

Abstract
Maf (c-Maf) and Mafb transcription factors (TFs) have compensatory roles in repressing somatostatin (SST+) interneuron (IN) production in medial ganglionic eminence (MGE) secondary progenitors in mice. Maf and Mafb conditional deletion (cDKO) decreases the survival of MGE-derived cortical interneurons (CINs) and changes their physiological properties. Herein, we show that (1) Mef2c and Snap25 are positively regulated by Maf and Mafb to drive IN morphological maturation; (2) Maf and Mafb promote Mef2c expression which specifies parvalbumin (PV+) INs; (3) Elmo1, Igfbp4 and Mef2c are candidate markers of immature PV+ hippocampal INs (HIN). Furthermore, Maf/Mafb neonatal cDKOs have decreased CINs and increased HINs, that express Pnoc, an HIN specific marker. Our findings not only elucidate key gene targets of Maf and Mafb that control IN development, but also identify for the first time TFs that differentially regulate CIN vs. HIN production.
Research organism: Mouse
Introduction
Pallial interneurons (INs) consist of cortical interneurons (CINs) and hippocampal interneurons (HINs). IN pathologies are hypothesized to underlie symptoms of neurological and neuropsychiatric disorders such as autism spectrum disorder (ASD), epilepsy and schizophrenia. One possible explanation is that IN pathology leads to disrupted circuit inhibition, resulting in circuit hyperexcitability and less efficient information processing (Lim et al., 2018; Rubenstein and Merzenich, 2003; Sohal and Rubenstein, 2019; Yizhar et al., 2011). Thus, understanding the genetic control of CIN and HIN development and function can have important clinical ramifications.
CINs are generated in three subpallial progenitor domains: the medial and caudal ganglionic eminences (MGE and CGE), and the preotic area (POA) (Gelman et al., 2011; Lim et al., 2018). MGE and POA-derived CINs express parvalbumin (PV) or somatostatin (SST), while CGE-derived CINs express either vasoactive intestinal peptide (VIP), cholecystokinin (CCK) or reelin [but lack SST]. HINs are generated by the same general progenitor pools and share similar molecular properties with CINs. However, so far there are no clear single markers that are known to define developing or mature HINs from CINs (Pelkey et al., 2017).
We have recently demonstrated that two Maf TFs, Maf (c-Maf) and Mafb, function together in the MGE to control the timing and quantity of SST+ CINs that are generated (Pai et al., 2019), schematically summarized in Schemes 1–3. Loss of Mafb and Maf in the Nkx2.1-Cre lineage (Maf cDKO) leads to the overproduction of SST+ CINs at the expense of PV+ CINs. Maf cDKOs also have changes in the quantity and distribution of the total Nkx2.1-cre lineage INs: reduced CINs (~30%) and increased HINs (~2 fold) at P0, and a progressive loss of INs that plateaued by adulthood at ~70% reduction (Schemes 1 and 2). Loss of Maf and Mafb also alters postnatal CIN properties, including process morphogenesis, synaptogenesis, intrinsic electrophysiology and circuit excitability (Pai et al., 2019; Schemes 2 and 3). However, the gene targets of Mafb and Maf that underlie these phenotypes remain to be elucidated.
Scheme 1. Schema depicting the prenatal roles of Maf and Mafb.

In Pai et al., 2019, we described that the expression of Maf and Mafb starts in the MGE SVZ. Deletion of Maf and Mafb using the Nkx2.1-Cre (Maf cDKO) leads to increased neurogenesis in the MGE SVZ and overproduction of somatostatin (SST) interneurons. In this paper, we aim to identify the downstream gene targets of Maf and Mafb that are involved in this process. SVZ: subventricular zone. MZ: mantle zone.
Scheme 3. Loss of Maf and Mafb leads to MGE-derived IN morphogenesis defect.

In Pai et al., 2019, we showed that cultured neonatal Maf cDKO interneurons have neurite outgrowth defect compared to the WT counterparts. In this paper, we aim to identify gene targets of Maf and Mafb that are involved in CIN morphological maturation.
Scheme 2. Schema depicting the postnatal roles of Maf and Mafb.

In Pai et al., 2019, we discovered that the adult Maf cDKO mice have drastic reduction of MGE-derived cortical interneurons (CINs) and a preferential loss of parvalbumin (PV) INs. In this paper, we aim to identify potential downstream gene targets of Maf and Mafb that are involved in PV CIN fate specification and differentiation. In this paper, we also provide data that suggest adult Maf cDKO animals have spontaneous epileptic activity in vivo.
Herein, we uncovered a set of Maf and Mafb regulated genes by performing single cell RNA-sequencing (scRNA-seq) of neonatal CINs and HINs from wild type (WT) and cDKO. These include Sst, neuropilin1 (Nrp1), and several genes that are implicated in neuropsychiatric disorders, including myocyte enhancer factor 2C (Mef2c) (ASD, Tu et al., 2017), synaptosome associated protein 25 (Snap25) (schizophrenia, Houenou et al., 2017), and C-X-C motif chemokine receptor 4 (Cxcr4) (22q11.2 deletion syndrome; López-Bendito et al., 2008). We provide evidence that Maf and Mafb promote Mef2c expression, which in turn promotes PV+ IN differentiation. We show that Maf and Mafb also have postnatal functions in CIN morphological maturation. Furthermore, we identified a secreted peptide encoding gene, prepronociceptin (Pnoc), as an immature HIN marker. Finally, we provide evidence that Maf and Mafb control the balance between CIN and HIN production.
Results
Single cell transcriptomic profiling of P0 WT and Maf cDKO
Deletion of both Maf (c-Maf) and Mafb using Nkx2.1-Cre (Maf cDKO) led to an overproduction of Sst+ CINs, a drastic loss of total MGE-derived CINs and abnormal electrophysiological properties (Pai et al., 2019). Furthermore, adult Maf cDKOs have spontaneous non-motor epileptic activity, likely associated with the interneuronopathy (Figure 1—figure supplement 1). The underlying gene targets of Maf and Mafb that may regulate these processes are explored below.
To identify dysregulated genetic targets in cDKO immature INs, we used scRNA-seq from P0 WT and cDKO neocortices (Figure 1A–D). We pooled the WT and cDKO datasets to perform unsupervised data analysis using Seurat pipeline (Butler et al., 2018; Ge et al., 2020). We identified 24 different clusters, including excitatory neurons, neural progenitors, microglia as well as MGE and CGE-derived INs, which were visualized by uniform manifold approximation and projection (UMAP) (Figure 1B and C; Becht et al., 2019). We assigned the cell identities to each cluster based on the expression of established marker genes (Figure 1—figure supplements 2 and 3, Figure 1—source data 1; Loo et al., 2019; Mi et al., 2018; Tasic et al., 2018). MGE-derived INs were in cluster 1, based on marker gene co-expression, including LIM homeobox 6 (Lhx6), Maf, Mafb and neurexophilin 1 (Nxph1) (Figure 1E, Figure 1—figure supplement 2, Figure 1—source data 1). We performed fluorescent in situ hybridization (FISH) to confirm the specificity of Nxph1 expression within the Nkx2.1-lineage by co-labeling Nxph1 and Nkx2.1-Cre driven tdTomato (Figure 1—figure supplement 4).
Figure 1. Single cell transcriptomic analysis of P0 WT and Maf cDKO neocortex.
(A) Experimental design for 10X genomics analysis of the P0 WT and Maf cDKO neocortex. Neocortical tissues were dissociated for single cell gel bead-in-emulsion (GEMs) particle preparation, followed by cDNA library preparation, sequencing and data analysis. (B) UMAP plot of WT and cDKO cells, color coded with cluster identities (see Figure 1—figure supplement 2). (C) UMAP plot of WT and cDKO cells, color coded with genotypes; cluster one are MGE INs. Note the genotype separation of the cells in cluster 1 (left bottom), but not other clusters, suggesting the lack of non-cell autonomous effects of the cDKO. (D) Heatmap showing cluster identities with corresponding marker genes. (E) Heatmap of cluster 1 cells from WT and cDKO showing average expression of selected MGE IN markers and pan-IN markers. Note the nearly full depletion of Mafb and Maf and increased expression of Sst, and the decreased expression of Mef2c, while other MGE-derived IN markers such as Dlx1/2/5, Gad2 and Lhx6 were not changed. This suggests that the deletion of Mafs in the MGE-lineage does not lead to a gross change in MGE-derived IN fate. (F–G) Enlarged feature plots showing the expression of Mef2c (F) and Sst (G) from WT and cDKO from cluster 1. (H–I) FISH in Sst together with EdU labeling showing the amount of E15.5 born MGE-derived CINs that became Sst+ in WTs and cDKOs. Arrows point to the cells that are EdU+;Sst+. (J) Quantification of Sst+ CINs at P2 WT and cDKO neocortex. (K) Quantification of EdU+;Sst+ CINs at P2 WT and cDKO neocortex. (L–M) Immunofluorescent staining of MEF2C colocalized with Nkx2.1-cre-driven tdTomato reporter. P2 Maf cDKOs have decreased MEF2C expression (M) compared to WTs (L). (N) Quantification of the proportion of tdTomato+ CINs that were MEF2C+ by bins. N = 3–4 animals per group and multiple brain sections were used for quantification. Scale bar in (I) = 100 um and in (M) = 50 um. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.

Figure 1—figure supplement 1. Adult Maf cDKO animals have spontaneous non-motor seizure activities.

Figure 1—figure supplement 2. Top 10 ranked marker genes for each cluster.

Figure 1—figure supplement 3. Dotplots of non-IN and IN population by selected marker genes.

Figure 1—figure supplement 4. Nxph1-tdTomato double FISH and Nxph2 ISH.

Figure 1—figure supplement 5. DEX gene list and Gene Ontology analysis on DEX genes Heatmap showing the DEX gene list of the MGE IN population (cluster 1) when compared cDKO to WT.

Figure 1—figure supplement 6. Downregulation of Cxcr4 expression at E15.5 and P2 in the Maf cDKO.

Figure 1—figure supplement 7. Maf cDKO has increased Sst+/Npy+/Nrp1+ expressing HINs.

