R. COVID-19 infection: origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020;24:91–98. doi: 10.1016/j.jare.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.te Velthuis A.J., van den Worm S.H., Snijder E.J. The SARS-coronavirus nsp7+nsp8 complex is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension. Nucleic Acids Res. 2012;40:1737–1747. doi: 10.1093/nar/gkr893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snijder E.J., Decroly E., Ziebuhr J. The nonstructural proteins directing coronavirus RNA synthesis and processing. Adv. Virus Res. 2016;96:59–126. doi: 10.1016/bs.aivir.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirza M.U., Froeyen M. 2020. Structural Elucidation of SARS-CoV-2 Vital Proteins: Computational Methods Reveal Potential Drug Candidates against Main Protease, Nsp 12 RNA-dependent RNA Polymerase and Nsp13 Helicase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elfiky A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020;248:117477. doi: 10.1016/j.lfs.2020.117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muralidharan N., Sakthivel R., Velmurugan D., Gromiha M.M. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. J. Biomol. Struct. Dyn. 2020:1–6. doi: 10.1080/07391102.2020.1752802. [DOI] [PubMed] [Google Scholar]
- 16.Kadioglu O., Saeed M., Johannes Greten H., Efferth T. Identification of novel compounds against three targets of SARS CoV-2 coronavirus by combined virtual screening and supervised machine learning. Bull. World Health Organ. 2020 doi: 10.2471/BLT.20.255943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study, J chem inf model. J. Chem. Inf. Model. 2020 doi: 10.1021/acs.jcim.0c00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weston S., Coleman C.M., Haupt R., Logue J., Matthews K., Frieman M. bioRxiv; 2020. Broad Anti-coronaviral Activity of FDA Approved Drugs against SARS-CoV-2 in Vitro and SARS-CoV in Vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caly L., Druce J.D., Catton M.G., Jans D.A., Wagstaff K.M. Antiviral research; 2020. The FDA-Approved Drug Ivermectin Inhibits the Replication of SARS-CoV-2 in Vitro; p. 104787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L.J.N.a.r. SWISS-MODEL: homology modelling of protein structures and complexes. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu F., Zhao S., Yu B., Chen Y., Wang W., Song Z., Hu Y. A novel coronavirus associated with a respiratory disease in Wuhan of hubei province, China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 23.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C.J.b. 2020. Structure of Mpro from COVID-19 Virus and Discovery of its Inhibitors. [DOI] [PubMed] [Google Scholar]
- 24.Labbé C.M., Rey J., Lagorce D., Vavruša M., Becot J., Sperandio O., Villoutreix B.O., Tufféry P., Miteva M.A. MTiOpenScreen: a web server for structure-based virtual screening. Nucleic Acids Res. 2015;43:W448–W454. doi: 10.1093/nar/gkv306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trott O., Olson A.J., Vina AutoDock. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sterling T., Irwin J.J. ZINC 15--ligand discovery for everyone. J. Chem. Inf. Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wishart D.S., Knox C., Guo A.C., Shrivastava S., Hassanali M., Stothard P., Chang Z., Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Online SMILES Translator and Structure File Generator. national cancer institute; 2020. https://cactus.nci.nih.gov/translate/ (last time visited on may 16th. [Google Scholar]
- 29.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools 4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laskowski R.A., Swindells M.B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 31.WHO Current status of amantadine and rimantadine as anti-influenza-A agents: Memorandum from a WHO meeting. Bull. World Health Organ. 1985;63:51–56. [PMC free article] [PubMed] [Google Scholar]
- 32.Xue X., Yu H., Yang H., Xue F., Wu Z., Shen W., Li J., Zhou Z., Ding Y., Zhao Q., Zhang X.C., Liao M., Bartlam M., Rao Z. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 2008;82:2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nitz R., Persch W., Schmidt A. Chemistry and anticonvulsive effect of new hydantoin derivatives. J Arzneimittel-Forschung. 1955;5:357. [PubMed] [Google Scholar]
- 34.Kunkel L. Treatment of childlike epilepsy with bagrosin, or bagrosin sodium. J Die Medizinische. 1956;10:1820–1821. [PubMed] [Google Scholar]
