

Abstract
The stress-induced susceptibility hypothesis, which predicts chronic stress weakens immune defences, was proposed to explain increasing infectious disease-related mass mortality and population declines. Previous work characterized wetland salinization as a chronic stressor to larval amphibian populations. Thus, we combined field observations with experimental exposures quantifying epidemiological parameters to test the role of salinity stress in the occurrence of ranavirus-associated mass mortality events. Despite ubiquitous pathogen presence (94%), populations exposed to salt runoff had slightly more frequent ranavirus related mass mortality events, more lethal infections, and 117-times greater pathogen environmental DNA. Experimental exposure to chronic elevated salinity (0.8–1.6 g l−1 Cl−) reduced tolerance to infection, causing greater mortality at lower doses. We found a strong negative relationship between splenocyte proliferation and corticosterone in ranavirus-infected larvae at a moderate elevation of salinity, supporting glucocorticoid-medicated immunosuppression, but not at high salinity. Salinity alone reduced proliferation further at similar corticosterone levels and infection intensities. Finally, larvae raised in elevated salinity had 10 times more intense infections and shed five times as much virus with similar viral decay rates, suggesting increased transmission. Our findings illustrate how a small change in habitat quality leads to more lethal infections and potentially greater transmission efficiency, increasing the severity of ranavirus epidemics.
Keywords: disease susceptibility, glucocorticoid, road ecology, wildlife disease, mass mortality event
1. Introduction
Mass mortality events due to infectious diseases in animals are increasing in frequency across taxa [1], many of which involve multiple environmental stressors [2–5]. The stress-induced susceptibility hypothesis posits that infectious disease-associated population declines are caused by persistent stress that increases host susceptibility to infection [5–7]. The presumed mechanism is that chronic activation of the neuroendocrine stress axis (hypothalamus–pituitary–adrenal/interrenal axis) causes negative feedback, elevating glucocorticoid levels that directly signal immune cell apoptosis [8,9]. Since the immunomodulatory effects of glucocorticoids depend on magnitude and timing [8,10] and can be enhancing at acute elevated levels [9,11], a key assumption of this hypothesis is that ecological change causes chronic stress, which is immunosuppressive. Other, glucocorticoid-independent mechanisms have been proposed, such as limited resources needed to fight infection [12], direct cytotoxicity (reviewed in [10]), or altered neuroendocrine-immune interactions causing homeostatic imbalance (e.g. endocrine disruptors [13]). A further challenge is translating individual-level effects to the population scale, since stressors can affect multiple, countervailing epidemiological factors [14]. Due to the complexity of host–pathogen–environment interactions, there are few examples in which causal links between ecological change, physiological stress and the likelihood of pathogen-induced mass mortality in wildlife have been established [3,15].
To investigate the effects of environmental change on pathogen-induced mass mortality events, we focused on amphibian populations affected by road salt runoff. In the USA, the yearly application of millions of tons of deicing salts (typically NaCl) has led to persistent salinization of freshwater systems (up to 25% of seawater) [16]. Salinization is also caused by increasing drought, mining activities, agriculture and sea-level rise [17], and has been associated with increased disease susceptibility in other systems (e.g. Streptococcus in freshwater tilapia [18]). Although some lineages have adapted to brackish water [19], even slight increases in salinity are energetically costly to evolutionarily naive amphibians, causing physiological stress evidenced by developmental retardation, gill inflammation and increased cell turnover [20–22]. At concentrations below the US EPA's recommended level for aquatic life (230 mg l−1 Cl−), salinization of roadside habitats in northeastern US (average of 150 mg l−1 Cl− in our study system) [23] reduces amphibian embryonic survival [24,25], larval growth and activity [26,27], and elevates plasma corticosterone concentrations in adults [26]. The evidence of salinization's influence on amphibian disease dynamics, however, is mixed. Experimental salt exposure increased trematode (Ribeiroia ondatrae) infection intensity, but reduced the number of infections, potentially through negative effects of salinity on the trematode [28]. Yet in a similar study, trematode infection was more prevalent with salt exposure [29]. On the other hand, elevated salinity reduces survival of the fungal pathogen Batrachochytridum dendrobatidis (Bd) outside its host, thus appears to reduce transmission [30]. Predicting the net effects of stressors on host–pathogen interactions is not straight forward [14], but there is an urgent need to clarify the potential outcomes as secondary salinization is rapidly changing freshwater ecosystems globally [17,31].
Here, we tested whether salinization of wetlands from salt runoff increases the frequency or severity of ranavirus epidemics in larval wood frog (Lithobates sylvaticus) populations. Ranaviruses are widely distributed, multi-host, often lethal viruses in the genus Ranavirus (Family Iridoviridae) [32]. In a range-wide survey of wood frogs, ranavirus was more prevalent in regions with greater host population density and road density [33]. Wood frog larvae are highly susceptible to ranavirus infection compared with other ranids (e.g. bullfrogs), and epidemics can vary from insubstantial mortality to the complete loss of a year class [34], with no clear explanation [35]. We predicted that if salinity stress plays a role increasing the severity or frequency of ranavirus epidemics, then more severe infections and greater mortality from infection would occur in populations affected by salt runoff. Following the stress-induced susceptibility hypothesis, we predicted the chronic stress of road salt exposure—which causes a reallocation of resources from body growth to coping with greater osmoregulatory demands [26]—will cause reduced immunocompetence through one or more of the aforementioned mechanisms. Since stress can reduce the probability of an epidemic through the loss of susceptible hosts [14], we would expect salinity stress to increase transmission, either by increasing the viral shedding or environmental persistence, both of which would lead to a greater chance of mass mortality in populations in salinized wetlands. We carried out field surveys to examine correlations with pathogen presence and mass mortality events, and laboratory experiments to test causal mechanisms through which salinity stress could increase the likelihood of a die-off through effects on host susceptibility and viral transmission.
