

Abstract
Glycogen shortage during fasting coincides with dramatic changes in hepatic adenine nucleotide levels. The aim of this work was to study the relevance of liver glycogen in the regulation of the hepatic energy state during food deprivation. To this end, we examined the response of mice with sustained increased liver glycogen content to prolonged fasting. In order to increase hepatic glycogen content, we generated mice that overexpress protein targeting to glycogen (PTG) in the liver (PTGOE mice). Control and PTGOE mice were fed ad libitum or fasted for 36 h. Upon fasting, PTGOE mice retained significant hepatic glycogen stores and maintained hepatic energy status. Furthermore, we show that liver glycogen controls insulin sensitivity, gluconeogenesis, lipid metabolism, and ketogenesis upon nutrient deprivation.
Keywords: glycogen, fasting, energy state, gluconeogenesis, AMPK, liver
Food deprivation in mammals results in significant changes to metabolism in order to sustain energy and the availability of metabolic substrates [1]. Hormones such as glucagon, insulin, catecholamines, fibroblast growth factor 21 (FGF21), and glucocorticoids play important roles in this process. To maintain blood glucose concentration during the early stage of fasting, liver glycogen is mobilized by glycogenolysis. Furthermore, during prolonged fasting and thus glycogen depletion, hepatocytes use lactate, pyruvate, glycerol, and amino acids to synthesize glucose through gluconeogenesis [2]. Moreover, fatty acids are mobilized from white adipose tissue to liver, where they are oxidized to acetyl-CoA, which is then used to synthesize ketone bodies [3].
Fasting provokes a reciprocal rise and fall in hepatic AMP and ATP concentrations [4,5], and it has been described that reductions in hepatic energy state due to metabolic stress require cytosolic phosphoenolpyruvate carboxykinase (PEPCK-C) [5]. Also, liver AMP-activated kinase (AMPK) is critical for stabilizing liver energy state during nutrient deprivation [6].
Liver glycogen is synthesized by the hepatic isoenzyme of glycogen synthase (LGS or GYS2). LGS activity is regulated by phosphorylation at multiple sites, which causes its inactivation. Conversely, LGS is activated by dephosphorylation. Protein phosphatase 1 (PP1) is regulated by a group of glycogen-targeting subunits (G subunits) that serve to localize PP1 to glycogen particles, leading to dephosphorylation and activation of LGS. In this regard, GL (PPP1R3B) and protein targeting to glycogen (PTG, also known as PPP1R5 or PPP1R3C) are the two main G subunits expressed in the liver, and overexpression of either one increases glycogen content [7,8].
In this study, we have analyzed the impact of liver glycogen in the regulation of hepatic energy state during food deprivation. To this end, we have generated mice that overexpress PTG in the liver and that maintain relatively high levels of hepatic glycogen even when fasted. Our results demonstrate that liver glycogen regulates the hepatic energy state upon nutrient deprivation.
Materials and Methods
Animals
All procedures were approved by the Barcelona Science Park Animal Experimentation Committee and carried out in accordance with the European Community Council Directive and the National Institute of Health guidelines for the care and use of laboratory animals. Mice that overexpress PTG in the liver (PTGOE) were generated as previously described [8]. Mice were housed with ad libitum access to water and a standard chow diet (Envigo, Indianapolis, IN, USA). When the animals were 6 months old, a cohort from each genotype had their food removed at the beginning of the dark cycle (19:00) and were sacrificed after a 36-h fast. A separate cohort of each genotype was allowed ad libitum access to food and was sacrificed at 08:00. Prior to sacrifice, whole blood was collected from the tail of the animals in EDTA-coated tubes. It was then centrifuged, and plasma was collected for analysis. After sacrifice, tissues were collected and stored at −80 °C until analysis.
Liver and skeletal muscle biochemical analysis
Glycogen was measured as previously described [9]. Briefly, frozen tissue was homogenized in 4 volumes of 30% KOH and heated at 100 °C for 15 min. Aliquots were then spotted on 31ET paper (Whatman, Sigma-Aldrich, St Louis, MO, USA). The papers were washed three times with 66% ethanol, which precipitates glycogen. The dried papers were incubated with amyloglucosidase (25 U L−1 Sigma) in 100 mM sodium acetate buffer at pH 4.8 to[C0] digest glycogen, and glucose release was determined by the reaction with hexokinase and glucose 6-phosphate dehydrogenase, following the original method described by Chan [10]. The intracellular concentration of ATP and AMP was measured in perchloric acid extracts by HPLC, as previously described [11]. Lactate was measured in perchloric acid extracts using a commercial spectrophotometric kit (Horiba, ABX, Montpellier, France). Triacylglycerols (TGs) in liver were quantified in 3 mol·L−1 KOH and 65% ethanol extracts based on the method described by Salmon and Flatt [12] using a kit (Sigma-Aldrich, St Louis, MO, USA).
Glycogen isolation
Liver samples were homogenized in 50 mM Tris/HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 50 mM NaF, 1% NP-40, 1 mM PMSF, and a protease inhibitor cocktail tablet (Roche, Basel, Switzerland) (buffer A). Homogenates were centrifuged at 16 000 g for 15 min at 4 °C. The resulting supernatant was ultracentrifuged at 100 000 g for 30 min (Optimax TX-rotor TLA 55). Glycogen was recovered in the pellet. The pellet was resuspended in the same volume as the supernatant.
Blood biochemical analysis
Blood glucose levels were measured using a glucometer (Ascensia Breeze 2; Bayer Healthcare, Leverkusen, Germany). Plasma insulin and FGF21 were determined by ELISA (Crystal Chem, Downers Grove, IL, USA; BioVendor—Laboratorni medicina a.s, Brno, Czech Republic). Plasma β-hydroxybutyrate was determined using a commercial kit (Sigma-Aldrich). Plasma free fatty acids (FFAs) were determined using a commercial kit (Abcam, Cambridge, UK).
Pyruvate tolerance test
For the pyruvate tolerance test, mice received an i.p. injection 2 g·kg−1 of sodium pyruvate (Sigma-Aldrich) dissolved in PBS. Blood glucose concentration was measured via tail bleed before and at times indicated after injection. All glucose measurements were made using an Ascensia Breeze 2 glucometer.
Western blot analysis
Western blots (WBs) were performed in liver samples homogenized in buffer A. Homogenates were centrifuged at 16 000 g for 15 min at 4 °C. Proteins were resolved in 10% or 15% acrylamide gels for SDS/PAGE and transferred to Immobilon membranes (Millipore, Sigma-Aldrich). The following antibodies were used: PEPCK (a kind gift from Dr. E. Beale); p-FoxO1 Thr24 (9464, Cell Signaling, Danvers, MA, USA); FoxO1 (9454, Cell Signaling); GK (peptide 414–428 raised by Sigma Genosys (Cambridge, UK)); p-AMPKβ Ser108 (4181, Cell Signaling); AMPKβ (4150, Cell Signaling); p-AMPKα Thr172 (2531, Cell Signaling); AMPKα (2532, Cell Signaling); p-AKT Thr308 (4056, Cell Signaling); p-AKT Ser473 (9271, Cell Signaling); AKT (9272, Cell Signaling); p-GSK3β Ser9 (9336, Cell Signaling); and GSK3β (sc7291, Santa Cruz, Dallas, TX, USA). Proteins were detected by the ECL method (Immobilon Western Chemiluminescent HRP Substrate, Millipore, Sigma-Aldrich). Loading control of the WB membrane was performed using the REVERT total protein stain.
RNA preparations and quantitative RT–PCR
Tissue preparation, RNA extraction, RT–PCR, and quantitative real-time PCR analyses were performed as described [13]. The following TaqMan primer/probe sets (Applied Biosystems, Madrid, Spain) were used for quantitative RT– PCR: BDH1 (Mm00558330_m1); BDH2 (Mm00459075_m1); HMGCS2 (Mm00550050_m1); CD36 (Mm01135202_g1); PPARα (Mm00440939_m1); CPT1α (Mm01231183_m1); PEPCK (Mm00440636_m1); PCX (Mm00500992_m1); FBP1 (Mm00490181_m1); FGF21 (Mm00840165_g1); and 18S ribosomal (18S) (Mm03928990_g1). 18S was used as a housekeeping gene.