There were no obvious transcriptomic changes in neural progenitors, excitatory neurons, oligodendrocytes, microglia and the CGE-derived IN population between WT and cDKO (Figure 1B and C). This suggests no gross non-cell autonomous effects exerted by loss of Maf and Mafb in the Nkx2.1-Cre-lineage. However, the MGE-derived IN population (cluster 1) showed clear differences between WT and cDKO (Figure 1C). We performed differential gene expression (DEX) analysis in this MGE-derived IN cluster, and identified 81 differentially expressed (DE) genes, with 47 upregulated (including Sst, Nrp1, Pnoc and Neuropeptide Y) and 34 down-regulated (including Maf, Mafb, Neurexophilin2, Mef2c, Cxcr4 and Snap25) in the Maf cDKO (Figure 1—figure supplement 5). We performed either immunohistochemistry or FISH to confirm the pattern of gene expression changes between control and cDKOs, including Mef2c, Sst, Neurexophilin2 (Nxph2), Cxcr4, Neuropeptide Y (Npy) and Nrp1 (Figure 1F–N, Figure 1—figure supplements 4–7). Gene ontology analysis, based upon cellular components, showed that most dysregulated genes are involved in synapse formation, neurite projection and neuronal maturation. Thus, the transcriptomic analysis provides molecular insights into the Maf cDKOs cellular phenotypes (Figure 1—figure supplement 5; Ge et al., 2020; Pai et al., 2019).
Loss of Maf and Mafb leads to decreased MEF2C expression and increased Sst+ CINs
Based on DEX analysis, Mef2c was the third most down-regulated gene, while Sst was the most upregulated gene (Figure 1F–G, S7). To validate the Mef2c result, we used immunofluorescence labeling; MEF2C expression was reduced in the tdTomato+ CINs across all cortical layers in the P2 cDKOs (Figure 1F and L–N) (Bin 3, 4, 7: p<0.05; Bin 5, 6: p<0.01; Density: p<0.01; Welch’s t test) (tdTomato fluorophore was generated by crossing Ai14 Rosa26-tdTomato Cre reporter with Nkx2.1-Cre; we will refer to the Cre reporter allele as RosaT and the expressed reporter as tdTomato throughout the paper)(Madisen et al., 2010). To validate the Sst result, we used FISH; we found both increased Sst ISH signal and increased numbers of Sst+ CINs at P2 in cDKOs as before (Figure 1G and H–J) (Sst density: p=0.0001, Welch’s t test) (Pai et al., 2019).
Since the Maf cDKO generated excessive Sst+ CINs at E13.5 (Pai et al., 2019), we also asked if excessive Sst+ CINs were generated at later developmental time points. Thus, we injected EdU in E15.5 pregnant mice and assessed the neocortical Sst+ INs at P2. There was a > 3 fold increase in Sst and EdU double labeled CINs in P2 cDKO neocortices (Figure 1K) (Sst-EdU density: p<0.0001; Welch’s t test). Thus, Maf cDKOs overproduce Sst+ CINs across development.
Maf and Mafb drive Mef2c expression to promote PV+ CIN generation
Prenatal loss of Mef2c reduces the number of PV+ CINs (Mayer et al., 2018). Given our finding that Mef2c expression is reduced in Maf cDKOs (Figure 1), it suggests that Maf and Mafb are genetically upstream of Mef2c. We thus hypothesized that deficits in Mef2c potentially underlie the mechanisms causal to the PV+ CIN loss in the Maf cDKO (this report; Pai et al., 2019). We tested this by increasing MEF2C levels in Maf cDKO MGE cells.
To this end, we used an MGE transplantation rescue assay (Vogt et al., 2015; Vogt et al., 2017), which previously showed that the Maf cDKO had reduced PV+ CINs, recapitulating the cDKO in vivo phenotype (Pai et al., 2019). E13.5 MGEs (RosaT+ or Maff/f; Mafbf/f; RosaT+) were dissociated and then transfected with a Cre-expressing plasmid to generate tdTomato+ WT or cDKO MGE-lineage cells. In parallel, some of the MGE cells were co-transfected with plasmids expressing Cre and Mef2c. The transfected cells were transplanted into WT P1 neocortices and were assessed 30 days later via SST and PV immunofluorescence (Figure 2A and C–K).
Figure 2. Mef2c rescues the deficit of PV CIN production in cDKOs.
(A) Schema showing the MGE transplantation assay. RosaT+ or Maff/f;Mafbf/f;RosaT+ MGE progenitors were dissected at E13.5, followed by transfection of Cre or Cre + Mef2c (expression vectors) to generate cDKO or cDKO + Mef2c MGE cells. We transplanted these cells into P1 WT neocortex and let them mature for 30 days before investigating CIN subtype markers. (B) Quantification of the proportion of transplanted WT, cDKO or cDKO with transfected Mef2c CINs that express either SST or PV. Note that the cDKO group recapitulated the phenotype of preferential loss of PV CINs; transfection with Mef2c rescued the production of PV CINs. Chi-square test was used for statistical analysis. (C–K) Representative singular cell images for group WT, cDKO and cDKO+Mef2c that are labeled with tdTomato and PV. Note that cDKO (F) has less complex neurite growing pattern compared to the WT (C). Also note the rescue of the PV expression in the cDKO+Mef2c (K) compared with the cDKO (H). Scale bar in (K) = 25 um. For SST IN comparison: WT-701 cells, cDKO-627 cells, cDKO+Mef2c- 304 cells; For PV IN comparison: WT- 670 cells, cDKO-587 cells, cDKO+Mef2c −236 cells. These numbers came from 6 WT animals, 4 cDKO animals and 3 cDKO + Mef2c animals. Cells analyzed were converted to contingency table to conduct Chi-square test. ****p<0.0001. Error bars represent standard errors of the mean.

Figure 2—figure supplement 1. Model.

The analysis confirmed the preferential loss of PV+ CINs in the cDKOs (Pai et al., 2019; Figure 2B). Importantly, we found that Mef2c transfection rescued this phenotype, and corrected the SST/PV ratio (Figure 2B and K) (PV population: WT vs. cDKO p<0.0001; cDKO vs. cDKO+Mef2c p<0.0001; Chi-square test). Together, these data suggest that Maf and Mafb promote Mef2c expression, which in turn promotes the PV+ IN fate (Figure 2—figure supplement 1. Model).
Identification of transcriptomic features of candidate immature PV+ INs
Studies have strived to identify transcriptional machinery that differentially controls PV vs. SST IN neurogenesis. Our current study has solidified a transcriptional cascade, Maf/Mafb → Mef2c → PV, that is involved in this process (Figure 2). However, there are no known early markers that specifically delineate immature PV+ INs before PV expression begins (del Rio et al., 1994).
We hypothesize that within MGE-derived INs, the Sst-negative (Sst-) group primarily comprises the future PV+ INs, in part since Sst-IRES-Cre lineage tracing showed that the majority of the SST+ INs remain SST+ (Pai et al., 2019; Figure 3—figure supplement 1). We computationally separated WT cluster 1 MGE-derived INs into Sst+ and Sst- subgroups and evaluated the mean expression of MGE IN markers (Figure 3A). We identified a group of genes that are enriched in the Sst- population, by comparing the IN marker expression between the two groups (Figure 3, Figure 3—figure supplement 2). From these, we selected six genes, ADP ribosylation factor like GTPase 4D (Arl4d), engulfment and cell motility protein 1 (Elmo1), insulin like growth factor binding protein 4 (Igfbp4), Mef2c, Sp9 and transcription factor 12 (Tcf12), with high-to-moderate expression to evaluate their RNA expression.
Figure 3. Multiplex RNA in situ hybridization validation of proposed immature PV IN markers.
(A) Heatmap of MGE IN marker genes for cluster 1 Sst- and Sst+ INs. (B–D’’) Lower magnification fluorescent images of multiplex RNA in situ hybridization for Mef2c (B–B’’), Sp9 (C–C’’) and Igfbp4 (D–D’’). (E–G’’’) Higher magnification fluorescent images of multiplex RNA in situ hybridization for Mef2c (E–E’’’), Sp9 (F–F’’’) and Igfbp4 (G–G’’’). White arrows point to INs that are marker (Mef2c, Sp9 or Igfbp4) and tdTomato positive but Sst negative. Violin plots showing the normalized expression value (Y-axis) of each cell analyzed in each group for Mef2c (H), Sp9 (J) and Igfbp4 (L). Quantification of the percentage of tdTomato+; Sst- and tdTomato+; Sst+ INs in the neocortex and in the hippocampus that are either Mef2c+(I), Sp9+(K) or Igfbp4+(M). Scale bar in (B) = 100 um, (B’’) = 200 um and (G) = 25 um. 2 WTs and multiple brain sections per animal were used for quantification. For statistical analysis, multiple independent t-tests without same standard deviation assumption were conducted to compare the expression of each gene in each brain region. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.

Figure 3—figure supplement 1. Schema depicting the hypothesis behind candidate PV IN marker discovery In Pai et al., 2019, we fate-mapped the Sst-IRES-Cres lineage and discovered that > 90% of Sst-IRES-Cre labeled CINs remain to be SST-expressing in adulthood.
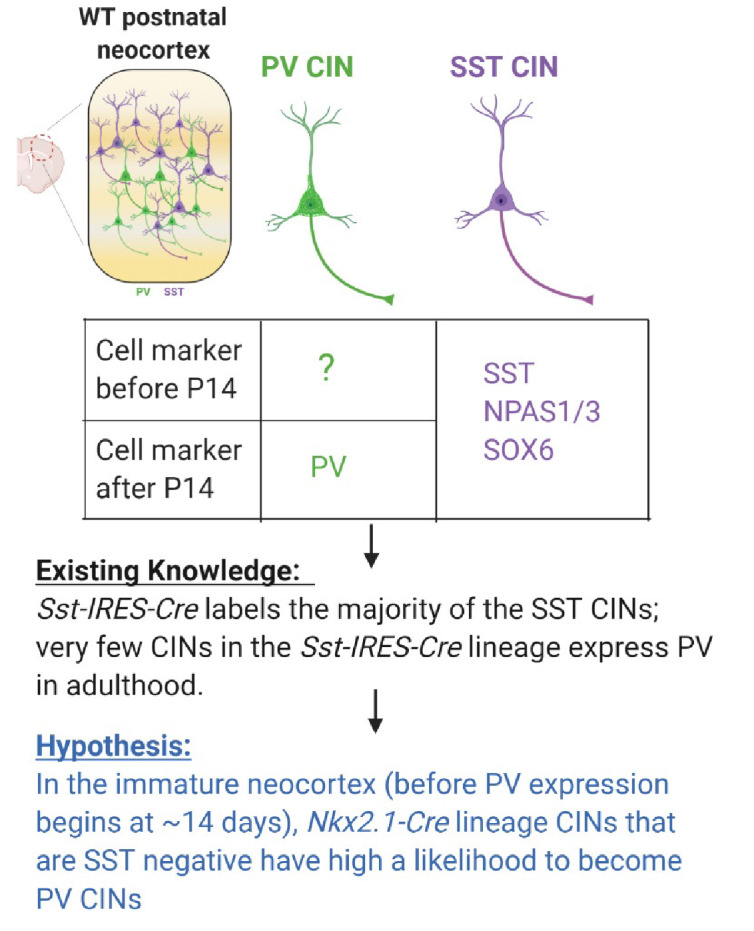
Figure 3—figure supplement 2. Multiplex RNA in situ hybridization validation of proposed immature PV IN markers.

Figure 3—figure supplement 3. Higher magnification view of HINs from multiplex fluorescent in situ hybridization.