2. Methods
(a). Observations of ranavirus-related mass mortality events
We surveyed 18 ephemeral ponds with similar wood frog larval densities that spanned a range of proximities to roads that receive salt application [24,36] in the mixed hardwood forests of Yale Myers Forest (YMF) in northeastern Connecticut, USA (figure 1a), where the primary disturbances are associated with road maintenance and rural housing. Roadside wetlands reach conductivity (a direct measure of salinity [37]) levels of 4000 µS cm−1 (approx. 2 g l−1 Cl−) due to deicing salt runoff [24,36], causing high levels of embryonic mortality in wood frogs [24,38], and thus decreased larval density, which would reduce our ability to detect mortality events. Selected ponds less than 250 m from paved roads had higher salinity (conductivity range: 30–360 µS cm−1; approx. 4–95 mg l−1 Cl−) than those further away (20–50 µS cm−1; approx. 2–5 mg l−1 Cl−; t-test: t11.44 = 2.38, p = 0.036); other abiotic factors measured in each pond were not correlated with conductivity (electronic supplementary material, table S1). We conducted weekly surveys to observe carcasses from the mid–late larval period, from 3 June to 10 July 2013, when ranavirus-related mortality events are most common [36,39]. Die-off events were defined as greater than 90% of larval amphibians found dead and ranavirus infections confirmed via qPCR. We also collected euthanized larvae (n = 5) and pond water samples to determine ranavirus abundance in the community (250 ml filtered water, n = 3 per pond; hereafter ‘eDNA’) at two points in larval development (see [35] for methods). We disinfected all equipment and waders with 10% bleach between ponds to prevent cross-pond contamination. All statistical analyses were performed in R v. 3.4.3 [40]. We applied an information-theoretic approach [41] (see electronic supplementary material, Methods) to estimate the importance of pond characteristics in predicting the observation of die-offs (logistic regression) and Ranavirus eDNA concentrations early and late in larval development (linear regressions).
Figure 1.

(a) Field survey of ranavirus occurrence and associated die-offs. (a) Map indicating locations of the 18 ephemeral ponds monitored weekly in June and July for ranavirus-associated die-offs of wood frog larvae in the vicinity of Yale Myers Forest in Northeastern CT, USA (top-left subset), a 32 km2 managed mixed-hardwood forest. Numbered sites on map refer to the ponds from which larvae were collected to observe the progression of naturally acquired ranavirus infections (11 ponds, d,e). Die-offs of greater than 90% were observed in seven ponds while die-offs were not observed in the remaining eleven ponds. (b) Proximity to major roads was correlated with the probability of observed die-offs. (c) Log10 ranavirus eDNA concentration by conductivity (measure of salinity) during early larval development. (mean ± s.e.m.) (d) Average days to death of naturally infected animals in captivity after collection with a line of predicted survival from Cox proportional hazards analysis. (e) Average larval ranavirus (RV) titre of infected animals collected in the field and monitored for mortality in the laboratory by pond salinity (11 sites numbered in a; n = 96; 3–11 per site). Lines and shaded areas are the logistic (b) or linear (c,e) regression lines and the 95% confidence envelope. In (a–e), black and grey points indicate ponds where die-offs were and were not observed, respectively. (Online version in colour.)
(b). The progression of naturally acquired ranavirus infections
We collected larvae (Gosner [42] stages 34–39, median = 37) from 11 of the 18 surveyed sites that had no observed mortality at the time (numbered in figure 1a, 10–13 per pond; 2–2236 m from a major road) within 3 days of the second eDNA sample, and monitored the progression of naturally acquired infections in the laboratory. Larvae were housed individually in containers of 525 ml of dechlorinated tap water (approx. 200 µS cm−1). With this design, we could examine lasting effects of road runoff exposure, which has been shown to continue even after returning larvae to freshwater [43], but we cannot rule out the confounding effect of exposure to a change in water chemistry when brought to the laboratory. We analysed survival to 17 d post-metamorphic climax by salinity using a mixed effects Cox proportional hazard (Cox PH) model with site as random variable (coxme function in coxme package [44]). We compared log10 ranavirus liver titres at death/euthanasia using a linear mixed model with site as a random variable (lmer in the lmerTest package).
(c). Effect of elevated salinity on laboratory-raised larvae
For this and subsequent experiments, we reared animals throughout their lives in freshwater or water with added road salt (NaCl collected from a Department of Transportation salt-shed in Union, CT, USA) to achieve desired conductivities. We collected wood frog eggs from a pond in Poughkeepsie, NY, where ranavirus die-offs had not been observed (150 µS cm−1 conductivity) and transported them to Washington State University where they were housed at 15°C with 15 L/9D light cycle. Eggs were rinsed with clean water and divided into three treatments: freshwater (dechlorinated water with a conductivity approx. 200 µS cm−1 due to calcium ions), average salinity (1500 µS cm−1; approx. 0.76 g l−1 Cl−), and near maximum salinity (3000 µS cm−1; approx. 1.6 g l−1 Cl−) for wood frog ponds in this region [24,26]). While the highest salinity treatment is more saline than those in our field survey, previous surveys have detected larvae surviving in ponds across the full range of treatment levels [24,45]. We euthanized 10 larvae and screened for ranavirus before the exposure experiment begun; none amplified.
First, we estimated the effects of road salt exposure on mortality from infection by conducting a dose-response experiment using the salinity treatments above crossed with four doses of an FV3 ranavirus: mock exposure, low, medium and high ranavirus doses (0, 3 × 103, 3 × 104, 3 × 105 plaque-forming units per ml [pfu ml−1], respectively; n = 20/dose × salinity level; see electronic supplementary material, Methods). Larvae were distributed into treatment groups to standardize developmental stage (same median: Gosner 34, and range: 30–38). We monitored survival until metamorphosis and compared case mortality (qPCR positives only) from infection in individually housed larvae using a logistic regression (glm with family bionmial, link = logit; in the package stats [40]) and a parametric survival analysis (survreg in the package survival [46]), with development stage at exposure as a covariate in both analyses.
We then exposed another sample of larvae from the same cohort (n = 20/salinity level, Gosner stages 33–36) to a moderate dose (3 × 104 pfu ml−1) or mock exposure to determine the effect of elevated salinity on infection intensity in gastrointestinal (GI) tissue, corticosterone and immune response, and shedding rate. At 6 days post-inoculation (dpi) we euthanized animals, dissected spleens (see below) and froze carcasses in liquid nitrogen. We also filtered 150 ml of water from each container to collect eDNA (following methods of [35]) to estimate viral shedding rate. Ranavirus eDNA is positively related to infectiousness in wood frog tadpoles [47], but this method does not measure concentrations of viable virions. We extracted DNA from dissected GI tissue and filters and assayed both using qPCR assays (n = 20). We measured corticosterone concentrations of dissected interrenal glands (n = 10/salinity × ranavirus level), where this hormone is synthesized [48], using ether lipid extractions and enzyme immunoassays. We measured splenocyte proliferation as an indicator of immune system activity by marking mitotic cells with a polycolonal antibody for phospho-histone H3 (PH3) using immunofluorohistochemistry methods. Prior research showed that elevated glucocorticoid signalling inhibits proliferation and stimulates apoptosis of splenocytes in amphibians [48]. See electronic supplementary material, Methods for details of each of these procedures.