Lean weight and fat weight
Lean weight and fat weight were measured using magnetic resonance imaging (EchoMRI System, EchoMRI LLC; Houston, TX, USA).
Statistics
Data are expressed as mean ± SEM. P values were calculated using two-way ANOVA with post hoc Tukey’s tests as appropriate. For pyruvate tolerance test (PTT) and area under the curve (AUC), Student’s t-test was performed.
Results
Effects on body weight, lean, and fat mass
In order to increase hepatic glycogen content, we generated mice that overexpress PTG in the liver (PTGOE mice) [8]. To study the role of liver glycogen during the adaptive response to fasting, we compared control and PTGOE mice that had been fed, or fasted for 36 h. Control and PTGOE mice lost about 6 g of body weight during fasting, accounting for approximately 20% of body weight loss. Of this loss, 2 g corresponded to fat and 4 g to lean weight (Fig. 1). This percentage of weight loss in control animals is in agreement with previous studies [14]. Fasting caused a significant decrease in blood glucose levels in control and PTGOE mice, although the decrease was attenuated in the latter (Fig. 1E). The plasma insulin concentration, which was already decreased in fed PTGOE mice, was greatly reduced in fasted control and PTGOE animals (Fig. 1F).
Fig. 1.
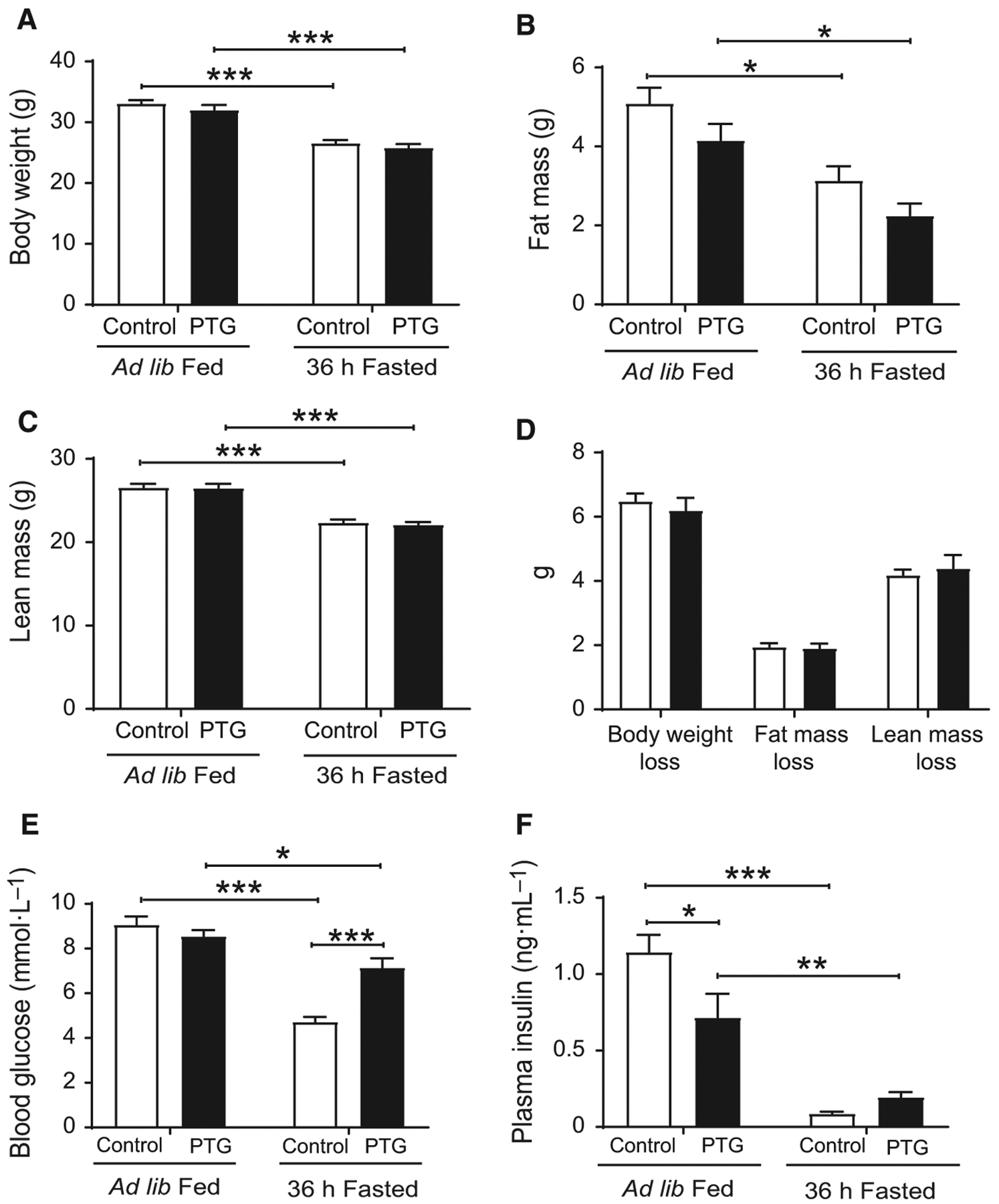
Body weight, lean, and fat mass of control (white bars) and PTGOE (black bars) mice under fed or fasted conditions. Animals were either allowed ad libitum access to food or were subjected to a 36-h fast. (A) Body weight. (B) Fat weight. (C) Lean weight. (D) Body weight loss, fat weight loss, and lean weight loss. (E) Blood glucose concentration. (F) Plasma insulin levels. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. n = 15 for body, fat, and lean weight, and n = 8 for glucose and insulin.
Effects on energy state
As we previously described [8], PTGOE mice overaccu-mulated hepatic glycogen under fed conditions. Following a 36-h fast, liver glycogen was completely depleted in control mice, while in PTGOE mice it was partially mobilized down to levels in the same range as those in fed control animals (Fig. 2A). Hepatic glucose 6-phosphate (G6P), the primary intracellular metabolite of glucose, was significantly reduced after fasting in control mice but not in PTGOE mice (Fig. 2B). Upon fasting, hepatic ATP and AMP levels in control mice decreased (Fig. 2C) and increased (Fig. 2D), respectively, resulting in an increase in the AMP/ATP ratio (Fig. 2E) compared with fed control counter-parts. Nonetheless, the changes in hepatic nucleotide levels mediated by fasting were abolished in PTGOE mice (Fig. 2C–E), thus preserving the energy state of the organ. Furthermore, low energy states under fasting conditions are associated with the activation of AMPK [2,15]. The phosphorylated AMPKβ/total AMPKβ ratio was decreased in fasted PTGOE mice compared with fasted control animals, indicating a lower activation state of the enzyme (Fig. 3A,G). However, the phosphorylated AMPKα/total AMPKα ratio was similar in all the experimental groups (Fig. 3B,H). Another interesting observation was that both total AMPKβ and total AMPKα were increased in the livers of PTGOE mice compared with control mice (Fig. 3A,E; B,F).
Fig. 2.