We used P2 WT tissues which were triple-labeled with Nkx2.1-Cre-driven tdTomato, Sst and an RNA of each of the six genes; the RNAs were detected using RNAscope (Figure 3B–G’’’, Figure 3—figure supplements 2 and 3). We quantified the proportion of tdTomato+; Sst+ or tdTomato+; Sst- INs that were positive for each gene (Figure 3I, K and M, Figure 3—figure supplement 2). Our results suggested that Mef2c is a very specific Sst- population marker, especially in the hippocampus (Cortex: p<0.05; Hippocampus: p<0.0001). Likewise, Elmo1, Igfbp4, and Sp9 appear to be good markers for Sst- MGE-derived INs in the hippocampus (Elmo1: p<0.001; Igfbp4: p<0.05; Tcf12: p<0.01; unpaired test). This likely is the first study to identify genes, such as Mef2c, that are almost exclusively expressed in Sst- MGE-derived immature HINs.
Maf and Mafb control CIN morphological maturation by regulating Mef2c and Snap25
Reduced expression of Mef2c and Snap25 was shown to impede neurite morphogenesis (Harrington et al., 2016; Houenou et al., 2017; Tu et al., 2017). Since Maf cDKOs have a neurite outgrowth defect in vitro and in vivo (Pai et al., 2019, Figure 4—figure supplement 1), we examined whether the decreased expression of Mef2c and Snap25 in the cDKOs underlies their neurite outgrowth defect (Pai et al., 2019).
To evaluate this, we tested whether transfection of these genes could rescue the Maf cDKO neurite phenotype using primary cell cultures of P1 neocortical neurons. Expression vectors encoding Gfp, Gfp-tagged Maf, Mef2c or Snap25 were transfected into Nkx2.1-Cre-lineage CINs (tdTomato+) the day after cell plating (DIV1). At DIV 14, CIN morphology was assessed (Figure 4A). As before, cDKO CINs had decreased numbers of neurites; Gfp transfection did not alter this phenotype (Figure 4B, C and G). Second, transfection of cDKO with Maf, Mef2c or Snap25 rescued neurite numbers, and the increased neurite numbers were comparable to WT (Figure 4B, D–F and H–I). These results provide evidence that reduced Mef2c and Snap25 expression are downstream of Maf and Mafb in regulating neurite complexity in neonatal CINs.
Figure 4. Mef2c and Snap25 are required downstream of Mafb and Maf to promote neurite outgrowth.
A) Schema showing the in vitro culture assay testing Maf and Mafb’s prenatal function on neurite outgrowth. P0 WT and cDKO neocortical tissues were dissociated (INs were labeled with Nkx2.1-cre-driven tdTomato) and diluted 5-fold with reporter negative dissociated P0 WT neocortical tissues before plating on culture slides. (B–C) Representative traces of cultured WT and cDKO INs. (D–F) Representative traces of cultured cDKO INs transfected with Maf, Mef2c or Snap25-expressing plasmids; each showed a rescue phenotype. (G–I) Sholl analysis showing the neurite complexity and the rescue effects of Maf (G), Mef2c (H) and Snap25 (I). (J) Schema showing the in vitro culture assay testing Maf and Mafb’s postnatal function on neurite outgrowth. (K–O) Representative traces of cultured INs using Maff/f;Mafbf/f; RosaT+ P0 neocortices to look at the effect of postnatal deletion of Maf and Mafb on neurite outgrowth and if Maf, Mef2c and Snap25 expression showed rescue effects. Thus, postnatal loss of Maf and Mafb (cDKOPOST) leads to defects in neurite outgrowth (K–L). (P–R) Sholl analysis showing the neurite complexity and the rescue effects of Maf (P), Mef2c (Q) and Snap25 (R) on cDKOPOST INs. Scale bar in (F) and (O) = 100 um. N = 3–4 per groups. The quantity of cells used for analysis was included in (G–I) and (P–R).

Figure 4—figure supplement 1. Individual CINs from transplanted WT and Maf cDKO MGE cells grown in vivo for 30 days.

Figure 4—figure supplement 2. Maf cDKOSst-IRES-Cre showed defects in neurite outgrowth We used in vitro culture assay to test Mafb and c-Maf’s post-mitotic function on neurite outgrowth.

In these experiments, the Maf and Mafb genes were deleted in the MGE at E10.5 using Nkx2-1-Cre. Thus, the CIN phenotypes could be the result of the continuous lack of Maf and Mafb throughout development and may not necessarily reflect Maf and Mafb post-mitotic functions. Therefore, we analyzed neuronal morphology in CINs generated from Maf cDKOs using Sst-IRES-Cre, a Cre line that turns on after IN cell cycle exit (Figure 4—figure supplement 2). We found that the Sst-IRES-Cre generated Maf cDKO had neurite defects similar to those found using Nkx2.1-Cre. Thus, Maf function in neurite outgrowth is also required post-mitotically.
To identify a postnatal function for Maf and Mafb, we deleted both in neonatal CINs using an in vitro primary culture assay. WT and Mafbf/f; Maff/f; RosaT+ cortical neurons were transfected at DIV1 with a Cre-expressing plasmid (driven by the Dlxi12b enhancer which is specifically active in forebrain GABAergic neurons). CRE mediated recombination generates tdTomato+ cDKOs CINs (cDKOPOST (POSTNATAL)) (Figure 4J). At DIV14, we performed Sholl analysis, which revealed that cDKOPOST INs had reduced neurite complexity, providing evidence that Maf and Mafb regulate IN morphology postnatally. We then tested whether Maf, Mef2c and Snap25 could rescue this phenotype, by co-transfecting Maff/f; Mafbf/f; RosaT+ cells at DIV1 with plasmids expressing Cre and either Maf, Mef2c or Snap25. We analyzed the neuronal morphology at DIV14. Maf, Mef2c and Snap25 each rescued the neurite outgrowth defect (Figure 4K–R). Thus, Maf and Mafb likely promote CIN neurite morphogenesis at postnatal stages through Mef2c and Snap25.
Prepronociceptin is a HIN marker
The DEX analysis revealed that prepronociceptin (Pnoc) was an upregulated gene in the Maf cDKO (Figure 1—figure supplement 5). A recent study focusing on adult CA1 HIN profiling suggested Pnoc is a marker of O-LM INs, a subtype that is mainly Sst+ (Harris et al., 2018; Asgarian et al., 2019). Thus, we investigated Pnoc expression by FISH in the neonatal hippocampus and cortex (Figure 5A–H). In P2 WTs, Pnoc+ cells were largely restricted to the hippocampus (Figure 5G).~40% of the Pnoc+ HINs are tdTomato+; very few of them are Htr3a+ (CGE marker), suggesting that the MGE/POA is the main origin of these HINs ((Figure 5I–M, Figure 5—figure supplement 1). In the Maf cDKO there was ectopic Pnoc expression in the neocortex (highest in the dorsomedial regions abutting the hippocampus), as well as increased Pnoc+ cells in the hippocampus, consistent with an increased density of HINs (Figure 5A–L) (WT vs cDKO: Neocortex p<0.01; dorsomedial Neocortex p<0.0001; Hippocampus p<0.01; t test). Indeed, Sst FISH and tdTomato+ IN quantifications both suggested increased HIN density in the Maf cDKO (Figure 1—figure supplement 7, Figure 5—figure supplement 1).
Figure 5. Maf cDKOs have preferential production of HINs over CINs that are Pnoc+.
(A–F) Multicolor FISH of tdTomato, Pnoc and Htr3a on P2 WT and cDKO neocortex (A–B) and hippocampus (C–F). (G–H) Pnoc expression in the WT and cDKO hippocampus. (I) Quantification of the Pnoc+ INs in the neocortex, dorsal-medial cortex (dmCortex) and hippocampus. (J) Quantification of the Pnoc+; tdTomato+ INs in the neocortex, dmCortex and hippocampus. (K) Quantification of the Pnoc+ HINs by regions. (L) Quantification of the Pnoc+; tdTomato+ HINs by regions. (M) Quantification of the Pnoc+; Htr3a+ HINs by regions. N = 3–4 per group and multiple sections were quantified per animal. Scale bar in (B) and (G) = 100 um and in (D) = 50 um. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Welch’s t test.

Figure 5—figure supplement 1. Quantification of Pnoc+ INs by region and by IN subgroups/Quantification of tdTomato+ HINs by hippocampal region.

Together, these data provide evidence that loss of Maf and Mafb leads to increased production of HINs, which are mainly Sst+ and/or Pnoc+. To further test this hypothesis, we studied HIN generation in the cDKO.
Maf and Mafb repress the generation of HINs
Since Maf cDKOs have increased density of Pnoc, a gene mainly expressed in the HINs, we postulated that Maf and Mafb together repress HIN generation. To validate the Nkx2-1-Cre tdTomato findings (Pai et al., 2019; Figure 5—figure supplement 1), we quantified CINs and HINs at P2 using Lhx6+ FISH. Lhx6 marks MGE-derived INs (Cobos et al., 2006; Liodis et al., 2007). Lhx6+ CIN density was decreased in Maf cDKOs as previously reported (Pai et al., 2019; Figure 6B). Consistent with our hypothesis, there was an increased density of Lhx6+ HINs across all hippocampal regions in the Maf cDKO (p<0.05; Welch’s t test) (Figure 6C). Furthermore, there was an increased density of Sst+ HINs cell density in all hippocampal regions (CA1:~1.5 fold; p<0.001, CA3 and DG:~2 fold; p<0.0001) (Figure 1—figure supplement 7).
Figure 6. Maf cDKOs overproduce INs that will become HINs.
(A) Schema depicting the BrdU/EdU pulse-chase experiment. Briefly, BrdU was injected into a pregnant mouse at E12.5 and EdU was injected at E15.5. Neonatal pups were harvested at P2 analyzed for Lhx6/BrdU, Lhx6/EdU co-expression. (B) Quantification of Lhx6+ IN cell density in the P2 neocortex. (C) Quantification of Lhx6+ IN cell density in the P2 hippocampus by region. (D–G) Lhx6 FISH with BrdU (D-E, early-born) or EdU (F-G, late-born) co-labeling in P2 WT and cDKO hippocampus. (H–I) Quantification of the Lhx6+; BrdU+ IN cell density in the hippocampus by region (H) and in total (I). (J–K) Quantification of the Lhx6+; EdU+ IN cell density in the hippocampus by region (J) and in total (K). N = 3 per group and multiple sections were quantified per animal. Scale bar in (E) = 100 um. Welch’s t test.

Figure 6—figure supplement 1. Analysis of early-born and late-born MGE-derived CINs in WT and Maf cDKO.