We analysed log10 GI and eDNA ranavirus titres and log10 interrenal corticosterone concentrations per mg of tissue by water treatments using univariate linear regressions for each response (lm analyzed using Anova in the package car [49]). We compared prevalence of infection using a χ2 test of proportions. We analysed the number of PH3 ir-cells (n = 6–10/salinity × ranavirus levels) using a Poisson regression (glm in the package stats), with main effects of ranavirus and salinity, their interaction, and the area of the spleen section as a covariate. We also used Poisson regression with a three-way interaction term between corticosterone or ranavirus titre, salinity and ranavirus exposure to determine if the relationships between these variables varied among treatments.
In a third sample of larvae from this cohort, we tested whether salinity affects contact rates to increase direct transmission [50], because elevated salinity reduces larval activity [26,28]. We counted the number of contacts between group-housed larvae (16 replicates of 10 l aquaria, 6 larvae each) that were either mock- or ranavirus-exposed (high dose, same as above) in either freshwater or 3000 µS cm−1 saltwater. After 7 days of acclimation, we tallied the number of contacts during a 10 min period 1 d before exposure, 2 dpi and 5 dpi (at same time of day). We analysed contact rate between water and ranavirus treatments using a linear mixed model with time and tank as random variables (lmer in the package lmerTest [51]).
To estimate the effects of salinity on viral persistence in the environment, we collected Gosner stages 33–38 wood frog larvae from YMF (Site 4 in figure 1a) when a die-off was occurring (ensuring they had high ranavirus titres). We set up eight replicate 10 l aquaria containing 6 larvae in either freshwater or high salinity (concentrations as above). Carcasses were removed daily with a disinfected net until day 7 when we removed all larvae. Water was sampled (150 ml; filtered and quantified as above) in each aquarium at eight timepoints. We analysed ranavirus eDNA titre using a linear regression before larvae were removed with the number of larvae remaining at d7 and timepoint as covariates, and after larvae were removed to compare decay rate between water treatments.
3. Results
(a). Observations of ranavirus-related mass mortality events
We observed ranavirus-related die-offs in seven (39%) of 18 ponds (figure 1a), five of which had elevated salinity (measured as conductivity) and were within 250 m of a paved road, and in three of which we observed ranavirus-related die-offs in 2011 (E.M.H., personal observation) and 2014 [36]. Distance to road was in all of the best-supported models, and salinity was in next best model that did not include distance to roads (electronic supplementary material, tables S2 and S3), which were the only two variables in the best-fit models that predicted the probability of a die-off. Specifically, the probability declined with distance (figure 1b; βsqrt(metres) = −0.132 ± 0.058, z = −2.272, p = 0.023) and marginally increased with salinity (βlog10(conductivity) = 4.300 ± 2.333, z = 1.843, p = 0.065). Our eDNA surveys revealed that ranavirus was ubiquitous (17/18 ponds) and ranavirus eDNA concentrations were positively related to salinity levels during early development (n = 18; βlog10(µs cm)−1 = 1.93 ± 0.690, t2,15 = 2.798, p = 0.014; figure 1c) and during late development and once die-offs began. Specifically, ponds with elevated salinity near major roads had approximately 117 times higher concentrations of ranavirus eDNA in pond water when larvae were nearing metamorphosis, which for some ponds coincided with or after observed die-offs (n = 17; βsqrt(metres) = −0.045 ± 0.017, t2,14 = −2.591, p = 0.021; βlog10(µs cm)−1 = 1.93 ± 0.823, t2,14 = 2.351, p = 0.034).
(b). The progression of naturally acquired ranavirus infections
All of the larvae collected had natural ranavirus infections at death or euthanasia, but larvae collected from ponds with higher salinity died at a faster rate from infection (Cox PH with pond as a random effect: βlog10(µs cm)−1 = 4.239 ± 1.943, z = 1.94, p = 0.029; figure 1d) and had greater larval titres at death or euthanasia (n = 96; 3–11 per site; βlog10(µs cm)−1 = 4.596 ± 1.911, t1,9.759 = 2.405, p = 0.038; figure 1e). Because salinity and eDNA concentrations were correlated, survival was also predicted by viral eDNA (βlog10(copies/ml) = 1.535 ± 0.540, z = 2.82, p = 0.005).
(c). Effects of elevated salinity on laboratory-raised larvae
Larvae had greater odds of dying from infection (positives only) with increasing virus dose and developmental stage at exposure (βlog10 RV dose = 0.899 ± 0.160, z = 5.637, p < 0.001; βstage = 0.271 ± 0.082, z = 3.310, p < 0.001), and with higher salinity (βsalinity = 3.737 ± 1.439 × 10−4, z = 2.598, p = 0.009; figure 2). Exposure to high salinity increased the odds of mortality by 2.85-fold relative to freshwater across ranavirus doses, and reduced the estimated LD50 (dose lethal to 50%) by nearly fourfold compared with freshwater (from 30 200 pfu ml−1 in freshwater to 7762 pfu ml−1). Larvae in the high-salt treatment also died faster from infections (parametric survival analysis: βsalinity = −1.23 ± 0.45 × 10−3, z = −2.84, p = 0.005; βlog10 RV dose = −0.851 ± 0.124, z = −6.88, p < 0.001; βstage = −0.179 ± 0.044, z = −4.08, p < 0.001). In the absence of ranavirus exposure, salinity had a marginal effect on mortality (βsalinity = 8.636 ± 5.157 × 10−4, z = 1.675, p = 0.094), and salinity did not affect the proportion infected (p = 0.62).
Figure 2.

Tolerance to ranavirus exposure of naive larvae reared in road salt-treated water in the laboratory. Salinity exposures reflect the average (1500 µS cm−1) and maximum (3000 µS cm−1) conductivities of roadside ponds surveyed in this region [24]. We standardized ranavirus exposures to the range of susceptible developmental stages for this species (Gosner 30–38; [52]). Points represent the proportion dead with a positive ranavirus titre (n = 20 per dose/group were exposed), and lines and 95% confidence envelope (shaded area) reflect the best-fit logistic model.