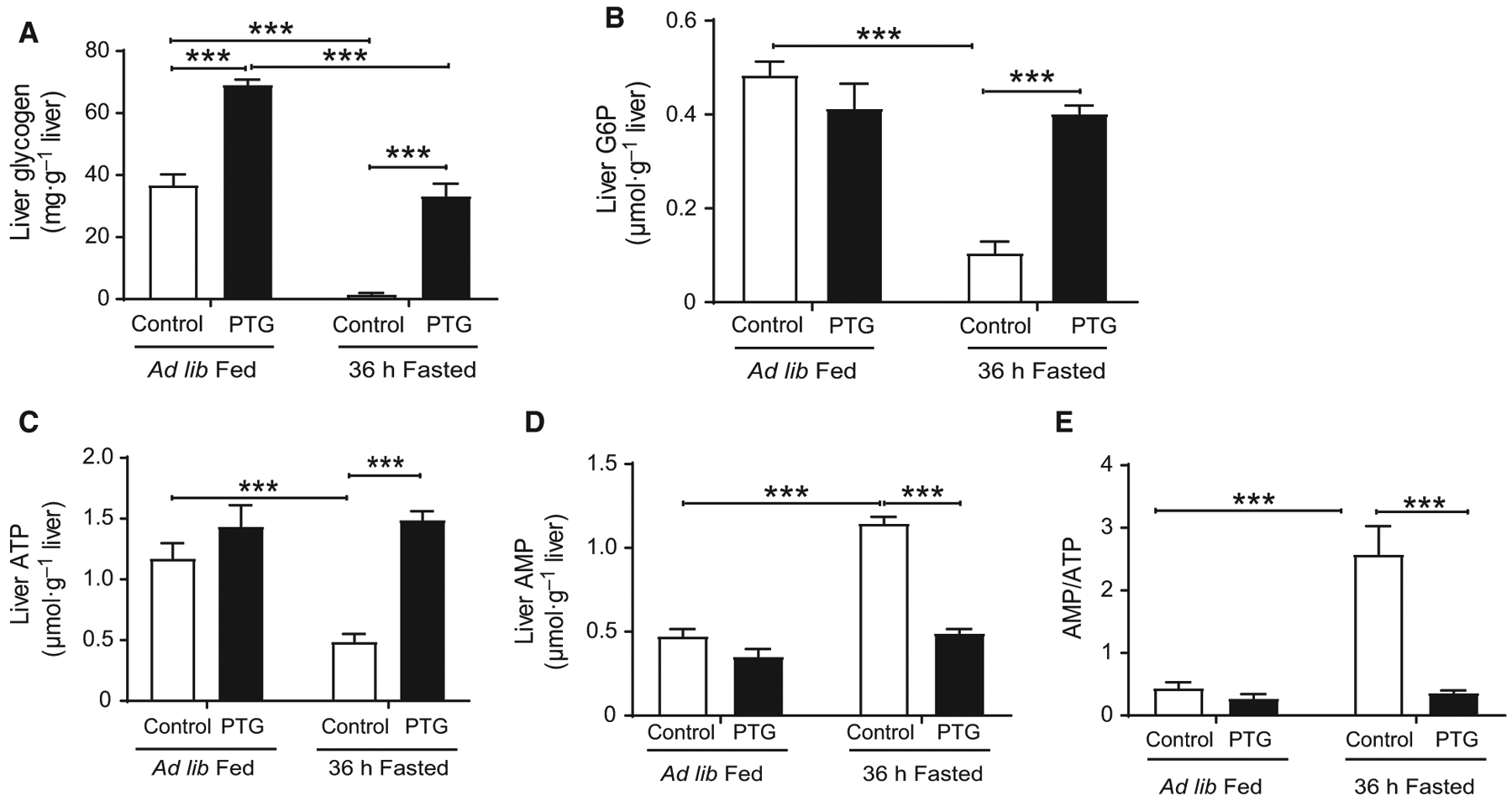
Hepatic energy state of control (white bars) and PTGOE (black bars) mice under fed or fasted conditions. (A) Glycogen content. (B) G6P content. (C) ATP concentration. (D) AMP concentration. (E) AMP/ATP ratio. Data are presented as mean ± SEM. ***P < 0.001. n = 6–8.
Fig. 3.
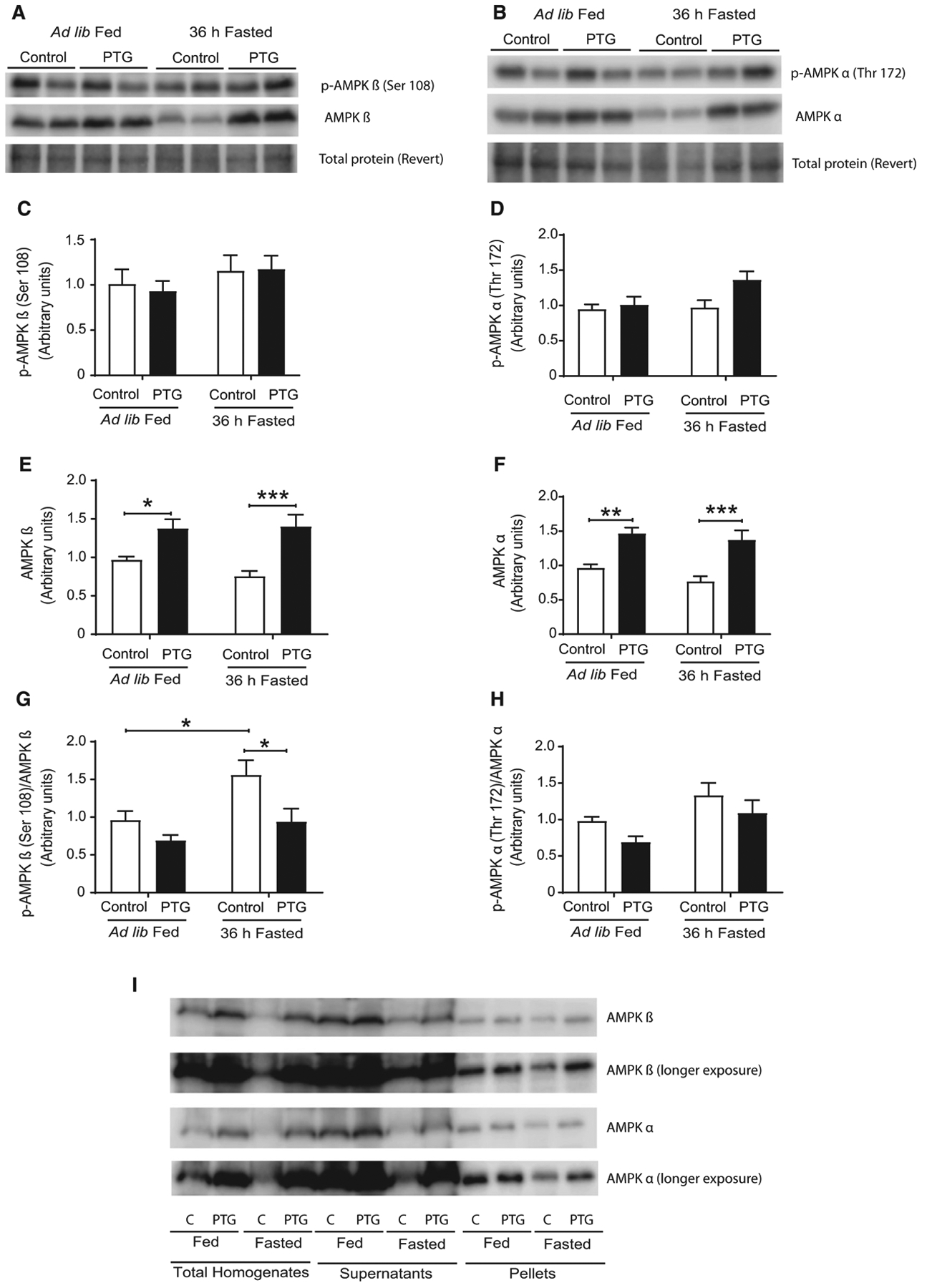
AMPK of control (white bars) and PTGOE (black bars) mice under fed or fasted conditions. (A, E) Total and (A, C) phosphorylated AMPKβ and (B, F) total and (B, D) phosphorylated AMPKα measured by western blot. (G) Phosphorylated to total AMPKα is provided as a ratio (A.U). (H) Phosphorylated to total AMPKα is provided as a ratio (A.U). (I) Amount of AMPKβ and AMPKα in homogenates, supernatant, and 100 000 g pellet fraction. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. n = 8.