Next, to test if Maf cDKOs generate more HINs at specific developmental stages, we conducted EdU/BrdU pulse-chase experiments. Pregnant mice were injected with BrdU at E12.5, and subsequently with EdU at E15.5. The animals were harvested at P2 to study co-expression of Lhx6 with BrdU and EdU (Figure 6A). We compared the generation of CINs and HINs.
At E12.5 (early-born; Lhx6+;BrdU+), we observed ~2 fold increase in HIN generation in the Maf cDKO (p<0.01; t test). The increase was similar across all hippocampal subregions (Figure 6D, E, H and I) (CA1: p=0.02; CA3: p<0.001; DG: p<0.01; t test). In contrast, Maf cDKOs had a trend toward decreased CIN generation at E12.5 (Figure 6—figure supplement 1). At E15.5 (late-born; Lhx6+;EdU+), we observed ~2.5 fold increase in HIN production (p<0.0001; t test). Similarly, the increase was across all hippocampal subregions (Figure 6F, G, J and K) (CA1: p<0.001; CA3: p<0.05; DG: p<0.01; t test). We further quantified the ratio of HIN density over CIN density by age and by genotypes. In WTs, the MGE tends to generate HINs/CINs in a 1:2 ratio at both E12.5 and E15.5. In cDKOs, the MGE overproduces HINs, especially at E12.5 (Figure 6—figure supplement 1). Together, these data support the hypothesis that loss of Maf and Mafb leads to HIN overproduction perhaps at the expense of generating CINs.
Discussion
Maf (c-Maf) and Mafb are co-expressed in the MGE and its lineages. Previously, to elucidate how Maf and Mafb regulated CIN and HIN development, we compared the transcriptomes of WT and Maf cDKO using bulk RNA-seq from E13.5 MGEs – this approach failed to identify differentially expressed genes (data not shown) probably because of the heterogeneity of the tissue. Here, we applied scRNA-seq to compare the transcriptomes of P0 WT and Maf cDKO neocortex and hippocampus. Through this approach, we successfully identified genes that were differentially expressed in pallial INs. Our experimental analyses provided evidence that some of the DEX genes contribute to Maf cDKO phenotypes: (1) reduced Mef2c expression contributed to overproduction of SST+ CINs and under production of PV+ CINs (Figure 1 and Figure 2); (2) reduced Snap25 and Mef2c contribute to defects in IN morphology (Figure 4); (3) reduced Cxcr4 expression led to defects in IN migration and lamination (Figure 1—figure supplement 6). We also discovered that the Maf cDKO MGE preferentially generates HINs over CINs (Figure 5 and Figure 6). To our knowledge, this may be the first identification of genes that control the differential production of HINs and CINs.
Identification of genes that are candidate markers of immature PV INs
Our scRNA-seq analysis not only identified DEX genes between WT and cDKOs, but also provided evidence for molecular markers of immature pallial INs, especially for the PV+ subtype, which has been elusive to identify before adolescence. Comparison of Sst+ and Sst- P2 INs identified sets of genes that were differentially expressed between the two groups, including Arl4d, distal-less homeobox 5 (Dlx5), Elmo1, Erb-B2 receptor tyrosine kinase 4 (Erbb4), Igfbp4, Mef2c, Sp9 and Tcf12 (Figure 3). Because Sst-IRES-Cre fate mapping into adulthood shows that Sst-IRES-Cre expression marks almost all SST+ INs and <10% of PV INs (Pai et al., 2019), we hypothesize that P2 Sst- INs primarily represent neurons that will become PV+ (del Rio et al., 1994). We used multicolor FISH to compare the expression of genes in P2 Nkx2.1-lineage Sst+ and Sst- INs. We validated that Elmo1, Igfbp4, Mef2c and Sp9 may be candidate markers for immature PV+ INs (Figure 3), especially for immature HINs that likely become PV+. This is particularly important and useful for future research that aims to identify different molecular mechanisms involved in PV versus SST HIN development.
Of note, here we report that ~22% of the Nkx2.1-Cre lineage CINs are Sp9 positive at P2. To our knowledge, this is the first report to show Sp9 expression in CINs in the Nkx2.1-Cre lineage. We agree that 22% may be an underestimate because of several reasons: (1) when we looked at the Sp9 expression in the hippocampus,~60% of the PV candidate INs are Sp9 positive; (2) the robustness of the Sp9 RNAscope probe seemed to be stronger in the hippocampus region; (3) the robustness and efficiency of the Sp9 RNAscope probe is not as strong as the traditional DIG-labeled Sp9 probe in the neocortex as reported by the Allen Brain Institute. Additionally, recent studies have emerged to study the role of Sp9 in IN development (Liu et al., 2019; Tao et al., 2019). Liu et al reported around 50% PV+ Nkx2.1-Cre lineage CIN loss in adulthood when comparing Nkx2.1-Cre-generated Sp9 conditional heterozygous and homozygous (cHet and cKO) mutants. However, it may be questionable to conclude that 50% of the PV CINs are Sp9+ due to the fact that Sp9 deletion also leads to 50% total loss of Nkx2.1-Cre lineage CINs (Liu et al., 2019). While more studies are needed to validate the roles of these candidate PV IN marker genes in PV IN development and maturation, recent reports on Sp9 and Mef2c have provided a genetic foundation for our discovery of early candidate PV IN markers (Liu et al., 2019; Mayer et al., 2018; Tao et al., 2019).
Mef2c expression driven by Maf and Mafb promotes PV IN production and morphological maturation
Mef2c is a high confidence risk gene for ASD, intellectual disability(ID) and schizophrenia (Tu et al., 2017). Notably, Mef2c has roles in promoting cortical excitatory neuron synapse formation and morphology maturation (Harrington et al., 2016). Furthermore, there is evidence that Mef2c marks PV IN precursors, and is required for PV IN development (Mayer et al., 2018).
Mef2c is one of the most down-regulated genes in Maf cDKO MGE derived INs, which we confirmed using immunofluorescence analysis in the P2 Maf cDKO neocortex (Figure 1, Figure 1—figure supplement 5). Mef2c rescued the IN neurite outgrowth defect of both prenatal and postnatal Maf cDKO INs (Figure 4). Thus, Mef2c promotes IN morphological maturation, similar to its functions in excitatory neurons (Harrington et al., 2016).
Deletion of Mef2c using Dlx5/6-Cre led to a preferential loss of PV CINs, similar to the Maf cDKO phenotype (Mayer et al., 2018; Pai et al., 2019). Here we found that transduction of Mef2c into Maf cDKO MGE cells largely rescued their deficit in generating PV CINs (Figure 2).
The Maf cDKO MGE generates ~2 fold increase in SST+ CINs born at E12.5 and at E15.5, probably at the expense of generating PV+ CINs (Figure 1; Pai et al., 2019; Scheme 1). Thus, in the absence of the Maf genes, the MGE has a persistent defect in generating the proper balance of SST/PV INs. We propose a transcriptional pathway in which Maf and/or Mafb drive Mef2c expression in MGE progenitors to promote PV IN production (Figure 2—figure supplement 1). This pathway may operate in conjunction with signaling hubs such as MTOR/MAPK/RYK, as perturbation in these pathways also lead to altered PV/SST expression (Malik et al., 2019; McKenzie et al., 2019; Angara et al., 2020). Thus, cellular signaling as well as the Mafs and Mef2c are critical components of the program driving PV IN development.
Maf, Mafb and Mef2c expression persist as INs migrate into and mature in the pallium. Neonatal deletion of Maf and Mafb led to reduced neurite outgrowth, a defect rescued by Mef2c transduction (Figure 4). Thus, our proposed Maf/Mafb→ Mef2c transcription pathway persists as INs mature postnatally. Future work should explore whether adult Maf/Mafb expression continues to control Mef2c and IN function (Figure 2—figure supplement 1. Model).
Mechanisms that underlie PV vs SST IN neurogenesis
While our results support that Maf and Mafb have important roles in regulating the balance of PV vs. SST IN neurogenesis in the SVZ of MGE (this report and Pai et al., 2019), other studies suggest a different hypothesis in which PV INs are primarily generated by the MGE SVZ progenitors, while SST INs are primarily generated by the MGE VZ progenitors (Glickstein et al., 2007; Lodato et al., 2011; Petros et al., 2015). In particular, Petros et al showed that more SST INs are produced when MGE progenitors stay in the VZ (basal progenitor), while more PV INs are produced when MGE progenitors stay longer in the SVZ phase (intermediate progenitor). While the experimental design and results are compelling, the study is based on the hypothesis that MGE-derived IN fates are determined by the progenitor cell cycle stage. Our data showed that Maf and Mafb expression starts in the MGE SVZ. Deletion of Maf and Mafb leads to overproduction of SST INs at the expense of PV INs, but not a total loss of PV INs. Therefore, we propose the hypothesis that MGE progenitors have the potential to generate different types of INs through distinct transcriptomic regulation. Future studies are certainly needed to better understand these two hypotheses.
Snap25 expression driven by Maf and Mafb promotes MGE-derived IN morphological maturation
Snap25 is part of the neuronal SNAP receptor (SNARE) complex which regulates vesicle fusion, receptor trafficking and neurite branching (Delgado-Martínez et al., 2007; Sudhof, 2004). Snap25 loss of function studies in hippocampal excitatory neurons causes defects in neurite outgrowth and neuronal survival (Delgado-Martínez et al., 2007).
Snap25 expression was decreased in the Maf cDKO INs (Figure 1—figure supplement 5). Expressing Snap25 in prenatal and postnatal Maf cDKO INs, rescued their neurite outgrowth defect, similar to what was observed with Mef2c (Figure 4). Thus, Maf and Mafb driven expression of both Snap25 and Mef2c are critical aspects of how the Maf genes regulate IN development. Since each gene could rescue these phenotypes, it is possible that they may regulate each other.
Maf and Mafb repress HIN production and thereby control the balance of CIN and HIN numbers
Maf cDKOs had increased numbers of HINs (Figure 5). BrdU and EdU birth-dating provide evidence that the cDKO generates more HIN at both early and late stages of MGE neurogenesis (Figure 6).
The molecular program that distinguishes CINs from HINs generated by the MGE remains largely unknown. Herein via scRNA-seq, we identified that Pnoc, a gene encoding prepronociceptin, may be a unique marker that distinguishes HINs from CINs. Pnoc was previously described as an IN marker (Russ et al., 2015; Spiegel et al., 2014). In a more recent scRNA-seq analysis from hippocampal tissue, Pnoc expression was found enriched in O-LM HINs (Harris et al., 2018). Our ISH analysis from P2 WTs demonstrated that Pnoc is widely and robustly expressed in HINs but not CINs (Figure 5). Furthermore, Pnoc expression is strongly upregulated in the hippocampus in the cDKO (Figure 5), consistent with evidence for increased HIN generation.
Altogether, our data suggests that Maf and Mafb control the CIN/HIN ratio by putting a brake on HIN generation. As a result, the cDKOs have a disproportionate number of HINs. Thus, in addition to controlling the SST/PV IN subtype fate decision, Maf and Mafb also control CIN/HIN regional fate determination.
Decreased Cxcr4 expression in the maf cDKO may alter IN laminar positioning