Larvae exposed to ranavirus had elevated resting interrenal corticosterone concentrations 6 dpi compared to unexposed larvae (d.f. = 1, F = 23.7, p < 0.001). Those reared in elevated salinity exhibited marginally elevated corticosterone (ANOVA; d.f. = 1, F = 3.576, p = 0.064; figure 3a), but the interaction with ranavirus exposure was not significant (p = 0.27). However, we detected main and interactive effects of salinity and ranavirus-exposure on proliferating splenocyte counts (GLM, salinity: χ2 = 16.483, d.f. = 1, p < 0.001; ranavirus: χ2 = 5.672, d.f. = 1, p = 0.017; interaction: χ2 = 4.210, d.f. = 1, p = 0.040; figure 3b). Salinity also increased GI viral titres of infected larvae (d.f. = 1, F = 7.97, p = 0.01; figure 3c). Splenocyte proliferation was negatively related to corticosterone levels (βCORT = −0.724 ± 0.099, z = −7.31, p < 0.001), but this relationship depended on salinity and ranavirus exposure (three-way interaction: χ2 = 8.343, d.f. = 1, p = 0.004, CORT–salinity interaction: χ2 = 8.594, d.f. = 1, p = 0.003, ranavirus–salinity interaction: χ2 = 3.188, d.f. = 1, p = 0.074; figure 3d). Specifically, salt-exposed animals had steeper slopes in the CORT–proliferation relationship compared to those raised in freshwater, except in high-salt and ranavirus-exposed larvae, which had a flatter slope with a lower intercept. Splenocyte proliferation was also negatively related to infection intensity, but this relationship depended on exposure to salinity (GI titre–salinity interaction: χ2 = 6.248, d.f. = 1, p = 0.012; GI titre: χ2 = 7.202, d.f. = 1, p = 0.007; electronic supplementary material, figure S1). Furthermore, greater salinity exposure slowed developmental rates (Kruskal–Wallis: χ2 = 7.532, d.f. = 2, p = 0.023), and splenocyte proliferation and developmental rate were negatively associated in elevated salinity but not in freshwater (developmental rate–salinity interaction: χ2 = 9.041, d.f. = 1, p = 0.003; development: p = 0.7374; salinity: p = 0.329, RV exposure = 0.329; electronic supplementary material, figure S1).
Figure 3.
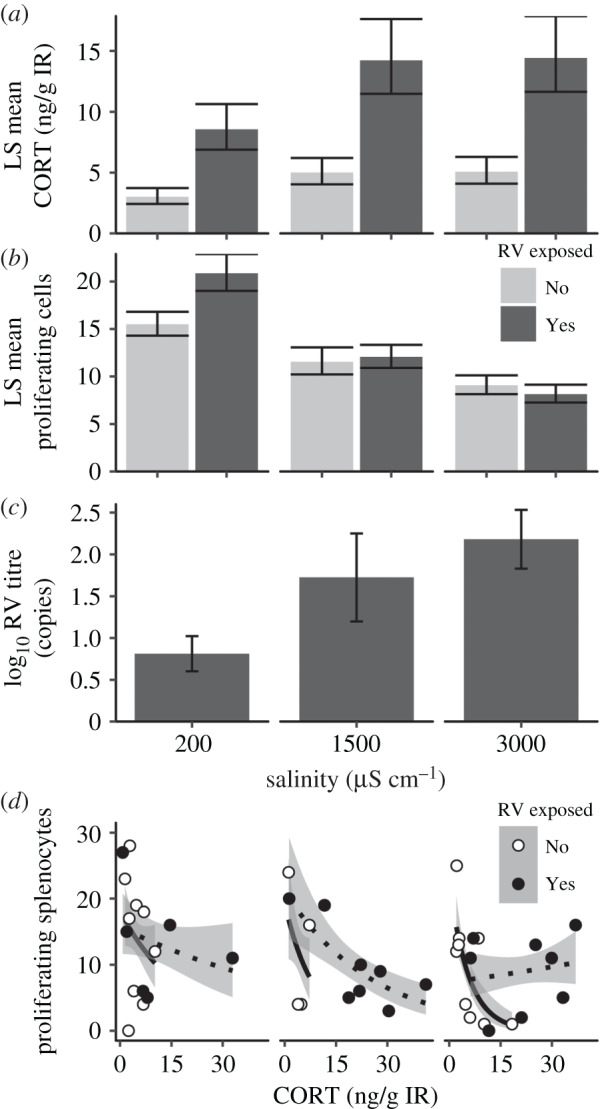
Stress and immune responses of naive larvae reared in road salt-treated water in a laboratory experiment at 6 d post-exposure (n = 20 per salinity/exposure group; exposed to 105 pfu ml−1: RV+; or mock exposed: RV−). (a) LS mean interrenal corticosterone (CORT) levels (±s.e.m., n = 10) accounting for developmental stage effects (b) LS mean proliferating splenocyte counts (±s.e.m., n = 6–10) accounting for spleen size and (c) positive log10 ranavirus titres in gastrointestinal (GI) tissue (n = 6–8). In (a–c), dark and light bars represent ranavirus-exposed and unexposed, respectively. (d) The relationship between proliferating splenocyte count and corticosterone levels across salinity (divided plots corresponding to x-axis of (a–c) and ranavirus treatments (n = 4–9). Lines represent best-fit Poisson regressions with white points and solid lines representing RV unexposed, and black points and dotted lines representing ranavirus exposed.
Our experiments designed to measure effects of salinity on routes of transmission showed that salinity treatments tended to have more extreme shedders (quantile regression ANOVA of 90th from 50th percentile: F = 4.879, p = 0.029; whiskers shown in figure 4). When we sum the ranavirus eDNA concentrations across individuals to estimate a population-level response, larvae in the high-salinity treatment shed 5.5 times more virus than the freshwater group (20 417 compared with 3715 copies total). Shed viral eDNA concentrations were correlated with GI titres (βlog10 GI titre = 0.954 ± 0.075, t1,58 = 12.737, p < 0.001). However, elevated salinity did not affect contact rates between larvae with or without ranavirus exposure (t21.18 = −1.175, p = 0.253).
Figure 4.

Effects of road salt on the shedding rate and persistence of ranavirus from infected larvae from two laboratory experiments. (a) The concentration of ranavirus DNA shed into housing water (eDNA) in points and box plots showing median, lower and upper quantiles of shed DNA, whiskers extend to 10th and 90th quantiles, representing the increase in ‘super-shedders’ in the salinity treatments (n = 20). (b) Average (±s.e.m., n = 4 aquaria) log10 ranavirus eDNA concentration in aquaria water after six ranavirus-infected wood frog larvae were introduced. Larvae were removed at 7 d (vertical dotted line), and eDNA sampling continued to examine decay rates.
We also observed an increase in viral shedding in high-salinity conditions in naturally infected larvae collected from a pond during a die-off. eDNA concentrations were more than an order of magnitude greater in the high-salinity treatment compared to freshwater (βsalinity = 1.411 ± 0.220, t1,6 = 6.407, p < 0.001), and remained higher for 12 d after we removed larvae, though there were no differences in decay rates between salinity treatments (t2,21 = 1.598, p = 0.125; figure 4).