To determine whether these increases were due to AMPK binding to glycogen, liver glycogen was precipitated by ultracentrifugation. AMPKα and AMPKβ were detected in the pellet, thereby indicating that a fraction of the enzyme was bound to glycogen (Fig. 3I). However, most of the enzyme was found in the supernatant.
Effects on gluconeogenesis
To study the impact of liver glycogen on the regulation of gluconeogenesis during fasting, the protein expression of hepatic PEPCK and the expression of several gluconeogenic genes were measured. After 36 h of fasting, hepatic PEPCK protein (Fig. 4A,C) and gene expression (Fig. 4D) were increased in control mice. However, this effect was attenuated in PTGOE mice (Fig. 4A,C,D). Fasting also increased the expression of the gluconeogenic genes pyruvate carboxylase (PCX) and fructose bisphosphatase 1 (FBP1) in control mice, but not in PTGOE counterparts (Fig. 4D). Fasted PTGOE mice showed an increase in liver lactate (Fig. 4E), indicative of less demand for lactate use for gluconeogenesis. Glucokinase expression was significantly higher in fasted PTGOE mice (Fig. 4A,B). To more directly assess the role of liver glycogen in gluconeogenesis, mice were challenged with the gluconeogenic precursor, pyruvate. An improved pyruvate tolerance was shown in PTGOE mice (Fig. 4F) as represented by a significant reduction in AUC (Fig. 4G), suggesting that liver glycogen attenuates gluconeogenesis in the liver. The PTT was performed after a 6-h fast and not after 36-h fast, like the rest of the experiments. At the later time point, the initial concentrations of blood glucose were different (Fig. 1E) and this may hamper the interpretation of the results.
Fig. 4.
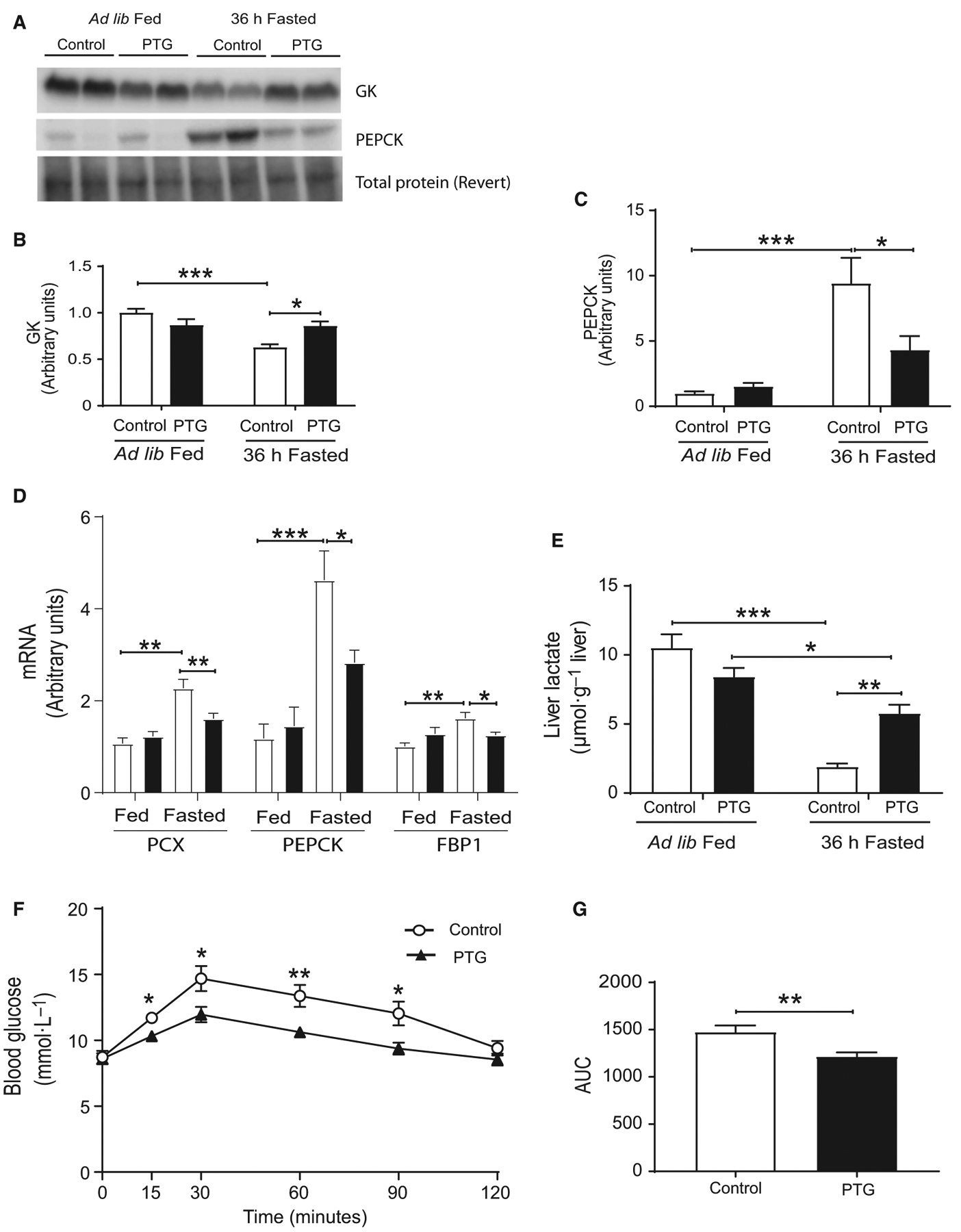
Hepatic gluconeogenesis of control (white bars) and PTGOE (black bars) mice under fed or fasted conditions. (A) Western blot of liver lysates. (B) Amount of GK measured by western blot. (C) Amount of PEPCK measured by western blot. (D) Expression of genes associated with gluconeogenesis (PCX, PEPCK, and FBP1). (E) Liver lactate content. (F) Pyruvate tolerance test after 6-h fasting. (G) AUC. AUC was calculated from blood glucose profiles during pyruvate tolerance test. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. n = 8 for western blot, n = 8–28 for gene expression, and n = 6–8 for pyruvate tolerance test.
Effects on hepatic insulin signaling
Hepatic insulin signaling was analyzed in fed and fasted mice of both genotypes. The phosphorylation of protein kinase B (AKT) (Fig. 5A–C) and glycogen synthase kinase 3β (GSK3β) (Fig. 5A,D) was increased in the liver of fasted PTGOE mice compared with fasted control animals, thereby indicating a higher activation state of the insulin signaling cascade. AKT signals through multiple downstream pathways, including the Forkhead box protein O1 (FoxO) family of transcription factors. Fasting decreased the phosphorylation of FoxO1 in the liver of control mice, but this effect was attenuated in PTGOE mice (Fig. 5A,E).
Fig. 5.