Our scRNA-seq data suggested that Cxcr4 expression was reduced in neonatal Maf cDKO MGE INs (Figure 1—figure supplement 5). Indeed, FISH at P2 showed clear reduction of Cxcr4 expression in Maf cDKO, especially in the hippocampus (CA1, CA3 and DG p=0.0159; Neocortex p<0.0001; t test) (Figure 1—figure supplement 6). Cxcr4 expression reduction starts as early as E15.5 (p=0.012; t test) (Figure 1—figure supplement 6). These provide evidence for a potential mechanism underlying premature IN entrance into the cortical plate, that we reported previously (Pai et al., 2019).
Note that Cxcr4 null mice did not show IN over-migration or over-production in the hippocampus (Sánchez-Alcañiz et al., 2011; Stumm et al., 2003; Wang et al., 2011; Li et al., 2008). While lacking Cxcr4 in the cDKO would lead to disruption in the IN chemotactic migration, reduced Cxcr4 alone probably does not explain why Maf cDKOs have more HINs.
Potential mechanisms that lead to an increased HIN/CIN ratio in the maf cDKO
DE analysis suggested that Nrp1 is upregulated in the cDKO (Figure 1—figure supplement 5). FISH showed higher Nrp1 expression in the P2 cDKO hippocampus (Figure 1—figure supplement 7). Semaphorin binding to the Nrp1 receptor is implicated in MGE-derived IN migration away from the striatum (Marín et al., 2001). Semaphorin/neuropilin signaling is also implicated the generation and migration of CINs (Andrews et al., 2017; Tamamaki et al., 2003). However, we are unaware of a study that focused on Nrp1’s role in promoting the production/migration of HINs vs CINs.
We hypothesized that the increase in Nrp1 expression might affect CIN/HIN migration or regional fate in the cDKO. We tested this idea by introducing Nrp1 expression into MGE WT progenitors at E12.5. We transplanted MGE immature INs carrying higher expression of Nrp1 into the P1 neocortex, and surveyed their final cortical destination at P21. The majority of these modified INs appear to remain as an aggregate at the transplantation site. Very few of them migrated within the neocortex or to the hippocampus (data not shown). These results suggest that an increase in Nrp1 expression alone is not sufficient to promote the increased HINs/CIN ratio.
Interestingly, Nrp1 is not only a receptor for semaphorins, but also for vascular endothelial growth factor (VEGF). There is evidence that VEGF is expressed in the developing cortex and may regulate IN migration (Barber et al., 2018). Mackenzie and Ruhrberg et al. extensively reviewed the functions of VEGF in the nervous system (Mackenzie and Ruhrberg, 2012), which includes regulating neurogenesis (Jin et al., 2002), neuronal migration (Schwarz et al., 2004) and axonal guidance (Erskine et al., 2011). In the developing mouse brainstem, VEGF guides somata migration of NRP1-expressing facial motor neurons (Schwarz et al., 2004). In the visual system, VEGF serves as a chemoattractant to NRP1-expressing retinal ganglionic cells to promote contralateral axonal extension (Erskine et al., 2011). Another study further suggested that VEGF-NRP1 binding works in conjunction with CXCR4-SDF1 to promote breast cancer cell invasion/metastasis (Bachelder et al., 2002).
Thus, while the change of Cxcr4 and Nrp1 expression in the Maf cDKO alone does not provide a clear mechanism for why the mutant generates more HINs, we propose that interactions between NRP1 and CXCR4 can guide IN migration, and that in this context, increases in NPR1 signaling may guide INs to the hippocampus.
Maf cDKOs exhibit seizures - disease implications of Maf and Mafb dysfunction
EEG recordings from freely behaving mice showed that Maf cDKOs exhibit frequent spike-and-wave-discharges (Figure 1—figure supplement 1) similar to those that have been previously described in mouse models of genetic absence type epilepsy (Crunelli and Leresche, 2002; Paz et al., 2011; Sorokin et al., 2016; Sorokin et al., 2017). The cellular and molecular mechanisms underlying this are undoubtedly multifaceted, and include the overall reductions in MGE-derived CINs and HINs in the adult Maf cDKO mutant (Pai et al., 2019). Furthermore, we identified several genes regulated by Maf and Mafb, including Mef2c, Snap25 and Npy, mutations of which could lead to seizures (Colmers and El Bahh, 2003; Harrington et al., 2016; Houenou et al., 2017). Moreover, human patients with Maf mutations exhibit seizures and ID (Niceta et al., 2015).
In summary, we identified genetic targets (e.g. Mef2c, Snap25, Cxcr4) that are downstream of Maf and Mafb. While Maf and Mafb themselves are not high confidence neuropsychiatric disease genes, they are key regulators of many disease genes that are responsible for balanced MGE SST and PV IN production (Mef2c), IN morphological maturation (Mef2c, Snap25), and balanced MGE CIN and HIN production (Cxcr4, Nrp1).
Materials and methods
Key resources table.
| Reagent type (species) or resource |
Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Genetic reagent (M. musculus) |
Mafbf/f | Yu et al., 2013 | Generous gift from Dr. Lisa Goodrich | |
| Genetic reagent (M. musculus) |
Maff/f | Wende et al., 2012 | Generous gift from Dr. Carmen Birchmeier | |
| Genetic reagent (M. musculus) |
Nkx2.1-Cre | Xu et al., 2008 | Jax labs #008661 | |
| Genetic reagent (M. musculus) |
Sst-IRES-Cre | Taniguchi et al., 2011 | Jax labs #013044 | |
| Genetic reagent (M. musculus) |
Ai14 Rosa26-tdTomato (RosaT) | Madisen et al., 2010 | Jax labs #007908 | |
| Antibody | Rabbit anti-PARVALBUMIN (polyclonal) |
Swant | PV27 RRID:AB_2631173 |
1:400 dilution; no need for antigen retrieval |
| Antibody | Rat anti-SOMATOSTATIN (monoclonal; clone YC7) |
Millipore Sigma | MAB354 RRID:AB_2255365 |
1:200 dilution; works well in low fixed tissue |
| Antibody | Rabbit anti-MEF2C (polyclonal) |
Proteintech | 10056–1-AP RRID:AB_513447 |
1:500 dilution; no need for antigen retrieval |
| Antibody | Rat anti-BrdU antibody (monoclonal [BU1/75(ICR1)] |
Abcam | Ab6326 RRID:AB_305426 |
1:500 dilution; BrdU was stained after FISH procedure (proteinase K-based antigen retrieval) |
| Antibody | Sheep anti-DIG-POD antibody (polyclonal) |
Millipore Sigma | 11207733910 RRID:AB_514500 |
1:1000 dilution |
| Antibody | Sheep anti-Fluorescein-POD antibody (polyclonal) |
Millipore Sigma | 11426346910 RRID:AB_840257 |
1:1000 dilution |
| Antibody | Chicken anti-GFP (polyclonal) | Aves | GFP-1020 RRID:AB_100000240 |
1:1000 dilution |
| Commercial assay or kit | Click-iT EdU kit | Life Technologies | Cat #C10340 | Follow the manufacturer’s instruction |
| Commercial assay or kit | RNAscope multiplex fluorescent kit (version 2) | ACD Bio | Cat #323120 | Follow the manufacturer’s instruction |
| Commercial assay or kit | Mm-Igfbp4 | ACD Bio | Cat #425711 | Follow the manufacturer’s instruction and used it with the RNAscope multiplex fluorescent kit (version 2) |
| Commercial assay or kit | Mm-Sst | ACD Bio | Cat #404631 | |
| Commercial assay or kit | Mm-Sp9 | ACD Bio | Cat #564531 | |
| Commercial assay or kit | Mm-Mef2c | ACD Bio | Cat #421011 | |
| Commercial assay or kit | Mm-Tcf12 | ACD Bio | Cat #504861 | |
| Commercial assay or kit | Mm-tdTomato | ACD Bio | Cat #317041 | |
| Commercial assay or kit | Mm-Pnoc | ACD Bio | Cat #437881 |
Animals
All procedures and animal care were approved and performed in accordance with the University of California San Francisco Laboratory Animal Research Center (LARC) guidelines. All mice strains have been previously published: Ai14 Rosa 26-tdTomato Cre-reporter (Madisen et al., 2010), Nkx2.1-Cre (Xu et al., 2008), Sst-IRES-Cre (Taniguchi et al., 2011), Mafb flox (Yu et al., 2013) and Maf flox (Wende et al., 2012). Mice were back-crossed onto a CD-1 background before analyses. For timed pregnancies, noon on the day of the vaginal plug was counted as embryonic day 0.5.
EdU/BrdU injections and analysis
For pulse-chase experiments, pregnant mice were pulsed with 5-Bromo-2’-deoxyuridine (BrdU, Sigma B5002, prepared at 20 mg/ml) at E12.5 at a dose of 50 mg BrdU/kg body weight and 5-Ethynyl-2′-Deoxyuridine (EdU, Thermo fisher Scientific E10187, prepared at 10 mg/ml) at E15.5 at a dose of 40 mg EdU/kg body weight. Progeny were sacrificed at P2. BrdU+ cells were visualized using anti-BrdU antibody (Abcam, #ab6326). EdU+ cells were visualized using standard procedures from the Clik-iT EdU plus kit (Thermo Fisher Scientific C10340). Of note, BrdU staining was done following Lhx6 FISH. Since the tissues were processed mainly for FISH, tissue permeabilization was done using proteinase K for 30 mins. There was no hydrogen chloride (HCl) treatment for the tissue. This step likely decreased the sensitivity of the BrdU antibody. Although the BrdU antibody we used has been reported previously for the cross-reactivity with EdU, since we skipped the HCl pre-treatment, we did not observe much cross-reactivity, especially in the Lhx6+ IN population.
Immunofluorescence/Immunohistochemistry
All P2 animals were perfused with 4% PFA, followed by 4% PFA overnight fixation. Tissues were transferred to 30% sucrose the next day for cryoprotection. Brains were embedded with OCT and tissues were sectioned coronally at 25 µm. Sections were all mounted onto glass slides before staining or ISH procedures. To perform immunofluorescence staining, all primary and secondary antibodies were diluted in PBS containing 2.5% BSA and 0.3% Triton X-100. For all experiments, primary antibodies were incubated with tissue overnight, while secondary antibodies were incubated for at least 2 hr in room temperature (RT). Immunofluorescent labeling was performed with the following primary antibodies: rabbit anti-parvalbumin (Swant PV27; 1:400), rat anti-somatostatin (Millipore MAB354; 1:400), rabbit anti-MEF2C (proteintech 10056–1-AP). The appropriate 488, 594 or 647 Alexa-conjugated secondary antibodies (1:800-1:1000) were from Life Technologies. After the staining, sections were cover slipped with Vectashield containing DAPI (Vector labs; H-1800).
Fluorescent In situ hybridization (FISH) and standard ISH
To generate riboprobes for target genes, cDNAs were PCR amplified from a homemade mouse cDNA library synthesized from P0 neocortex using Superscript II. The 5’ and 3’ primers while containing sequences for cDNA synthesis, also introduced ClaI and XbaI restriction enzymes sites. The PCR products and the vector, pSP73 (Promega Cat # P2221), were digested with ClaI and XbaI, and then ligated using T4 ligase. The RNA anti-sense fluorescein-labeled/Digoxigenin-labeled probe was generated by T7 RNA polymerase (Roche) and Fluorescein labeling kit (Roche)/DIG labeling kit (Roche) from a NdeI linearized vector, with the size of the probe usually between ~600–800 bp. Briefly, 40 um sections were digested with proteinase K for 20 mins followed by 4% PFA post-fixation and 3% H2O2 incubation to block endogenous peroxidase. Hybridization with DIG and/or Fluorescein-labeled probes were performed in prehybridization buffer at 65°C overnight. Standard ISH was amplified with BM purple after anti-DIG-AP labeling, while FISH signals were performed following TSA plus kit (Akoya Biosciences) protocol after anti-DIG-POD or anti-Fluorescein-POD labeling. Detailed materials and steps can be found as previously published (Duan et al., 2018).
RNAscope multi-color fluorescent in situ hybridization