4. Discussion
Since first proposed to explain infectious disease-linked amphibian population declines [7], the stress-induced susceptibility hypothesis has received support linking environmental change to physiological stress and greater mortality from infection (e.g. agricultural runoff, but not natural stressors; see review [53]). Yet few studies causally link immunosuppression observed in the laboratory to prevalence of infection on the landscape [54], and none we know of link to mass mortality events. Since salinization is a persistent, energetically demanding stressor [22,26] we hypothesized reduced immune function via glucocorticoid-dependent and -independent pathways at the individual level; and immunocompromised populations with increased transmission and mortality from infection would pass the tipping point of more frequent and severe ranavirus epidemics (electronic supplementary material, figure S2). Though our field observations were necessarily biased towards robust populations in order to observe mass mortality (since higher salinity roadside ponds had much lower larval densities we selected roadside populations with lower than typical salinity [26,36]), we discovered a correlation between the probability of observed ranavirus-associated die-offs and proximity to roads and a weak correlation with salinity. This observation was corroborated with more than 100 times greater concentration of viral eDNA—which is tightly associated with larval titres [35]—and greater mortality and viral loads in larvae with naturally acquired infections from more saline wetlands. Perhaps, at the maximum tolerable salinity levels, embryonic mortality reduces the probability of an epidemic through the loss of susceptible individuals [14]; thus, future work is needed to more strongly link salt stress and mass mortality events through broader surveys. Altogether, these correlations set the stage for experimentally testing whether salinity stress plays a role in the severity and outcome of infections in wood frog larvae.
Our experimental exposures at environmentally relevant road salt concentrations supported several components of the stress-induced susceptibility hypothesis and revealed additional nuances. Larvae reared in elevated salinity were more likely to die from virus exposure and exhibited suppressed immune function. Previous work also found that ranavirus infection induces a stress response [52] similar to other infections [55]. However, contrary to our expectations for the involvement of the HPI axis, salinity only marginally increased resting glucocorticoid levels. Although salinity and ranavirus exposure did not interact to predict resting interrenal corticosterone concentrations, we observed a strong negative relationship between splenocyte proliferation and corticosterone that depended on salinity and ranavirus exposure. Thus, glucocorticoid-mediated immunosuppression explains in part reduced tolerance (i.e. greater infection intensity) due to salt exposure. Splenocyte proliferation is negatively affected by glucocorticoid signalling in experimental studies in anurans [48] and is an essential acute inflammatory response that ranavirus is likely capable of inhibiting [56], offering a potential explanation to the negative relationship we found between infection loads and proliferation. Additional work is needed to determine if this splenocyte response coincides with the inhibition of other antiviral responses that occurs in other vertebrates via increased GR signalling [57] to cause greater viral loads observed here. While the increased ranavirus titres and ranavirus-induced mortality of field-caught larvae is consistent with these experimental responses, future work is still needed to determine whether wild amphibian populations exposed to deicing salt also experience a rise in glucocorticoid levels and immunosuppression similar to laboratory findings to cause more frequent mortality events.
Our findings also show that elevated salinity suppressed splenocyte proliferation more than what could be explained by corticosterone levels. Specifically, we observed a dose-dependent reduction of splenocyte proliferation with elevated salinity in ranavirus-unexposed larvae, and lower proliferation across corticosterone levels at high salinity in ranavirus-exposed larvae. These patterns suggest that elevated salinity is suppressing immunity through glucocorticoid-independent mechanisms. Since we observed reduced feeding activity in a prior study [26], salinity may affect resource acquisition or cause a delay in developmental processes that affect susceptibility to ranavirus infection. Our findings support evidence of a trade-off between the significant energetic demands of osmoregulation [19] and mounting an immune response, specifically, salt exposed larvae exhibited lower splenocyte proliferation when maintaining a comparable developmental rate, whereas the freshwater individuals exhibited no such trade-off. Similarly, migratory waterbirds experienced a trade-off between mounting an immune response and maintaining osmotic homeostasis in response to elevated salinity [58]. Additional experiments are also needed to test whether elevated internal osmolality is cytotoxic to the spleen, or whether salt changes interrenal function in ranavirus-infected larvae. This finding highlights that the stress-induced susceptibility hypothesis operates via mechanisms beyond glucocorticoid-mediated immunosuppression.
At the population level, these individual-level effects of salinity on ranavirus infection appear to drive more severe epidemics through increased disease-induced mortality rate combined with potentially greater transmission efficiency. The increase in cumulative viral shedding we observed in high salinity would be expected to increase rates of transmission through the water [47], which corroborates our finding of greater pathogen eDNA levels in higher salinity ponds. Contact rates were not affected by salinity, but we would expect the greater than 10-fold more intense infections are more transmissible given a contact. Elevated salinity could also increase water-borne transmission through its negative effect survival of zooplankton [59], which consume infectious ranaviruses in the water [60]; or the accumulation of infected carcasses resulting from greater mortality could increase ranavirus transmission either by contact or via necrophagy [61]. Thus, small changes in habitat quality can affect multiple mechanisms that concertedly push host–pathogen systems beyond the tipping point towards a rapid, mass mortality event [1]. Such interactions are increasingly recognized as critical to understanding and perhaps mitigating the impact of infectious disease on host populations [62]. Recent work with integral projection models of dose-dependent host–parasite interactions show that small changes in host resistance or tolerance, akin to those we observed with salinity, can lead to large changes in the population-level outcomes [63].
Mass mortality events caused by infectious diseases are often associated with environmental changes [1,2,5], yet we are just beginning to clarify generalizable patterns and mechanisms to explain the rise of emerging infectious diseases in wildlife [64]. In addition to salinity, numerous environmental changes are known to act on the neuroendocrine stress axis in amphibians (e.g. low pH [65]; see review [53]). Although some cause increased mortality from infection (e.g. atrazine [66]; see review [67]), further research is needed to determine whether these individual-level effects manifest as more common or severe epidemics. Our research suggests that the mechanisms relating stressors to disease outcomes at the individual and population level will be complex, and experiments need to be designed to test multiple hypotheses. Considering salinization has been proposed as a management strategy to provide refuge from another amphibian disease, Bd [30], our study suggests a holistic approach is required to successfully mitigate the impact of stress and disease on amphibian populations.
Supplementary Material
Acknowledgements
We thank the anonymous reviewers for helpful comments. We thank Yale Myers Forest for research sites and usage of facilities, J. Schwarz for collection and shipment of wood frog eggs used in lab experiments, and C. Goldberg for extracting eDNA samples, and her funding from Department of Defense Environmental Security Technology Certification Program. J. Cundiff grew and cultured ranavirus isolates, R. Reeve completed the microscopy and imaging, and A. Storfer, J. Owen, M. Lambert, M. Atwood and D. Skelly provided key project advice.