- 35.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grum-Tokars V., Ratia K., Begaye A., Baker S.C., Mesecar A.D. Evaluating the 3C-like protease activity of SARS-Coronavirus: recommendations for standardized assays for drug discovery. Virus Res. 2008;133:63–73. doi: 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L., Lin D. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alric L., Bonnet D. Grazoprevir+ elbasvir for the treatment of hepatitis C virus infection. J Expert opinion on pharmacotherapy. 2016;17:735–742. doi: 10.1517/14656566.2016.1161028. [DOI] [PubMed] [Google Scholar]
- 39.Romano K.P., Ali A., Aydin C., Soumana D., Ozen A., Deveau L.M., Silver C., Cao H., Newton A., Petropoulos C.J., Huang W., Schiffer C.A. The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soumana D.I., Kurt Yilmaz N., Prachanronarong K.L., Aydin C., Ali A., Schiffer C.A. Structural and thermodynamic effects of macrocyclization in HCV NS3/4A inhibitor MK-5172. ACS Chem. Biol. 2016;11:900–909. doi: 10.1021/acschembio.5b00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruhlmann C., Herrstedt J. Casopitant: a novel NK1-receptor antagonist in the prevention of chemotherapy-induced nausea and vomiting. J Therapeutics clinical risk management. 2009;5:375. doi: 10.2147/tcrm.s4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jia Z., Yan L., Ren Z., Wu L., Wang J., Guo J., Zheng L., Ming Z., Zhang L., Lou Z. Delicate structural coordination of the severe acute respiratory syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019;47:6538–6550. doi: 10.1093/nar/gkz409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huppertz L.M., Bisel P., Westphal F., Franz F., Auwärter V., Moosmann B. Characterization of the four designer benzodiazepines clonazolam, deschloroetizolam, flubromazolam, and meclonazepam, and identification of their in vitro metabolites. Forensic Toxicol. 2015;33:388–395. [Google Scholar]
- 44.Naveed H., Hameed U.S., Harrus D., Bourguet W., Arold S.T., Gao X. An integrated structure- and system-based framework to identify new targets of metabolites and known drugs. Bioinformatics. 2015;31:3922–3929. doi: 10.1093/bioinformatics/btv477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leelananda S.P., Lindert S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016;12:2694–2718. doi: 10.3762/bjoc.12.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson D.K., Karanicolas J. Computational screening and design for compounds that disrupt protein-protein interactions. Curr. Top. Med. Chem. 2017;17:2703–2714. doi: 10.2174/1568026617666170508153904. [DOI] [PubMed] [Google Scholar]
- 47.Seidel T., Schuetz D.A., Garon A., Langer T. The pharmacophore concept and its applications in computer-aided drug design. Prog. Chem. Org. Nat. Prod. 2019;110:99–141. doi: 10.1007/978-3-030-14632-0_4. [DOI] [PubMed] [Google Scholar]
- 48.Ganesan A., Barakat K. Applications of computer-aided approaches in the development of hepatitis C antiviral agents. Med. Res. Rev. 2017;12:407–425. doi: 10.1080/17460441.2017.1291628. [DOI] [PubMed] [Google Scholar]
- 49.Ashraf M.U., Iman K., Khalid M.F., Salman H.M., Shafi T., Rafi M., Javaid N., Hussain R., Ahmad F., Shahzad-Ul-Hussan S., Mirza S., Shafiq M., Afzal S., Hamera S., Anwar S., Qazi R., Idrees M., Qureshi S.A., Chaudhary S.U. Evolution of efficacious pangenotypic hepatitis C virus therapies. Med. Res. Rev. 2019;39:1091–1136. doi: 10.1002/med.21554. [DOI] [PubMed] [Google Scholar]
- 50.Zheng Y.X., Ma S.J., Xiong Y.H., Fan X.G. The efficacy and safety of direct-acting antiviral regimens for HCV/HIV Co-infection: a systematic review and network meta-analysis. J. Gastroenterol. Hepatol. 2020 doi: 10.1111/jgh.14462. 2020. [DOI] [PubMed] [Google Scholar]
- 51.Par A., Par G. [Three decades of the hepatitis C virus from the discovery to the potential global elimination: the success of translational researches] Orv. Hetil. 2018;159:455–465. doi: 10.1556/650.2018.30997. [DOI] [PubMed] [Google Scholar]
- 52.Qadir A., Riaz M., Saeed M., Shahzad-Ul-Hussan S. Potential targets for therapeutic intervention and structure based vaccine design against Zika virus. Eur. J. Med. Chem. 2018;156:444–460. doi: 10.1016/j.ejmech.2018.07.014. [DOI] [PubMed] [Google Scholar]
- 53.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S. An overview of severe acute respiratory syndrome–coronavirus (SARS-CoV) 3CL protease inhibitors. Peptidomimetics and Small Molecule Chemotherapy. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alves Galvao M.G., Rocha Crispino Santos M.A., Alves da Cunha A.J. Amantadine and rimantadine for influenza A in children and the elderly. Cochrane Database Syst. Rev. 2012;1 doi: 10.1002/14651858.CD002745.pub2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.