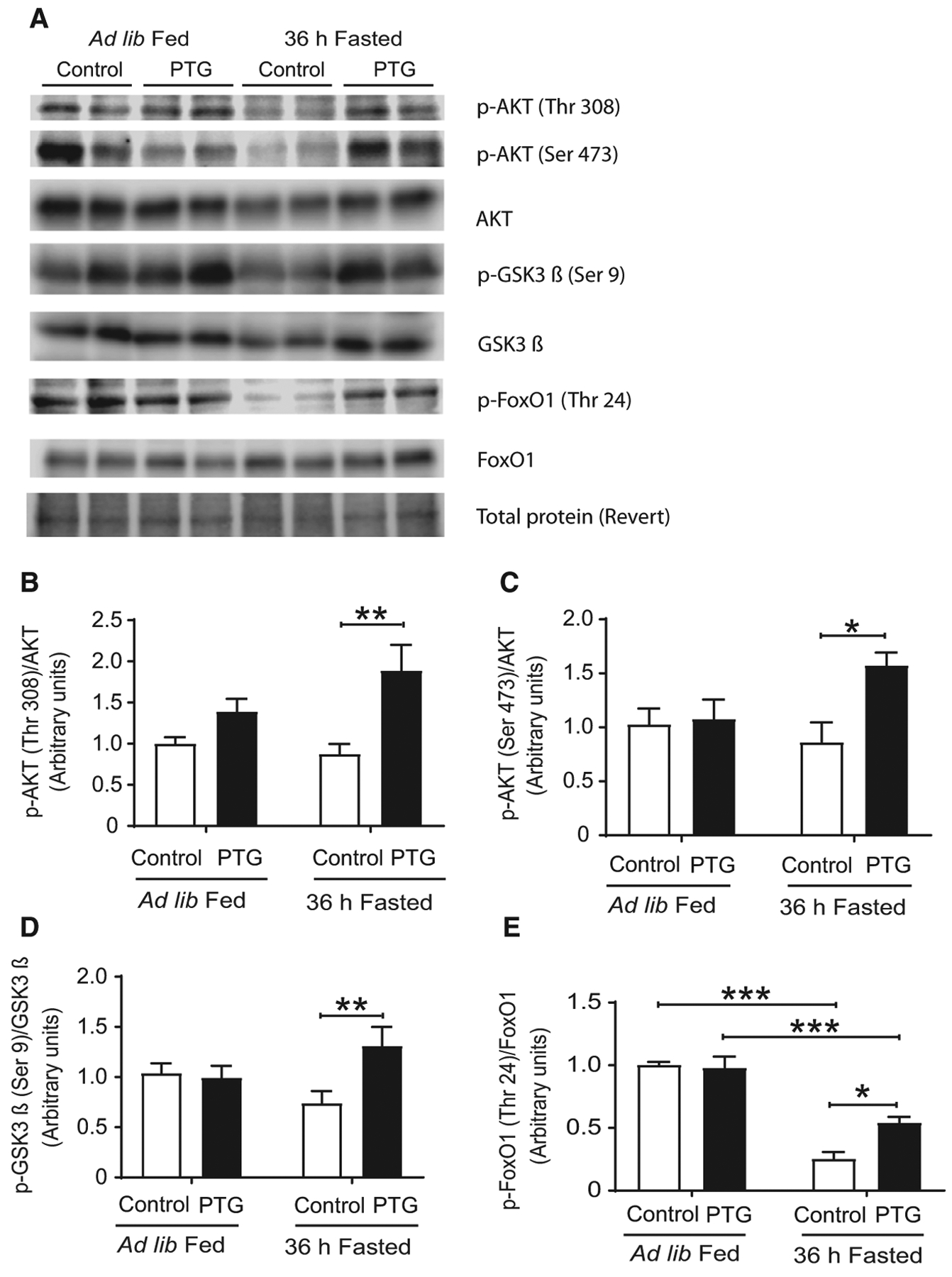
Hepatic insulin signaling of control (white bars) and PTGOE (black bars) mice under fed or fasted conditions. (A) Western blot of liver lysates. (B) Amount of total and phosphorylated AKT in threonine 308 measured by western blot. (C) Amount of total and phosphorylated AKT in serine 473 assessed by western blot. (D) Amount of total and phosphorylated GSK3β assessed by western blot. (E) Amount of total and phosphorylated FoxO1 assessed by western blot. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. n = 8.
Effects on lipid metabolism and ketogenesis
Fasting has a significant impact on lipid metabolism [16]. Prolonged fasting elicited increased hepatic TG accumulation in control mice, but this increase was attenuated in PTGOE mice (Fig. 6A). Fasted control mice and PTGOE mice showed an increased concentration of FFAs, but no significant differences between the genotypes were found (Fig. 6B). The liver is the primary source of ketone bodies. Fasting increased plasma β-hydroxybutyrate concentration in control mice; however, as previously described for a shorter period of fasting [8], the increase was attenuated in PTGOE animals (Fig. 6C). The expression of genes associated with ketogenesis was examined next. The expression of BDH1 increased in the liver of fasted control mice but not in that of fasted PTGOE animals (Fig. 6E). BDH2 expression decreased after 36 h of food deprivation (Fig. 6E) and HMGCS2 expression increased upon fasting in both genotypes, although the increase was less marked in PTGOE mice (Fig. 6E). Given that hepatic FFA β-oxidation/ketogenesis increases during fasting [17], we examined the impact of liver glycogen on the expression of genes involved in fatty acid oxidation, namely CD36, PPARα, CPT1α, and FGF21. The expression of all these genes was attenuated in PTGOE animals (Fig. 6F). FGF21, a key regulator of the metabolic response to fasting, is secreted by the liver in nutrient-deficient states [18–20]. Control mice, but not PTGOE animals, showed a significant increase in plasma levels of FGF21 after a 36-h fast (Fig. 6D). This parameter correlated with the hepatic expression of FGF21 (Fig. 6F). This result is consistent with recent findings that demonstrate that the liver is the primary source of circulating FGF21 [21].
Fig. 6.
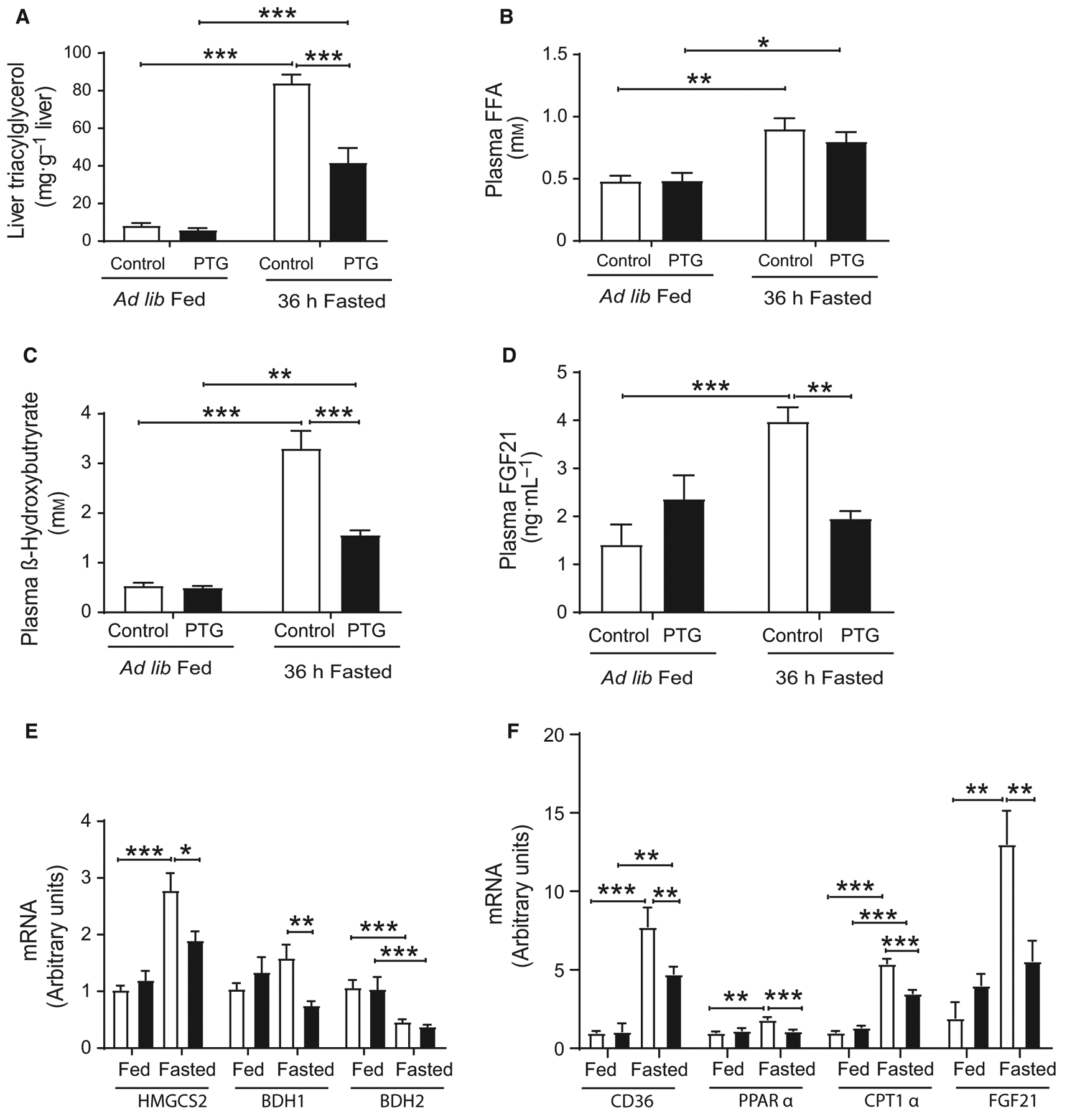
Hepatic TG, liver β-oxidation, and ketogenesis of control (white bars) and PTGOE (black bars) mice under fed or fasted conditions. (A) Hepatic TG concentration. (B) Plasma FFA levels. (C) Plasma β -hydroxybutyrate levels. (D) Plasmα FGF21 concentration. (E) Expression of genes associated with ketogenesis (HMGCS2, BDH1, and BDH2). (F) Expression of genes associated with β-oxidation (CD36, PPARα, CPT1a, and FGF21). Data are presented as mean ± SEM.*P < 0.05, **P < 0.01, and ***P < 0.001. n = 6 for hepatic TG, n = 7–8 for plasma metabolites and n = 8–28 for gene expression.