Data accessibility
Raw data from the environmental survey and laboratory experiments are available from the Dryad Digital Repository: https://dx.doi.org/10.5061/dryad.ffbg79cr5 [68].
Authors' contributions
E.M.H., J.L.B. and E.J.C. contributed to study design, data analysis and writing the manuscript. E.M.H. and B.H. contributed to data collection.
Competing interests
The authors declare no competing interests.
Funding
This publication was developed under STAR Fellowship Assistance Agreement no. 91767901 awarded by the US Environmental Protection Agency (EPA). It has not been formally reviewed by EPA. The views expressed in this publication are solely those of E.M.H. and co-authors, and EPA does not endorse any products or commercial services mentioned in this publication. This research was approved by the Animal Care and Use Committee (Protocol no. 04366-002) of Washington State University. Collections were approved by the Connecticut Department of Energy and Environmental Protection (Scientific collections permit no. 1114003) and Yale Myers Forest, and New York Department of Environmental Conservation (Collection Permit no.1481). This work was supported by an EPA STAR Fellowship, Elling Endowment at Washington State University and the Sigma-Xi Grants in Aid of Research to E.M.H.; NSF-DEB 1139199 to J.L.B., NSF-DEB 1754474 to J.L.B. and E.J.C. and an Undergraduate Summer Research Minigrant from the College of Arts and Sciences at Washington State University to B.H.
References
- 1.Fey SB, Siepielski AM, Nusslé S, Cervantes-Yoshida K, Hwan JL, Huber ER, Fey MJ, Catenazzi A, Carlson SM. 2015. Recent shifts in the occurrence, cause, and magnitude of animal mass mortality events. Proc. Natl Acad. Sci. USA 112, 1083–1088. ( 10.1073/pnas.1414894112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daszak P, Cunningham A, Hyatt A.. 2001. Anthropogenic environmental change and the emergence of infectious diseases in wildlife. Acta Trop. 78, 103–116. [DOI] [PubMed] [Google Scholar]
- 3.Brearley G, Rhodes J, Bradley A, Baxter G, Seabrook L, Lunney D, Liu Y, McAlpine C.. 2013. Wildlife disease prevalence in human-modified landscapes. Biol. Rev. Camb. Phil. Soc. 88, 427–442. ( 10.1111/brv.12009). [DOI] [PubMed] [Google Scholar]
- 4.Marcogliese DJ, Pietrock M.. 2011. Combined effects of parasites and contaminants on animal health: parasites do matter. Trends Parasitol. 27, 123–130. ( 10.1016/j.pt.2010.11.002) [DOI] [PubMed] [Google Scholar]
- 5.Patz JA, Graczyk TK, Geller N, Vittor AY.. 2000. Effects of environmental change on emerging parasitic diseases. Int. J. Parasitol. 30, 1395–1405. [DOI] [PubMed] [Google Scholar]
- 6.Smith KF, Sax DF, Lafferty KD.. 2006. Evidence for the role of infectious disease in species extinction and endangerment. Conserv. Biol. 20, 1349–1357. ( 10.1111/j.1523-1739.2006.00524.x) [DOI] [PubMed] [Google Scholar]
- 7.Carey C, Cohen N, Rollins-Smith L.. 1999. Amphibian declines: an immunological perspective. Dev. Comp. Immunol. 23, 459–472. ( 10.1016/s0145-305x(99)00028-2). [DOI] [PubMed] [Google Scholar]
- 8.Sapolsky RM, Romero LM, Munck AU.. 2000. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 21, 55–89. ( 10.1210/er.21.1.55). [DOI] [PubMed] [Google Scholar]
- 9.Dhabhar FS, Mcewen BS.. 1997. Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: a potential role for leukocyte trafficking. Brain Behav. Immun. 11, 286–306. [DOI] [PubMed] [Google Scholar]
- 10.Martin LB, Hopkins WA, Mydlarz LD, Rohr JR.. 2010. The effects of anthropogenic global changes on immune functions and disease resistance. Ann. NY Acad. Sci. 1195, 129–148. [DOI] [PubMed] [Google Scholar]
- 11.Bauer ME, Perks P, Lightman SL, Shanks N.. 2001. Restraint stress is associated with changes in glucocorticoid immunoregulation. Physiol. Behav. 73, 525–532. ( 10.1016/s0031-9384(01)00503-0) [DOI] [PubMed] [Google Scholar]
- 12.McEwen BS, Wingfield JC.. 2003. The concept of allostasis in biology and biomedicine. Horm. Behav. 43, 2–15. ( 10.1016/s0018-506x(02)00024-7). [DOI] [PubMed] [Google Scholar]
- 13.Ahmed SA. 2000. The immune system as a potential target for environmental estrogens (endocrine disrupters): a new emerging field. Toxicology 150, 191–206. ( 10.1016/S0300-483X(00)00259-6) [DOI] [PubMed] [Google Scholar]
- 14.Lafferty KD, Holt RD.. 2003. How should environmental stress affect the population dynamics of disease? Ecol. Lett. 6, 654–664. ( 10.1046/j.1461-0248.2003.00480.x) [DOI] [Google Scholar]
- 15.Plowright RK, Sokolow SH, Gorman ME, Daszak P, Foley JE.. 2008. Causal inference in disease ecology: investigating ecological drivers of disease emergence. Front. Ecol. Environ. 6, 420–429. ( 10.1890/070086) [DOI] [Google Scholar]
- 16.Jackson RB, Jobbagy EG.. 2005. From icy roads to salty streams. Proc. Natl Acad. Sci. USA 102, 14 487–14 488. ( 10.1073/pnas.0507389102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herbert ER, Boon P, Burgin AJ, Neubauer SC, Franklin RB, Ardón M, Hopfensperger KN, Lamers LP, Gell P.. 2015. A global perspective on wetland salinization: ecological consequences of a growing threat to freshwater wetlands. Ecosphere 6, 1–43. ( 10.1890/es14-00534.1) [DOI] [Google Scholar]
- 18.Chang P, Plumb J.. 1996. Effects of salinity on Streptococcus infection of Nile tilapia, Oreochromis niloticus. J. Appl. Aquaculture 6, 39–45. ( 10.1300/J028v06n01_04) [DOI] [Google Scholar]
- 19.Hopkins GR, Brodie JED. 2015. Occurrence of amphibians in saline habitats: a review and evolutionary perspective. Herpetological Monogr. 29, 1–27. ( 10.1655/HERPMONOGRAPHS-D-14-00006) [DOI] [Google Scholar]