Tissues were all sectioned at 25 um and mounted onto microscope slides before the experiment. The day before the experiment, tissues were defrosted and baked at 60°C for an hour for better attachment followed by PFA post-fixation at 4°C overnight. The in situ hybridization was performed following ACD protocol for the multiplex fluorescent kit (version 2). The RNAscope probes were as follows: Mm-Igfbp4 (Cat. 425711), Mm-Sst (Cat. 404631), Mm-Sp9 (Cat. 564531), Mm-Mef2c (Cat. 421011), Mm-Tcf12 (Cat. 504861), tdTomato (Cat. 317041) and Pnoc (Cat. 437881). Slides were mounted with Vectashield mounting media containing DAPI (Cat. H-1800). For the PV marker discovery experiment, we used multiple sections from 2 WT animals for validation and quantification.
Single cell RNA-seq sample and library preparation
For single cell dissociation, primary neocortex tissues were dissected (1 P0 WT and 1 P0 cDKO; age and gender matched), and incubated with a pre-warmed solution of Papain (BrainBits) prepared according to manufacturer’s instructions for 15 min at 37°C. After incubation, tissues were washed with DMEM/10% FBS (Thermo Fisher) solution multiple times, followed by gentle trituration. Samples were diluted to approximately 2,000 cells per microliter. Cell concentration was confirmed using a CountessII automatic hemocytometer before loading onto a 10X genomics single cell preparation platform following manufacturer’s instruction to target 10,000 cell recovery per sample. Library preparation was performed by UCSF Institute of Human Genetics Core (IHG), using 10X Chromium Single 3’ Reagent Kits Version 2 (10X Genomics). The library length and concentration were quantified using a Bioanalyzer (Agilent) according to manufacturer’s protocol. Paired-end sequencing was performed on the Illumina NovaSeq.
Processing of sequencing reads
Raw base call (BCL) files were converted into FASTQ files and demultiplexed to align cells with their own barcodes using Cell Ranger V.2. Transcriptome alignment (mm10), cell filtering, barcode counting, UMI counting as well as matrix data output for downstream clustering and differential expression analyses were also performed using Cell Ranger V.2 following manufacturer’s manual. Up to this point, the pipeline was completely ran by UCSF IHG computational core.
Cell clustering, t-SNE/UMAP visualization and marker-gene identification
A digital gene expression matrix was constructed from the raw sequencing data as described using Cell Ranger. Cells with fewer than 500 UMIs or over 20,000 UMIs were discarded. Downstream analyses were performed with Seurat v.2 and the R package (Butler et al., 2018; Stuart et al., 2018). First, cells that have genes with very few counts or cells that were with high mitochondrial genes were filtered out for the downstream analyses. Gene expression value was normalized based on Seurat pipeline suggestion. Briefly, the ‘LogNormalize’ command normalizes the feature barcode (UMI) expression measurements by the total batch UMI counts; the outputs were multiplied by a scale factor of 10,000 and log-transformed. After normalization, data were scaled based on cell-cell variation regression (we called these numbers normalized expression level) to minimize the unwanted variations likely coming from technical noise or batch effects. The top ~1000 genes with the highest variance were selected to perform principal component analysis (PCA) for dimensionality reduction. 20 PC components were used to perform Louvain clustering with resolution = 1.0 to generate t-SNE plot for cluster visualization. 24 clusters were identified. We then performed FindAllMarker function from Seurat to identify markers for each cluster. Clusters were assigned to known cell types based upon cluster-specific markers. DE genes between WT and cDKO were performed per cluster basis using MarkerComp function. DE genes with adjusted p-value less than 0.05 and |LogFC| > 0.3 were selected to perform ISH for validation and functional studies. Of note, we also performed unsupervised clustering, marker gene identification, DEX analysis and UMAP visualization using Scanpy with Python (Wolf et al., 2018). We found consistent results between the two unbiased analyses.
MGE transplantation
A detailed protocol for this procedure is available in a methods format (Vogt et al., 2015; Vogt et al., 2017). We bred male mice that would yield embryos carrying Ai14 and either WT (AiFlox/Flox) or homozygous (MafFlox/Flox; MafbFlox/Flox; RosaT+) for both of the Maf alleles. The embryos were collected at E13.5, dissociated and then transfected either with a DlxI12b-Cre expressing vector or both DlxI12b-Cre expressing and target gene expressing vectors (Vogt et al., 2015; Vogt et al., 2017) in DMEM supplemented with 10% Fetal Bovine Serum (FBS) at 37°C and at pH ~7.2 for 30 min. The successful delivery of the Cre DNA will deletes the Maf genes and activates tdTomato expression from the Ai14 allele. The cells were then washed several times with DMEM/FBS and pelleted. Next, the cells were loaded into the front of a beveled glass needle. P1 WT pups were anesthetized on ice and injected with ~300 nL of cells over 3–5 sites in the right hemisphere. Pups were warmed until able to move and then put back with their mom. They were aged to 30 days and then perfused. Note that we section whole brains of all tissues into 25 um sections, and we used all brain sections for PV and SST staining followed by cell quantification.
Primary neuronal culture for analysis of dendritic arborization (Sholl) using neonatal cortex
Primary cortical neuron cultures were prepared as described (Shepherd et al., 2006). Briefly, we bred MafFlox/Flox, MafbFlox/Flox RosaT+ females with MafFlox/+, MafbFlox/+, Nkx2-1 Cre+ males to generate P0 Maf cKO, Mafb cKO and cDKOs. Control P0 animals were generated either through the same crossing or through Ai14fFlox/Flox females bred with Nkx2-1 Cre+ males. tdTomato+ P0 pups were pre-screened using fluorescence dissection microscope. We also collected some tdTomato- P0 pups for cell preparation. Cortical tissues were dissected in cold EBSS, followed by trypsin (Thermo Fisher Scientific 25200056) treatment for 15 min at 37°C. Trypsinization was inhibited using 10% FBS containing DMEM. Cells were washed once with DMEM, then resuspended in 10% FBS containing Neuralbasal-A medium (Thermo Fisher Scientific 12348017) with B27 (Thermo Fisher Scientific 17504044). Cell density was quantified using hemocytometer. tdTomato+ cell preparations were diluted using tdTomato- P0 cell preparation roughly at a ratio of 1:10. Cells were plated onto poly-D-lysine and laminin coated coverslips (Corning 08-774-385) preloaded in 24-well plates and then cultured in a 37°C incubator for 14 days. Serum free Neuralbasal-A medium with B27 and Glutamax (Thermo Fisher Scientific 35050061) was used to maintain the cell growth. After 14 days in vitro, cell culture medium was removed and replaced with freshly made 4% PFA for 15 min fixation. PFA was washed off several times with 1X PBS followed by a regular immunostaining protocol. For Sholl analysis,≥3 animals and 15–20 neurons were analyzed per genotype. Images were processed and analyzed using FIJI software based on previously described protocol (Pai et al., 2019; Ferreira et al., 2014; Martin et al., 2018).
EEG recordings and analysis
EEG recordings and analysis were done as previously described (Clemente-Perez et al., 2017; Ritter-Makinson et al., 2019). The devices for EEG recordings in freely behaving mice were all custom made in the Paz lab. Briefly, we designed devices containing multiple screws for acquisition of the EEG signals implanted in primary somatosensory (S1), primary visual (V1), and prefrontal cortices. S1: −0.5 mm posterior from Bregma,±3.25 mm lateral. V1: −2.9 mm posterior from Bregma,±2.5 mm lateral. PFC: 1 mm anterior from Bregma, midline. Mice were allowed to recover for at least 1 week before recording. EEG signals were recorded using RZ5 (TDT) and sampled at 1221 Hz. A video camera that was synchronized to the signal acquisition was used to continuously monitor the animals. Each recording trial lasted 30–180 min. To control for circadian rhythms, we housed our animals using a regular light/dark cycle, and performed recordings between roughly 11:00 AM and 6:00 PM. All the recordings were performed during wakefulness.
Image acquisition and processing
Immunohistochemistry images were taken using 10X or 20X objectives under Nikon Ti microscope with DS-Qi2 color camera together with NIS Elements acquisition software. Image brightness and contrast were adjusted and merged for publication using FIJI software.
Sholl analysis and excitatory synapse images were taken using the Nikon Ti inverted fluorescence microscope with CSU-W1 large field of view confocal. Images for Sholl analysis were taken using 40X oil objective. Open source micromanager 2.0 beta was used to acquire images. Brightness/contrast adjustment, z-stack image and binary image processing and Sholl analysis were all conducted using the image calculator and Sholl analysis plugins in FIJI software.
Schema and Model Illustration
All schemas were drafted using the BioRender online platform.
Quantification and statistical analyses
All bar graphs were shown as mean ± SEM. All statistical analyses were performed using Graphpad Prism (version 7) and a p value of < 0.05 was considered significant. For RNA expression comparison shown in Figure 3, we used python to conduct non-parametric analysis, and a p value of < 0.05 was considered significant. The specific n for each experiment as well as the post-hoc test and corrected p-values, can be found in the Results section, in the Figure legends or in supplementary tables.
For all cell counts performed for immunohistochemistry and in situ hybridization, we used the cell counter plug-in in FIJI software. For P2 hippocampus immunofluorescence and FISH cell counts, we focused on the central part of the hippocampi (exclude very rostral and very caudal region). Note that for P2 HIN Nrp1 FISH, we excluded the pyramidal cell layer of hippocampi for quantification. The Nrp1 expression is very high in the hippocampal excitatory neurons (pyramidal layer) and we think that might confound the analysis.
For statistical analyses, we mainly used two methods depending on whether data were parametric or nonparametric in distribution. For parametric data, we utilized a Welch’s t test. For data that was normalized (eg. proportions), we used the non-parametric Mann-Whitney test. For MGE transplantation, we used non-parametric Chi-square test for comparison.
Acknowledgements
This work was supported by the following research grants. JLRR is supported by Nina Ireland, NIMH R01 MH081880, NIMH R37/R01 MH049428, NIH/NIDDK P30DK098722 (UCSF NORC); DV is funded by Spectrum Health-MSU Alliance Corporation. ELLP is funded by the UCSF neuroscience graduate program and NIMH R01 MH081880. Jin Chen is funded by the Jane Coffin Childs Memorial Fund for Medical Research and the NIH K99/R00 Pathway to Independence Award (GM134154); SFD is funded by NINDS R01 NS34661 and Simons Foundation (SFARI A133320); MFP and JSC are funded by NIH/NINDS K08NS091537; JTP is supported by NIH/NINDS R01NS096369, Gladstone Institutes, and NSF # 1608236. FSC is supported by the National Science Foundation Graduate Research Fellowship Award (NSF #1144247), NIH F31 NS111819-01A1 and the UCSF neuroscience graduate program; Jiapei Chen is supported by the UCSF biomedical sciences graduate program. We thank John Askins for assistance with blinded seizure analysis. We thank UCSF Institute of Human Genomics (IHG) for the assistance with 10X single-cell RNA sequencing library preparation and sequencing.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