Effects on skeletal muscle metabolism upon fasting
To address the potential role of liver glycogen on skeletal muscle metabolism during fasting, we measured various metabolites in the gastrocnemius muscle. Thirty-six hours of food deprivation did not alter the concentration of glycogen, lactate, or ATP in the gastrocnemius (Table 1) in either genotype. However, the concentration of AMP and the AMP/ATP ratio were increased to the same extent in the muscle in both genotypes after fasting (Table 1).
Table 1.
Skeletal muscle metabolism of control and PTGOE mice. Glycogen, lactate, ATP, AMP, and AMP/ATP ratio were measured in the gastrocnemius muscle. Data are presented as mean ± SEM.
| Fed | Fasted | |||
|---|---|---|---|---|
| Control | PTGOE | Control | PTGOE | |
| Glycogen (mg per g tissue) | 1.4 ± 0.1 | 1.4 ± 0.1 | 1.3 ± 0.1 | 1.3 ± 0.1 |
| Lactate (μmol per g tissue) | 7.7 ± 0.6 | 8.4 ± 0.3 | 7.8 ± 0.5 | 7.4 ± 0.5 |
| ATP (μmol per g tissue) | 5.2 ± 0.2 | 4.9 ± 0.3 | 5.0 ± 0.1 | 4.8 ± 0.2 |
| AMP (μmol per g tissue) | 0.033 ± 0.003 | 0.026 ± 0.003 | 0.073 ± 0.018* | 0.077 ± 0.014* |
| AMP/ATP | 0.007 ± 0.001 | 0.005 ± 0.001 | 0.015 ± 0.004* | 0.016 ± 0.003* |
P < 0.05 between fed and fasted mice from the same genotype. n = 4–8.
Discussion
Here, we report that liver glycogen is a regulator of the hepatic energy state of the liver in response to long-term fasting. We show that glycogen controls the hepatic energy state, gluconeogenesis, lipid metabolism, ketogenesis, and insulin sensitivity upon nutrient deprivation.
In order to increase liver glycogen stores, we generated mice overexpressing PTG specifically in this organ. These animals show a doubling of glycogen concentration in the fed state. Our results show that, in spite of the long period of fasting, PTGOE mice experienced only a 50% decrease in hepatic glycogen stores. An explanation for this observation is that, although glycogenolysis is active, glycogen stores are constantly replenished by glycogen synthase. Remarkably, PTGOE mice showed an increased capacity to maintain plasma glucose under a 36-h fast, while the percentage of body weight loss upon long-term fasting was similar to that of control mice.
The hepatic energy state is defined by adenine nucleotide levels and particularly by the AMP/ATP ratio [5]. Adenine nucleotide concentrations in the mouse liver are highly sensitive to acute physiological stressors, as well as to metabolic disease, and the hepatic energy state is substantially reduced following fasting [5]. Previous studies from our laboratory have demonstrated that liver glycogen levels regulate hepatic ATP content in mice fed a high-fat diet (HFD) [8,11]. In the present study, we show that liver glycogen contributes to maintaining the hepatic energy charge after long-term fasting (36 h). As expected, fasted control mice presented dramatic changes in hepatic adenine nucleotide levels. However, fasted PTGOE mice maintained ATP and AMP concentrations in the same range as those in fed control mice, thus abolishing the decrease in the hepatic energy state associated with fasting. Remarkably, PTGOE mice also showed reduced PEPCK expression. It has been proposed that higher hepatic AMP/ATP ratios correlate with increased total PEPCK protein content. Mice with liver-specific deletion of PEPCK preserved hepatic energy state in response to an overnight fast, thereby supporting the notion that PEPCK-mediated gluconeogenesis is needed to induce changes in the hepatic energy state [5].
Given that the gluconeogenesis pathway consumes ATP, a decrease in PEPCK, as occurred in PTGOE mice, could partly explain the higher levels of ATP found in the livers of these animals upon fasting. Hepatic lactate, a precursor of gluconeogenesis, was higher in fasted PTGOE mice, indicative of less demand for lactate use for gluconeogenesis. Accordingly, PTGOE mice showed improved pyruvate tolerance in the PTT.
Remarkably, AMPK was less active in fasted PTGOE mice. However, the total levels of the enzyme were increased in these animals. Research over the past two decades has revealed that the regulation of AMPK is influenced by glycogen availability [22]. A significant body of evidence demonstrates that AMPK binds glycogen [23,24]. Therefore, the increase in this enzyme in the fasted PTGOE mice could be due to the higher levels of glycogen. To verify this hypothesis, we precipitated glycogen by ultracentrifugation and found that indeed a fraction of AMPK was bound to glycogen particles, especially in PTGOE mice. Nevertheless, we found that most of the AMPK was in the non-glycogen-bound fraction. Therefore, the lesser activation but higher levels of AMPK cannot be attributed solely to a physical interaction between AMPK and glycogen. Our results support the idea that AMPK senses cellular energy in the form of AMP/ATP but may also be able to sense the availability of energy reserves in the form of glycogen, as previously described [24].
Hepatic insulin signaling is mediated by the liver insulin receptor, which signals through the downstream kinase AKT, to coordinate hepatic metabolism [25,26]. AKT signals through multiple downstream pathways, including the FoxO family of transcriptional factors and GSK3. Fasting PTGOE mice showed an increase in the hepatic insulin signaling cascade (increased phosphorylation of AKT, GSK3β, and FoxO1), in spite of maintaining low insulin levels. Therefore, the liver of these animals became more sensitive to insulin. FoxO1 is a substrate of AKT and it contains three AKT phosphorylation sites (Thr24, Ser256, and Ser319) [27]. During the fasting state, FoxO1 is activated by dephosphorylation and translocated into the nucleus, which leads to the transcriptional induction of PEPCK [28]. The livers of fasted PTGOE mice showed an increase in the activation of AKT, which in turn phosphorylates FoxO1. The phosphorylation of FoxO1 induces its nuclear exclusion and cytoplasmic retention, thereby suppressing PEPCK gene expression and attenuating gluconeogenesis. In this regard, it has been described that LGS knockout mice show an increase in the liver gluconeogenic enzyme PEPCK [29] and decreased phosphorylation of components of the hepatic insulin signaling pathway, as well as phosphorylation of FoxO1 [30]. Conversely, the liver of streptozotozin diabetic rats, in which hepatic glycogen had been increased by the expression of an active form of LGS, also show a reduction in PEPCK and other gluconeogenic enzymes [31].