- 20.Gomez-Mestre I, Tejedo M, Ramayo E, Estepa J.. 2004. Developmental alterations and osmoregulatory physiology of a larval anuran under osmotic stress. Physiol. Biochem. Zool. 77, 267–274. ( 10.1086/378143) [DOI] [PubMed] [Google Scholar]
- 21.Uchiyama M, Yoshizawa H. 1992. Salinity tolerance and structure of external and internal gills in tadpoles of the crab-eating frog, Rana cancrivora. Cell Tissue Res. 267, 35–44. ( 10.1007/bf00318689). [DOI] [PubMed] [Google Scholar]
- 22.Bernabò I, Bonacci A, Coscarelli F, Tripepi M, Brunelli E.. 2013. Effects of salinity stress on Bufo balearicus and Bufo bufo tadpoles: tolerance, morphological gill alterations and Na+/K+-ATPase localization. Aquat. Toxicol. 132, 119–133. ( 10.1016/j.aquatox.2013.01.019) [DOI] [PubMed] [Google Scholar]
- 23.Findlay S.EG, Kelly VR. 2011. Emerging indirect and long-term road salt effects on ecosystems. Ann. NY Acad. Sci. 1223, 58–68. [DOI] [PubMed] [Google Scholar]
- 24.Brady SP. 2013. Microgeographic maladaptive performance and deme depression in response to roads and runoff. PeerJ 1, e163 ( 10.7717/peerj.163). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karraker NE, Gibbs JP.. 2011. Road deicing salt irreversibly disrupts osmoregulation of salamander egg clutches. Environ. Pollut 159, 833–835. ( 10.1016/j.envpol.2010.11.019). [DOI] [PubMed] [Google Scholar]
- 26.Hall EM, Brady SP, Mattheus NM, Earley RL, Diamond M, Crespi EJ.. 2017. Physiological consequences of exposure to salinized roadside ponds on wood frog larvae and adults. Biol. Conserv. 209, 98–106. ( 10.1016/j.biocon.2017.02.013) [DOI] [Google Scholar]
- 27.Squires ZE, Bailey PC, Reina RD, Wong BB.. 2008. Environmental deterioration increases tadpole vulnerability to predation. Biol. Lett. 4, 392–394. ( 10.1098/rsbl.2008.0144). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milotic D, Milotic M, Koprivnikar J.. 2017. Effects of road salt on larval amphibian susceptibility to parasitism through behavior and immunocompetence. Aquat. Toxicol. 189, 42–49. ( 10.1016/j.aquatox.2017.05.015) [DOI] [PubMed] [Google Scholar]
- 29.Buss N, Hua J.. 2018. Parasite susceptibility in an amphibian host is modified by salinization and predators. Environ. Pollut 236, 754–763. ( 10.1016/j.envpol.2018.01.060) [DOI] [PubMed] [Google Scholar]
- 30.Stockwell M, Clulow J, Mahony M.. 2015. Evidence of a salt refuge: chytrid infection loads are suppressed in hosts exposed to salt. Oecologia 177, 901–910. ( 10.1007/s00442-014-3157-6) [DOI] [PubMed] [Google Scholar]
- 31.Hintz WD, Relyea RA.. 2019. A review of the species, community, and ecosystem impacts of road salt salinisation in fresh waters. Freshwater Biol. 64, 1081–1097. ( 10.1111/fwb.13286) [DOI] [Google Scholar]
- 32.Miller D, Gray M, Storfer A.. 2011. Ecopathology of ranaviruses infecting amphibians. Viruses 3, 2351–2373. ( 10.3390/v3112351) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crespi EJ, Rissler LJ, Mattheus NM, Engbrecht K, Duncan SI, Seaborn T, Hall EM, Peterson JD, Brunner JL.. 2015. Geophysiology of wood frogs: landscape patterns of prevalence of disease and circulating hormone concentrations across the eastern range. Integr. Comp. Biol. 55, 602–617. ( 10.1093/icb/icv096) [DOI] [PubMed] [Google Scholar]
- 34.Brunner JL, Barnett KE, Gosier CJ, McNulty SA, Rubbo MJ, Kolozsvary MB.. 2011. Ranavirus infection in die-offs of vernal pool amphibians in New York, USA. Herpetol. Rev. Herpetological Rev. 42, 76–79. [Google Scholar]
- 35.Hall EM, Crespi EJ, Goldberg CS, Brunner JL.. 2016. Evaluating environmental DNA-based quantification of ranavirus infection in wood frog populations. Mol. Ecol. Res. 16, 423–433. ( 10.1111/1755-0998.12461). [DOI] [PubMed] [Google Scholar]
- 36.Hall EM, Goldberg CS, Brunner JL, Crespi EJ.. 2018. Seasonal dynamics and potential drivers of ranavirus epidemics in wood frog populations. Oecologia 188, 1253–1262. ( 10.1007/s00442-018-4274-4) [DOI] [PubMed] [Google Scholar]
- 37.Granato GE, Smith KP.. 1999. Estimating concentrations of road-salt constituents in highway-runoff from measurements of specific conductance. Northborough, MA: US Department of the Interior, US Geological Survey. [Google Scholar]
- 38.Dananay KL, Krynak KL, Krynak TJ, Benard MF.. 2015. Legacy of road salt: apparent positive larval effects counteracted by negative postmetamorphic effects in wood frogs. Environ. Toxicol. Chem. 34, 2417–2424. ( 10.1002/etc.3082). [DOI] [PubMed] [Google Scholar]
- 39.Green DE, Converse KA, Schrader AK.. 2002. Epizootiology of sixty-four amphibian morbidity and mortality events in the USA, 1996–2001. Ann. NY Acad. Sci. 969, 323–339. [DOI] [PubMed] [Google Scholar]
- 40.R Core Team. 2015. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; See http://www.R-project.org/. [Google Scholar]
- 41.Burnham KP, Anderson DR.. 2004. Multimodel inference: understanding AIC and BIC in model selection. Sociol. Methods Res. 33, 261–304. [Google Scholar]
- 42.Gosner KL. 1960. A simplified table for staging anuran embryos and larvae with notes on identification. Herpetologica 16, 183–190. ( 10.2307/3890061). [DOI] [Google Scholar]
- 43.Wu C-S, Gomez-Mestre I, Kam YC. 2012. Irreversibility of a bad start: early exposure to osmotic stress limits growth and adaptive developmental plasticity. Oecologia 169, 15–22. ( 10.1007/s00442-011-2170-2) [DOI] [PubMed] [Google Scholar]
- 44.Therneau TM, Therneau MTM. 2018. Package ‘coxme’. Mixed effects cox models R package version2.