John LR Rubenstein, Email: John.rubenstein@ucsf.edu.
Joseph G Gleeson, Howard Hughes Medical Institute, The Rockefeller University, United States.
Marianne E Bronner, California Institute of Technology, United States.
Funding Information
This paper was supported by the following grants:
National Institute of Mental Health MH081880 to Emily Ling-Lin Pai, John LR Rubenstein.
National Institute of Mental Health MH049428 to John LR Rubenstein.
National Institute of Diabetes and Digestive and Kidney Diseases P30DK098722 to Emily Ling-Lin Pai, John LR Rubenstein.
NIH Office of the Director GM134154 to Jin Chen.
National Institute of Neurological Disorders and Stroke NS34661 to Siavash Fazel Darbandi, John LR Rubenstein.
Simons Foundation SFARI A133320 to Siavash Fazel Darbandi, John LR Rubenstein.
National Institute of Neurological Disorders and Stroke K08NS091537 to Julia S Chu, Mercedes F Paredes.
Spectrum Health-MSU Alliance Corporation to Daniel Vogt.
National Institute of Neurological Disorders and Stroke R01NS096369 to Frances S Cho, Jiapei Chen, Jeanne T Paz.
National Science Foundation 1608236 to Frances S Cho, Jeanne T Paz.
National Science Foundation 1144247 to Frances S Cho.
National Institute of Neurological Disorders and Stroke F31 NS111819-01A1 to Frances S Cho.
The Roberta and Oscar Gregory Endowment in Stroke and Brain Research to Mercedes F Paredes.
University of California, San Francisco Neuroscience Graduate Program to Emily Ling-Lin Pai, Frances S Cho.
Jane Coffin Childs Memorial Fund for Medical Research to Jin Chen.
Gladstone Institutes to Jeanne T Paz.
University of California, San Francisco Biomedical Sciences Graduate Program to Jiapei Chen.
Additional information
Competing interests
No competing interests declared.
is cofounder, stockholder, and currently on the scientific board of Neurona, a company studying the potential therapeutic use of interneuron transplantation.
Author contributions
Conceptualization, Data curation, Formal analysis, Supervision, Validation, Methodology, Writing - original draft, Writing - review and editing.
Data curation, Formal analysis, Validation, Investigation, Methodology, Writing - review and editing.
Validation, Methodology, Writing - review and editing.
Formal analysis, Investigation, Methodology, Writing - original draft, Writing - review and editing.
Formal analysis, Investigation, Methodology, Writing - review and editing.
Investigation, Methodology, conducted experiments, which generated data that we used to address some of referees' questions.
Supervision, Validation, Methodology, Writing - review and editing.
Data curation, Formal analysis, Funding acquisition, Methodology, Writing - original draft, Writing - review and editing.
Conceptualization, Validation, Investigation, Methodology, Writing - review and editing.
Conceptualization, Resources, Supervision, Investigation, Methodology, Writing - review and editing.
Resources, Supervision, Funding acquisition, Investigation, Writing - original draft, Project administration, Writing - review and editing.
Ethics
Animal experimentation: All procedures and animal care were approved and performed in accordance with the University of California San Francisco Laboratory Animal Research Center (LARC) guidelines. All animals were handled based on the approved institutional animal care and use committee (IACUC) protocol (AN180174-01B) at the University of California San Francisco.
Additional files
Data availability
We submitted the original source data that was used for Seurat pipeline analysis to GEO under the accession number GSE144222. Readers can utilize these datasets for reanalysis and new analysis using Seurat pipeline or other customized codes for more data mining.
The following dataset was generated:
Pai ELL, Chen J, Darbandi SF, Cho FS, Lindtner S, Chu JS, Paz JT, Vogt D, Paredes MF, Rubenstein JLR. 2020. Function of Mafb and c-Maf in MGE-derived interneuron development. NCBI Gene Expression Omnibus. GSE144222
References
- Andrews WD, Barber M, Nemitz M, Memi F, Parnavelas JG. Semaphorin3A-neuropilin1 signalling is involved in the generation of cortical interneurons. Brain Structure and Function. 2017;222:2217–2233. doi: 10.1007/s00429-016-1337-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angara K, Pai EL, Bilinovich SM, Stafford AM, Nguyen JT, Li KX, Paul A, Rubenstein JL, Vogt D. Nf1 deletion results in depletion of the Lhx6 transcription factor and a specific loss of parvalbumin+ cortical interneurons. PNAS. 2020;117:6189–6195. doi: 10.1073/pnas.1915458117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgarian Z, Magno L, Ktena N, Harris KD, Kessaris N. Hippocampal CA1 somatostatin interneurons originate in the embryonic MGE/POA. Stem Cell Reports. 2019;13:793–802. doi: 10.1016/j.stemcr.2019.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachelder RE, Wendt MA, Mercurio AM. Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Research. 2002;62:7203–7206. [PubMed] [Google Scholar]
- Barber M, Andrews WD, Memi F, Gardener P, Ciantar D, Tata M, Ruhrberg C, Parnavelas JG. Vascular-Derived vegfa promotes cortical interneuron migration and proximity to the vasculature in the developing forebrain. Cerebral Cortex. 2018;28:2577–2593. doi: 10.1093/cercor/bhy082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, Newell EW. Dimensionality reduction for visualizing single-cell data using UMAP. Nature Biotechnology. 2019;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente-Perez A, Makinson SR, Higashikubo B, Brovarney S, Cho FS, Urry A, Holden SS, Wimer M, Dávid C, Fenno LE, Acsády L, Deisseroth K, Paz JT. Distinct thalamic reticular cell types differentially modulate normal and pathological cortical rhythms. Cell Reports. 2017;19:2130–2142. doi: 10.1016/j.celrep.2017.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos I, Long JE, Thwin MT, Rubenstein JL. Cellular patterns of transcription factor expression in developing cortical interneurons. Cerebral Cortex. 2006;16:i82–i88. doi: 10.1093/cercor/bhk003. [DOI] [PubMed] [Google Scholar]
- Colmers WF, El Bahh B. Neuropeptide Y and epilepsy. Epilepsy Currents. 2003;3:53–58. doi: 10.1046/j.1535-7597.2003.03208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nature Reviews Neuroscience. 2002;3:371–382. doi: 10.1038/nrn811. [DOI] [PubMed] [Google Scholar]
- del Rio J, de Lecea L, Ferrer I, Soriano E. The development of parvalbumin-immunoreactivity in the neocortex of the mouse. Developmental Brain Research. 1994;81:247–259. doi: 10.1016/0165-3806(94)90311-5. [DOI] [PubMed] [Google Scholar]
- Delgado-Martínez I, Nehring RB, Sørensen JB. Differential abilities of SNAP-25 homologs to support neuronal function. Journal of Neuroscience. 2007;27:9380–9391. doi: 10.1523/JNEUROSCI.5092-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X, Krishnaswamy A, Laboulaye MA, Liu J, Peng Y-R, Yamagata M, Toma K, Sanes JR. Cadherin combinations recruit dendrites of distinct retinal neurons to a shared interneuronal scaffold. Neuron. 2018;99:1145–1154. doi: 10.1016/j.neuron.2018.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erskine L, Reijntjes S, Pratt T, Denti L, Schwarz Q, Vieira JM, Alakakone B, Shewan D, Ruhrberg C. VEGF signaling through neuropilin 1 guides commissural axon crossing at the optic chiasm. Neuron. 2011;70:951–965. doi: 10.1016/j.neuron.2011.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira TA, Blackman AV, Oyrer J, Jayabal S, Chung AJ, Watt AJ, Sjöström PJ, van Meyel DJ. Neuronal morphometry directly from bitmap images. Nature Methods. 2014;11:982–984. doi: 10.1038/nmeth.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge SX, Jung D, Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics. 2020;36:2628–2629. doi: 10.1093/bioinformatics/btz931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman D, Griveau A, Dehorter N, Teissier A, Varela C, Pla R, Pierani A, Marín O. A wide diversity of cortical GABAergic interneurons derives from the embryonic preoptic area. Journal of Neuroscience. 2011;31:16570–16580. doi: 10.1523/JNEUROSCI.4068-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickstein SB, Moore H, Slowinska B, Racchumi J, Suh M, Chuhma N, Ross ME. Selective cortical interneuron and GABA deficits in cyclin D2-null mice. Development. 2007;134:4083–4093. doi: 10.1242/dev.008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AJ, Raissi A, Rajkovich K, Berto S, Kumar J, Molinaro G, Raduazzo J, Guo Y, Loerwald K, Konopka G, Huber KM, Cowan CW. MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. eLife. 2016;5:ee20059. doi: 10.7554/eLife.20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD, Hochgerner H, Skene NG, Magno L, Katona L, Bengtsson Gonzales C, Somogyi P, Kessaris N, Linnarsson S, Hjerling-Leffler J. Classes and continua of hippocampal CA1 inhibitory neurons revealed by single-cell transcriptomics. PLOS Biology. 2018;16:e2006387. doi: 10.1371/journal.pbio.2006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houenou J, Boisgontier J, Henrion A, d'Albis MA, Dumaine A, Linke J, Wessa M, Daban C, Hamdani N, Delavest M, Llorca PM, Lançon C, Schürhoff F, Szöke A, Le Corvoisier P, Barau C, Poupon C, Etain B, Leboyer M, Jamain S. A multilevel functional study of a SNAP25 At-Risk Variant for Bipolar Disorder and Schizophrenia. The Journal of Neuroscience. 2017;37:10389–10397. doi: 10.1523/JNEUROSCI.1040-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. PNAS. 2002;99:11946–11950. doi: 10.1073/pnas.182296499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Adesnik H, Li J, Long J, Nicoll RA, Rubenstein JL, Pleasure SJ. Regional distribution of cortical interneurons and development of inhibitory tone are regulated by Cxcl12/Cxcr4 signaling. Journal of Neuroscience. 2008;28:1085–1098. doi: 10.1523/JNEUROSCI.4602-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L, Mi D, Llorca A, Marín O. Development and functional diversification of cortical interneurons. Neuron. 2018;100:294–313. doi: 10.1016/j.neuron.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liodis P, Denaxa M, Grigoriou M, Akufo-Addo C, Yanagawa Y, Pachnis V. Lhx6 activity is required for the normal migration and specification of cortical interneuron subtypes. Journal of Neuroscience. 2007;27:3078–3089. doi: 10.1523/JNEUROSCI.3055-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhang Z, Lindtner S, Li Z, Xu Z, Wei S, Liang Q, Wen Y, Tao G, You Y, Chen B, Wang Y, Rubenstein JL, Yang Z. Sp9 regulates medial ganglionic Eminence-Derived cortical interneuron development. Cerebral Cortex. 2019;29:2653–2667. doi: 10.1093/cercor/bhy133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato S, Tomassy GS, De Leonibus E, Uzcategui YG, Andolfi G, Armentano M, Touzot A, Gaztelu JM, Arlotta P, Menendez de la Prida L, Studer M. Loss of COUP-TFI alters the balance between caudal ganglionic eminence- and medial ganglionic eminence-derived cortical interneurons and results in resistance to epilepsy. Journal of Neuroscience. 2011;31:4650–4662. doi: 10.1523/JNEUROSCI.6580-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo L, Simon JM, Xing L, McCoy ES, Niehaus JK, Guo J, Anton ES, Zylka MJ. Single-cell transcriptomic analysis of mouse neocortical development. Nature Communications. 2019;10:134. doi: 10.1038/s41467-018-08079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Bendito G, Sánchez-Alcañiz JA, Pla R, Borrell V, Picó E, Valdeolmillos M, Marín O. Chemokine signaling controls intracortical migration and final distribution of GABAergic interneurons. Journal of Neuroscience. 2008;28:1613–1624. doi: 10.1523/JNEUROSCI.4651-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie F, Ruhrberg C. Diverse roles for VEGF-A in the nervous system. Development. 2012;139:1371–1380. doi: 10.1242/dev.072348. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput cre reporting and characterization system for the whole mouse brain. Nature Neuroscience. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Pai EL, Rubin AN, Stafford AM, Angara K, Minasi P, Rubenstein JL, Sohal VS, Vogt D. Tsc1 represses parvalbumin expression and fast-spiking properties in somatostatin lineage cortical interneurons. Nature Communications. 2019;10:4994. doi: 10.1038/s41467-019-12962-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín O, Yaron A, Bagri A, Tessier-Lavigne M, Rubenstein JL. Sorting of striatal and cortical interneurons regulated by semaphorin-neuropilin interactions. Science. 2001;293:872–875. doi: 10.1126/science.1061891. [DOI] [PubMed] [Google Scholar]