Another interesting finding was that GK protein expression did not decrease in the liver of PTGOE mice upon fasting, despite low insulin levels. The expression of GK in this organ is highly dependent on the presence of insulin, and several lines of evidence support the notion that AKT activation is involved in the induction of hepatic GK [32–34]. The increase in AKT phosphorylation in the liver of PTGOE mice upon fasting could explain the higher levels of GK found in these animals. Furthermore, the increased GK, combined with the higher blood glucose concentration, may account for the observed increase in G6P.
Fasting increases hepatic TGs in rodents [35]. This fasting-induced hepatic steatosis results from an excessive uptake of circulating fatty acids derived from adipose tissue lipolysis. The amount of hepatic TG content was lower in fasted PTGOE mice than control mice. We propose that the impaired hepatic fatty acid uptake caused by downregulation of CD36 expression in the liver of fasted PTGOE mice is responsible for the reduced TG content in these animals. CD36 is an important regulator of hepatic fatty acid uptake under conditions of elevated FFAs [36]. Hepatic CD36 disruption in various models of hepatic steatosis significantly improves steatosis by lowering TG, diacylglycerol, and cholesterol ester content [36,37]. It could be argued that that lower CD36 expression in the liver of fasted PTGOE mice would cause an increase in FFAs. In this regard, Wilson CG et al. [36] addressed the role of CD36 in regulating liver lipid content by specifically deleting CD36 in two steatosis models, one genetic (JAK2L) and another nutritional (HFD feeding) in which the elevated CD36 expression was associated with profound lipid accumulation in liver. Those authors conclusively showed that hepatocyte-specific CD36 disruption significantly reduced liver TGs without affecting FFA concentration. Only mice with global CD36 deletion showed a significant increase in FFAs, caused by severely impaired fatty acid uptake into peripheral tissues (muscle, heart, and adipose tissue) [38]. However, we are unable to explain what happens to the FFAs not taken up in fasted PTGOE mice.
In liver, the intracellular accumulation of diacylglycerol triggers the activation of novel protein kinases C, with subsequent impairments in insulin signaling [39]. There is an inverse relationship between hepatic TG stores and hepatic insulin sensitivity [30,40,41]. The higher sensitivity to insulin found in fasted PTGOE mice could be due to the lower hepatosteatosis found in these animals.
PPARα, a nuclear hormone receptor activated by FFAs, is another transcription factor that is central to the metabolic shift—from glucose to fat utilization— initiated by fasting [42]. Upon activation of PPARα, fatty acid oxidation is stimulated and increased amounts of acetyl-CoA are produced. A large proportion of acetyl-CoA derived from fatty acid β-oxidation is utilized in ketogenesis [43,44]. Fasted PTGOE mice presented decreased expression of genes related to ketogenesis and β-oxidation, resulting in lower levels of β-hydroxybutyrate. Our results are in agreement with those of Mehta et al., who reported that enhanced glycogen storage buffers the onset of the fasting-induced metabolic shift to β-oxidation [45]. AMPK regulates cellular lipid metabolism mostly through an increase in CPT1 activity and the subsequent activation of fatty acid oxidation [46,47]. Fasted PTGOE mice showed lower fatty acid oxidation compared with control mice. This observation could be related to the lower activation state of AMPK in these animals. Of particular relevance in the regulation of ketogenesis and fatty acid oxidation is the ‘hormone-like’ FGF21 [20]. The hepatic expression of this molecule is PPARα-dependent, and it is induced by fasting [19,20]. An additional important finding of this work is that, upon fasting, PTGOE mice did not show an increase in FGF21 in plasma or liver. These observations thus suggest that liver glycogen contributes to regulating the secretion of FGF21.
In summary, our data indicate that, during fasting, hepatic glycogen contributes to the control of adenine nucleotide in the liver through the decrease in gluconeogenesis. In the presence of glycogen, the liver is less predisposed to the fasting state. On the basis of our findings, we conclude that glycogen is more than just a simple storage form of glucose and that it plays an active role as a gauge of liver energy metabolism.
Acknowledgments
The authors thank Emma Veza, Vanessa Hernandez, and Laura Alcaide for technical assistance. This study was supported by grants from the Spanish MINECO (BFU2017-84345-P), Fundació La Marató de TV3 (201613-10), and the CIBER de Diabetes y Enfermedades Metabólicas Asociadas (ISCIII, Ministerio de Ciencia e Innovación). None of the supporting agencies had any role in performing the work or in writing the manuscript. IRB Barcelona is recipient of a Severo Ochoa excellence award.
Abbreviations
- AKT
protein kinase B
- AMP
adenosine monophosphate
- AMPK
AMP kinase
- ATP
adenosine triphosphate
- AUC
area under the curve
- CPT1α
carnitine palmitoyltransferase 1α
- FFA
free fatty acid
- FBP1
fructose bisphosphatase 1
- FGF21
fibroblast growth factor 21
- FoxO1
forkhead box protein O1
- G subunits
glycogen-targeting regulatory subunits
- G6P
glucose 6-phosphate
- GK
glucokinase
- GSK3
glycogen synthase kinase 3
- LGS
liver glycogen synthase
- PCX
pyruvate carboxylase
- PEPCK
phosphoenolpyruvate carboxykinase
- PKA
protein kinase A
- PP1
protein phosphatase 1
- PPAR α
peroxisome proliferator-activated receptor α
- PTG
protein targeting to glycogen
- PTT
pyruvate tolerance test
- TG
triacylglycerol
Footnotes
Data accessibility
The data are available on request from the authors.
References
- 1.Cahill GJ Jr, Owen OE and Morgan AP (1968) The consumption of fuels during prolonged starvation. Adv Enzyme Regul 6, 143–150. [DOI] [PubMed] [Google Scholar]
- 2.Rui L (2014) Energy metabolism in the liver. Compr Physiol 4, 177–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fukao T, Lopaschuk GD and Mitchell GA (2004) Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids 70, 243–251. [DOI] [PubMed] [Google Scholar]
- 4.Start C and Newsholme EA (1968) The effect of starvation and alloxan-diabetes on the contents of citrate and other metabolic intermediates in rat liver. Biochem J 107, 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berglund ED, Lee-Young RS, Lustig DG, Lynes SE, Donahue EP, Camacho RC, Meredith ME, Magnuson MA, Charron MJ and Wasserman DH (2009) Hepatic energy state is regulated by glucagon receptor signaling in mice. J Clin Invest 119, 2412–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hasenour CM, Ridley DE, James FD, Hughey CC, Donahue EP, Viollet B, Foretz M, Young JD and Wasserman DH (2017) Liver AMP-activated protein kinase is unnecessary for gluconeogenesis but protects energy state during nutrient deprivation. PLoS ONE 12, e0170382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gasa R, Jensen PB, Berman HK, Brady MJ, DePaoli-Roach AA and Newgard CB (2000) Distinctive regulatory and metabolic properties of glycogen-targeting subunits of protein phosphatase-1 (PTG, GL, GM/RGl) expressed in hepatocytes. J Biol Chem 275, 26396–26403. [DOI] [PubMed] [Google Scholar]
- 8.Lopez-Soldado I, Zafra D, Duran J, Adrover A, Calbo J and Guinovart JJ (2015) Liver glycogen reduces food intake and attenuates obesity in a high-fat diet–fed mouse model. Diabetes 64, 796–807. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Rocha M, Roca A, De La Iglesia N, Baba O, Fernandez-Novell JM, Ferrer JC and Guinovart JJ (2001) Intracellular distribution of glycogen synthase and glycogen in primary cultured rat hepatocytes. Biochem J 357, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan TM and Exton JH (1976) A rapid method for the determination of glycogen content and radioactivity in small quantities of tissue or isolated hepatocytes. Anal Biochem 71, 96–105. [DOI] [PubMed] [Google Scholar]