- 45.Karraker NE, Gibbs JP, Vonesh JR.. 2008. Impacts of road deicing salt on the demography of vernal pool-breeding amphibians. Ecol. Appl. 18, 724–734. ( 10.1890/07-1644.1). [DOI] [PubMed] [Google Scholar]
- 46.Therneau TM, Lumley T.. 2015. Package ‘survival’. R. Top. Doc. 128 See https://CRAN.R-projct.org/package=survival. [Google Scholar]
- 47.Araujo A, Kirschman L, Warne RW.. 2016. Behavioural phenotypes predict disease susceptibility and infectiousness. Biol. Lett. 12, 20160480 ( 10.1098/rsbl.2016.0480) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rollins-Smith LA. 1998. Metamorphosis and the amphibian immune system. Immunol. Rev. 166, 221–230. ( 10.1111/j.1600-065X.1998.tb01265.x) [DOI] [PubMed] [Google Scholar]
- 49.Fox J, et al. 2012. Package ‘car’. Vienna: R Foundation for Statistical Computing. [Google Scholar]
- 50.Brunner JL, Schock DM, Collins JP.. 2007. Transmission dynamics of the amphibian ranavirus Ambystoma tigrinum virus. Dis. Aquat. Organ. 77, 87–95. ( 10.3354/dao01845) [DOI] [PubMed] [Google Scholar]
- 51.Kuznetsova A, Brockhoff PB, Christensen RHB. 2017. lmerTest package: tests in linear mixed effects models. J. Stat. Softw. 82, 1–26. ( 10.18637/jss.v082.i13) [DOI] [Google Scholar]
- 52.Warne RW, Crespi EJ, Brunner JL.. 2011. Escape from the pond: stress and developmental responses to ranavirus infection in wood frog tadpoles. Funct. Ecol. 25, 139–146. ( 10.1111/j.1365-2435.2010.01793.x). [DOI] [Google Scholar]
- 53.Rollins-Smith LA. 2017. Amphibian immunity–stress, disease, and climate change. Dev. Comp. Immunol. 66, 111–119. ( 10.1016/j.dci.2016.07.002) [DOI] [PubMed] [Google Scholar]
- 54.Rohr JR, et al. 2008. Agrochemicals increase trematode infections in a declining amphibian species. Nature 455, 1235–1239. ( 10.1038/nature07281) [DOI] [PubMed] [Google Scholar]
- 55.O'Dwyer K, Dargent F, Forbes MR, Koprivnikar J. 2019. Parasite infection leads to widespread glucocorticoid hormone increases in vertebrate hosts: a meta-analysis. J. Anim. Ecol. 89, 519–529. ( 10.1111/1365-2656.13123) [DOI] [PubMed] [Google Scholar]
- 56.De Jesús Andino F, Chen G, Li Z, Grayfer L, Robert J.. 2012. Susceptibility of Xenopus laevis tadpoles to infection by the ranavirus Frog-Virus 3 correlates with a reduced and delayed innate immune response in comparison with adult frogs. Virology 432, 435–443. ( 10.1016/j.virol.2012.07.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sheridan JF, Dobbs C, Jung J, Chu X, Konstantinos A, Padgett D, Glaser R.. 1998. Stress-induced neuroendocrine modulation of viral pathogenesis and immunity. Ann. NY Acad. Sci. 840, 803–808. ( 10.1111/j.1749-6632.1998.tb09618.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gutiérrez JS, Abad-Gómez JM, Villegas A, Sánchez-Guzmán JM, Masero JA.. 2013. Effects of salinity on the immune response of an ‘osmotic generalist’ bird. Oecologia 171, 61–69. ( 10.1007/s00442-012-2405-x) [DOI] [PubMed] [Google Scholar]
- 59.Van Meter RJ, Swan CM.. 2014. Road salts as environmental constraints in urban pond food webs. PLoS ONE 9, e90168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson A, Brunner J.. 2014. Persistence of an amphibian ranavirus in aquatic communities. Dis. Aquat. Organ. 111, 129–138. [DOI] [PubMed] [Google Scholar]
- 61.Le Sage MJ, Towey BD, Brunner JL.. 2019. Do scavengers prevent or promote disease transmission? The effect of invertebrate scavenging on ranavirus transmission. Funct. Ecol. 33, 1342–1350. ( 10.1111/1365-2435.13335) [DOI] [Google Scholar]
- 62.Kock RA, et al. 2018. Saigas on the brink: multidisciplinary analysis of the factors influencing mass mortality events. Sci. Adv. 4, eaao2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilber MQ, Knapp RA, Toothman M, Briggs CJ.. 2017. Resistance, tolerance and environmental transmission dynamics determine host extinction risk in a load-dependent amphibian disease. Ecol. Lett. 20, 1169–1181. ( 10.1111/ele.12814) [DOI] [PubMed] [Google Scholar]
- 64.Hing S, Narayan EJ, Thompson RA, Godfrey SS.. 2016. The relationship between physiological stress and wildlife disease: consequences for health and conservation. Wildlife Res. 43, 51–60. ( 10.1071/wr15183) [DOI] [Google Scholar]
- 65.Chambers DL, Wojdak JM, Du P, Belden LK.. 2013. Pond acidification may explain differences in corticosterone among salamander populations. Physiol. Biochem. Zool. 86, 224–232. ( 10.1086/669917) [DOI] [PubMed] [Google Scholar]
- 66.Forson DD, Storfer A.. 2006. Atrazine increases ranavirus susceptibility in the tiger salamander, Ambystoma tigrinum . Ecol. Appl. 16, 2325–2332. ( 10.1890/1051-0761(2006)016[2325:airsit]2.0.co;2) [DOI] [PubMed] [Google Scholar]
- 67.Blaustein A, et al. 2018. Effects of emerging infectious diseases on amphibians: a review of experimental studies. Diversity 10, 81. [Google Scholar]
- 68.Hall EM, Brunner JL, Hutzenbiler B, Crespi EJ. 2020. Data from: Salinity stress increases the severity of ranavirus epidemics in amphibian populations Dryad Digital Repository. ( 10.5061/dryad.ffbg79cr5) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Hall EM, Brunner JL, Hutzenbiler B, Crespi EJ. 2020. Data from: Salinity stress increases the severity of ranavirus epidemics in amphibian populations Dryad Digital Repository. ( 10.5061/dryad.ffbg79cr5) [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
Raw data from the environmental survey and laboratory experiments are available from the Dryad Digital Repository: https://dx.doi.org/10.5061/dryad.ffbg79cr5 [68].