- Martin PM, Stanley RE, Ross AP, Freitas AE, Moyer CE, Brumback AC, Iafrati J, Stapornwongkul KS, Dominguez S, Kivimäe S, Mulligan KA, Pirooznia M, McCombie WR, Potash JB, Zandi PP, Purcell SM, Sanders SJ, Zuo Y, Sohal VS, Cheyette BNR. DIXDC1 contributes to psychiatric susceptibility by regulating dendritic spine and glutamatergic synapse density via GSK3 and wnt/β-catenin signaling. Molecular Psychiatry. 2018;23:467–475. doi: 10.1038/mp.2016.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C, Hafemeister C, Bandler RC, Machold R, Batista Brito R, Jaglin X, Allaway K, Butler A, Fishell G, Satija R. Developmental diversification of cortical inhibitory interneurons. Nature. 2018;555:457–462. doi: 10.1038/nature25999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie MG, Cobbs LV, Dummer PD, Petros TJ, Halford MM, Stacker SA, Zou Y, Fishell GJ, Au E. Non-canonical wnt signaling through ryk regulates the generation of somatostatin- and Parvalbumin-Expressing cortical interneurons. Neuron. 2019;103:853–864. doi: 10.1016/j.neuron.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi D, Li Z, Lim L, Li M, Moissidis M, Yang Y, Gao T, Hu TX, Pratt T, Price DJ, Sestan N, Marín O. Early emergence of cortical interneuron diversity in the mouse embryo. Science. 2018;360:81–85. doi: 10.1126/science.aar6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niceta M, Stellacci E, Gripp KW, Zampino G, Kousi M, Anselmi M, Traversa A, Ciolfi A, Stabley D, Bruselles A, Caputo V, Cecchetti S, Prudente S, Fiorenza MT, Boitani C, Philip N, Niyazov D, Leoni C, Nakane T, Keppler-Noreuil K, Braddock SR, Gillessen-Kaesbach G, Palleschi A, Campeau PM, Lee BH, Pouponnot C, Stella L, Bocchinfuso G, Katsanis N, Sol-Church K, Tartaglia M. Mutations impairing GSK3-Mediated MAF phosphorylation cause cataract, deafness, intellectual disability, seizures, and a down Syndrome-like facies. The American Journal of Human Genetics. 2015;96:816–825. doi: 10.1016/j.ajhg.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai EL, Vogt D, Clemente-Perez A, McKinsey GL, Cho FS, Hu JS, Wimer M, Paul A, Fazel Darbandi S, Pla R, Nowakowski TJ, Goodrich LV, Paz JT, Rubenstein JLR. Mafb and c-Maf have prenatal compensatory and postnatal antagonistic roles in cortical interneuron fate and function. Cell Reports. 2019;26:1157–1173. doi: 10.1016/j.celrep.2019.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Bryant AS, Peng K, Fenno L, Yizhar O, Frankel WN, Deisseroth K, Huguenard JR. A new mode of corticothalamic transmission revealed in the Gria4(-/-) model of absence epilepsy. Nature Neuroscience. 2011;14:1167–1173. doi: 10.1038/nn.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, McBain CJ. Hippocampal GABAergic inhibitory interneurons. Physiological Reviews. 2017;97:1619–1747. doi: 10.1152/physrev.00007.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros TJ, Bultje RS, Ross ME, Fishell G, Anderson SA. Apical versus basal neurogenesis directs cortical interneuron subclass fate. Cell Reports. 2015;13:1090–1095. doi: 10.1016/j.celrep.2015.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter-Makinson S, Clemente-Perez A, Higashikubo B, Cho FS, Holden SS, Bennett E, Chkhaidze A, Eelkman Rooda OHJ, Cornet MC, Hoebeek FE, Yamakawa K, Cilio MR, Delord B, Paz JT. Augmented reticular thalamic bursting and seizures in Scn1a-Dravet syndrome. Cell Reports. 2019;26:54–64. doi: 10.1016/j.celrep.2018.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes, Brain and Behavior. 2003;2:255–267. doi: 10.1034/j.1601-183X.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ JB, Borromeo MD, Kollipara RK, Bommareddy PK, Johnson JE, Kaltschmidt JA. Misexpression of ptf1a in cortical pyramidal cells in vivo promotes an inhibitory peptidergic identity. The Journal of Neuroscience. 2015;35:6028–6037. doi: 10.1523/JNEUROSCI.3821-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Alcañiz JA, Haege S, Mueller W, Pla R, Mackay F, Schulz S, López-Bendito G, Stumm R, Marín O. Cxcr7 controls neuronal migration by regulating chemokine responsiveness. Neuron. 2011;69:77–90. doi: 10.1016/j.neuron.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Schwarz Q, Gu C, Fujisawa H, Sabelko K, Gertsenstein M, Nagy A, Taniguchi M, Kolodkin AL, Ginty DD, Shima DT, Ruhrberg C. Vascular endothelial growth factor controls neuronal migration and cooperates with Sema3A to pattern distinct compartments of the facial nerve. Genes & Development. 2004;18:2822–2834. doi: 10.1101/gad.322904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Rubenstein JLR. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Molecular Psychiatry. 2019;24:1248–1257. doi: 10.1038/s41380-019-0426-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin JM, Paz JT, Huguenard JR. Absence seizure susceptibility correlates with pre-ictal β oscillations. Journal of Physiology-Paris. 2016;110:372–381. doi: 10.1016/j.jphysparis.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin JM, Davidson TJ, Frechette E, Abramian AM, Deisseroth K, Huguenard JR, Paz JT. Bidirectional control of generalized epilepsy networks via rapid Real-Time switching of firing mode. Neuron. 2017;93:194–210. doi: 10.1016/j.neuron.2016.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel I, Mardinly AR, Gabel HW, Bazinet JE, Couch CH, Tzeng CP, Harmin DA, Greenberg ME. Npas4 regulates excitatory-inhibitory balance within neural circuits through cell-type-specific gene programs. Cell. 2014;157:1216–1229. doi: 10.1016/j.cell.2014.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Stoeckius M, Peter Smibert RS. Comprehensive integration of single cell data. bioRxiv. 2018 doi: 10.1101/460147. [DOI] [PMC free article] [PubMed]
- Stumm RK, Zhou C, Ara T, Lazarini F, Dubois-Dalcq M, Nagasawa T, Höllt V, Schulz S. CXCR4 regulates interneuron migration in the developing neocortex. The Journal of Neuroscience. 2003;23:5123–5130. doi: 10.1523/JNEUROSCI.23-12-05123.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annual Review of Neuroscience. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Fujimori K, Nojyo Y, Kaneko T, Takauji R. Evidence that Sema3A and Sema3F regulate the migration of GABAergic neurons in the developing neocortex. The Journal of Comparative Neurology. 2003;455:238–248. doi: 10.1002/cne.10476. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Kvitsani D, Fu Y, Lu J, Lin Y, Miyoshi G, Shima Y, Fishell G, Nelson SB, Huang ZJ. A resource of cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71:995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao G, Li Z, Wen Y, Song X, Wei S, Du H, Yang Z, Xu Z, You Y. Transcription factors Sp8 and Sp9 regulate medial ganglionic Eminence-Derived cortical interneuron migration. Frontiers in Molecular Neuroscience. 2019;12:75. doi: 10.3389/fnmol.2019.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, Penn O, Bakken T, Menon V, Miller J, Fong O, Hirokawa KE, Lathia K, Rimorin C, Tieu M, Larsen R, Casper T, Barkan E, Kroll M, Parry S, Shapovalova NV, Hirschstein D, Pendergraft J, Sullivan HA, Kim TK, Szafer A, Dee N, Groblewski P, Wickersham I, Cetin A, Harris JA, Levi BP, Sunkin SM, Madisen L, Daigle TL, Looger L, Bernard A, Phillips J, Lein E, Hawrylycz M, Svoboda K, Jones AR, Koch C, Zeng H. Shared and distinct transcriptomic cell types across neocortical Areas. Nature. 2018;563:72–78. doi: 10.1038/s41586-018-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu S, Akhtar MW, Escorihuela RM, Amador-Arjona A, Swarup V, Parker J, Zaremba JD, Holland T, Bansal N, Holohan DR, Lopez K, Ryan SD, Chan SF, Yan L, Zhang X, Huang X, Sultan A, McKercher SR, Ambasudhan R, Xu H, Wang Y, Geschwind DH, Roberts AJ, Terskikh AV, Rissman RA, Masliah E, Lipton SA, Nakanishi N. NitroSynapsin therapy for a mouse MEF2C haploinsufficiency model of human autism. Nature Communications. 2017;8:1488. doi: 10.1038/s41467-017-01563-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt D, Wu P-R, Sorrells SF, Arnold C, Alvarez-Buylla A, Rubenstein JLR. Viral-mediated labeling and transplantation of medial ganglionic eminence (MGE) Cells for <em>in vivo</em> studies. Journal of Visualized Experiments. 2015;23:52740. doi: 10.3791/52740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt D, Cho KKA, Shelton SM, Paul A, Huang ZJ, Sohal VS, Rubenstein JLR. Mouse Cntnap2 and human CNTNAP2 ASD alleles cell autonomously regulate PV+ cortical interneurons. Cerebral Cortex. 2017;28:3868–3879. doi: 10.1093/cercor/bhx248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li G, Stanco A, Long JE, Crawford D, Potter GB, Pleasure SJ, Behrens T, Rubenstein JL. CXCR4 and CXCR7 have distinct functions in regulating interneuron migration. Neuron. 2011;69:61–76. doi: 10.1016/j.neuron.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wende H, Lechner SG, Cheret C, Bourane S, Kolanczyk ME, Pattyn A, Reuter K, Munier FL, Carroll P, Lewin GR, Birchmeier C. The transcription factor c-Maf controls touch receptor development and function. Science. 2012;335:1373–1376. doi: 10.1126/science.1214314. [DOI] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biology. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Tam M, Anderson SA. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. The Journal of Comparative Neurology. 2008;506:16–29. doi: 10.1002/cne.21529. [DOI] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WM, Appler JM, Kim YH, Nishitani AM, Holt JR, Goodrich LV. A Gata3-Mafb transcriptional network directs post-synaptic differentiation in synapses specialized for hearing. eLife. 2013;2:e01341. doi: 10.7554/eLife.01341. [DOI] [PMC free article] [PubMed] [Google Scholar]