- 11.Lopez-Soldado I, Fuentes-Romero R, Duran J and Guinovart JJ (2017) Effects of hepatic glycogen on food intake and glucose homeostasis are mediated by the vagus nerve in mice. Diabetologia 60, 1076–1083. [DOI] [PubMed] [Google Scholar]
- 12.Salmon DM and Flatt JP (1985) Effect of dietary fat content on the incidence of obesity among ad libitum fed mice. Int J Obes 9, 443–449. [PubMed] [Google Scholar]
- 13.Ros S, Zafra D, Valles-Ortega J, Garcia-Rocha M, Forrow S, Dominguez J, Calbo J and Guinovart JJ (2010) Hepatic overexpression of a constitutively active form of liver glycogen synthase improves glucose homeostasis. J Biol Chem 285, 37170–37177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jensen TL, Kiersgaard MK, Sorensen DB and Mikkelsen LF (2013) Fasting of mice: a review. Lab Anim 47, 225–240. [DOI] [PubMed] [Google Scholar]
- 15.Dite TA, Ling NXY, Scott JW, Hoque A, Galic S, Parker BL, Ngoei KRW, Langendorf CG, O’Brien MT, Kundu M et al. (2017) The autophagy initiator ULK1 sensitizes AMPK to allosteric drugs. Nat Commun 8, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishikawa S, Doi K, Nakayama H and Uetsuka K (2008) The effect of fasting on hepatic lipid accumulation and transcriptional regulation of lipid metabolism differs between C57BL/6J and BALB/cA mice fed a high-fat diet. Toxicol Pathol 36, 850–857. [DOI] [PubMed] [Google Scholar]
- 17.Cahill GF Jr (2006) Fuel metabolism in starvation. Ann Rev Nutr 26, 1–22. [DOI] [PubMed] [Google Scholar]
- 18.Lundasen T, Hunt MC, Nilsson LM, Sanyal S, Angelin B, Alexson SE and Rudling M (2007) PPARa is a key regulator of hepatic FGF21. Biochem Biophys Res Commun 360, 437–440. [DOI] [PubMed] [Google Scholar]
- 19.Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V et al. (2007) Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab 5, 415–425. [DOI] [PubMed] [Google Scholar]
- 20.Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS and Maratos-Flier E (2007) Hepatic fibroblast growth factor 21 is regulated by PPARa and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab 5, 426–437. [DOI] [PubMed] [Google Scholar]
- 21.Markan KR, Naber MC, Ameka MK, Anderegg MD, Mangelsdorf DJ, Kliewer SA, Mohammadi M and Potthoff MJ (2014) Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 63, 4057–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janzen NR, Whitfield J and Hoffman NJ (2018) Interactive roles for AMPK and glycogen from cellular energy sensing to exercise metabolism. Int J Mol Sci 19, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Polekhina G, Gupta A, Michell BJ, van Denderen B, Murthy S, Feil SC, Jennings IG, Campbell DJ, Witters LA, Parker MW et al. (2003) AMPK β subunit targets metabolic stress sensing to glycogen. Curr Biol 13, 867–871. [DOI] [PubMed] [Google Scholar]
- 24.McBride A, Ghilagaber S, Nikolaev A and Hardie DG (2009) The glycogen-binding domain on the AMPK β subunit allows the kinase to act as a glycogen sensor. Cell Metab 9, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Titchenell PM, Quinn WJ, Lu M, Chu Q, Lu W, Li C, Chen H, Monks BR, Chen J, Rabinowitz JD and et al. (2016) Direct hepatocyte insulin signaling is required for lipogenesis but is dispensable for the suppression of glucose production. Cell Metab 23, 1154–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu M, Wan M, Leavens KF, Chu Q, Monks BR, Fernandez S, Ahima RS, Ueki K, Kahn CR and Birnbaum MJ (2012) Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med 18, 388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, Gan L, Pan H, Guo S, He X, Olson ST, Mesecar A, Adam S and Unterman TG (2002) Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. J Biol Chem 277, 45276–45284. [DOI] [PubMed] [Google Scholar]
- 28.Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, Klotsas A, Matika R, Xiao X, Franks R et al. (2006) FoxO1 regulates multiple metabolic pathways in the liver. J Biol Chem 281, 10105–10117. [DOI] [PubMed] [Google Scholar]
- 29.Irimia JM, Meyer CM, Peper CL, Zhai L, Bock CB, Previs SF, McGuinness OP, DePaoli-Roach A and Roach PJ (2010) Impaired glucose tolerance and predisposition to the fasted state in liver glycogen synthase knock-out mice. J Biol Chem 285, 12851–12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Irimia JM, Meyer CM, Segvich DM, Surendran S, DePaoli-Roach AA, Morral N and Roach PJ (2017) Lack of liver glycogen causes hepatic insulin resistance and steatosis in mice. J Biol Chem 292, 10455–10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ros S, Garcia-Rocha M, Calbo J and Guinovart JJ (2011) Restoration of hepatic glycogen deposition reduces hyperglycaemia, hyperphagia and gluconeogenic enzymes in a streptozotocin-induced model of diabetes in rats. Diabetologia 54, 2639–2648. [DOI] [PubMed] [Google Scholar]
- 32.Iynedjian PB (2009) Molecular physiology of mammalian glucokinase. Cell Mol Life Sci 66, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ribaux PG and Iynedjian PB (2003) Molecular physiology of mammalian glucokinase. Biochem J 376, 697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agius L (2016) Hormonal and metabolite regulation of hepatic glucokinase. Ann Rev Nutr 36, 389–415. [DOI] [PubMed] [Google Scholar]
- 35.Hashimoto T, Cook WS, Qi C, Yeldandi AV, Reddy JK and Rao MS (2000) Defect in peroxisome proliferator-activated receptor a-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem 275, 28918–28928. [DOI] [PubMed] [Google Scholar]
- 36.Wilson CG, Tran JL, Erion DM, Vera NB, Febbraio M and Weiss EJ (2016) Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-Fed mice. Endocrinology 157, 570–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Angulo P (2002) Nonalcoholic fatty liver disease. N Engl J Med 346, 1221–1231. [DOI] [PubMed] [Google Scholar]
- 38.Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF and Silverstein RL (1999) A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem 274 19055–19062. [DOI] [PubMed] [Google Scholar]
- 39.Samuel VT, Petersen KF and Shulman GI (2010) Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375, 2267–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Voshol PJ, Haemmerle G, Ouwens DM, Zimmermann R, Zechner R, Teusink B, Maassen JA, Havekes LM and Romijn JA (2003) Increased hepatic insulin sensitivity together with decreased hepatic triglyceride stores in hormone-sensitive lipase-deficient mice. Endocrinology 144, 3456–3462. [DOI] [PubMed] [Google Scholar]
- 41.Reitman ML (2002) Metabolic lessons from genetically lean mice. Ann Rev Nutr 22, 459–482. [DOI] [PubMed] [Google Scholar]
- 42.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B and Wahli W (1999) Peroxisome proliferator–activated receptor α mediates the adaptive response to fasting. J Clin Invest 103, 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Satapati S, Sunny NE, Kucejova B, Fu X, He TT, Mendez-Lucas A, Shelton JM, Perales JC, Browning JD and Burgess SC (2012) Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J Lipid Res 53, 1080–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sunny NE, Satapati S, Fu X, He T, Mehdibeigi R, Spring-Robinson C, Duarte J, Potthoff MJ, Browning JD and Burgess SC (2010) Progressive adaptation of hepatic ketogenesis in mice fed a high-fat diet. Am J Physiol Endocrinol Metab 298, E1226–E1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mehta MB, Shewale SV, Sequeira RN, Millar JS, Hand NJ and Rader DJ (2017) Hepatic protein phosphatase 1 regulatory subunit 3B (Ppp1r3b) promotes hepatic glycogen synthesis and thereby regulates fasting energy homeostasis. J Biol Chem 292, 10444–10454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Foretz M and Viollet B (2011) Regulation of hepatic metabolism by AMPK. J Hepatol 54, 827–829. [DOI] [PubMed] [Google Scholar]
- 47.Dobrzyn P, Dobrzyn A, Miyazaki M, Cohen P, Asilmaz E, Hardie DG, Friedman JM and Ntambi JM (2004) Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. Proc Natl Acad Sci USA 101, 6409–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]